

Abstract
Tooth agenesis (TA) is one of the most common developmental anomalies that affects the number of teeth. An extensive analysis of publicly accessible databases revealed 15 causative genes responsible for non-syndromic TA, along with their signaling pathways in Wnt/β-catenin, TGF-β/BMP and Eda/Edar/NF-κB. However, genotype-phenotype correlation analysis showed that most of the causal genes are also responsible for syndromic TA or other conditions. In a total of 198 different mutations of the 15 genes responsible for non-syndromic TA, 182 mutations (91.9%) are derived from 7 genes (AXIN2, EDA, LRP6, MSX1, PAX9, WNT10A and WNT10B) compared to the remaining 16 mutations (8.1%) identified in the remaining 8 genes (BMP4, DKK1, EDAR, EDARADD, GREM2, KREMEN1, LTBP3 and SMOC2). Furthermore, specificity analysis in terms of the ratio of non-syndromic TA mutations verses syndromic mutations in each of the aforementioned 7 genes showed a 98.2% specificity rate in PAX9, 58.9% in WNT10A, 56.6% in MSX1, 41.2% in WNT10B, 31.4% in LRP6, 23.8% in AXIN2, and 8.4% in EDA. These findings underscore an important role of the Wnt and Wnt-associated pathways in the genetic etiology of this heterozygous disease and shed new lights on the discovery of novel molecular mechanisms associated with tooth agenesis.
Keywords: Tooth Agenesis, Oligodontia, Hypodontia, Craniofacial Genetics, Wnt Pathway
1. Introduction
Tooth agenesis (TA), the absence of teeth due to developmental failure, is one of the most common developmental malformations. TA may present in a syndromic form with the involvement of other organs or tissues, or in a non-syndromic form that only affects the dentition. Epidemiological studies indicate that the prevalence of non-syndromic TA ranges from 1.6% to 9.6% in different geographical areas and races (Galluccio et al., 2012, Zhang et al., 2015). Based on the number of missing teeth in permanent dentition, TA can be further classified into three categories: hypodontia (less than 6 missing teeth), oligodontia (6 or more missing teeth) and anodontia (complete agenesis of the dentition). The congenital loss of teeth consequently leads to masticatory, speech, esthetic and psychological problems, thus placing a heavy burden on the affected individuals and associated societies.
Although many factors may contribute to the etiology of TA (e.g., epigenetic and environmental effects), there is compelling evidence proving that genetic factors play a predominant role in the pathogenesis of the disease (Ye & Attaie, 2016). Since a mutation of MSX1 was first discovered in affected individuals with selective TA (Vastardis et al., 1996), various mutations in several dozens of genes have been identified in affected individuals with syndromic and non-syndromic TA, thus reflecting the allelic and genetic heterogeneities of these conditions. Notably, mutated genes encoding the components in the canonical Wnt/β-catenin pathway and Wnt-associated genes account for the highest genetic risk for isolated TA compared to mutated genes involved in several other pathways (van den Boogaard et al., 2012, Yin & Bian, 2015).
In this review, we aim to summarize all causal genes responsible for non-syndromic TA (Supplementary Table 1), which are either curated in publicly accessible databases or recently discovered by whole-exome sequencing (WES) analysis. We also illustrate the pathogenic effects of Wnt and other associated signaling pathways on TA and related dental anomalies. Additionally, potential molecular and cellular mechanisms associated with the alteration of Wnt and other pathways in non-syndromic TA are briefly discussed.
2. Wnt is a major pathway responsible for TA
Tooth development is a dynamic process that includes the bud, cap and bell stages, root development and tooth eruption (Huang & Chai, 2012). The Wnt/β-catenin signaling pathway is involved in many aspects of embryonic development and is spatiotemporally activated in tooth-forming regions at all stages of tooth development, thereby implying its essential role in the process of odontogenesis (Thesleff & Sharpe, 1997). The machinery of the Wnt pathway comprises extracellular secreted glycoproteins (19 Wnt ligands at human level), seven-transmembrane-spanning receptors (Frizzled and LRP5/6), cytoplasmic proteins (DVL, APC, AXIN, GSK3β and β-catenin, etc.), nuclear transcription factors (TCF/LEF), and several Wnt-associated molecules (MSX1, DKK1, KREMEN1, and ANTXR1) (Figure 1, left panel).
Figure 1.
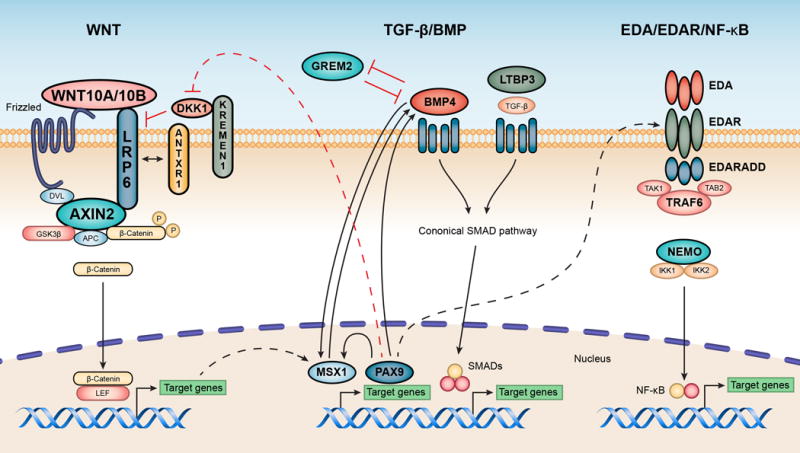
Three tooth agenesis-associated signaling pathways. Mutated components of these pathways that underlie non-syndromic tooth agenesis in affected human subjects are shown in bolded gene symbols. Genetic interactions between different pathways are briefly discussed in the context. Left panel: the Wnt/β-catenin signaling pathway. Middle panel: the TGF-β/BMP pathway. Right panel: the Eda/Edar/NF-κB pathway. The MSX1 encoded transcriptional repressor is involved in the Wnt (www.genecards.org) as well as the BMP4 pathway.
The genetic link between TA and the Wnt pathway was first evidenced by the identification of a mutation of the AXIN2 gene in an oligodontia family (OMIM: 608615). To date, 9 different mutations in AXIN2 have been identified in non-syndromic TA, while 12 mutations were identified in syndromic TA and other conditions (Supplementary Table 1). AXIN2 encodes an intracellular inhibitor of Wnt/β-catenin signal and is highly expressed in the enamel knot and mesenchymal odontoblasts during tooth formation. AXIN2 missense mutants were found to enhance β-catenin degradation and reduced Wnt activation, whereas the truncated mutants seemed to heighten the activation of Wnt/β-catenin (Yue et al., 2016).
A large amount of mutations in genes encoding Wnt ligands (e.g., WNT10A and WNT10B) and associated receptors (LRP6 and KREMEN1) were recently discovered by whole exome and Sanger sequencing. WNT10A is mapped on chromosome 2q35 and is preferentially expressed in dental epithelium and enamel knots during tooth development (He et al., 2013). Since the first recessive mutations in WNT10A were identified in individuals with odonto-onycho-dermal dysplasia (OMIM 257980), 73 different mutations have been identified to be responsible for 14 different diseases/phenotypes based on data obtained from the Human Gene Mutation Database (HGMD). 43 of the mutations (58.9%, Supplementary Table 1) resulted in the non-syndromic TA (HGMS), which are categorized into six conditions (tooth agenesis, non-syndromic hypodontia, hypodontia, maxillary lateral incisor agenesis, oligodontia and non-syndromic selective tooth agenesis) (van den Boogaard et al., 2012). This makes WNT10A as the second most frequently mutated gene in individuals with non-syndromic TA (Figure 2). Clinical analysis showed a more severe TA condition in affected individuals carried with biallelic WNT10A mutations compared to those with monoallelic mutations (Ye & Attaie, 2016).
Figure 2.
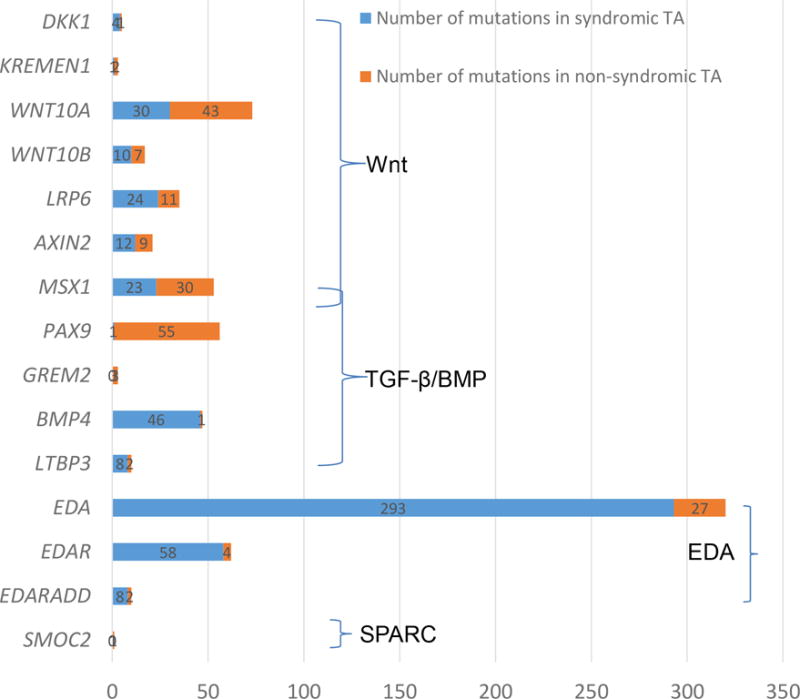
Spectrum of causal mutations in 15 genes responsible for non-syndromic TA. Numbers of mutations in each of the causal genes in affected individuals with non-syndromic TA (in red bar) versus syndromic TA or other conditions (in blue bar) are shown based on the data curated in Human Gene Mutation Database (HGMD), which also are summarized in more details in Supplementary Table 1. More specific genes for non-syndromic TA are clustered, including four Wnt genes (WNT10A, WNT10B, LRP6, and AXIN2) and two genes (MSX1 and PAX9) in the loop of Wnt and BMP4 pathways.
WNT10B is mapped to chromosome 12q13.12, and at protein level, shares 60% identity and 72% similarity to WNT10A. During tooth development in mice, Wnt10b is also expressed in the dental epithelium at the bud and cap stages. Using the WES technique, four deleterious mutations in WNT10B were identified in an affected family, as well as three unrelated individuals with non-syndromic oligodontia (Yu et al., 2016), which is designated as the autosomal dominant tooth agenesis, selective 8, STHAG8 (OMIM 617073). In a separate study, two additional novel mutations in WNT10B were identified in 5 families with isolated dental anomalies, including oligodontia and several other types of isolated TA (Kantaputra et al., 2018). Functional studies showed that WNT10B mutants are insufficient at activating Wnt signaling, thus resulting in an impaired odontoblastic differentiation and vasculogenesis of dental pulp stem cells (DPSCs) (Yu et al., 2016). This finding provides evidence that odontoblasts as well as vascular endothelial cells play a critical role in success of dental pulp tissue engineering. Further research on how the Wnt pathway regulates the decision between odontoblastic fate and vasculogenic fate of DPSCs may shed a light on TA pathogenesis and tooth regeneration.
LRP6 encodes a key component of the co-receptor complex for the transmission of Wnt/β-catenin signaling cascade for cell differentiation and proliferation. While LRP6 mutations are believed to be associated with coronary artery disease (OMIM: 610947) and several other conditions, 11 different mutations (Supplementary Table 1) were identified in affected families and individuals with non-syndromic TA (Massink et al., 2015). The transfected LRP6 mutants in mammalian cells failed to activate β-catenin due to their abnormal glycosylation and immature high-mannose form in the endoplasmic reticulum, thereby leading to the abolishment of Wnt activation (Massink et al., 2015). Recently, LRP6 in the Wnt signaling was also shown to be necessary for the vasculogenic differentiation of human DPSCs (Silva et al., 2017).
Interestingly, mutations in MSX1, encoding a transcriptional repressor in the loop of both Wnt and BMP4 pathways, have been repeatedly found to cause non-syndromic TA (Wong et al., 2014). Among 53 different mutations in MSX1 (HGMD), 30 of them (56.6%, Supplementary Table 1) are associated with isolated TA, often missing second premolars and third molars. In contrast, loss-of-function mutations of MSX1 were reported to cause more severe conditions, such as Witkop syndrome, Wolf-Hirschhorn syndrome, and oligodontia with cleft lip/palate (HGMD). Deletion of Msx1 in mice was found to cause an arrested tooth development, deficiency of alveolar bones and cleft palate (Jumlongras et al., 2001).
It is worthwhile to mention that rare mutations of DKK1 and KREMEN1 (Supplementary Table 1) were recently identified by WES analysis to be implicated in isolated TA or oligodontia accompanied with ectodermal dysplasia (Dinckan et al., 2018b, Issa et al., 2016). DKK1 encodes the Dickkopf Wnt signaling pathway inhibitor 1, which binds to its transmembrane receptor KREMEN as well as LRP6 co-receptor to inhibit Wnt/β-catenin signaling in embryonic and vascular development. KREMEN1 encodes a transmembrane receptor that functionally cooperates with DKK1 to block Wnt/β-catenin signaling. Further studies are required to demonstrate the functional significance of DKK1 and KREMEN1 variants in TA (Dinckan et al., 2018b). In addition, recessive mutations of the ANTXR1 gene (OMIM: 230740) have been identified in 1 case with syndromic TA (Dinckan et al., 2018a) and 11 cases with GAPO syndrome and vascular anomalies (HGMD). Of note, ANTXR1 encodes a transmembrane protein directly interacting with LRP6 (Figure 1, left panel) for beta-catenin stabilization and modulating Wnt signaling during normal vascular development.
3. Gene mutations in the TGF-β/BMP pathway contribute to TA
The TGF-β/BMP pathway plays an important role in embryonic development (Figure 1, middle panel). Mutations in BMP4, which encodes a secreted ligand of the TGF-β family of proteins, result in more than 20 different conditions, including autosomal dominant inherited syndromic microphthalmia (OMIM 607932), multiple oral and craniofacial development-related disorders, and also non-syndromic TA in one case (HGMD).
The LTBP3 gene encodes a ligand that forms a complex with TGF-β, which is involved in the assembly, secretion and targeting of TGF-β molecules. Mutations in LTBP3 were shown to cause inherited dental anomalies and short stature as well as geleophysic dysplasia 3 (OMIM: 602090). In 10 different reported mutations, 2 were found to cause isolated oligodontia (Supplementary Table 1). Experimentally, Ltbp3−/− mice exhibit abnormal enamel and root morphogenesis (Huckert et al., 2015).
GREM2 encodes an antagonist protein to BMP4, which participates in the regulation of TGF-β signaling in tooth development. Three mutations in GREM2 identified so far (OMIM: 608832) are associated with isolated TA, microdontia, short tooth roots and taurodontism (Kantaputra et al., 2015). Similarly, Grem2−/− mice manifested small deformed incisors (Kantaputra et al., 2015).
Lastly, it is worthwhile to emphasize that almost all 56 mutations of the PAX9 gene are specific (Supplementary Table 1), making PAX9 the most prevalent gene for non-syndromic TA (Figure 2). Only one allele (640A>G, S214G, rs375436662) in the exon 3 of PAX9 was detected in two siblings with cleft lip and their phenotypically normal mother (HGMD). However, this SNP is not an extremely rare deleterious mutation (MAF= 0.001084 in East Asian, ExAC database), as predicted by MutationTaster.
PAX9 encodes a member of the paired box family of transcription factors and is expressed in the presumptive dental mesenchyme to induce odontogenic signals and initiate dental development (Wong et al., 2018). Most of the mutations are clustered in the exon 2, which encodes the paired box DNA binding domain involving protein-DNA interactions, often affecting the second molars (Wong et al., 2018). Pax9-deficient mice exhibit arrests of tooth and taste bud development, and cleft palate (Peters et al., 1998). Interestingly, deletion of Pax9 in mice was found to upregulate the expression of Wnt pathway genes including two Wnt signaling antagonists DKK1 and DKK2 (Jia et al., 2017).
4. Gene mutations in Eda/Edar/NF-κB pathway underlie TA
The EDA gene-encoded ligand, Ectodysplasin A, functions to bind to its receptor EDAR to activate the IKBKG-NF-κB signaling for the development of ectodermal organs and teeth (Figure 1, right panel). In the past two decades, 293 different mutations in EDA were identified to cause the X-linked hypohidrotic ectodermal dysplasia compared to 27 additional mutations in non-syndromic TA (Han et al., 2008), reaching 8.4% of the specificity (Supplementary Table 1). The effects of EDA on ectodermal dysplasia and tooth development were confirmed in the Eda mutant mice (OMIM: 300451).
Similarly, a majority of EDAR mutations were found to result in ectodermal dysplasia, compared to four cases with non-syndromic TA (6.5%, Supplementary Table 1). Out of 10 different mutations in EDARADD, two were associated with non-syndromic TA and eight with ectodermal dysplasia (Supplementary Table 1). In 149 different mutations of IKBKG (a.k.a., NEMO), most of them are responsible for incontinentia pigmenti and ectodermal dysplasia (HGMD). In addition, two mutations in the TRAF6 gene, which encodes a TNF receptor associated factor (TRAF) protein (OMIM: 602355), were identified in a family with ectodermal dysplasia and TA (HGMD). Mutations in the above-mentioned genes are expected to affect the NF-κB activity, thereby resulting in ectodermal dysplasia, non-syndromic TA and other conditions.
5. Network of tooth agenesis-associated signaling pathways
Tooth morphogenesis is orchestrated by a complex development signaling network, which mainly includes the Wnt, TGF-β/BMP and Eda/Edar/NF-κB pathways. Extensive studies have uncovered cross-link loops among these pathways, which are interconnected and mutually-dependent (Figure 1).
PAX9 appears to be at the top hierarchy of above-mentioned odontogenic pathways, as it has been shown to induce activation of both Wnt and TGF-β/BMP signaling for organogenesis (Jia et al., 2017). Conversely, Pax9 deficiency resulted in a decreased activation of Wnt/β-catenin in vivo, whereas small-molecule Wnt agonists were able to rescue the craniofacial defect in Pax9 knockout mice (Jia et al., 2017). Interestingly, expression level of Edar was also observed to be significantly decreased in Pax9−/− mice, suggesting potential interactions between PAX9 and the Eda/Edar/NF-κB pathway.
As a direct target of Wnt/β-catenin, increased expression of MSX1 will then up-regulate Bmp4 expression and also activates TGF-β/BMP signaling for odontogenesis (Yin & Bian, 2015). Furthermore, MSX1 can physically interact with PAX9 to synergistically augment its own and Bmp4 expression.
Wnt/β-catenin and Eda/Edar/NF-κB pathways also display reciprocal controls, as evidenced by their spatio-temporal co-expression in developing tooth. It has been shown that Wnt/β-catenin activates the Eda/Edar/NF-κB pathway by inducing the expression of Eda, which in turn is required for maintaining of the sustained Wnt/β-catenin activities. Clinical evidence showed that digenic mutations in EDA and WNT10A modified the phenotypic severity in affected individuals with TA (He et al., 2013).
In addition, a causal mutation in the SMOC2 gene, which seems not linked to above signaling pathways, has also been identified in two cousins with severe non-syndromic TA (Bloch-Zupan et al., 2011). This gene encodes a secreted protein, which promotes matrix assembly and stimulates endothelial cell proliferation and migration, as well as angiogenic activity. Knockdown of Smoc2 in zebrafish showed pharyngeal teeth that had abnormalities reminiscent of the human phenotype (Bloch-Zupan et al., 2011).
6. Summary and future direction
Enormous advancements have been made to decode the genetic complexity and heterogeneity of TA over the last few decades, especially during the past several years. Majority of the mutations (91.9%) responsible for affected individuals with non-syndromic TA are identified only from 7 genes (i.e., PAX9, WNT10A, MSX1, EDA, LRP6, WNT10B, and AXIN2). Specificity analysis in terms of the ratio of non-syndromic TA mutations verses syndromic mutations of these genes showed a dramatic difference from 98.2% to 8.4% (as listed in descending order: PAX9, WNT10A, MSX1, WNT10B, LRP6, AXIN2, and EDA). These findings demonstrate a major role of the Wnt and Wnt-associated pathways in the molecular pathogenesis of the non-syndromic TA. It is conceivable that high-throughput sequencing techniques, such as WES and post-genomic bioinformatics, will markedly enhance the detection of extremely rare mutations in known and novel causal genes.
For affected individuals with more complicated non-syndromic and syndromic TA, renewed efforts shall be made to establish large databases for in-depth analysis of genotype-phenotype correlation, single- or multiple-allele traits, epigenetic effectors and genetic modifiers. Gene function studies should be performed in mammalian cells and animal models to uncover molecular and cellular mechanisms of TA. For instance, whether the above-mentioned Wnt-associated components play a crucial role in determining of odontoblastic and vasculogenic fate of DPSCs in affected individuals with non-syndromic TA during tooth development. Based on the understanding of genetic defects, on-going studies utilizing small molecular inhibitors in the Wnt pathway have shown a great potential in the treatment of developmental disturbances in experimental animals and ultimately in clinical trials.
Supplementary Material
Supplementary Table 1. Analyses of 15 causative genes for non-syndromic tooth agenesis
Acknowledgments
We thank Professor Emeritus Hailan Feng for her academic supports and leadership in the field of dental genetics. This work was supported in part by National Natural Science Foundation of China (81670949 to DH), NIDCR (R90DE022527 to SWW). This research was supported in part by the Intramural Research Program of the NIH, NIDCR. We apologize to those authors whose research about tooth agenesis were not discussed or cited because of space limitation.
Footnotes
DR TAO CAI (Orcid ID : 0000-0002-2877-8637)
CONFLICT OF INTEREST
The authors declare no competing financial interests.
AUTHOR CONTRIBUTIONS
MY, SWW and DH participated the writing. SWW and TC made the figures and tables. TC supervised the work and edited the manuscript.
References
- Bloch-Zupan A, Jamet X, Etard C, et al. Homozygosity mapping and candidate prioritization identify mutations, missed by whole-exome sequencing, in SMOC2, causing major dental developmental defects. Am J Hum Genet. 2011;89:773–81. doi: 10.1016/j.ajhg.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinckan N, Du R, Akdemir ZC, et al. A biallelic ANTXR1 variant expands the anthrax toxin receptor associated phenotype to tooth agenesis. Am J Med Genet A. 2018a;176:1015–1022. doi: 10.1002/ajmg.a.38625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinckan N, Du R, Petty LE, et al. Whole-Exome Sequencing Identifies Novel Variants for Tooth Agenesis. J Dent Res. 2018b;97:49–59. doi: 10.1177/0022034517724149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluccio G, Castellano M, La Monaca C. Genetic basis of non-syndromic anomalies of human tooth number. Arch Oral Biol. 2012;57:918–30. doi: 10.1016/j.archoralbio.2012.01.005. [DOI] [PubMed] [Google Scholar]
- Han D, Gong Y, Wu H, et al. Novel EDA mutation resulting in X-linked non-syndromic hypodontia and the pattern of EDA-associated isolated tooth agenesis. Eur J Med Genet. 2008;51:536–46. doi: 10.1016/j.ejmg.2008.06.002. [DOI] [PubMed] [Google Scholar]
- He H, Han D, Feng H, et al. Involvement of and Interaction between WNT10A and EDA Mutations in Tooth Agenesis Cases in the Chinese Population. Plos One. 2013;8:e80393. doi: 10.1371/journal.pone.0080393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang XF, Chai Y. Molecular regulatory mechanism of tooth root development. Int J Oral Sci. 2012;4:177–81. doi: 10.1038/ijos.2012.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huckert M, Stoetzel C, Morkmued S, et al. Mutations in the latent TGF-beta binding protein 3 (LTBP3) gene cause brachyolmia with amelogenesis imperfecta. Hum Mol Genet. 2015;24:3038–49. doi: 10.1093/hmg/ddv053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issa YA, Kamal L, Rayyan AA, et al. Mutation of KREMEN1, a modulator of Wnt signaling, is responsible for ectodermal dysplasia including oligodontia in Palestinian families. Eur J Hum Genet. 2016;24:1430–5. doi: 10.1038/ejhg.2016.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia S, Zhou J, Fanelli C, et al. Small-molecule Wnt agonists correct cleft palates in Pax9 mutant mice in utero. Development. 2017;144:3819–3828. doi: 10.1242/dev.157750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumlongras D, Bei M, Stimson JM, et al. A nonsense mutation in MSX1 causes Witkop syndrome. Am J Hum Genet. 2001;69:67–74. doi: 10.1086/321271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantaputra PN, Hutsadaloi A, Kaewgahya M, et al. WNT10B mutations associated with isolated dental anomalies. Clin Genet. 2018;93:992–999. doi: 10.1111/cge.13218. [DOI] [PubMed] [Google Scholar]
- Kantaputra PN, Kaewgahya M, Hatsadaloi A, et al. GREMLIN 2 Mutations and Dental Anomalies. J Dent Res. 2015;94:1646–52. doi: 10.1177/0022034515608168. [DOI] [PubMed] [Google Scholar]
- Massink MP, Creton MA, Spanevello F, et al. Loss-of-Function Mutations in the WNT Co-receptor LRP6 Cause Autosomal-Dominant Oligodontia. Am J Hum Genet. 2015;97:621–6. doi: 10.1016/j.ajhg.2015.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters H, Neubuser A, Kratochwil K, et al. Pax9-deficient mice lack pharyngeal pouch derivatives and teeth and exhibit craniofacial and limb abnormalities. Genes Dev. 1998;12:2735–47. doi: 10.1101/gad.12.17.2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva GO, Zhang Z, Cucco C, et al. Lipoprotein Receptor-related Protein 6 Signaling is Necessary for Vasculogenic Differentiation of Human Dental Pulp Stem Cells. J Endod. 2017;43:S25–S30. doi: 10.1016/j.joen.2017.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thesleff I, Sharpe P. Signalling networks regulating dental development. Mech Dev. 1997;67:111–23. doi: 10.1016/s0925-4773(97)00115-9. [DOI] [PubMed] [Google Scholar]
- van den Boogaard MJ, Creton M, Bronkhorst Y, et al. Mutations in WNT10A are present in more than half of isolated hypodontia cases. J Med Genet. 2012;49:327–31. doi: 10.1136/jmedgenet-2012-100750. [DOI] [PubMed] [Google Scholar]
- Vastardis H, Karimbux N, Guthua SW, et al. A human MSX1 homeodomain missense mutation causes selective tooth agenesis. Nat Genet. 1996;13:417–21. doi: 10.1038/ng0896-417. [DOI] [PubMed] [Google Scholar]
- Wong SW, Han D, Zhang H, et al. Nine Novel PAX9 Mutations and a Distinct Tooth Agenesis Genotype-Phenotype. J Dent Res. 2018;97:155–162. doi: 10.1177/0022034517729322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong SW, Liu HC, Han D, et al. A novel non-stop mutation in MSX1 causing autosomal dominant non-syndromic oligodontia. Mutagenesis. 2014;29:319–23. doi: 10.1093/mutage/geu019. [DOI] [PubMed] [Google Scholar]
- Ye X, Attaie AB. Genetic Basis of Nonsyndromic and Syndromic Tooth Agenesis. J Pediatr Genet. 2016;5:198–208. doi: 10.1055/s-0036-1592421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin W, Bian Z. The Gene Network Underlying Hypodontia. J Dent Res. 2015;94:878–85. doi: 10.1177/0022034515583999. [DOI] [PubMed] [Google Scholar]
- Yu P, Yang W, Han D, et al. Mutations in WNT10B Are Identified in Individuals with Oligodontia. Am J Hum Genet. 2016;99:195–201. doi: 10.1016/j.ajhg.2016.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue H, Liang J, Yang K, et al. Functional analysis of a novel missense mutation in AXIN2 associated with non-syndromic tooth agenesis. Eur J Oral Sci. 2016;124:228–33. doi: 10.1111/eos.12273. [DOI] [PubMed] [Google Scholar]
- Zhang J, Liu HC, Lyu X, et al. Prevalence of tooth agenesis in adolescent Chinese populations with or without orthodontics. Chin J Dent Res. 2015;18:59–65. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. Analyses of 15 causative genes for non-syndromic tooth agenesis