

Abstract
The activity and stability of the adapter protein MAVS (also known as VISA, Cardif and IPS-1), which critically mediates cellular antiviral responses, are extensively regulated by ubiquitination. However, the process whereby MAVS is deubiquitinated is unclear. Here, we report that the ovarian tumor family deubiquitinase 4 (OTUD4) targets MAVS for deubiquitination. Viral infection leads to the IRF3/7-dependent upregulation of OTUD4 which interacts with MAVS to remove K48-linked polyubiquitin chains, thereby maintaining MAVS stability and promoting innate antiviral signaling. Knockout or knockdown of OTUD4 impairs RNA virus-triggered activation of IRF3 and NF-κB, expression of their downstream target genes, and potentiates VSV replication in vitro and in vivo. Consistently, Cre-ER Otud4fl/fl or Lyz2-Cre Otud4fl/fl mice produce decreased levels of type I interferons and proinflammatory cytokines and exhibit increased sensitivity to VSV infection compared to their control littermates. In addition, reconstitution of MAVS into OTUD4-deficient cells restores virus-induced expression of downstream genes and cellular antiviral responses. Together, our findings uncover an essential role of OTUD4 in virus-triggered signaling and contribute to the understanding of deubiquitination-mediated regulation of innate antiviral responses.
Subject terms: Innate immunity, Cell signalling, Ubiquitylation
Introduction
Detection of pathogen-associated molecular patterns (PAMPs) associated with invading pathogens by host pattern-recognition receptors (PRRs) is the first step to initiate the innate immune responses.1 Viral nucleic acids are classical PAMPs and are detected by retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs), Toll-like receptors (TLRs), cytosolic DNA sensors and nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), which trigger signal transduction leading to the induction of type I interferons (IFNs) and proinflammatory cytokines.2 For example, cytoplasmic 5′ triphosphorylated single-stranded RNA (5′pppssRNA), panhandle-like RNA or double-stranded RNA (dsRNA) is recognized by RLRs,3–7 whereas cytoplasmic AT-rich DNA is detected by RNA polymerase III and transcribed into 5′pppssRNA to be recognized by RLRs.8,9 After binding to the viral RNAs, RLRs undergo conformational changes to expose N-terminal CARD domains and interact with and induce the oligomerization of the downstream adapter protein mitochondrial antiviral signaling protein (MAVS, also known as VISA, Cardif and IPS-1).10–15 MAVS further recruits multiple TNF receptor-associated factors (TRAFs) and downstream kinases TBK1 or IKKε or the kinase complex IKKα/β/γ.16,17 TBK1 or IKKε phosphorylate IRF3 and IRF7 to activate the transcription of type I IFNs,18,19 whereas IKKα/β/γ phosphorylates IκBα leading to the release of NF-κB into the nucleus and subsequent expression of proinflammatory cytokines.
Studies with MAVS deficient mice suggest that MAVS is the sole adapter protein downstream of RLRs required for defense against various RNA viruses.20,21 Therefore, the activity of MAVS should be finely tuned to exert sufficient protective immune responses and so avoid excessive harmful immune pathology. Various posttranslational modifications have been reported to regulate the activity and availability of MAVS. For example, VSV infection induces TBK1 and IKKε-mediated phosphorylation of MAVS, thereby providing a platform to recruit and activate IRF3,22 whereas PPM1A targets MAVS for dephosphorylation and dampens innate immune responses against RNA viruses.23 TRIM31 catalyzes K63-linked ubiquitination of MAVS which promotes the aggregation of MAVS and the recruitment of TBK1 and IKKε after viral infection.24 TRIM25 mediates K48-linked ubiquitination of MAVS resulting in partial degradation of MAVS and the release of the signalosome into the cytoplasm to activate innate antiviral signaling.25 PCBP1, PCBP2 and TAX1BP1 recruit the E3 ubiquitin ligase Itch to induce K48-linked ubiquitination and degradation of MAVS.26–28 RNF5, PSMA7, Smurf1/2 and MARCH5 have been reported to catalyze K48-linked ubiquitination and proteasome-dependent degradation of MAVS and thereby shut down the immune responses after viral infections.29–33 The E3 ubiquitin ligase MARCH8 catalyzes addition of K27-linked ubiquitin chains and promotes the autophagic degradation of MAVS.34 More recently, it has been reported that OTUD1 induces the upregulation of Smurf1 to promote ubiquitination and proteasomal degradation of MAVS,35 whereas iRhom2 promotes ubiquitination and degradation of RNF5 and MARCH5 to antagonize MAVS degradation.36 However, the process whereby MAVS is deubiquitinated is unclear and whether and how its deubiquitination might regulate the innate antiviral responses remain to be investigated.
Deubiquitinating enzymes (DUBs) cleave poly- or mono-ubiquitin chains from their targets, and thereby counteract E3s-mediated ubiquitination. To date, six families of DUBs have been reported, including the ubiquitin-specific proteases (USPs), the ovarian tumor proteases (OTUs), the ubiquitin C-terminal hydrolases (UCHs), the Josephin family, the JAB1/MPN/MOV34 metalloproteases (JAMMs), and the motif interacting with Ub-containing novel DUB family (MINDYs).37 Ovarian tumor family deubiquitinase 4 (OTUD4) belongs to the OTU family and is implicated to be critical for embryonic development of zebrafish and is associated with the syndrome of hypogonadotropic hypogonadism, ataxia, and dementia in humans.38–40 It has been reported that OTUD4 preferentially cleaves K48-linked poly ubiquitin chains.41,42 However, phosphorylation at the catalytic domain of OTUD4 at Ser202/204 activates its K63-specific DUB activity, which targets MyD88 for deubiquitination and thereby downregulates TLR-mediated NF-κB activation.43 In addition, OTUD4 promotes the repair of DNA alkylation damage in a manner independent of its DUB activity.41 The K48-specific DUB activity of OTUD4 and the target(s) and the related physiological or pathological significance of such an activity remain to be elucidated.
In this study, we report that MAVS is an OTUD4 substrate and that OTUD4-mediated deubiquitination of MAVS positively regulates innate antiviral signaling. Viral infection induces the upregulation of OTUD4 in an IRF3/7-dependent manner. Knockout or knockdown of OTUD4 impairs RNA virus-triggered activation of IRF3 and NF-κB and expression of downstream genes and potentiates VSV replication. In addition, Cre-ER Otud4fl/fl or Lyz2-Cre Otud4fl/fl mice are more susceptible to lethal VSV infection than their control littermates. Mechanistically, OTUD4 interacts with MAVS to remove K48-linked polyubiquitin chains thereby inhibiting the proteasome-dependent degradation of MAVS and promoting innate antiviral signaling. Together these findings indicate that OTUD4 plays an essential role in innate antiviral immune responses.
Results
Identification of OTUD4 as a MAVS-interacting DUB
MAVS is a critical adapter protein required for innate immune responses against RNA viruses and its activity and availability are heavily regulated by E3s-mediated ubiquitination. We thus hypothesized that DUBs-mediated deubiquitination of MAVS is equally important as ubiquitination and requires to be properly regulated. To test this hypothesis, we screened for DUBs that interact with MAVS by cotransfecting individual FLAG-tagged DUBs and HA-MAVS into HEK293 cells followed by coimmunoprecipitation and immunoblot assays.44,45 This led to the identification of multiple DUBs that interacted with MAVS, including OTUD4, CYLD and USP35 (Fig. 1a and Supplementary information, Fig. S1a). Interestingly, out of the eight OTU we could test, only OTUD4 interacted with MAVS and potentiated SeV-induced activation of the IFN-β promoter (Supplementary information, Fig. S1b, c). We next examined the association of endogenous MAVS and OTUD4 and found that they interacted in mouse embryonic fibroblasts (MEFs) and mouse bone marrow-derived dendritic cells (BMDCs) after SeV or VSV infection (Fig. 1b). Domain mapping analyses suggested that the N terminal OTU motif (aa1-245) and the C terminus containing the transmembrane domain of MAVS (aa361–540) were responsible for their associations (Fig. 1c, d). Thus OTUD4 interacts with MAVS after viral infection.
Fig. 1.

Identification of OTUD4 as a MAVS-interacting DUB. a Immunoprecipitation (IP, with anti-FLAG) and immunoblot (IB, with anti-FLAG and anti-HA and goat anti-mouse IgG(H+L) antibody) analysis of HEK293 cells that were transfected with plasmids encoding HA-MAVS and FLAG-tagged DUBs for 24 h. Cell lysates were analyzed by immunoblot with anti-FLAG or anti-HA. b Immunoprecipitation (with control IgG or anti-OTUD4) and immunoblot (with anti-OTUD4 and anti-MAVS and goat anti-mouse IgG F(ab’)2 fragment specific antibody) of MEFs (upper panels) and BMDCs (lower panels) that were left uninfected or infected with SeV or VSV for 5–10 h. Cell lysates were analyzed by immunoblot with antibodies against the indicated proteins. The graphs show relative intensities of OTUD4 obtained by normalizing the intensities of OTUD4 to the intensities of β-Actin. c, d Immunoblot of HEK293 cells that were transfected with plasmids encoding HA-MAVS and FLAG-tagged OTUD4 or mutants (c) or with plasmids encoding GFP-OTUD4 and FLAG-tagged MAVS or truncates (d), lysed and immunoprecipitated with anti-FLAG. Cell lysate was analyzed by immunoblot with anti-FLAG or anti-FLAG-HRP, anti-GFP or anti-HA. Data are representative of three (a) or two (b–d) independent experiments (mean ± S.D. in b)
We observed that the protein levels of OTUD4 were increased in MEFs or BMDCs after SeV or VSV infection (Fig. 1b). Results from quantitative RT-PCR (qRT-PCR) analysis showed that SeV infection induces a 2–3 fold upregulation of the mRNA levels of Otud4 but not other OTU DUB-encoding genes (Supplementary information, Fig. S2a), indicating that viral infection promotes transcriptional activation of Otud4. Sequence analysis identified an IRF3/7-binding site on the promoter of Otud4 gene (−298 to −274 bp) and reconstitution of IRF3 or IRF7 into Irf3−/−Irf7−/− MEFs restored the SeV-induced upregulation of OTUD4 (Supplementary information, Fig. S2b). In contrast, deficiency of p65 did not affect the upregulation of OTUD4 after SeV infection (Supplementary information, Fig. S2c). Chromatin immunoprecipitation (ChIP) demonstrated that IRF3 or IRF7 bound directly to the IRF binding sites of the Otud4 promoter (Supplementary information, Fig. S2d). Together, these data suggest that viral infection induces the IRF3/7-dependent upregulation of OTUD4 which interacts with MAVS.
Knockdown of OTUD4 impairs RNA virus-triggered signaling
Because OTUD4 was upregulated by viral infection and interacted with MAVS, we next investigated the role of OTUD4 in virus-triggered signaling. We designed two siRNAs targeting OTUD4, both of which potently downregulated the levels of OTUD4 protein and inhibited the SeV-induced activation of the ISRE, NF-κB and IFN-β promoters assessed through a luciferase assay in HEK293 cells (Fig. 2a). Results from qRT-PCR analysis suggested that knockdown of OTUD4 inhibited SeV- or VSV-induced expression of IFNB, CXCL10 and IL6 in THP-1 cells and the SeV-induced expression of IFNB, ISG56, CXCL1 and CCL5 in HeLa cells (Fig. 2b and Supplementary information, Fig. S3a). Consistent with the gene induction analysis, SeV- or VSV-induced phosphorylation of IRF3 and IκBα was substantially impaired by the knockdown of OTUD4 in THP-1 or HeLa cells (Fig. 2c and Supplementary information, Fig. S3b). These data collectively suggest that OTUD4 promotes RNA virus-triggered signaling in human cell lines.
Fig. 2.

Knockdown of OTUD4 impairs RNA virus-triggered signaling. a Immunoblot analysis (with anti-OTUD4 or anti-β-Actin) of HEK293 cells transfected with control siRNA (Con) or siRNA targeting OTUD4 (#1 or #2) for 36 h (left panels). Luciferase reporter assays analyzing ISRE, NF-κB and IFN-β promoter activity of HEK293 cells transfected with siRNA (Con, #1 or #2) for 36 h followed by infection with SeV for 8 h (right graphs). b Quantitative reverse-transcription PCR (qRT-PCR) analysis of IFNB, CXCL10 and IL-6 mRNA in THP-1 cells transfected with control siRNA (Con) or siRNA targeting OTUD4 (siOTUD4#1 or siOTUD4#2) for 36 h followed by infection with VSV or SeV for 0–12 h. c Immunoblot analysis of total and phosphorylated (p-)IκBα, IRF3, total OTUD4, β-Actin in THP-1 cells transfected with siRNA (Con or siOTUD4#1) for 36 h followed by infection with VSV or SeV for 0–12 h. *P < 0.05, **P < 0.01 and ***P < 0.001 (analysis of two-way ANOVA followed by Bonferroni post-test). Data are representative of four (a) or three (b, c) independent experiments (mean ± S.D. in a, b)
Knockout of OTUD4 inhibits RNA virus-triggered signaling
A role for OTUD4 has been implicated in the embryonic development of zebrafish and in the syndromes of ataxia, dementia, and hypogonadotropism in human.38–40 Germline deletion of OTUD4 in mice might affect development and growth. To further investigate the role of OTUD4 in antiviral signaling in vivo, we generated Otud4fl/+ mice by CRISPR/Cas9-mediated genome editing (Supplementary information, Fig. S4a). Southern blot analysis indicated that the targeting vector successfully recombined with the wild-type allele (Supplementary information, Fig. S4b). Cre recombinase-mediated deletion of the exon 4 between flanking loxp sites would result in an early translational termination of OTUD4 to generate a 115 amino-acid peptide or could lead to Otud4 mRNA instability (Supplementary information, Fig. S4c). The Otud4fl/+ mice were crossed with ROSA26-CreERT2 mice (here referred to as Cre-ER) to obtain Cre-ER Otud4fl/+ and Cre-ER Otud4fl/fl mice.46 The intraperitoneal injection of tamoxifen (Tam) led to efficient deletion of OTUD4 in several organs including the brain, lung, heart and spleen (Supplementary information, Fig. S4d). Thus, the strategy to knockout OTUD4 in mice was reliable and successful. Flow cytometry analysis suggested that knockout of OTUD4 in mice by Tamoxifen did not affect lymphocyte numbers or proportions in thymus, spleen or peripheral lymph nodes (Supplementary information, Fig. S4e-g). In addition, knockout of OTUD4 by Cre-ER did not affect the differentiation of BMDMs or BMDCs in M-CSF or GM-CSF cultures, respectively (Supplementary information, Fig. S4j). These data indicate that OTUD4 is not required for the development and homeostasis of lymphocytes in vivo.
We next examined RNA virus-triggered induction of downstream genes in 4-hydroxytamoxifen (4-OH Tam)-treated Cre-ER Otud4fl/+ and Cre-ER Otud4fl/fl cells. As expected, the expression of Otud4 was efficiently diminished in 4-OH Tam-treated Cre-ER Otud4fl/fl mouse lung fibroblasts (MLFs) and BMDMs compared to the Cre-ER Otud4fl/+ counterparts (Fig. 3a and Supplementary information, Fig. S5a). qRT-PCR analyses suggested that knockout of OTUD4 significantly impaired SeV-, VSV-, EMCV- or transfected poly(I:C)-induced expression of Ifnb, Ifna4, Ccl5, Tnf, and Il6 in MLFs and BMDMs (Fig. 3a and b and Supplementary information, Fig. S5a). In addition, the production of IFN-β and IL-6 was also significantly compromised by knockout of OTUD4 in MLFs and BMDMs (Fig. 3c and Supplementary information, Fig. S5b). Consistent with these observations, knockout of OTUD4 substantially impaired SeV-, VSV- or EMCV-induced phosphorylation of TBK1, IRF3 and IκBα in MLFs and BMDMs (Fig. 3d and Supplementary information, Fig. S5c). The virus-triggered expression of downstream genes plays critical roles in restricting the replication of viruses. Interestingly, VSV or VSV-GFP replication was significantly potentiated in Cre-ER Otud4fl/fl cells compared to Cre-ER Otud4fl/+ cells, as monitored by the expression of the VSV-N gene or GFP intensity (Fig. 3e, f and Supplementary information, Fig. S5d, e and f), indicating that OTUD4-mediated promotion of virus-triggered signaling plays an essential role in restricting virus replication.
Fig. 3.

Knockout of OTUD4 inhibits RNA virus-triggered signaling in MLFs. a, b qRT-PCR analysis of Ifnb, Ifna4, Ccl5, Il6, Tnf, or Otud4 mRNA in Cre-ER Otud4fl/+ and Cre-ER Otud4fl/fl MLFs treated with 4-hydroxytamoxifen (4OH Tam, 1 μM) for 3 days followed by infection with SeV or VSV for 0–12 h or transfection with poly(I:C) for 3 h. c ELISA analysis of IFN-β and IL-6 in the supernatants of Cre-ER Otud4fl/+ and Cre-ER Otud4fl/fl MLFs treated with 4-OH Tam (1 μM) for 3 days followed by infection with SeV or VSV for 12–24 h, or transfection with poly(I:C) for 6 h. d Immunoblot analysis of total and phosphorylated (p-)TBK1, IκBα, IRF3, total OTUD4, β-Actin in Cre-ER Otud4fl/+ and Cre-ER Otud4fl/fl MLFs treated with 4-OH Tam (1 μM) followed by infection with SeV or VSV for 0–8 h. e Flow cytometric analysis (left) and microscopy imaging (right) of the replication of GFP-tagged VSV (MOI = 0.5) in Cre-ER Otud4fl/+ and Cre-ER Otud4fl/fl MLFs treated with 4-OH Tam left uninfected or infected with VSV-tagged-GFP for 1 h followed by two times PBS wash and culture in full medium for 24 h. Numbers adjacent to the outlined areas indicate percentages of GFP+ MLFs. f qRT-PCR analysis (upper graphs) of VSV-N and Otud4 mRNA and immunoblot analysis (lower panels) with anti-VSV-N and anti-GAPDH of the replication of VSV in Cre-ER Otud4fl/+ and Cre-ER Otud4fl/fl MLFs treated with 4-OH Tam infected with VSV-GFP for 1 h followed by two times PBS wash and culture in full medium for 12 or 24 h. *P < 0.05, **P < 0.01 and ***P < 0.001 (analysis of two-way ANOVA followed by Bonferroni post-test). Data are representative of three (a–c) or two (d–f) independent experiments (mean ± S.D. in a–c)
We also obtained Lyz2-Cre Otud4fl/+ and Lyz2-Cre Otud4fl/fl mice by crossing Otud4fl/+ mice and Lyz2-Cre mice and generated Lyz2-Cre Otud4fl/+ and Lyz2-Cre Otud4fl/fl BMDMs. qRT-PCR analysis suggested that the expression of Otud4 in Lyz2-Cre Otud4fl/fl BMDMs was downregulated to 10–30% compared to Lyz2-Cre Otud4fl/+ BMDMs with or without of viral infection (Fig. 4a). Lyz2-Cre-mediated deletion of OTUD4 neither altered the percentiles or numbers of CD11c+CD11blo, CD11c+CD11b+, CD11cloCD11b+, or CD11b+F4/80+ cells in the spleen or peripheral lymph nodes nor affected BMDC or BMDM differentiation in vitro (Supplementary information, Fig. S4h–j). Similar to the findings in Cre-ER Otud4fl/fl cells, Lyz2-Cre-mediated deletion of OTUD4 significantly impaired SeV-, VSV-, EMCV-, or transfected poly(I:C)-induced expression of type I IFNs and proinflammatory cytokines in BMDMs (Fig. 4a, b). Consistently, the phosphorylation of TBK1, IRF3 and IκBα was substantially inhibited and VSV-GFP replication was potentiated in Lyz2-Cre Otud4fl/fl BMDMs compared to Lyz2-Cre Otud4fl/+ BMDMs (Fig. 4c, d). Together, these data suggest that OTUD4 is required for optimal activation of virus-triggered signaling and cellular antiviral responses.
Fig. 4.

OTUD4 deficiency in myeloid-derived cells impairs RNA virus-triggered signaling. a qRT-PCR analysis of Ifnb, Ccl5, Il6 and Otud4 mRNA in Lyz2-Cre Otud4fl/+ and Lyz2-Cre Otud4fl/fl BMDMs infected with SeV or VSV or EMCV for 0–12 h. b ELISA analysis of IFN-β and IL-6 in the supernatants of Lyz2-Cre Otud4fl/+ and Lyz2-Cre Otud4fl/fl BMDMs left uninfected or infected with SeV or VSV for 12–24 h, or transfected with poly(I:C) for 6 h. c Immunoblot analysis of total and phosphorylated (p-)TBK1, IκBα, IRF3, total OTUD4, β-Actin in Lyz2-Cre Otud4fl/+ and Lyz2-Cre Otud4fl/fl BMDMs infected with SeV or VSV or EMCV for 0–8 h. d Flow cytometry analysis (upper graphs) and microscopy imaging (lower images) of the replication of VSV-GFP in Lyz2-Cre Otud4fl/+ and Lyz2-Cre Otud4fl/fl BMDMs infected with VSV-GFP (MOI = 1). *P < 0.05, **P < 0.01 and ***P < 0.001 (analysis of two-way ANOVA followed by Bonferroni post-test). Data are representative of three (a, b) or two (c, d) independent experiments (mean ± S.D. in a, b)
We next determined the role of OTUD4 in cytoplasmic dsDNA- or HSV-1 triggered signaling and found comparable expression of Ifnb, Ifna4, Ccl5, Il6 or Tnf in Cre-ER Otud4fl/+ and Cre-ER Otud4fl/fl MLFs after transfection of DNA90 or HSV-1infection (Supplementary information, Fig. S6a). Consistently, knockout of OTUD4 either by Cre-ER in MLFs or by Lyz2-Cre in BMDMs did not affect the HSV-1-induced phosphorylation of TBK1, IRF3 or IκBα (Supplementary information, Fig. S6b) Thus, OTUD4 is not essential for the HSV-1-triggered STING-dependent pathway.
OTUD4 deficiency leads to increased susceptibility to lethal VSV infection
To characterize the role of OTUD4 in RNA virus infection in vivo, we infected Cre-ER Otud4fl/fl mice or Lyz2-Cre Otud4fl/fl mice and respective control animals with VSV by tail vein injection and monitored their survival. As shown in Fig. 5a, b, Cre-ER Otud4fl/fl mice (injected intra-peritoneally with Tam before intra venous injection of VSV) and Lyz2-Cre Otud4fl/fl mice were more susceptible to lethal VSV infection than their control littermates. In addition, the levels of IFN-β and IL-6 were significantly decreased in the sera of Cre-ER Otud4fl/+ or Lyz2-Cre Otud4fl/fl mice compared to their respective control littermates 12 h after VSV infection (Fig. 5c, d). The expression of Ifnb was severely impaired and the replication of VSV (as monitored by expression of the VSV-N gene) was exacerbated in lungs or brains from Cre-ER Otud4fl/fl mice compared to Cre-ER Otud4fl/+ mice 24 h or 4 days after VSV infection, respectively (Fig. 5e, f). Plaque assays further confirmed that OTUD4 deficiency led to increased VSV titers in the brains of Cre-ER Otud4fl/fl mice compared to Cre-ER Otud4fl/+ mice (Fig. 5g). These results suggest that OTUD4 positively regulates virus-induced expression of downstream genes and is essential for host defense against RNA viruses in vivo.
Fig. 5.

OTUD4 deficiency increases susceptibility to lethal VSV infection. a Survival (Kaplan–Meier curve) of Cre-ER Otud4fl/+ (n = 11) and Cre-ER Otud4fl/fl (n = 12) mice intraperitoneal injected with tamoxifen (80 μg/g dissolved in corn oil) for five consecutive days and seven days later intravenously injected with VSV (2 × 107 PFUper mouse). b Survival (Kaplan–Meier curve) of Lyz2-Cre Otud4fl/+ (n = 8) and Lyz2-Cre Otud4fl/fl (n = 7) mice intravenously injected with VSV (2 × 107 PFUper mouse). c, d ELISA analysis of IFN-β and IL-6 in the sera of Cre-ER Otud4fl/+ and Cre-ER Otud4fl/fl (c) treated as in (a) or Lyz2-Cre Otud4fl/+ and Lyz2-Cre Otud4fl/fl mice (d) (n = 4) and intravenously injected with VSV (2 × 107 PFUper mouse) for 12 h. e, f qRT-PCR analysis of VSV-N, Ifnb or Otud4 mRNA in the lungs (d, n = 3) and brains (e, n = 3) from Cre-ER Otud4fl/+ and Cre-ER Otud4fl/fl mice treated as in (a) and intravenously injected with VSV (2 × 107 PFUper mouse) for 24 h or 4 days. g Plaque assays analyzing VSV titers in the brains from Cre-ER Otud4fl/+ and Cre-ER Otud4fl/fl mice treated as in (a) and intraperitoneally injected with VSV (2 × 107 PFUper mouse) for 4 days. *P < 0.05; **P < 0.01; ***P < 0.001 (unpaired student’s t-test, c–e). Data are representative of two independent experiments (mean ± S.D. in c–f)
The DUB activity of OTUD4 is required for activation
OTUD4 has been reported to promote the repair of DNA alkylation damage independently of its DUB activity.41 To examine whether the DUB activity of OTUD4 is involved in the regulation of virus-triggered signaling, we performed luciferase reporter assays with wild-type OTUD4 and enzymatically inactive mutant OTUD4(C45A) and found that the latter failed to potentiate SeV-induced activation of IFN-β promoter (Supplementary information, Fig. S7a), indicating that the DUB activity is indispensable for promoting antiviral signaling. To further substantiate this conclusion, we introduced the empty vector, or reconstituted OTUD4 or OTUD4(C45A) in Cre-ER Otud4fl/fl cells and subjected them to RNA virus infection. qRT-PCR and ELISA analyses showed that SeV-induced expression of Ifnb, Ccl20 or Il6 and production of IFN-β and IL-6 were substantially rescued in Cre-ER Otud4fl/fl BMDCs reconstituted with OTUD4 but not in those with OTUD4(C45A) (Fig. 6a, b). In addition, SeV-induced phosphorylation of TBK1, IRF3 or IκBα was increased by the reconstitution of OTUD4 but not OTUD4(C45A) in Cre-ER Otud4fl/fl BMDCs (Fig. 6c). Consistent with these data, reconstitution of OTUD4 but not OTUD4(C45A) in Cre-ER Otud4fl/fl BMDCs substantially inhibited the replication of VSV-GFP as monitored by GFP signals (Fig. 6d). OTUD4 can hydrolyze both K48- and K63-linked polyubiquitin chains and its K63-specific DUB activity requires phosphorylation at Ser202/204.41–43 Interestingly, however, reconstitution of OTUD4(S202/204 A) into Lyz2-Cre Otud4fl/fl BMDCs rescued SeV-induced expression of Ifnb and Ccl20 and phosphorylation of TBK1, IRF3 and IκBα equally as did reconstitution of wild-type OTUD4 (Supplementary information, Fig. S7b, c). Thus, K63-specific DUB activity of OTOD4 is not required for its regulation of antiviral signaling. Together, these data suggest that OTUD4-mediated potentiation of RNA virus-triggered signaling requires its deubiquitinating enzymatic activity but is independent of its K63-specific DUB activity.
Fig. 6.

The DUB activity of OTUD4 is required for cellular antiviral response. a, b qRT-PCR analysis of Ifnb, Ccl20, or Otud4 mRNA (a) or ELISA analysis of IFN-β and IL-6 (b) in Cre-ER Otud4fl/fl BMDCs treated with 4OH-Tam for three days and reconstituted with the empty vector (Vec), OTUD4, or OTUD4(C45A) followed by infection with SeV for 0–6 h (a) or with VSV or EMCV for 24 h (b). c Immunoblot analysis of total and phosphorylated (p-)TBK1, IκBα, IRF3, FLAG-OTUD4 or FLAG-OTUD4(C45A), or β-Actin in cells obtained in (a) infected with VSV for 0–8 h. d Flow cytometry analysis (left) and microscopy imaging (right images) of the replication of GFP-VSV in cells obtained in (a) infected with VSV-GFP (MOI = 1). *P < 0.05; **P < 0.01; ***P < 0.001 (analysis of two-way ANOVA followed by Bonferroni post-test). Data are representative of three (a) or two (b–d) independent experiments (mean ± S.D. in a, b)
OTUD4 deubiquitinates and stabilizes MAVS
Our luciferase reporter assays analyzing IFN-β promoter activity in HEK293 cells suggested that overexpression and knockdown of OTUD4 potentiated and inhibited RIG-I-mediated activation of the IFN-β promoter, respectively. In contrast, neither overexpression nor knockdown of OTUD4 had any effect on TBK1- or IRF3-mediated activation of the IFN-β promoter (Supplementary information, Fig. S7d, e). Thus, OTUD4 functions downstream of RIG-I and upstream of TBK1. Since OTUD4 interacted with MAVS and the DUB activity was required for its function, we investigated whether OTUD4 functions by deconjugating polyubiquitin chains from MAVS. As expected, we found that OTUD4 but not the enzymatically inactive mutant OTUD4(C45A) catalyzed deubiquitination of MAVS but not TRAF3 or TRAF6 in HEK293 cells or in vitro (Fig. 7a, b and Supplementary information, Fig. S8a). In addition, both OTUD4(S202/204 A) and OTUD4(aa1-245) containing the OTU domain were capable of deubiquitinating MAVS (Supplementary information, Fig. S8b). Knockout of OTUD4 potentiated VSV- or SeV-induced-K48-linked ubiquitination of MAVS (Fig. 7c and Supplementary information, Fig. S8c), whereas reconstitution of OTUD4 but not OTUD4(C45A) in Cre-ER Otud4fl/fl BMDCs impaired SeV-induced K48-linked ubiquitination of MAVS (Supplementary information, Fig. S8d), indicating that OTUD4 removes K48-linked polyubiquitin chains from MAVS after viral infection and may control the protein stability of MAVS. Consistent with this notion, knockout of OTUD4 promoted degradation of MAVS in the absence or presence of CHX (100 μg/ml) (half-life of MAVS: 5.47 h vs. 9.47 h and 2 h vs. 5.61 h, respectively) (Fig. 7d). The degradation of MAVS in OTUD4 deficient cells was completely blocked by the proteasome inhibitor MG132 and partially blocked by the autophagy inhibitor bafilomycin-A1 (BafA1) (Fig. 7e). In addition, reconstitution of OTUD4 but not OTUD4(C45A) inhibited the degradation of MAVS after VSV infection (Fig. 7f). These collective data suggest that OTUD4 deconjugates K48-linked polyubiquitin chains from MAVS and protects MAVS from proteasome-dependent degradation, thereby facilitating innate antiviral signaling.
Fig. 7.

OTUD4 deubiquitinates and stabilizes MAVS. a Denature-immunoprecipitation (Denature-IP) (with anti-FLAG) and immunoblot analysis (with anti-FLAG, anti-HA or anti-OTUD4) of HEK293 cells transfected with plasmids encoding FLAG-MAVS, HA-Ubiquitin and empty vector, OTUD4, or OTUD4(C45A) for 24 h. b In vitro deubiquitination analysis of ubiquitin-modified MAVS eluted from anti-FLAG precipitates by FLAG peptide of HEK293 cells transfected with FLAG-MAVS and HA-ubiquitin incubated with in vitro generated OTUD4 or OTUD4 C45A obtained from an in vitro transcription and translation kit. c Denature-IP (with anti-MAVS) and immunoblot analysis (with anti-K48 linkage polyubiquitin, anti-MAVS, anti-OTUD4 or anti-Tubulin) of Cre-ER Otud4fl/+ and Cre-ER Otud4fl/fl MLFs treated with 4-OH Tam (1 μM) for 3 days followed by MG132 treatment for 2 h prior to VSV infection for 0–6 h. d Immunoblot analysis of MAVS, OTUD4 and GAPDH in Cre-ER Otud4fl/+ and Cre-ER Otud4fl/fl MLFs treated with 4-OH Tam infected with VSV for 0–12 h in the presence or absence of cycloheximide (100 μg/ml). The relative intensities of MAVS were determined by normalizing the intensities of MAVS by the respective intensities of GAPDH. e Immunoblot analysis of MAVS, OTUD4 and GAPDH in Cre-ER Otud4fl/+ and Cre-ER Otud4fl/fl BMDMs treated with 4-OH Tam followed by infection with VSV for 8 h in the presence of MG132 or Baf A1 for 4 h. f Immunoblot analysis of MAVS, OTUD4 and GAPDH in Cre-ER Otud4fl/fl BMDMs treated with 4-OH Tam and reconstituted with Vec, OTUD4 or OTUD4(C45A) followed by infection with VSV for 0–8 h. The relative intensities of MAVS were determined by normalizing the intensities of MAVS by the respective intensities of GAPDH. g qRT-PCR analysis of Cre-ER Otud4fl/+ and Cre-ER Otud4fl/fl BMDCs treated with 4-OH Tam and reconstituted with Vec, or MAVS followed by infection with SeV for 0–6 h. h Immunoblot analysis of total and phosphorylated (p-)TBK1, IκBα, IRF3, FLAG-VISA, OTUD4 or β-Actin in cells obtained in (g) infected with SeV for 0–6 h. i Flow cytometry analysis (left) and microscopy imaging (right images) of the replication of GFP-VSV in cells obtained in (g) infected with VSV-GFP (MOI = 1). Data are representative of two or three independent experiments
Because the degradation of MAVS is accelerated in OTUD4 deficient cells, we hypothesized that supplementation of MAVS in OTUD4 knockout cells would restore virus-triggered expression of downstream genes. As anticipated, reconstitution of MAVS in Cre-ER Otud4fl/fl BMDCs rescued the expression of Ifnb and Il6 and the phosphorylation of TBK1, IRF3 and IκBα after SeV infection (Fig. 7g, h). Consistently, reconstitution of MAVS inhibited VSV-GFP replication in Cre-ER Otud4fl/fl BMDCs (Fig. 7i). In contrast, reconstitution of RIG-I in Otud4fl/fl BMDCs did not fully rescue SeV-induced expression of Ifnb, Ccl5 and Ip10 (Supplementary information, Fig. S9). Together, these data suggest that MAVS is the major target of OTUD4 in response to RNA virus infection.
Discussion
MAVS is an essential adapter protein for the innate immune defense against RNA viruses. The activity and availability of MAVS are strictly controlled by several posttranslational modifications to exert sufficient protective immunity and avoid excessive harmful immunity. Well-documented examples include the TBK1- and PPM1A-mediated phosphorylation and dephosphorylation of MAVS that respectively turns on and off MAVS’ ability to recruit downstream IRF3.22,23 Although around a half dozen E3s have been reported to catalyze K48-linked ubiquitination and degradation of MAVS, it remained unknown how deubiquitination of MAVS is regulated and whether such a regulation modulates innate immune responses against RNA viruses. Through an unbiased screening by cotransfection and immunoprecipitation assays that we have previously established,44,45 we identified OTUD4 as a MAVS-interacting DUB (Fig. 1 and Supplementary information, Fig. S1a). Viral infection induced upregulation of OTUD4 in a manner dependent on IRF3/7 but not p65 and catalyzed deubiquitination of MAVS (Supplementary information, Fig. S2b–d and Fig. 7a, b). In contrast, knockout of OTUD4 led to increased K48-linked ubiquitination and accelerated degradation of MAVS after viral infection (Fig. 7c, d and Supplementary information, Fig. S8c, d). In accord with these observations, OTUD4 deficiency resulted in potentiated VSV replication in vitro and in vivo and increased hypersensitivity to lethal VSV infection (Fig. 5), suggesting an essential role of OTUD4 in protecting the host cell against viral infection by preventing excessive degradation of MAVS (Fig. 8). Our findings have, for the first time, identified the DUB responsible for catalyzing deubiquitination of MAVS and have revealed the physiological significance of such regulation in mediating the innate immune responses against RNA viruses.
Fig. 8.
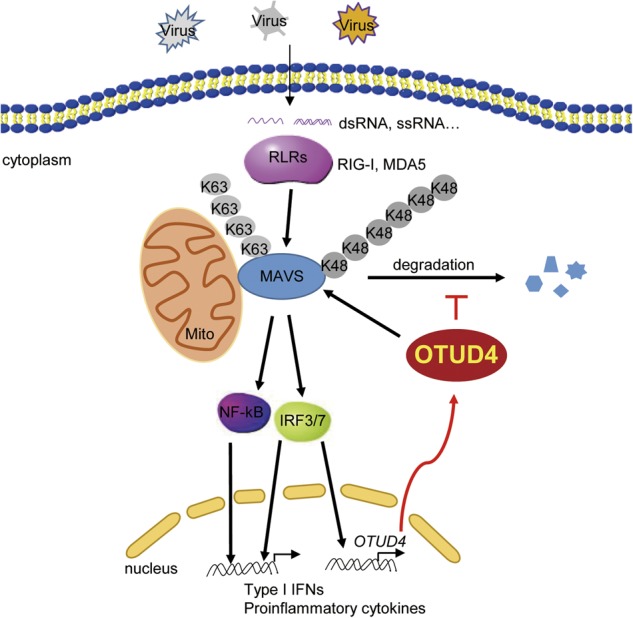
Illustration of a model on OTUD4-mediated regulation of innate antiviral signaling. RNA virus infection triggers RIG-I-MAVS signaling which activates NF-κB and IRF3/7 and subsequent induction of type I IFNs and proinflammatory cytokines and the upregulation of OTUD4. Constant infection also induces K48-linked ubiquitination and degradation of MAVS. OTUD4 interacts with MAVS and removes the K48-linked polyubiquitin chains from MAVS to prevent excessive degradation of MAVS, thereby keeping cellular antiviral response at a certain level to restrict replication of invading viruses
It has been reported that OTUD4 is required for deubiquitination of two DNA demethylases ALKBH2 and ALKBH3, promoting the stability of ALKBH2 and ALKBH3 proteins and the repair of DNA alkylation damage. This process is independent of the DUB activity of OTUD4.41 However, the DUB activity of OTUD4 is responsible for the deubiquitination of MAVS and the promotion of cellular antiviral responses, as was evident from the findings first that OTUD4 but not OTUD4(C45A) could deubiquitinate MAVS in vivo and in vitro; and second, that the reconstitution of wild-type OTUD4, but not OTUD4(C45A), in OTUD4 knockout cells impaired the ubiquitination of MAVS, potentiated virus-triggered signaling and inhibited VSV-GFP replication (Fig. 6, Fig. 7a, b and Supplementary information, Fig. S8d). It has been shown that viral infection leads to the generation of reactive oxygen species and that such oxidative stress contributes to viral pathogenesis, at least in part, through induction of aberrant DNA alkylation and damage-associated inflammation and cell death.47–49 The DUB-dependent and independent functions of OTUD4 might complement each other in response to viral infection by turning on MAVS-mediated signaling and turning down DNA damage-associated inflammation and cell death, respectively.
It has been previously demonstrated that the OTU domain of OTUD4 selectively hydrolyzes K48-linked diUb molecules and that OTUD4 purified from bacteria exhibits a preference against K48-linked polyubiquitin chains.41,42 A very recent study has shown that OTUD4 purified from HEK293 cells is phosphorylated at Ser202/204 which activates its dormant K63-specific DUB activity.43 In our study, we found that purified OTUD4 expressed in vitro, which supposedly lacked phosphorylation on its Ser202/204 residues could catalyze deubiquitination of MAVS (Fig. 7b). The OTU domain of OTUD4 (amino acids 1-245), which has intact K48- but not K63-speficic DUB activity, interacted with and impaired the ubiquitination of MAVS in cells43 (Fig. 1c and Supplementary information, Fig. S8b). Moreover, OTUD4(S202/204A) efficiently deubiquitinated MAVS in HEK293 cells in our overexpression system and reconstitution of OTUD4(S202/204A) restored SeV-induced expression of downstream genes and phosphorylation of downstream signaling molecules (Supplementary information, Fig. S7b, c and Fig. S8b). Thus, the K63-specific DUB activity of OTUD4 is not involved in its regulation of RLR signaling. In contrast, OTUD4 deficiency potentiated virus-induced K48-linked ubiquitination and degradation of MAVS, indicating that OTUD4 exhibits the K48-specific DUB activity against MAVS (Fig. 7c and Supplementary information, Fig. S8c, d). In support of this notion, virus-triggered degradation of MAVS was inhibited by the proteasome inhibitor MG132 and supplementing MAVS but not RIG-I into OTUD4 knockout cells rescued virus-triggered signaling (Fig. 7e–i and Supplementary information, Fig. S9). Thus, the K63- and K48-specific DUB activity of OTUD4 inhibits TLR- or IL-1-triggered MyD88-dependent inflammatory responses and promotes virus-triggered MAVS-dependent innate immune responses, respectively. This is not surprising for a DUB that has dual enzymatic specificities in different pathways. In this context, the E3 ubiquitin ligase Nrdp1 catalyzes K48-linked ubiquitination and degradation of MyD88 and K63-linked ubiquitination and activation of TBK1, thereby turning down TLR-triggered inflammation and turning up virus-triggered signaling.50
To date, MAVS recruits or interacts with downstream signaling components through at least three mechanisms, namely in an N terminal domain-, TRAF-interacting motif (TIM)- or transmembrane (TM) domain-dependent manner. MAVS interacts with TRIM31 through its N terminal CARD and Pro-rich domain.24 MAVS contains multiple TIMs which mediate the recruitment of TRAF2, TRAF6 and TRAF3 in a manner independent of its transmembrane domain.13,51 Most reported MAVS-interacting proteins associate with MAVS in a manner dependent on its TM domain. These include OTUD4, PCBP2, RNF5, Tetherin, NS1 of Duck Tembusu Virus, iRhom2, ECSIT and GP73.27,29,34,36,52–54 The TM domain of MAVS is also crucial for its oligomerization after viral infection.11,55 Interestingly, the truncated MAVS(aa361–540) used in this study is sufficient to activate ISRE or NF-κB in reporter assays.13 The simplest explanation for this is that TM-mediated subcellular localization or structural conformation of MAVS is critical for its recruitment of downstream proteins. Further investigations are required to fully address this point.
OTUD4 has been implicated in the embryonic development of zebrafish and it is associated with neurodegeneration and reproductive dysfunction in humans.39,40 Although the Otud4−/− mice are viable and exhibit normal growth, they are born in a sub-Mendelian ratio, indicating a critical role of OTUD4 in embryonic development.43 In this study, we generated OTUD4 conditional knockout mice and found that acute deletion of OTUD4 did not affect their survival and growth (Supplementary information, Fig. S3). Immune cell numbers and in vitro differentiation of BMDCs or BMDMs were not affected by deletion of OTUD4 induced by 4-OH Tam treatment. Moreover, deletion of OTUD4 in myeloid cells by Lyz2-Cre mediated recombination did not affect the survival and development of mice and Lyz2-Cre Otud4fl/fl mice were born in Mendelian ratio. Thus, OTUD4 is dispensable in myeloid cells for mouse development. Other unknown targets and cell-type specific roles of OTUD4 therefore remain to be characterized in regard of embryonic development, neurodegeneration and reproductive dysfunction. The OTUD4 conditional knockout mice will provide a powerful tool to address these questions in the future.
Materials and methods
Mice
The Otud4fl/+ mice were generated by Nanjing Biomedical Research Institute of Nanjing University through CRISPR/Cas9-mediated gene editing. In brief, guide RNAs (5′-TTGAGAACTCACCTATAGCAAGG-3′ and 5′-CTGGCCCAGGAACAGAGTTCAGG-3′) were obtained through in vitro transcription and purification. The gRNAs were incubated with purified Cas9 protein and injected into the fertilized eggs (at the one-cell stage) together with the targeting vector with two loxp sites flanking the exon 4 of the Otud4 gene. The injected fertilized eggs were cultured to the two-cell stage followed by transplantation into pseudopregnant mice. The targeted genomes of F0 mice were amplified by PCR and sequenced and the chimeras were crossed with wild-type C57BL/6 mice to obtain F1 Otud4fl/+ mice. Southern blot analysis was conducted with the tail DNA from F1 mice to confirm correct recombination and exclude random insertions of the targeting vector. Lyz2-Cre mice were purchased from the Nanjing Biomedical Research Institute of Nanjing University. Cre-ER mice (B6.129-Gt(ROSA)26Sortm1(cre/ERT2)Tyj) were from the Jackson Laboratory and were kindly provided by Dr. Chen Dong (Tsinghua University). Otud4fl/+ mice were crossed with Cre-ER or Lyz2-Cre mice to obtain Cre-ER Otud4fl/+ or Lyz2-Cre Otud4fl/+ mice, respectively. The Cre-ER Otud4fl/+ and Cre-ER Otud4fl/fl littermates or the Lyz2-Cre Otud4fl/+ and Lyz2-Cre Otud4fl/fl littermates were used throughout the study. The genotyping primers are listed in Supplementary information, Table S1. All mice were housed in the specific pathogen-free animal facility at Wuhan University and all animal experiments were carried out in accordance with protocols approved by the Institutional Animal Care and Use Committee of Wuhan University.
Tamoxifen-mediated knockout of OTUD4
To achieve consitional knockout of OTUD4, 9–10 week-old Cre-ER Otud4fl/+ and Cre-ER Otud4fl/fl mice were injected intraperitoneally with tamoxifen (80 μg per g bodyweight, dissolved in corn oil) (Sigma, #T5648) for five successive days. After 7 days without treatment, mice were either subjected to euthanasia to test the knockout efficiency or infected with VSV. To knockout OTUD4 in cultured cells, Cre-ER Otud4fl/+ and Cre-ER Otud4fl/fl cells were treated with 4-hydroxytamoxifen (1 μM) (Sigma, H6278) for three days. Cells were then re-seeded into culture dishes or plated in 4-hydroxytamoxifen free medium and rested for 24 h followed by infection with SeV, VSV, EMCV or HSV-1 or transfection with poly(I:C) or dsDNA.
Reagents, antibodies, and constructs
Poly(I:C) was used as described previously.56 Cycloheximide (CHX), MG132 and bafilomycin A1 were purchased from Sigma.44 Immuno-reagents were obtained as follows: Mouse control IgG (Santa Cruz Biotechnology, sc-2025); rabbit control IgG (Millipore, 12–370); HRP-conjugated goat-anti mouse or rabbit IgG (Thermo Scientific, PA1-86717 and SA1-9510); HRP-conjugated mouse anti-FLAG (Sigma, A8592); HRP-conjugated goat anti-mouse IgG, F(ab′)2 fragment specific (Jackson Immuno Research,115-035-006); HRP-conjugated goat anti-rabbit IgG, F(ab′)2 fragment specific (Jackson Immuno Research,111-035-006); mouse anti-FLAG (Sungene, KM8002); anti-GFP (Sungene, KM8009); anti-β-Actin (KM9001); anti-GAPDH (KM9002); anti-Myc (KM8003); anti-Tubulin (KM9003); anti-HA (COVANCE, MMS-101R); anti-Ubiquitin (sc-8017); anti-pIκBα (Cell Singling Technologies, 9246L); anti-mouse MAVS (sc-365333) and anti-IRF3 (sc-33641); Rabbit anti-ubiquitin K48-specific linkage (Millipore, 05-1307); anti-ubiquitin K63-specific linkage (Millipore, 05-1308); anti-TBK1 (Abcam, 96328-11); anti-p-TBK1 (Abcam, 109272), anti-IRF3 (sc-9082); anti-p-IRF3 (Cell Singling Technologies, 4947S); anti-IκBα (sc-371); anti-mouse MAVS (CST 4983s) and anti-human MAVS (BETHYL, A300-782A). Anti-mouse-OTUD4 antisera were raised against recombinant mouse OTUD4 (aa239-550). The DUB expression library, ISRE and IFN-β promoter luciferase reporter constructs, mammalian expression plasmids for MAVS, MAVS truncations, p65, MAVS, TBK1, IRF3, IRF7, RIG-I, ubiquitin and anti-VSV were kindly provided by Dr. Hong-Bing Shu (Wuhan University) and have been previously described.44,45,56 Mammalian expression plasmids for OTUD4 and OTUD4 mutants and truncations were constructed by standard molecular biology techniques.
Quantitative RT-PCR and ELISA
Total RNA was extracted from cells using TRIzol (Invitrogen) and the first-strand cDNA was synthesized using All-in-One cDNA Synthesis SuperMix (Biotool). Gene expression was examined with a Bio-Rad CFX Connect system using a fast two-step amplification program with 2x SYBR Green Fast qPCR Master Mix (Biotool). The value obtained for each gene was normalized to that of the β-actin gene. Gene-specific primers have been described previously57,58 or are listed in Supplementary information, Table S2. ELISA kits for IFN-β and IL-6 (BioLegend) were used to detect the indicated cytokines in the sera.
Co-immunoprecipitation and immunoblot analysis
The experiments were performed as previously described.44,45,56,58,59 In brief, cells were lysed in Nonidet P-40 lysis buffer containing 150 mM NaCl, 1 mM EDTA, 1% Nonidet P-40, and 1% protease and phosphatase inhibitor cocktail (Biotool). Cell lysates were subjected to SDS-PAGE and immunoblot analysis was performed with the appropriate antibodies. For immunoprecipitation assays, the lysates were immunoprecipitated with IgG or the appropriate antibodies, and the precipitates were washed three times with lysis buffer containing 500 mM NaCl, followed by immunoblot analysis. The antibodies were diluted in 3–5% (wt/vol) fat-free milk (BD Biosciences) or 1% BSA (Sigma) in TBS (1:500–1:2000).
In vivo and in vitro deubiquitination assays
These experiments were performed as previously described.44,45,58 For deubiquitination assays in cultured cells, the cells were lysed with the lysis buffer (100 μl) and the supernatants were denatured at 95 °C for 5 min in the presence of 1% SDS. The denatured lysates were diluted with lysis buffer to reduce the concentration of SDS below 0.1% followed by immunoprecipitation (denature-IP) with the indicated antibodies. The immunoprecipitates were subjected to immunoblot analysis with anti-ubiquitin, or anti-K48-linked ubiquitin chains. For in vitro deubiquitination assays, FLAG-tagged MAVS and HA-tagged ubiquitin were cotransfected into HEK 293 cells. Denature-IP was performed and the precipitates were eluted by addition of 3x FLAG peptide (sigma) to obtain ubiquitin-modified MAVS. OTUD4 and OTUD4(C45A) were obtained using a coupled in vitro transcription/translation kit (Promega). The ubiquitinated MAVS were incubated with in vitro synthesized proteins at 37 °C for 2 h followed by an overnight incubation at 16 °C in the presence of 1 μM ATP. The mixture was analyzed by immunoblot with the indicated antibodies.
Transfection and luciferase reporter assay
HEK293 cells were co-transfected with the IFN-β promoter firefly luciferase reporter plasmid and a TK-Renilla luciferase reporter, together with vector alone or various DUB constructs, or control siRNA or siRNA targeting various DUBs. Twenty-four hours later, cells were infected with SeV for 8 h. Luciferase activity was measured with a Dual-Luciferase Reporter Assay System (Promega).
Cell culture
Mouse embryonic fibroblasts (MEFs) were prepared from E14.5 embryos and mouse lung fibroblasts (MLFs) isolated as previously described.44,45,58 Bone marrow cells were isolated from mouse femur. The cells were cultured in DMEM containing 10% fetal bovine serum, 1% streptomycin and penicillin, and 10 μM β-mercaptoethanol, with M-CSF (10 ng/ml, Peprotech) for BMDM differentiation or GM-CSF (20 ng/ml, Peprotech) for BMDC differentiation, respectively. On day 3, 4-hydroxyltamoxifen (1 μM) was added to the culture medium for 3 days. On day 6, the cells were re-seeded and cultured in 4-hydroxyltamoxifen-free medium for 24 h before subsequent experiments.
siRNA
The siRNAs were synthesized and transfected into cells using Lipofectamine 2000 according to the manufacturer’s manual. Thirty-six hours after transfection, cells were harvested or stimulated followed by immunoblot, qPCR or deubiquitination assays. siRNA sequences were as follows:
Control: 5′-UUCUCCGAACGUGUCACGUTT-3′;
siOTUD4#1: 5′-GGAACUAGACACGUUGGAATT-3′;
siOTUD4#2: 5′-CCAGCAGAACAUGUGUCUUTT-3′.
Chromatin immunoprecipitation (ChIP) assays
These experiments were performed as previously described.56,60 Cells were fixed with 1% formaldehyde and quenched by glycine and then washed three times with PBS and then harvested in ChIP lysis buffer (50 mM Tris·HCl pH 8.0, 1% SDS, 5 mM EDTA) followed by sonication to give DNA fragments of 400–600 bp. The lysate was centrifuged at 4 °C for 15 min and ChIP dilution buffer (20 mM Tris·HCl, pH 8.0, 150 mM NaCl, 2 mM EDTA, 1% Triton X-100) was added to the supernatant (4:1 volume). The resulting lysate was then incubated with protein G beads and anti-FLAG or control IgG at 4 °C for 4 h. DNA was eluted using ChIP elution buffer (0.1 M NaHCO3, 1% SDS, 30 μg/mL proteinase K) by incubation at 65 °C overnight, and the DNA was purified with a DNA purification kit (TIANGEN). The purified DNA was assayed by quantitative PCR using the SFX connect system with a SYBR Green One Step Real-Time PCR kit (Biotool). The promoter qPCR primers sequences were as follows:
Ifnb promoter F, 5′-ATTCCTCTGAGGCAGAAAGGACC-3′; Ifnb promoter R, 5′-GCAAGATGAGGCAAAGGCTGTCA-3′; Otud4 promoter 1F, 5′-AACTACAACTCCCGTCGTCC-3′, Otud4 promoter 1R, 5′-GAGGAAACGCGGAGAGAAAG-3′, Otud4 promoter 2F, 5′-CCGCGGCTTAAACTACAACT-3′, Otud4 promoter 2R, 5′-CTGAGATTAAGCGAGGCCTG-3′, Control promoter F, 5′-TAGTGATCATGCCCATTAAC-3′, Control promoter R, 5′-ATCTTCTAAAGTCTCAAGGT-3′.
Viral infection
For qRT-PCR or immunoblot analysis, cells were seeded into 24-well plates (2–5 × 105 cells per well) or six-well plates (106–107 cells per well) and infected with the specified viruses for the indicated time points. For viral replication assays, cells (2–5 × 105) were infected with VSV-GFP. One hour later, the supernatants were removed and cells were washed twice with 1 ml/plate of prewarmed PBS followed by culture in full medium for 24 h. Viral replication was analyzed by flow cytometry, fluorescent microscopy or qRT-PCR analysis. For the infection of mice, age- and sex-matched Cre-ER Otud4fl/+ and Cre-ER Otud4fl/fl (tamoxifen treated) or Lyz2-Cre Otud4fl/+ and Lyz2-Cre Otud4fl/fl littermates were injected with VSV (2 × 107 PFU/per mouse) and the survival of animals was monitored every day. The lungs or brains were collected for qRT-PCR analysis or plaque assays at 24 h or 4 days after infection, respectively.
Plaque assay
Homogenates of brains (or the serial dilutions) from infected Cre-ER Otud4fl/+ and Cre-ER Otud4fl/fl mice (tamoxifen treated) were used to infect monolayers of Vero cells. One hour later, the homogenates or dilutions were removed and the infected Vero cells were washed with pre-warmed PBS twice followed by incubation with DMEM containing 2% methylcellulose for 48 h. Cells were fixed with 4% paraformaldehyde for 15 min and stained with 1% crystal violet for 30 min before counting the plaques.
Lentivirus-mediated gene transfer
HEK293 cells were transfected with phage-6tag-p65, phage-6tag-IRF3, phage-6tag-IRF7, phage-6tag-OTUD4, phage-6tag-OTUD4(C45A), phage-6tag-OTUD4(S202/204A), phage-6tag-RIG-I, phage-6tag-MAVS or the empty vector along with the packaging vectors pSPAX2 and pMD2G. The medium was changed for fresh full medium (10% FBS, 1% streptomycin-penicillin and 10 μM β-mercaptoethanol) after 8 h. Forty hours later, supernatants were harvested to infect 4-hydroxyltamoxifen-treated Cre-ER Otud4fl/fl MLFs or BMDCs followed by various analyses.
Statistical analysis
Differences between experimental and control groups were tested using Student’s t-test or two-way ANOVA with Bonferroni post-test. P values less than 0.05 were considered statistically significant. For animal survival analysis, the Kaplan–Meier method was adopted to generate graphs, and the survival curves were analyzed with log-rank analysis.
Electronic supplementary material
Acknowledgements
We would like to thank Drs. Hong-Bing Shu (Wuhan University) and Chen Dong (Tsinghua University) for reagents, members of Zhong lab and the core facilities of Medical Research Institute for technical help. This study was supported by grants from the Ministry of Science and Technology of China (2014CB542601), Natural Science Foundation of China (31521091, 31601131, 31671454 and 31622036), National Natural Science Foundation of Hubei Province (2018CFA016), Wuhan University (2042017kf0199 and 2042017kf0242) and State Key Laboratory of Veterinary Etiological Biology (SKLVEB2017KFKT004).
Author contributions
B.Z. designed and supervised the study. T.L. designed and performed the major experiments; K.Y., L.Y., M.Z. and Z.C. helped with the animal studies; Z.Z. and Y.R. performed the ChIP analysis; Q.Z. provided reagents; B.Z., D.L. and T.L. wrote the paper. All authors analyzed data.
Competing interests
The authors declare no competing interests.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41422-018-0107-6.
References
- 1.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 2.Wu J, Chen ZJ. Innate immune sensing and signaling of cytosolic nucleic acids. Annu. Rev. Immunol. 2014;32:461–488. doi: 10.1146/annurev-immunol-032713-120156. [DOI] [PubMed] [Google Scholar]
- 3.Pichlmair A, et al. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5 ‘-phosphates. Science. 2006;314:997–1001. doi: 10.1126/science.1132998. [DOI] [PubMed] [Google Scholar]
- 4.Hornung V, et al. 5 ‘-triphosphate RNA is the ligand for RIG-I. Science. 2006;314:994–997. doi: 10.1126/science.1132505. [DOI] [PubMed] [Google Scholar]
- 5.Myong S, et al. Cytosolic viral sensor RIG-I is a 5 ‘-triphosphate-dependent translocase on double-stranded RNA. Science. 2009;323:1070–1074. doi: 10.1126/science.1168352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schlee M, et al. Recognition of 5 ‘triphosphate by RIG-I helicase requires short blunt double-stranded RNA as contained in panhandle of negative-strand virus. Immunity. 2009;31:25–34. doi: 10.1016/j.immuni.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kato H, et al. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J. Exp. Med. 2008;205:1601–1610. doi: 10.1084/jem.20080091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chiu YH, MacMillan JB, Chen ZJ. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell. 2009;138:576–591. doi: 10.1016/j.cell.2009.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ablasser A, et al. RIG-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III-transcribed RNA intermediate. Nat. Immunol. 2009;10:1065–72. doi: 10.1038/ni.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seth RB, Sun LJ, Ea CK, Chen ZJJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappa B and IRF3. Cell. 2005;122:669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 11.Hou F, et al. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell. 2011;146:448–461. doi: 10.1016/j.cell.2011.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cai X, et al. Prion-like polymerization underlies signal transduction in antiviral immune defense and inflammasome activation. Cell. 2014;156:1207–1222. doi: 10.1016/j.cell.2014.01.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu LG, et al. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol. Cell. 2005;19:727–740. doi: 10.1016/j.molcel.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 14.Meylan E, et al. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437:1167–1172. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- 15.Kawai T, et al. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 2005;6:981–988. doi: 10.1038/ni1243. [DOI] [PubMed] [Google Scholar]
- 16.Liu S, et al. MAVS recruits multiple ubiquitin E3 ligases to activate antiviral signaling cascades. eLife. 2013;2:eLife.00785. doi: 10.7554/eLife.00785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fang R, et al. MAVS activates TBK1 and IKK epsilon through TRAFs in NEMO dependent and independent manner. PLoS. Pathog. 2017;13:e1006720. doi: 10.1371/journal.ppat.1006720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hemmi H, et al. The roles of two IkappaB kinase-related kinases in lipopolysaccharide and double stranded RNA signaling and viral infection. J. Exp. Med. 2004;199:1641–1650. doi: 10.1084/jem.20040520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perry AK, Chow EK, Goodnough JB, Yeh WC, Cheng G. Differential requirement for TANK-binding kinase-1 in type I interferon responses to toll-like receptor activation and viral infection. J. Exp. Med. 2004;199:1651–1658. doi: 10.1084/jem.20040528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kumar H, et al. Essential role of IPS-1 in innate immune responses against RNA viruses. J. Exp. Med. 2006;203:1795–1803. doi: 10.1084/jem.20060792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun QM, et al. The specific and essential role of MAVS in antiviral innate immune responses. Immunity. 2006;24:633–642. doi: 10.1016/j.immuni.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 22.Liu S, et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science. 2015;347:1217–U1217. doi: 10.1126/science.aaa2630. [DOI] [PubMed] [Google Scholar]
- 23.Xiang W, et al. PPM1A silences cytosolic RNA sensing and antiviral defense through direct dephosphorylation of MAVS and TBK1. Sci. Adv. 2016;2:e1501889. doi: 10.1126/sciadv.1501889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu B, et al. The ubiquitin E3 ligase TRIM31 promotes aggregation and activation of the signaling adaptor MAVS through Lys63-linked polyubiquitination. Nat. Immunol. 2017;18:214–224. doi: 10.1038/nrm.2017.17. [DOI] [PubMed] [Google Scholar]
- 25.Castanier C, et al. MAVS ubiquitination by the E3 ligase TRIM25 and degradation by the proteasome is involved in type I interferon production after activation of the antiviral RIG-I-like receptors. BMC Biol. 2012;10:44. doi: 10.1186/1741-7007-10-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou X, You F, Chen H, Jiang Z. Poly(C)-binding protein 1 (PCBP1) mediates housekeeping degradation of mitochondrial antiviral signaling (MAVS) Cell Res. 2012;22:717–727. doi: 10.1038/cr.2011.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.You F, et al. PCBP2 mediates degradation of the adaptor MAVS via the HECT ubiquitin ligase AIP4. Nat. Immunol. 2009;10:1300–U1310. doi: 10.1038/ni.1815. [DOI] [PubMed] [Google Scholar]
- 28.Choi YB, Shembade N, Parvatiyar K, Balachandran S, Harhaj EW. TAX1BP1 restrains virus-induced apoptosis by facilitating Itch-mediated degradation of the mitochondrial adaptor MAVS. Mol. Cell. Biol. 2017;37:pii: e00422–16. doi: 10.1128/MCB.00422-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhong B, et al. The E3 ubiquitin ligase RNF5 targets virus-induced signaling adaptor for ubiquitination and degradation. J. Immunol. 2010;184:6249–6255. doi: 10.4049/jimmunol.0903748. [DOI] [PubMed] [Google Scholar]
- 30.Jia Y, et al. Negative regulation of MAVS-mediated innate immune response by PSMA7. J. Immunol. 2009;183:4241–4248. doi: 10.4049/jimmunol.0901646. [DOI] [PubMed] [Google Scholar]
- 31.Wang Y, Tong X, Ye X. Ndfip1 negatively regulates RIG-I-dependent immune signaling by enhancing E3 ligase Smurf1-mediated MAVS degradation. J. Immunol. 2012;189:5304–5313. doi: 10.4049/jimmunol.1201445. [DOI] [PubMed] [Google Scholar]
- 32.Pan Y, et al. Smurf2 negatively modulates RIG-I-dependent antiviral response by targeting VISA/MAVS for ubiquitination and degradation. J. Immunol. 2014;192:4758–4764. doi: 10.4049/jimmunol.1302632. [DOI] [PubMed] [Google Scholar]
- 33.Yoo YS, et al. The mitochondrial ubiquitin ligase MARCH5 resolves MAVS aggregates during antiviral signalling. Nat. Communs. 2015;6:8910. doi: 10.1038/ncomms9910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jin S, et al. Tetherin suppresses type I interferon signaling by targeting MAVS for NDP52-mediated selective autophagic degradation in human cells. Mol. Cell. 2017;68:308–322. doi: 10.1016/j.molcel.2017.09.005. [DOI] [PubMed] [Google Scholar]
- 35.Zhang L, et al. Induction of OTUD1 by RNA viruses potently inhibits innate immune responses by promoting degradation of the MAVS/TRAF3/TRAF6 signalosome. Plos Pathog. 2018;14:e1007067. doi: 10.1371/journal.ppat.1007067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Luo WW, et al. iRhom2 is essential for innate immunity to RNA virus by antagonizing ER- and mitochondria-associated degradation of VISA. Plos Pathog. 2017;13:e1006693. doi: 10.1371/journal.ppat.1006693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mevissen TET, Komander D. Mechanisms of deubiquitinase specificity and regulation. Annu. Rev. Biochem. 2017;86:159–192. doi: 10.1146/annurev-biochem-061516-044916. [DOI] [PubMed] [Google Scholar]
- 38.Tse WKF, et al. Genome-wide loss-of-function analysis of deubiquitylating enzymes for zebrafish development. BMC Genom. 2009;10:637. doi: 10.1186/1471-2164-10-637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tse WKF, Jiang YJ, Wong CKC. Zebrafish transforming growth factor-beta-stimulated clone 22 domain 3 (TSC22D3) plays critical roles in Bmp-dependent dorsoventral patterning via two deubiquitylating enzymes Usp15 and Otud4. Biochim Biophys. Acta1. 2013;830:4584–4593. doi: 10.1016/j.bbagen.2013.05.006. [DOI] [PubMed] [Google Scholar]
- 40.Margolin DH, et al. Ataxia, dementia, and hypogonadotropism caused by disordered ubiquitination. N. Engl. J. Med. 2013;368:1992–2003. doi: 10.1056/NEJMoa1215993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao Y, et al. Noncanonical regulation of alkylation damage resistance by the OTUD4 deubiquitinase. EMBO J. 2015;34:1687–1703. doi: 10.15252/embj.201490497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mevissen TET, et al. OTU deubiquitinases reveal mechanisms of linkage specificity and enable ubiquitin chain restriction analysis. Cell. 2013;154:169–184. doi: 10.1016/j.cell.2013.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao Y, et al. OTUD4 is a phospho-activated K63 deubiquitinase that regulates MyD88-dependent signaling. Mol. Cell. 2018;69:505–516. doi: 10.1016/j.molcel.2018.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang M, et al. USP18 recruits USP20 to promote innate antiviral response through deubiquitinating STING/MITA. Cell Res. 2016;26:1302–1319. doi: 10.1038/cr.2016.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sun H, et al. USP13 negatively regulates antiviral responses by deubiquitinating STING. Nat. Communs. 2017;8:15534. doi: 10.1038/ncomms15534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hameyer D, et al. Toxicity of ligand-dependent Cre recombinases and generation of a conditional Cre deleter mouse allowing mosaic recombination in peripheral tissues. Physiol. Genom. 2007;31:32–41. doi: 10.1152/physiolgenomics.00019.2007. [DOI] [PubMed] [Google Scholar]
- 47.Aas PA, et al. Human and bacterial oxidative demethylases repair alkylation damage in both RNA and DNA. Nature. 2003;421:859–863. doi: 10.1038/nature01363. [DOI] [PubMed] [Google Scholar]
- 48.Camini FC, da Silva Caetano CC, Almeida LT, de Brito Magalhaes CL. Implications of oxidative stress on viral pathogenesis. Arch. Virol. 2017;162:907–917. doi: 10.1007/s00705-016-3187-y. [DOI] [PubMed] [Google Scholar]
- 49.Allen EK, et al. SNP-mediated disruption of CTCF binding at the IFITM3 promoter is associated with risk of severe influenza in humans. Nat. Med. 2017;23:975–983. doi: 10.1038/nm.4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang C, et al. The E3 ubiquitin ligase Nrdp1 ‘preferentially’ promotes TLR-mediated production of type I interferon. Nat. Immunol. 2009;10:744–752. doi: 10.1038/ni.1742. [DOI] [PubMed] [Google Scholar]
- 51.Paz S, et al. A functional C-terminal TRAF3-binding site in MAVS participates in positive and negative regulation of the IFN antiviral response. Cell Res. 2011;21:895–910. doi: 10.1038/cr.2011.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lei CQ, et al. ECSIT bridges RIG-I-like receptors to VISA in Signaling events of innate antiviral responses. J. Innate Immun. 2015;7:153–164. doi: 10.1159/000365971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang X, et al. GP73 represses host innate immune response to promote virus replication by facilitating MAVS and TRAF6 degradation. PLoS. Pathog. 2017;13:e1006321. doi: 10.1371/journal.ppat.1006321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang J, et al. Duck Tembusu virus nonstructural protein 1 antagonizes IFN-beta signaling pathways by targeting VISA. J. Immunol. 2016;197:4704–4713. doi: 10.4049/jimmunol.1502317. [DOI] [PubMed] [Google Scholar]
- 55.Zhong B, et al. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity. 2008;29:538–550. doi: 10.1016/j.immuni.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 56.Lu B, et al. Induction of INKIT by viral infection negatively regulates antiviral responses through inhibiting phosphorylation of p65 and IRF3. Cell. Host. Microbe. 2017;22:86–98. doi: 10.1016/j.chom.2017.06.013. [DOI] [PubMed] [Google Scholar]
- 57.Zhong B, et al. Negative regulation of IL-17-mediated signaling and inflammation by the ubiquitin-specific protease USP25. Nat. Immunol. 2012;13:1110–1117. doi: 10.1038/ni.2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lin D, et al. Induction of USP25 by viral infection promotes innate antiviral responses by mediating the stabilization of TRAF3 and TRAF6. Proc. Natl Acad. Sci. USA. 2015;112:11324–113291. doi: 10.1073/pnas.1509968112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhao Y, et al. USP2a supports metastasis by tuning TGF-beta signaling. Cell Rep. 2018;22:2442–2454. doi: 10.1016/j.celrep.2018.02.007. [DOI] [PubMed] [Google Scholar]
- 60.Ren Y, et al. The type I interferon-IRF7 axis mediates transcriptional expression of Usp25 gene. J. Biol. Chem. 2016;291:13206–13215. doi: 10.1074/jbc.M116.718080. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.