

Abstract
NETosis is a regulated form of neutrophil cell death that contributes to the host defense against pathogens and was linked to various diseases soon after its first description in 2004. During NETosis, neutrophils release neutrophil extracellular traps (NETs), which can capture and kill bacteria and other pathogens to prevent them from spreading. Although substantial progress has been made in our understanding of NETosis, the precise mechanism underlying NETosis is still a matter of debate. Research continues to elucidate the molecular pathways involved in NETosis. In recent years, interactions with the complement and coagulation systems have become increasingly apparent. Activated complement proteins can stimulate NET formation, and NETs, in turn, can serve as a platform for complement activation. In addition, NETs can act as a scaffold for thrombus formation during coagulation. While crosstalk between the coagulation and complement systems has been previously described, NETosis appears to be a third important player in this consortium to protect the host against pathogens. This review summarizes our current knowledge on the mutual interactions between NETosis, the complement system and the coagulation system, with an emerging description of their complex triangular relationship.
Keywords: coagulation, complement, NETosis, neutrophils, thrombosis
Introduction
Neutrophils make up approximately 70% of the white blood cells in humans and serve as a first line of defense against invading pathogens. Two mechanisms of action, the release of cytotoxic molecules from granules, named degranulation, and phagocytosis, the engulfment and intracellular killing of pathogens, have been established for quite some time, but in 2004, a new antibacterial strategy of neutrophils was described. This strategy involves the extrusion of neutrophilic chromatin together with antibacterial proteins originating from the neutrophil granules.1 This process is called NETosis, and this name refers to the neutrophil extracellular trap (NET) that is formed during this process. After its initial discovery, an extensive number of investigators not only addressed the mechanism(s) of NET formation but also the role of NETs as initiators of diseases, such as autoimmune diseases, diabetes, atherosclerosis, and vasculitis.2 More recently, interactions with other host systems have been studied, including the complement and coagulation systems.
While NETosis was first described not much more than a decade ago, research on the complement system commenced a century ago. The system consists of approximately 30 serum-associated proteins and is classified as part of the humoral innate immune system. The name ‘‘complement’’ was given because the cascade reaction of these proteins ‘‘complement’’, or support, the antibacterial effects of antibodies. Three distinct pathways of complement activation have emerged over the years, and it was recently discovered that NETs can play a role in their initiation.
An injury will induce coagulation of the blood to quickly close the wound, but uncontrolled clot formation might lead to thrombosis. Activated coagulation factors induce the activation of the complement system as a side effect, and activated complement, in turn, alerts the immune system to recruit neutrophils. It is generally assumed that these neutrophils are recruited to the site of injury to quickly respond to any opportunistic attacks of invading pathogens. Recently, it was posed that NET structures released by neutrophils can also serve as a scaffold for clot formation, shining new light on the role of neutrophils and NETosis in coagulation-mediated diseases.
This review focuses on NETosis and its interactions with both the complement and coagulation systems, placing the three interconnected processes within a broader perspective.
NETosis
NETosis, such as phagocytosis and degranulation, is a component of the neutrophil arsenal that is available for fighting pathogens.1 During NETosis, the neutrophil expels its chromatin, which is decorated with antimicrobial proteins,3 resulting in a sticky NET that can catch and kill pathogens.1, 4 This self-sacrificing strategy has initiated a research field that has advanced in the direction of autoimmunity, since defective clearance of NETs can result in an immune response against DNA, histones, and other intracellular proteins that are associated with NETs.5
The activity of several proteins is thought to be indispensable for the formation of NETs. Neutrophil elastase (NE), myeloperoxidase (MPO), and the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex are all involved in producing reactive oxygen species (ROS) and/or DNA decondensation.6–9 Upon induction of NETosis, the NADPH oxidase complex is activated, leading to the production of superoxide anions, which are converted to hydrogen peroxide. Hydrogen peroxide is a substrate of MPO and induces the release of NE from neutrophil granules. NE subsequently migrates to the nucleus to induce histone degradation, which in turn, leads to DNA decondensation.10 On its way to the nucleus, NE has also been shown to degrade actin filaments, which inhibits the movement of the neutrophils. At later stages, MPO also travels to the nucleus to assist NE in the decondensation of chromatin, but the precise mechanism of action remains unclear.10
When the nuclear membrane disintegrates, the decondensed chromatin can mix with cytosolic and granular proteins of the neutrophil, such as NE and MPO. These components will decorate the chromatin networks once the NET is expelled after cell membrane rupture. The proteins aid in the stability of the NETs11 as well as give the NETs their antibacterial properties.12 MPO produces hypochlorous acid that can kill pathogens, and the serine-protease NE can degrade bacterial proteins. The most abundant protein components of NETs are histones, which are also known to have antibacterial properties.1, 13, 14 However, the antimicrobial properties of NETs are controversial since NETosis-deficient mice and patients deficient in MPO (and thus NETosis) are not more vulnerable to infections than healthy subjects.15, 16
The whole process of NET formation takes approximately 3 h and is lethal to neutrophils. However, in addition to this “suicidal” form of NETosis, “vital” NETosis has also been described. In this process, NETs are formed within half an hour, and the cytoplasmic neutrophils are able to crawl along and retain their antimicrobial functions.17 Vital NETosis is mostly induced by bacterial stimulation and does not require the activity of NADPH oxidase or the formation of ROS species to form NETs.18 This division is only the beginning of discerning the different forms of NETosis, as it appears that each stimulus show different characteristics with respect to chromatin decondensation, requirements for protein activity and protein content of the resulting NET.3, 19–21 All types of NETs share the following features: they are built from chromatin and antibacterial proteins, which are able to interact with the complement and coagulation systems.
In vivo, NETs are degraded by plasma DNases and subsequently cleared by macrophages.22–24 The importance of NET clearance is exemplified by mice that are deficient in DNase 1 and DNase 3, which die within a few days after neutrophil activation due to blood vessel occlusion by the large amount of NETs.22 Defective clearance of NETs has been implicated in the evolution of autoimmune diseases due to exposure to intracellular antigens on NETs.5 In systemic lupus erythematosus (SLE) patients, decreased activity of DNases correlate with the amount of NET complexes in the blood and disease severity.23 In NETosis research, dismantling of NETs with DNase treatment is often used to demonstrate the NET dependency of a process.
The complement system
The complement system, which is the humoral part of the innate immune system, consists of approximately thirty serum proteins. The system is made up of serine-protease cascade reactions involving consecutive cleavages of complement proteins, eventually leading to the formation of the membrane attack complex (MAC). The MAC creates a pore in the cellular membrane where the complement system was activated. Through this pore, metabolites and small proteins can diffuse freely, resulting in lysis of the target cell.25, 26 This process is meant to kill pathogens, but it can also affect host cells if the complement system is not properly controlled.
There are three pathways that can initiate the complement cascade, all of which culminate in the generation of a C3-convertase that cleaves the central component of the complement system, C3.25, 27 The classical pathway is dependent on target-bound antibodies that are bound by C1q binds, which leads to activation of the C3-convertase C2bC4b. The lectin pathway functions in the same way, but it is dependent on the binding of lectins to polysaccharide structures on pathogens. In both pathways, the activated C3-convertase C2bC4b cleaves C3 into C3a and C3b. This cleavage reaction is amplified by a third pathway, called the alternative pathway, which is triggered when the C3b breakdown product is deposited on the surface of a microbe and, together with cleaved Factor B, forms C3bBb, another C3-convertase. The complement Factor properdin or Factor P is the only known complement-stabilizing factor and functions by stabilizing this C3-convertase C3bBb, thereby boosting the alternative pathway.25 The classical, lectin and alternative pathways converge when an additional C3b protein associates with either C2bC4b or C3bBb, which creates a C5-convertase. This convertase can cleave C5 into C5a and C5b, the latter being the starting point for the terminal stages of complement activation. C5b recruits C6 followed by C7 and C8, which insert into the target membrane together. Finally, multiple C9 proteins are recruited to form the pore in the membrane to induce cell lysis.25, 26 The complex of C5b, C6, C7, C8, and C9 is called the MAC or the terminal complement complex, and it is also shortened to the C5b–C9 complex.
In addition to the formation of the MAC complex, the complement system has other functionalities that are used to fight pathogens. C3b, for example, is important in the process of opsonization.25, 27 When a pathogen is covered with deposited C3b (i.e., is opsonized with C3b), it will be recognized by complement receptor 1 (CR1) on neutrophils and other phagocytes, which will subsequently ingest and degrade this pathogen or stimulate NETosis.28 Furthermore, cleavage products C3a and C5a are so-called anaphylatoxins, or danger signals of the immune system. These anaphylatoxins are potent chemoattractants for different types of immune cells, such as neutrophils, and are able to activate them to initiate an immune response at the site of infection.25, 29
Many complement-inhibiting proteins are known to ensure protection of host cells against complement activation. Two of the main complement inhibitors that have gained a great deal of attention in current research are Factor H and Factor I. Factor H acts as a cofactor of Factor I in the inhibition of alternative pathway complement deposition by degrading C3b into iC3b and accelerating the decay of C3bBb.30 Another example is the C4b-binding protein, which can sequester C4b and thereby inhibit the C3-convertases of the classical- and lectin pathways. Additionally, proteins that do not belong to the complement system can inhibit its activation, such as vitronectin. Vitronectin is a protein that binds C5bC6C7 complexes and prevents the formation of an active MAC complex.25 Furthermore, different receptors on host cells have been shown to reduce C3b deposition, such as CD35, CD46, CD55, and CD59.31
The coagulation pathway
After vessel injury, primary hemostasis involves the aggregation of platelets at the damaged tissue, creating a platelet plug. This plug is subsequently strengthened by the coagulation cascade in a process referred to as secondary hemostasis, which entails the formation of fibrin networks around the platelets. The platelet-fibrin clot forms the thrombus that seals the blood vessel to prevent further blood loss and opportunistic infections. Similar to the complement system, the coagulation cascade consists of multiple serine proteases, which ultimately initiate the formation of fibrin fibers. The coagulation cascade can start via two distinct pathways that lead to the cleavage of prothrombin into thrombin.32 In the extrinsic pathway, this is achieved via the exposure of tissue factor (TF), a protein that is normally expressed on cells that are not directly in contact with the blood flow. TF becomes exposed to the blood when a blood vessel is damaged and then can act as a receptor for Factor VII present in blood. Altogether they form a complex that is able to cleave Factor X (FX). In the intrinsic pathway, FX cleavage is accomplished by the consecutive cleavage of factors FXII, FXI, and FIX, with Factor FVIII serving as a cofactor. The initiator of this pathway is Factor FXII, a protein that is activated upon binding to negatively charged surfaces, such as collagen. Factor X, once cleaved and activated (referred to as FXa) via either pathway, is able to cleave prothrombin into thrombin. Thrombin can then cleave fibrinogen into the active fibrin, which can form the strong fibrin fibers that make up a clot. As a negative feedback loop, excess thrombin can activate protein C, which subsequently inhibits thrombin formation and further fibrin cleavage.33 Furthermore, plasmin can degrade fibrin and is, therefore, an important regulator of clotting; it can prevent excessive build-up of fibrin fibers and thereby counteract thrombosis.
The initiator of the intrinsic pathway, FXII, can also bind to negatively charged surfaces on bacteria, such as those rich in lipopolysaccharide (LPS), where it activates the plasma enzyme kallikrein to, in turn, induce the release of the pro-inflammatory peptide bradykinin and together activate FXII as a feed-forward loop.25 As LPS is the major component of the bacterial cell wall, it is thought that bacterial surfaces might induce clot formation. Indeed, fibrin formation has been detected on bacterial surfaces.34 Moreover, some bacteria, such as Staphylococcus aureus, have evolved to induce coagulation to shield themselves from recognition by the immune system.35 These observations suggest that coagulation might also play a role in host defense. Coagulation to entrap pathogens is an evolutionarily conserved mechanism, as clot formation is a well-known host-defense mechanism in invertebrates. In a sense, fibrin clots might resemble NETs because they can capture bacteria and prevent them from spreading. It has been shown that NETs and fibrin fiber structures indeed show some similarities, although NETs have a smaller and finer structure.36 However, bacterial killing via so-called “immunothrombosis” still depends on coagulation-independent mechanisms, such as phagocytosis by immune cells. In contrast to fibrin clots, NETs do possess intrinsic antimicrobial properties, which enable them to not only prevent the spreading of pathogens but also induce cell death.12, 37, 38
It remains to be determined whether coagulation induced by bacteria is biologically relevant or simply reflects bystander effects.25 Furthermore, its biological relevance in vertebrates is unclear, considering the development of the cellular and humoral immune systems.37 There seems to be a large amount of redundancy between the antimicrobial properties of the coagulation system and innate immune system, which might be explained by the long course of evolution.39 It has been proposed that the antimicrobial properties of the coagulation system still exist because they are useful at sites of injury, where coagulation can play a dual role: to seal the wound and to defend against environmental pathogens with easy access to the body at the site of injury. However, for pathogens that have evolved to infect mammals, more sophisticated antibacterial defense mechanisms, such as phagocytosis or NETosis, are needed.
Interplay of the complement system and NETosis
One of the first indications that the complement system influenced NETosis was the finding that neutrophils of C3 knock-out mice did not form NETs.17 Similarly, neutrophils from C3a receptor (C3aR)-deficient mice also did not form NETs.40 When C3 knock-out mice were given exogenous C3-containing serum from healthy mice, the NETotic ability of the knock-out mice was restored, substantiating the importance of the complement system for NETosis.17
Complement activation stimulates NETosis
In response to the defense mechanisms of the complement system, pathogens have evolved strategies to evade the complement system, such as the recruitment of complement Factor H on their surface to prevent C3b opsonization. It was recently shown that complement opsonization not only induces phagocytosis but also aids in inducing NETosis.28 Bacteria that are opsonized with serum IgG are more potent NETosis inducers than non-opsonized bacteria, and effect that is reinforced when the pathogen is pre-incubated with serum instead of IgG only, suggesting that complement C3b opsonization (via classical- or alternative pathway activation) also facilitates NET formation (Fig. 1a). This hypothesis was corroborated by the finding that addition of an antagonist of CR1, which prevents the detection of C3b-opsonized microbes by neutrophils, decreases NETosis.28 Likewise, blocking of complement receptor 3 (CR3), which binds the opsonin iC3b, inhibits NETosis in response to certain pathogens.41, 42 Since this effect was not observed with other pathogens,28 it is likely that multiple complement receptors are needed to detect different pathogens. The ability of a pathogen to induce NETosis might inversely correlate with its ability to evade complement activation and opsonization. A pathogen that can preclude complement deposition is probably a less efficient NET inducer.28
Fig. 1.

Schematic overview of interactions between NETosis and the complement system. a C3b and iC3b opsonization affects NET formation. b C5a induces the upregulation of complement receptors. c C3, Factor B and properdin produce and stabilize C3-convertase on the neutrophilic membrane and in NETs. d Factor H can be recruited to the neutrophil membrane to prevent complement activation. e NETs form a platform on which complement activation can occur. f NET proteins can generate anaphylatoxins C3a and C5a to alarm the immune system
It is not only the opsonizing capacity of the complement system that can enhance NETosis but also the anaphylatoxin C5a (Fig. 1b). C5a can recruit and subsequently prime neutrophils, which results in the upregulation of immune receptors, such as toll-like receptors (TLRs) and/or complement receptors. This process can, in turn, lead to a more vigorous NET response. To exemplify this phenomenon, the induction of NETosis by immune complexes and antibodies is greatly enhanced when neutrophils are first primed with C5a or tumor-necrosis factor (TNF)α.42–44 C5a has also been demonstrated to be a NETosis stimulus itself when interferon gamma is used as a priming cytokine.45
Direct interactions between neutrophils and the complement system are difficult to study in vitro. Serum is mostly used as a source of complement, but it also contains active DNases.46 If neutrophils are stimulated in medium supplemented with (autologous) serum, the endogenous DNases will degrade the formed NETs. Interestingly, binding of C1q to NETs prevents them from being degraded by DNase I. C1q probably shields the DNA from DNases, or alternatively, C1q inhibits DNase I directly.46 This activity of C1q might be relevant for the involvement of NETs in autoimmune diseases, as autoimmune patients often have antibodies against NET components to which C1q can bind. As a result, NETs formed in these patients will be protected from degradation and thus will sustain the pro-inflammatory loop that is characteristic of autoimmune disease. Although this is an attractive model, experiments with C1q, autoantibodies against NETs and DNase I have shown no direct evidence for this concept to date.46
NETs activate the complement pathway
Neutrophils and the complement system act hand in hand to defend the host against invading pathogens. Neutrophils produce complement factors themselves as a weapon in their antimicrobial arsenal, and the release of these factors might activate the complement system on invading pathogens. Activated neutrophils have been shown to express C3, Factor B, and properdin, the three components of the alternative pathway needed to produce and stabilize C3-convertase, which was demonstrated to be functionally active and able to induce the complement cascade47 (Fig. 1c). Treatment of neutrophils with LPS, TNFα, or PMA induces the release of small amounts of properdin, which is deposited on the neutrophil membrane. As a consequence, the cleaved form of C3, C3b, was also found on the surface of neutrophils, indicating complement activation by properdin.30
Conversely, the neutrophil membrane has also been shown to bind Factor H, a well-known inhibitor of the alternative pathway, as described above.48 Factor H selectively binds to CR3 on the neutrophil membrane and remains active once bound because it inhibits C3b deposition48 (Fig. 1d). It is likely that there is a balance between the amounts of Factor H and properdin recruited to the neutrophilic membrane to either activate or inhibit complement, but further research is needed to provide more insight into this issue. It is possible that Factor H is recruited to prevent aberrant complement activation and subsequent MAC complex formation on the neutrophil membrane. The MAC complex has been shown to lyse neutrophils in the presence of antibodies against neutrophilic proteins,49 and, therefore, Factor H may protect neutrophil from this fate. Lysis of neutrophils may also occur in patients with antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV), who often have antibodies against neutrophilic proteins, which in theory can induce complement activation via the classical pathway and subsequently lead to MAC complex activity.50
Complement activation occurs not only on the neutrophil membrane but also on released NETs. Properdin, Factor B and C3 have been found to be deposited on PMA-induced NETs.44, 46, 47 Because MPO and, to a lesser extent, cathepsin G and proteinase 3 can bind to and activate properdin, NETs might form a platform on which complement activation can occur51 (Fig. 1e). Indeed, when isolated NETs were incubated with serum, complement components were consumed and levels of C5a were increased.46 Furthermore, C3b, as well as the assembled MAC complex, were found to be deposited on NETs.47, 48 In all cases, complement activation by and on NETs was strongly decreased when DNase I was added to disrupt the NET structures.46–48
Additionally, Factor H is recruited to NETs, but only when C3b is deposited on the NETs.52 Factor H has been shown to remain active on NETs because it is able to decrease C3b deposition. In addition, Factor H has been shown to inhibit PMA-stimulated NET formation.48, 52 Further research has shown that the deposition of complement proteins on NETs is only partly diminished following the addition of EGTA to inhibit classical and lectin pathway activation,44 indicating that the alternative pathway is also important for complement deposition. Similarly, MAC formation on NETs is also partially inhibited in the presence of antibody against properdin, suggesting the involvement of both the alternative and classical /lectin pathways.47
The biological relevance of complement activation on NETs might be the formation of the anaphylatoxins C3a and C5a, which can further alarm the immune system. However, a more direct effect has also been shown in experiments in which the bacterium Pseudomonas aeruginosa was used to stimulate NETosis in vitro. In this experiment, properdin was not only found on neutrophils and NETs but also on bacteria, which suggests that NETosis can target bacteria for complement activation. However, deposition of the complement proteins C3 and C3b and the MAC complex on the bacterial surface was not observed.47 It is possible that P. aeruginosa is capable of evading complement activation, whereas other bacteria might become opsonized by NET activate complement. Further research should thus be designed to elucidate the biological relevance of properdin binding to the pathogen.
It is not only complement proteins secreted by neutrophils that are responsible for complement activation but also some granular proteins that are present on NETs.30, 51 As early as the seventies, cationic proteins purified from azurophilic granules of neutrophils were shown to cleave C3 and C5 to generate resulting products that can induce the chemotaxis of other leukocytes.53 It was later described that MPO is able to cleave C5 into active fragments, despite probably yielding C5 fragments other than the canonical C5a and C5b.54 Similarly, cathepsin G and possibly also neutrophil elastase can cleave C3 in C3a- and C3b-like proteins.55 This finding strengthens the hypothesis that NETs can activate the complement system to alarm the immune system (Fig. 1f).
The interplay of coagulation and NETosis
Neutrophils have been found abundantly within thrombi of injured mice as well as in thrombi isolated from patients who have had a heart attack, suggesting a role for neutrophils in coagulation in addition to their antibacterial function.56–59 Moreover, neutrophils are the first cells to arrive at sites of endothelial damage, even before platelets, which contribute to the initiation of coagulation. In addition, when neutrophil homing to endothelial cells is inhibited by ICAM-1 or LFA-1 inhibitors, thrombus formation is reduced in mice.4, 60 NETs appear to stimulate thrombosis in a platelet-dependent manner, but they can also stimulate the coagulation cascade directly.61
Interactions of platelets with neutrophils and NETs
Neutrophils and platelets are known to interact and adhere to each other via the glycoprotein Ibα, which can also mediate the adherence of platelets to endothelium and other leukocytes.62 Once activated, platelets expose P-selectin from their α-granules on their membranes, which provides additional means for interactions with leukocytes. Neutrophils recognize P-selectin via the P-selectin glycoprotein ligand-1 (PSGL-1) receptor,63 and this platelet-neutrophil interaction has been shown to facilitate NET formation by neutrophils64, 65 (Fig. 2a). However, in these types of experiments, blocking of P-selectin did not lead to the inhibition of NET formation by neutrophils.58 Further research has identified a factor secreted by platelets that is responsible for the enhancement of NET formation, namely high mobility group box 1 protein (HMGB1), which is involved in nucleosome stabilization and gene expression.58 Attachment of activated platelets to neutrophils might bring them in such close proximity that the secreted HMGB1 is able to activate neutrophils via RAGE, TLR2, and TLR4 receptors, leading to the induction of NETosis66 (Fig. 2b). HMGB1 has also been reported to be present on NETs and thereby to stimulate macrophages.67
Fig. 2.

Schematic overview of interactions between NETosis, platelets, and coagulation. a The interaction between activated platelets and neutrophils. b Platelet-derived HMGB1 can induce NETosis. c Platelets can interact with C3b and histones on NETs, which stimulate the excretion of polyP. d Multiple factors can cause thrombin cleavage on NETs. e NE is able to generate thrombin-derived immune modulatory peptides. f Fibrin fibers strengthened by NETs are less prone to degradation by plasmin
In a simple experiment in which whole blood was perfused over pre-formed NETs, platelets were shown to be recruited to NETs specifically.68 Platelets might bind to C3b deposits on NETs because platelets express CR1 on their membrane.30, 69 The results of other experiments have shown that platelet binding to NETs is dependent on histones.68 To show the effect of histones in vivo, mice were injected with calf thymus histones before injury, and these mice had larger thrombi than those without histone pre-treatment.56 Platelet binding to NETs might lead to platelet aggregation, which is an important step in the formation of clots. Moreover, the histones present on NETs, especially H4, can activate platelets, which in turn, may stimulate NET formation via HMGB1, creating a positive feedback loop (Fig. 2c). Additionally, histones induce platelets to secrete short-chain polyP from α-granules, a compound that activates the extrinsic pathway of blood clotting by activating FXII70, 71 (Fig. 2c). Interestingly, the anticoagulant protein C is able to cleave histones and has been shown to inhibit the cytotoxic effects of histones,72 but whether protein C can also degrade histones on NETs remains unknown. Overall, NETs decorated with platelets may form scaffolds on which thrombus formation can occur. This hypothesis is corroborated by the fact that citrullinated histone 3, a marker for NETosis, has been found in the thrombi of mice and humans.56, 58 Similarly, experiments in mice have demonstrated the formation of smaller thrombi when mice are treated with DNase.56, 62, 73
Activation of the coagulation cascade by NETs
When NETs are perfused with recalcified blood, a gradual increase in fibrinogen attached to the NETs has been seen over time.68, 74 This effect is abrogated following the addition of a thrombin inhibitor, suggesting that NETs can actively form a scaffold on which fibrin formation can occur.68 The exact mechanism by which the coagulation pathway is activated on NETs remains unknown; it has been reported that only the separate components of NETs (DNA and histones) can induce thrombin activation, whereas the same components in complex (nucleosomes and NETs) cannot.70 The selective exposure of TF, the initiator of the extrinsic coagulation pathway, on NETs may provide an explanation.62 Neutrophils treated with cytokines have been shown to upregulate TF mRNA and release TF on their NETs, resulting in the induction of thrombin cleavage.75 However, not all NETs display TF, which may explain why not all experiments show the ability of NETs to initiate coagulation. Whether TF is exposed on NETs seems to be dependent on the stimulus used to induce NETosis. For example, stimulation of neutrophils with E. coli or RA patient sera does not yield TF-decorated NETs, in contrast to stimulation with AAV patient sera.76
Another explanation for the direct activation of coagulation by NETs is that the negatively charged NETs can bind and activate FXII, the starting point of the intrinsic coagulation pathway62 (Fig. 2d). This finding is corroborated by an in vitro study in which thrombin generation in the presence of NETs was reduced upon inhibition of FXII or FXI.61 FXII on NETs has also been shown to induce the activity of kallikrein.77 As fibrinogen has been shown to colocalize on NETs and FX and prothrombin on activated neutrophils,74 it is tempting to conclude that fibrin formation can occur on NETs after initiation of coagulation via either intrinsic or extrinsic pathway activation.
The NET-associated protein NE and, to a minor extent, cathepsin G may also contribute to fibrin formation on NETs since they have been shown to degrade TFPI, the major extrinsic coagulation pathway inhibitor.78 Nucleosomes found at the site of injury are able to recruit TFPI regardless of the presence of NE or cathepsin G, but recruited TFPI is only degraded when these enzymes are present78 (Fig. 2d). Further research should elucidate the source of the TFPI that is recruited to NETs and whether the local concentration of TFPI on NETs is sufficient to induce coagulation in vivo.
Furthermore, NE has been shown to cleave prothrombin directly, resulting in the release of small peptides that exert antibacterial and immunomodulatory effects (Fig. 2e). These prothrombin-derived peptides even exhibit therapeutic properties when administered to mice infected with bacteria.38 These results provide a rationale for the induction of coagulation by NETs: the release of antibacterial molecules as a side effect of coagulation could aid in the elimination of invading pathogens.
The thrombin/fibrin fibrils may also reinforce the NET structure that cages pathogens to prevent their spread. Scanning electron microscopy images of fibrin clots formed in the presence of NETs have shown that both structures are interwoven.35 The NETs form a finer structure within the pores of the larger fibrin structure, which could prevent pathogen escape. However, fibrin clots mixed with NETs are more resistant to fibrinolysis by plasmin, which might be a critical issue in thrombosis-related diseases35 (Fig. 2f). A mouse model of sepsis showed that these NET/thrombin structures stuck to vessel walls in the liver and inhibited perfusion resulting in organ damage, emphasizing the potential downside of the interwoven NETs/clot structures.73
Interplay of the complement system and coagulation
The crosstalk between the complement and coagulation systems has been recognized for years, and there are extensive review articles on this topic.25, 79–81 During the last decade, it has become generally accepted that complement and coagulation cannot be considered separate and independent entities and that they are interconnected and display mutual fine-tuning.37, 82 The crosstalk even extends to proteins that play a role in the regulation of both processes.81 However, most interactions were found in the eighties, and since most experiments were performed in vitro, the in vivo relevance has not been consistently clear.25 The following section focuses on research examining the factors involved in the interplay of the complement and coagulation systems that also affect NETosis.
In C3 knock-out mice, C5a levels were unexpectedly similar to those of wild-type mice,83 although C3b forms the main component of the C5-convertase. Further research has identified thrombin as the main C5-convertase in these knock-out mice because C5a levels are diminished when the knock-out mice are treated with a thrombin inhibitor. Furthermore, these mice exhibit elevated levels of prothrombin and higher thrombin activity in plasma. These findings suggest that thrombin is able to compensate for the C3 deficiency, although the underlying regulatory mechanism is still unknown.83 Indeed, thrombin is able to convert C3 and C5 into the biologically active C3a and C5a fragments in vitro, and these fragments are able to induce chemotaxis and activation of neutrophils.82, 83 Another study has demonstrated that thrombin cleaves C5 at a different site than normal C5 convertases and that the resulting unconventional C5b fragment induces the formation of a more potent MAC complex than the canonical C5b.84 In addition to thrombin, the active forms of FX, FXI, and plasmin, can also generate C3a and C5a, which contributes to the recruitment of neutrophils.82 Plasmin has been shown to degrade C5b, which counteracts C5b deposition and MAC formation.85
Conversely, the complement system also seems to play a role in fibrin formation. Experiments using C3 knock-out mice have revealed that these mice require longer to cease bleeding after cutaneous injury than wild-type mice.86 This effect was abrogated when the C3 knock-out mice were rescued with serum of wild-type mice.87 C5 knock-out mice had normal ceasing times (i.e., the time required to stop initial bleeding), but just like the C3 knock-outs, the frequency of re-bleeding was higher than that in wild-type mice, suggesting a weaker thrombus.86 Furthermore, markedly fewer immune cells were found in the ulcers of C5 knock-out compared with wild-type mice.87 These findings can be explained by reduced complement activation, which leads to decreased recruitment of immune cells and platelets to the site of injury, thus impairing wound healing.86, 87
Platelets can also activate the complement system on their own membranes, either via the alternative or the classical pathway.88 The activated pathway is dependent on the type of stimulation because platelets also contain a C1q inhibitor in their α-granules. When platelet activation induces the exposure of P-selectin from α-granules, C1q inhibitor is also released, leading to inhibition of the classical pathway.63, 88 The biological relevance of complement activation might be related to the clearance of platelets because opsonization with complement factors attracts neutrophils and other phagocytes that can clear the platelets.88
Bacteria have also recognized the importance of both systems and evolved several strategies to evade or (mis)use them.25 The recruitment of complement-inhibiting proteins to avoid complement opsonization or MAC complex formation is a well-known defense mechanism of bacteria.21 Furthermore, bacteria can inhibit clotting to prevent entrapment, or induce clotting to hide from the immune system.34, 36 Interestingly, bacteria also have strategies to avoid being captured in NETs, as many bacteria express DNases on their cell walls, which are able to degrade NETs.13
The triangular relationship between NETosis, complement, and coagulation
At first glance, NETs, coagulation, and complement seem to have their own ‘‘field of expertize.’’ Neutrophils and NET formation are mainly responsible for defense against pathogens, platelets and the coagulation cascade are most important for clot formation and the complement system mainly triggers the immune system to take further action. However, recent research has identified extensive crosstalk between these systems, and it appears that they can act hand in hand to reinforce each other (Fig. 3). Mutual links are known to exist between the coagulation and complement cascades. Their connection with NETosis is a new concept that adds to the complexity of these systems. The NETs can serve as a direct scaffold for both thrombogenesis and complement activation. As a result, NETs are strengthened with fibrin networks to capture pathogens, and the antibacterial properties of NETs are reinforced by the opsonizing and lytic activities of the complement system. Despite being associated with distinct processes, NETs, complement proteins, and coagulation factors can function in a large consortium that protects the host against both hemorrhage and infections. This cooperation is not limited to a site of injury but also occurs within the bloodstream. When the complex interplay is not carefully balanced, complications may arise, such as sepsis, deep vein thrombosis (DVT), autoimmunity, and even cancer.
Fig. 3.
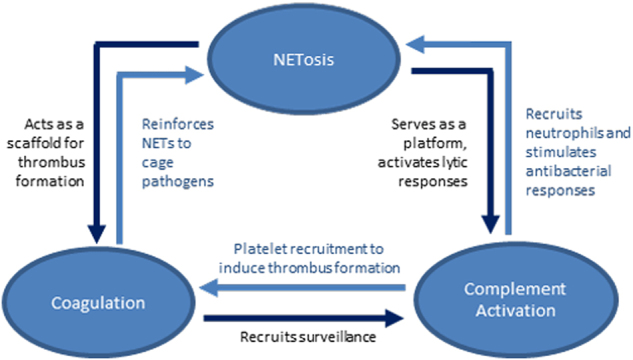
Summary of the interplay between NETosis, complement activation, and coagulation. See text for explanation
In patients with sepsis, complement proteins are almost completely consumed, indicating a tremendous immune activation, which results in the elevated cytokine levels observed in these patients, referred to as the cytokine storm.89 Furthermore, high levels of fibrinogenesis are also found in such patients, but it remains unclear why and how the coagulation cascade is activated.90 Sepsis is mostly induced by microbial infection, which is mainly fought off by neutrophils. Emerging knowledge concerning the neutrophil–complement–coagulation interplay may contribute to our understanding of the phenotype observed in septic patients.
The neutrophil–complement–coagulation system has been shown to play a critical role in DVT, as shown in a newly developed mouse model of DVT in which the interior vena cava of mice is stenosed, leading to a 90% reduction of blood flow that results in hypoxia of the downstream endothelium.62 The epithelium responds by upregulating and exposing P-selectin, which in turn, attracts neutrophils. The neutrophils become activated and release NETs, which recruit platelets and additional immune cells. Furthermore, thrombogenesis is initiated on the NETs, and clots are consequently formed that can initiate thrombosis further downstream in the circulation, very similarly to thrombosis observed in human DVT patients. Less or smaller thrombi are formed when neutrophil homing to the endothelium is blocked, or when NETs are degraded by DNase treatment.56, 62 The complement system is also likely to be activated in this model by both NETs and coagulation cascade products, which might contribute to uncontrolled clot formation.
In a mouse model of spontaneous small intestinal tumorigenesis, the neutrophil-complement-coagulation system was shown to be responsible for tumor growth.39 In this model, the formation of polyps and ulcers compromises the intestinal epithelium, leading to elevated levels of LPS originating from the gut in the bloodstream of these mice. LPS can activate neutrophils directly, but additionally activates the complement system, which in turn, aids in stimulating neutrophils to form NETs via the C3a-C3aR axis. Subsequently, fibrinogenesis is induced on the NETs, leading to thrombus formation. The formed thrombi induce neutrophils to enter a protumorigenic (N2) state, which favors the outgrowth of polyps in the small intestine. Moreover, the N2 neutrophils have reduced effector functions and a propensity to form NETs spontaneously, increasing both hypercoagulation and complement activation, further exacerbating the disease.39 This differential activation of neutrophils in the tumor environment might explain why thromboembolic disease shows a high correlation with the incidence of cancer in the clinic.91
Concluding remarks
It has become clear that NETosis, the complement system and coagulation are not separate and independent functional entities but that intensive interactions between these processes exist. More insight into the interplay among NETosis, complement and coagulation is beginning to emerge, which can facilitate the understanding of the complex processes of wound healing, host defense against pathogens and thrombosis. Furthermore, this interplay provides new insights into the molecular mechanisms of diseases as NET formation has been implicated in various diseases; this also holds true for certain types of tumors that misuse neutrophils and coagulation to create a protumorigenic environment. However, the importance of these NET-related mechanisms in disease is not yet clear, and further research should clarify whether the in vitro findings demonstrating the triangular relationship of NETs, coagulation, and complement are also relevant in vivo. For example, (pre-)clinical studies using DNase as a treatment for diseases, such as sepsis or thrombosis, might support the involvement of NETs in coagulopathies.
Acknowledgements
We would like to acknowledge Johan van der Vlag and Nils Rother for their useful feedback on the manuscript. This work was supported in part by the Dutch Technology Foundation STW.
Competing interests
The authors declare no competing interests.
References
- 1.Brinkmann V, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–1535. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 2.Jorch SK, Kubes P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat. Med. 2017;23:279–287. doi: 10.1038/nm.4294. [DOI] [PubMed] [Google Scholar]
- 3.Urban CF, et al. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog. 2009;5:e1000639. doi: 10.1371/journal.ppat.1000639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McDonald B, Urrutia R, Yipp BG, Jenne CN, Kubes P. Intravascular neutrophil extracellular traps capture bacteria from the bloodstream during sepsis. Cell Host Microbe. 2012;12:324–333. doi: 10.1016/j.chom.2012.06.011. [DOI] [PubMed] [Google Scholar]
- 5.Pinegin B, Vorobjeva N, Pinegin V. Neutrophil extracellular traps and their role in the development of chronic inflammation and autoimmunity. Autoimmun. Rev. 2015;14:633–640. doi: 10.1016/j.autrev.2015.03.002. [DOI] [PubMed] [Google Scholar]
- 6.Fuchs TA, et al. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007;176:231–241. doi: 10.1083/jcb.200606027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Metzler KD, et al. Myeloperoxidase is required for neutrophil extracellular trap formation: implications for innate immunity. Blood. 2011;117:953–959. doi: 10.1182/blood-2010-06-290171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 2010;191:677–691. doi: 10.1083/jcb.201006052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Palmer LJ, et al. Hypochlorous acid regulates neutrophil extracellular trap release in humans. Clin. Exp. Immunol. 2012;167:261–268. doi: 10.1111/j.1365-2249.2011.04518.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Metzler KD, Goosmann C, Lubojemska A, Zychlinsky A, Papayannopoulos V. A myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell Rep. 2014;8:883–896. doi: 10.1016/j.celrep.2014.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pires RH, Felix SB, Delcea M. The architecture of neutrophil extracellular traps investigated by atomic force microscopy. Nanoscale. 2016;8:14193–14202. doi: 10.1039/c6nr03416k. [DOI] [PubMed] [Google Scholar]
- 12.Urban CF, Reichard U, Brinkmann V, Zychlinsky A. Neutrophil extracellular traps capture and kill Candida albicans yeast and hyphal forms. Cell. Microbiol. 2006;8:668–676. doi: 10.1111/j.1462-5822.2005.00659.x. [DOI] [PubMed] [Google Scholar]
- 13.Li P, et al. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J. Exp. Med. 2010;207:1853–1862. doi: 10.1084/jem.20100239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brinkmann V, Zychlinsky A. Neutrophil extracellular traps: is immunity the second function of chromatin? J. Cell Biol. 2012;198:773–783. doi: 10.1083/jcb.201203170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martinod K, et al. PAD4-deficiency does not affect bacteremia in polymicrobial sepsis and ameliorates endotoxemic shock. Blood. 2015;125:1948–1956. doi: 10.1182/blood-2014-07-587709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sorensen OE, et al. Papillon-Lefevre syndrome patient reveals species-dependent requirements for neutrophil defenses. J. Clin. Invest. 2014;124:4539–4548. doi: 10.1172/JCI76009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yipp BG, et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat. Med. 2012;18:1386–1393. doi: 10.1038/nm.2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pilsczek FH, et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J. Immunol. 2010;185:7413–7425. doi: 10.4049/jimmunol.1000675. [DOI] [PubMed] [Google Scholar]
- 19.Kenny EF, et al. Diverse stimuli engage different neutrophil extracellular trap pathways. eLife. 2017;6:e24437. doi: 10.7554/eLife.24437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoppenbrouwers T, et al. In vitro induction of NETosis: Comprehensive live imaging comparison and systematic review. PLoS ONE. 2017;12:e0176472. doi: 10.1371/journal.pone.0176472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khandpur R, et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci. Transl. Med. 2013;5:178ra40. doi: 10.1126/scitranslmed.3005580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jimenez-Alcazar M, et al. Host DNases prevent vascular occlusion by neutrophil extracellular traps. Science. 2017;358:1202–1206. doi: 10.1126/science.aam8897. [DOI] [PubMed] [Google Scholar]
- 23.Hakkim A, et al. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc. Natl Acad. Sci. USA. 2010;107:9813–9818. doi: 10.1073/pnas.0909927107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Farrera C, Fadeel B. Macrophage clearance of neutrophil extracellular traps is a silent process. J. Immunol. 2013;191:2647–2656. doi: 10.4049/jimmunol.1300436. [DOI] [PubMed] [Google Scholar]
- 25.Berends ET, Kuipers A, Ravesloot MM, Urbanus RT, Rooijakkers SH. Bacteria under stress by complement and coagulation. FEMS Microbiol. Rev. 2014;38:1146–1171. doi: 10.1111/1574-6976.12080. [DOI] [PubMed] [Google Scholar]
- 26.Morgan BP. The membrane attack complex as an inflammatory trigger. Immunobiology. 2016;221:747–751. doi: 10.1016/j.imbio.2015.04.006. [DOI] [PubMed] [Google Scholar]
- 27.Joiner KA, Brown EJ, Frank MM. Complement and bacteria: chemistry and biology in host defense. Annu. Rev. Immunol. 1984;2:461–491. doi: 10.1146/annurev.iy.02.040184.002333. [DOI] [PubMed] [Google Scholar]
- 28.Palmer LJ, Damgaard C, Holmstrup P, Nielsen CH. Influence of complement on neutrophil extracellular trap release induced by bacteria. J. Periodontal Res. 2016;51:70–76. doi: 10.1111/jre.12284. [DOI] [PubMed] [Google Scholar]
- 29.Sayegh ET, Bloch O, Parsa AT. Complement anaphylatoxins as immune regulators in cancer. Cancer Med. 2014;3:747–758. doi: 10.1002/cam4.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van der Maten E, et al. Alternative pathway regulation by factor H modulates Streptococcus pneumoniae induced proinflammatory cytokine responses by decreasing C5a receptor crosstalk. Cytokine. 2016;88:281–286. doi: 10.1016/j.cyto.2016.09.025. [DOI] [PubMed] [Google Scholar]
- 31.Camous L, et al. Complement alternative pathway acts as a positive feedback amplification of neutrophil activation. Blood. 2011;117:1340–1349. doi: 10.1182/blood-2010-05-283564. [DOI] [PubMed] [Google Scholar]
- 32.Geddings JE, Mackman N. New players in haemostasis and thrombosis. Thromb. Haemost. 2014;111:570–574. doi: 10.1160/TH13-10-0812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Esmon CT. The regulation of natural anticoagulant pathways. Science. 1987;235:1348–1352. doi: 10.1126/science.3029867. [DOI] [PubMed] [Google Scholar]
- 34.Herwald H, et al. Activation of the contact-phase system on bacterial surfaces--a clue to serious complications in infectious diseases. Nat. Med. 1998;4:298–302. doi: 10.1038/nm0398-298. [DOI] [PubMed] [Google Scholar]
- 35.Thammavongsa V, Kim HK, Missiakas D, Schneewind O. Staphylococcal manipulation of host immune responses. Nat. Rev. Microbiol. 2015;13:529–543. doi: 10.1038/nrmicro3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Varju I, et al. DNA, histones and neutrophil extracellular traps exert anti-fibrinolytic effects in a plasma environment. Thromb. Haemost. 2015;113:1289–1298. doi: 10.1160/TH14-08-0669. [DOI] [PubMed] [Google Scholar]
- 37.Pfeiler S, Stark K, Massberg S, Engelmann B. Propagation of thrombosis by neutrophils and extracellular nucleosome networks. Haematologica. 2017;102:206–213. doi: 10.3324/haematol.2016.142471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schulz C, Engelmann B, Massberg S. Crossroads of coagulation and innate immunity: the case of deep vein thrombosis. J. Thromb. Haemost. 2013;11:233–241. doi: 10.1111/jth.12261. [DOI] [PubMed] [Google Scholar]
- 39.Papareddy P, et al. Proteolysis of human thrombin generates novel host defense peptides. PLoS Pathog. 2010;6:e1000857. doi: 10.1371/journal.ppat.1000857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guglietta S, et al. Coagulation induced by C3aR-dependent NETosis drives protumorigenic neutrophils during small intestinal tumorigenesis. Nat. Commun. 2016;7:11037. doi: 10.1038/ncomms11037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Neeli I, Dwivedi N, Khan S, Radic M. Regulation of extracellular chromatin release from neutrophils. J. Innate Immun. 2009;1:194–201. doi: 10.1159/000206974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Behnen M, et al. Immobilized immune complexes induce neutrophil extracellular trap release by human neutrophil granulocytes via FcgammaRIIIB and Mac-1. J. Immunol. 2014;193:1954–1965. doi: 10.4049/jimmunol.1400478. [DOI] [PubMed] [Google Scholar]
- 43.Huang YM, Wang H, Wang C, Chen M, Zhao MH. Promotion of hypercoagulability in antineutrophil cytoplasmic antibody-associated vasculitis by C5a-induced tissue factor-expressing microparticles and neutrophil extracellular traps. Arthritis Rheumatol. 2015;67:2780–2790. doi: 10.1002/art.39239. [DOI] [PubMed] [Google Scholar]
- 44.Wang H, Wang C, Zhao MH, Chen M. Neutrophil extracellular traps can activate alternative complement pathways. Clin. Exp. Immunol. 2015;181:518–527. doi: 10.1111/cei.12654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Martinelli S, et al. Induction of genes mediating interferon-dependent extracellular trap formation during neutrophil differentiation. J. Biol. Chem. 2004;279:44123–44132. doi: 10.1074/jbc.M405883200. [DOI] [PubMed] [Google Scholar]
- 46.Leffler J, et al. Neutrophil extracellular traps that are not degraded in systemic lupus erythematosus activate complement exacerbating the disease. J. Immunol. 2012;188:3522–3531. doi: 10.4049/jimmunol.1102404. [DOI] [PubMed] [Google Scholar]
- 47.Yuen J, et al. NETosing neutrophils activate complement both on their own NETs and bacteria via alternative and non-alternative pathways. Front. Immunol. 2016;7:137. doi: 10.3389/fimmu.2016.00137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schneider AE, Sandor N, Karpati E, Jozsi M. Complement factor H modulates the activation of human neutrophil granulocytes and the generation of neutrophil extracellular traps. Mol. Immunol. 2016;72:37–48. doi: 10.1016/j.molimm.2016.02.011. [DOI] [PubMed] [Google Scholar]
- 49.Romero V, et al. Immune-mediated pore-forming pathways induce cellular hypercitrullination and generate citrullinated autoantigens in rheumatoid arthritis. Sci. Transl. Med. 2013;5:209ra150. doi: 10.1126/scitranslmed.3006869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kessenbrock K, et al. Netting neutrophils in autoimmune small-vessel vasculitis. Nat. Med. 2009;15:623–625. doi: 10.1038/nm.1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.O’Flynn J, Dixon KO, Faber Krol MC, Daha MR, van Kooten C. Myeloperoxidase directs properdin-mediated complement activation. J. Innate Immun. 2014;6:417–425. doi: 10.1159/000356980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Halder LD, et al. Factor H binds to extracellular DNA traps released from human blood monocytes in response to Candida albicans. Front. Immunol. 2016;7:671. doi: 10.3389/fimmu.2016.00671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Venge P, Olsson I. Cationic proteins of human granulocytes. VI. Effects on the complement system and mediation of chemotactic activity. J. Immunol. 1975;115:1505–1508. [PubMed] [Google Scholar]
- 54.Vogt W. Complement activation by myeloperoxidase products released from stimulated human polymorphonuclear leukocytes. Immunobiology. 1996;195:334–346. doi: 10.1016/S0171-2985(96)80050-7. [DOI] [PubMed] [Google Scholar]
- 55.Maison CM, Villiers CL, Colomb MG. Proteolysis of C3 on U937 cell plasma membranes. Purification of cathepsin G. J. Immunol. 1991;147:921–926. [PubMed] [Google Scholar]
- 56.Brill A, et al. Neutrophil extracellular traps promote deep vein thrombosis in mice. J. Thromb. Haemost. 2012;10:136–144. doi: 10.1111/j.1538-7836.2011.04544.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.de Boer OJ, et al. Neutrophils, neutrophil extracellular traps and interleukin-17 associate with the organisation of thrombi in acute myocardial infarction. Thromb. Haemost. 2013;109:290–297. doi: 10.1160/TH12-06-0425. [DOI] [PubMed] [Google Scholar]
- 58.Maugeri N, et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J. Thromb. Haemost. 2014;12:2074–2088. doi: 10.1111/jth.12710. [DOI] [PubMed] [Google Scholar]
- 59.Grasso S, et al. Interaction of factor VII activating protease (FSAP) with neutrophil extracellular traps (NETs) Thromb. Res. 2017;161:36–42. doi: 10.1016/j.thromres.2017.11.012. [DOI] [PubMed] [Google Scholar]
- 60.Darbousset R, et al. Tissue factor-positive neutrophils bind to injured endothelial wall and initiate thrombus formation. Blood. 2012;120:2133–2143. doi: 10.1182/blood-2012-06-437772. [DOI] [PubMed] [Google Scholar]
- 61.Gould TJ, et al. Neutrophil extracellular traps promote thrombin generation through platelet-dependent and platelet-independent mechanisms. Arterioscler. Thromb. Vasc. Biol. 2014;34:1977–1984. doi: 10.1161/ATVBAHA.114.304114. [DOI] [PubMed] [Google Scholar]
- 62.von Bruhl ML, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J. Exp. Med. 2012;209:819–835. doi: 10.1084/jem.20112322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ma AC, Kubes P. Platelets, neutrophils, and neutrophil extracellular traps (NETs) in sepsis. J. Thromb. Haemost. 2008;6:415–420. doi: 10.1111/j.1538-7836.2007.02865.x. [DOI] [PubMed] [Google Scholar]
- 64.Clark SR, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007;13:463–469. doi: 10.1038/nm1565. [DOI] [PubMed] [Google Scholar]
- 65.Pieterse E, Rother N, Yanginlar C, Hilbrands LB, van der Vlag J. Neutrophils discriminate between lipopolysaccharides of different bacterial sources and selectively release neutrophil extracellular traps. Front. Immunol. 2016;7:484. doi: 10.3389/fimmu.2016.00484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stark K, et al. Disulfide HMGB1 derived from platelets coordinates venous thrombosis in mice. Blood. 2016;128:2435–2449. doi: 10.1182/blood-2016-04-710632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Peng HH, et al. Mineral particles stimulate innate immunity through neutrophil extracellular traps containing HMGB1. Sci. Rep. 2017;7:16628. doi: 10.1038/s41598-017-16778-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fuchs TA, et al. Extracellular DNA traps promote thrombosis. Proc. Natl Acad. Sci. USA. 2010;107:15880–15885. doi: 10.1073/pnas.1005743107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hamzeh-Cognasse H, et al. Platelets and infections—complex interactions with bacteria. Front. Immunol. 2015;6:82. doi: 10.3389/fimmu.2015.00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Noubouossie DF, et al. In vitro activation of coagulation by human neutrophil DNA and histone proteins but not neutrophil extracellular traps. Blood. 2017;129:1021–1029. doi: 10.1182/blood-2016-06-722298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Semeraro F, et al. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood. 2011;118:1952–1961. doi: 10.1182/blood-2011-03-343061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Xu J, et al. Extracellular histones are major mediators of death in sepsis. Nat. Med. 2009;15:1318–1321. doi: 10.1038/nm.2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McDonald B, et al. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood. 2017;129:1357–1367. doi: 10.1182/blood-2016-09-741298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Healy LD, et al. Colocalization of neutrophils, extracellular DNA and coagulation factors during NETosis: Development and utility of an immunofluorescence-based microscopy platform. J. Immunol. Methods. 2016;435:77–84. doi: 10.1016/j.jim.2016.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kambas K, et al. Autophagy mediates the delivery of thrombogenic tissue factor to neutrophil extracellular traps in human sepsis. PLoS ONE. 2012;7:e45427. doi: 10.1371/journal.pone.0045427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kambas K, et al. Tissue factor expression in neutrophil extracellular traps and neutrophil derived microparticles in antineutrophil cytoplasmic antibody associated vasculitis may promote thromboinflammation and the thrombophilic state associated with the disease. Ann. Rheum. Dis. 2014;73:1854–1863. doi: 10.1136/annrheumdis-2013-203430. [DOI] [PubMed] [Google Scholar]
- 77.Oehmcke S, Morgelin M, Herwald H. Activation of the human contact system on neutrophil extracellular traps. J. Innate Immun. 2009;1:225–230. doi: 10.1159/000203700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Massberg S, et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat. Med. 2010;16:887–896. doi: 10.1038/nm.2184. [DOI] [PubMed] [Google Scholar]
- 79.Markiewski MM, Nilsson B, Ekdahl KN, Mollnes TE, Lambris JD. Complement and coagulation: strangers or partners in crime? Trends Immunol. 2007;28:184–192. doi: 10.1016/j.it.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 80.Lupu F, Keshari RS, Lambris JD, Coggeshall KM. Crosstalk between the coagulation and complement systems in sepsis. Thromb. Res. 2014;133:S28–S31. doi: 10.1016/j.thromres.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Conway EM. Reincarnation of ancient links between coagulation and complement. J. Thromb. Haemost. 2015;13:S121–S132. doi: 10.1111/jth.12950. [DOI] [PubMed] [Google Scholar]
- 82.Amara U, et al. Molecular intercommunication between the complement and coagulation systems. J. Immunol. 2010;185:5628–5636. doi: 10.4049/jimmunol.0903678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Huber-Lang M, et al. Generation of C5a in the absence of C3: a new complement activation pathway. Nat. Med. 2006;12:682–687. doi: 10.1038/nm1419. [DOI] [PubMed] [Google Scholar]
- 84.Krisinger MJ, et al. Thrombin generates previously unidentified C5 products that support the terminal complement activation pathway. Blood. 2012;120:1717–1725. doi: 10.1182/blood-2012-02-412080. [DOI] [PubMed] [Google Scholar]
- 85.Barthel D, Schindler S, Zipfel PF. Plasminogen is a complement inhibitor. J. Biol. Chem. 2012;287:18831–18842. doi: 10.1074/jbc.M111.323287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Subramaniam S, et al. Distinct contributions of complement factors to platelet activation and fibrin formation in venous thrombus development. Blood. 2017;129:2291–2302. doi: 10.1182/blood-2016-11-749879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rafail S, et al. Complement deficiency promotes cutaneous wound healing in mice. J. Immunol. 2015;194:1285–1291. doi: 10.4049/jimmunol.1402354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Peerschke EI, Yin W, Ghebrehiwet B. Complement activation on platelets: implications for vascular inflammation and thrombosis. Mol. Immunol. 2010;47:2170–2175. doi: 10.1016/j.molimm.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.de Jong HK, van der Poll T, Wiersinga WJ. The systemic pro-inflammatory response in sepsis. J. Innate Immun. 2010;2:422–430. doi: 10.1159/000316286. [DOI] [PubMed] [Google Scholar]
- 90.Levi M, Schultz M, van der Poll T. Sepsis and thrombosis. Semin. Thromb. Hemost. 2013;39:559–566. doi: 10.1055/s-0033-1343894. [DOI] [PubMed] [Google Scholar]
- 91.Demers M, et al. Cancers predispose neutrophils to release extracellular DNA traps that contribute to cancer-associated thrombosis. Proc. Natl Acad. Sci. USA. 2012;109:13076–13081. doi: 10.1073/pnas.1200419109. [DOI] [PMC free article] [PubMed] [Google Scholar]