

Abstract
Mesangial proliferative glomerulonephritis (MsPGN), one of the most common glomerulonephritis pathological types, often leads to end-stage renal disease over a prolonged period. But the current treatment of MsPGN is non-specific and causes serious side effects, thus novel therapeutics and targeting strategies are urgently demanded. By combining the advantages of PEG-PLGA nanoparticles and the size selection mechanism of renal glomerulus, we designed and developed a novel PEG-PLGA nanoparticle delivery system capable of delivering dexamethasone acetate (A-DEX) into glomerular mesangium. We determined that 90 nm was the optimum size to encapsulate A-DEX for glomerular mesangium targeting based on the size-selection mechanism of glomerulus. After intravenous administration in rats, 90 nm DiD-loaded NPs were found to accumulate to a greater extent in the kidney and kidney cortex compared with the free DiD solution. The 90 nm A-DEX NPs are also more stable at room temperature and showed a sustained release pattern. In rat glomerular mesangial cells (HBZY-1) in vitro, we found that the uptake of 90 nm A-DEX NPs was both temperature-dependent and energe-dependent, and they were mostly engulfed via clathrin-dependent endocytosis pathways. In summary, we have successfully developed a glomerular mesangium-targeted PEG-PLGA NPs, which is potential for the treatment of MsPGN.
Keywords: Glomerulonephritis, Mesangium, Mesangial cells, PEG-PLGA nanoparticles, Dexamethasone acetate
Introduction
Glomerulonephritis is a collection of primary renal disorders and disorders secondary to a systemic disease. It is associated with increased morbidity and mortality. Mesangial proliferative glomerulonephritis (MsPGN) is the most common pathological type of glomerulonephritis and is linked primarily to the mesangium. Because of the underlying immunologic nature of these disorders, consisting of immune reactions and immune factors, common clinical therapies often use high doses of glucocorticoid in combination with cytotoxic agents [1]. These therapeutic regimes are non-specific and have serious side effects, so new and effective targeted therapeutic strategies are highly demanded [2].
The glomerulus resides within the renal corpuscle, the most proximal portion of the nephron within the kidney cortex, and consists of a tuft of blood capillaries and support tissue (the mesangium). The blood vessel endothelium of the glomerulus is fenestrated, with pores that are 80–150 nm in diameter. The glomerular basement membrane (GBM), which is next to the endothelium, is a 300 nm thick connective-tissue membrane rich in heparan sulfate (with a pore size of 3 nm) [3]. Behind the GBM lie the podocytes, which form filtration slits that are 32 nm wide [4]. Altogether, the glomerular endothelium, the GBM, and podocytes form the glomerular filtration apparatus, which possesses an effective size cutoff of 10 nm and facilitates the rapid renal clearance of small molecules and nanoparticles (NPs) [5]. Inside of the glomerulus, without the blockade of the GBM and podocytes, the mesangium (mesangial cells and extracellular matrix) is connected directly to the fenestrated endothelium (Fig. 1).
Fig. 1.
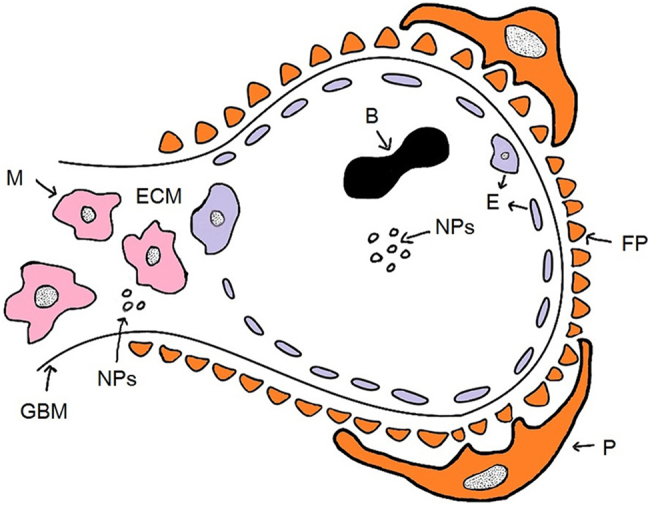
A schematic overview of the composition of the glomerulus and the anticipated pathway of NPs. B blood cell, NPs nanoparticles, GBM glomerular basement membrane, E endothelium, P podocyte, FP foot processes, M mesangial cell, ECM extracellular matrix
Because of this special renal anatomy, NPs of a certain size can accumulate in the mesangium after they pass the endothelium through the pores. Shimizu et al. discovered that PIC nanocarriers 10–20 nm in diameter, in contrast to HVJ-E vectors (200–500 nm in diameter), accumulated most in glomeruli, particularly in the mesangium [6]. Choi et al. examined the distribution of 10–150 nm poly (ethylene glycol) (PEG) gold NPs in the kidney after a systemic injection and found an optimal size of 80 nm for bulk particle uptake in the mesangium [7]. Pollinger et al. reported that PEG-coated Qdots (hydrodynamic diameter of 20 nm) showed a pronounced enrichment in the kidney mesangium upon intravenous application [8]. These results all confirm that the size of NPs is indeed critical for achieving mesangium targeting.
The FDA-approved poly (D, L-lactic-co-glycolic acid) (PLGA)-derived NPs are excellent choices for drug delivery with controlled release ability. The particle matrix will slowly degrade in vivo, and the by-products, such as lactic acid and glycolic acid, can be quickly metabolized and excreted [9]. On the other hand, functionalizing PLGA NPs with PEG is often beneficial, as a protective PEG layer can prevent opsonization and reduce systemic clearance [10]. PEG-PLGA NPs are thus widely used to encapsulate various cancer-related drugs [11, 12]. However, their usage in kidney disease has seldom been reported.
In this study, we aimed to combine the advantages of PEG-PLGA NPs and the size selection mechanism of glomerulus. We designed and developed a novel PEG-PLGA NP delivery system capable of delivering drugs into glomerular mesangium. We prepared PEG-PLGA NPs loaded with dexamethasone acetate (A-DEX) as a model drug and identified the optimal particle size. Biodistribution experiments showed that our A-DEX NPs targeted the kidney and kidney cortex efficiently. We also investigated the release behavior of our A-DEX NPs and further analyzed their uptake using cultured glomerular mesangial cells (HBZY-1).
Materials and methods
Materials
Dexamethasone acetate was purchased from Dalian Melone Biological Technology Co. Ltd. (Dalian, China). Poly (ethylene glycol)-poly (D, L-lactide-co-glycolide 50/50) of molecular weight 45,000–75,000 Da was purchased from the University of Electronic Science and Technology of China (Chengdu, China). Deoxycholic acid sodium salt was provided by Amresco Co. (Boise, ID, USA). Solutol HS15 was obtained from BASF Aktiengesellschaft (Ludwigshafen, Germany). 1, 1-Dioctadecyl-3, 3, 3, 3-tetramethylindodicarbocyanine (DiD) was provided by Sigma-Aldrich (St. Louis, MO, USA). DMEM (low glucose) medium was purchased from HyCloneTM Co. (Logan, UT, USA). Penicillin/streptomycin was obtained from Solarbio Science & Technology Co. Ltd. (Beijing, China). Fetal bovine serum and trypsin-EDTA were provided by Gibco BRL (Carlsbad, CA, USA). The BCA kit was from Thermo Fisher Scientific Inc. (Waltham, MA, USA). Double-distilled water was purified using a Millipore Simplicity System (Bedford, MA, USA). All other chemicals used were of reagent grade.
Cell lines and animals
The glomerular mesangial cell line HBZY-1 was purchased from Jennio Biotech Co. Ltd. (Guangzhou, China). HBZY-1 cells were cultured in DMEM supplemented with 10% FBS and 1% (v/v) penicillin/streptomycin at 37 °C in a 5% CO2 environment.
Sprague–Dawley (SD) rats (200 ± 20 g) were obtained from the Experimental Animal Center of Sichuan University (Chengdu, China). The rats were fasted overnight with free access to water before experiments. All animal experiments met the requirements of the National Act on the Use of Experimental Animals (People’s Republic of China) and were approved by Sichuan University’s animal ethics committee (Chengdu, China).
Sample preparation and LC–MS/MS analysis
Liquid–liquid extraction was used for sample pretreatment. Cell digests, tissue homogenates, and plasma samples (0.1 mL) were vortexed with 1 mL ethyl acetate for 5 min and then centrifuged at 7104 × g for 10 min. The supernatant was transferred to a disposable tube and evaporated to dryness under a stream of nitrogen at 45 °C. The residue was redissolved in 0.1 mL methanol, vortexed for 5 min to facilitate the complete dissolution of the residues, and filtered with a 0.22 μm filter membrane before sample injection.
Samples were analyzed by LC-MS/MS (Agilent Technologies Co. Ltd.; Palo Alto, CA, USA). The analysis was carried on an Agilent 1200 series RRLC system with Agilent triple-quadrupole MS. A Diamonsil ODS column (1.8 μm, 50 mm × 4.6 mm) with the corresponding guard column (ODS, 5 μm) was used to separate particles, and it was maintained at 30 °C. The mobile phase consisted of 0.1% formic acid and methanol (35:65, v/v), and the flow rate was 0.4 mL/min. The injection volume was 1 μL. Quantification was performed using selective ion monitoring and multiple reaction monitoring for dexamethasone acetate and dexamethasone, respectively, combined with positive ion modes. The [M+K]+ m/z 473.05 was for dexamethasone acetate and [M+H]+ m/z 393.3 → 354.0 for dexamethasone.
Preparation of dexamethasone acetate-loaded PEG-PLGA NPs
A-DEX NPs were prepared with an emulsion/solvent evaporation method as described previously [13]. First, 20 mg PEG-PLGA copolymer and 2 mg dexamethasone acetate were dissolved in 1 mL chloroform, forming the oil phase, which was then added into an aqueous solution of 10 mL 0.5% deoxycholic acid sodium salt and HS 15. To obtain different sized NPs, the mixture was then emulsified by sonication with a probe sonicator (Xinyi Ultrasonic Equipment Co. Ltd.; Ningbo, China) at varying power in an ice-water bath. Later, chloroform was evaporated at low pressure at 40 °C using a Büchi Rotavapor R-114 (Büchi, Switzerland). NPs of 70, 90, and 110 nm in diameter were obtained using 0.45 μm filter membranes. DiD-loaded NPs were prepared with the same procedure but with the addition of 0.005% (w/v) DiD for encapsulation. The obtained NPs were subjected to a 1.5 × 20 cm Sephadex G50 column and eluted with distilled water to remove the unentrapped DiD.
Characterization of the NPs
Particle size
The particle sizes of A-DEX NPs were measured in water by photon correlation spectroscopy (Malvern Zetasizer Nano ZS90, UK) at 25 °C, using dynamic light scattering (DLS) and electrophoretic light scattering technology.
Surface morphology
A transmission electron microscope (H-600, Hitachi, Japan) was used for the morphological examination of the NPs. The A-DEX NPs were stained with 2% (w/v) phosphotungstic acid for 30 s, and TEM was used to examine the morphology of the NPs.
Colloidal stability
To compare the stability of NPs of 70, 90, and 110 nm at room temperature, variations in the size of the NPs were assessed by DLS at predetermined time points (0.5, 1, 2, 4, 8, and 12 h) at 25 °C.
NP localization in rat kidney
We prepared DiD-NPs of different sizes (70, 90, and 110 nm in diameter). The optimum size of NPs for accumulation in rat kidney was selected by an in vivo imaging system (IVIS) (Caliper; Hopkinton, MA, USA). The SD rats were randomly divided into four groups (n = 5) and intravenously (i.v.) administered free DiD solution or DiD-NPs of different sizes at a dose of 50 μg/kg DiD. After 1 h, blood samples were collected from the oculi chorioideae vein into 0.5 mL heparinized Eppendorf tubes and centrifuged at 2000 × g for 5 min to obtain plasma. Rats were scarified to collect tissues (heart, liver, spleen, lung, and kidney). We further investigated the renal distribution of the optimal NPs at different time points (15 min, 30 min, 1 h, 2 h, and 4 h).
Immunofluorescence
The optimal NPs, loaded with DiD, were administered to SD rats (n = 5) for immunofluorescence of renal tissues. The renal tissues were obtained at 1 h after tail vein injection and fixed in 10% formaldehyde. Sections (3 μm) were stained with the OX-7 monoclonal antibody (Bio-Rad, Hercules, CA, USA) to the mesangial marker thymic antigen 1 (Thy1), which were then detected by secondary antibody (Thermo Fisher Scientific Inc.), and were observed under a confocal laser scanning microscope (Leica, Germany).
Tissue biodistribution
To determine whether i.v.-administered 90 nm A-DEX NPs could accumulate in the kidney, rats were used to study the distribution of 90-nm A-DEX NPs and A-DEX solution. Rats were randomly divided into ten groups (n = 5) and injected with A-DEX solution or 90 nm A-DEX NPs at an A-DEX dose of 1 mg/kg. At the prescribed times (0.25, 0.5, 1, 2, and 4 h), we obtained blood samples, and then rats were scarified to collect tissues (heart, liver, spleen, lung, kidney, and kidney cortex). The blood was collected to obtain plasma as above, and tissues were homogenized with normal saline (0.9% NaCl) (of twice the weight of tissues) for quantitative analysis of A-DEX by LC-MS/MS.
In vitro drug release
After the most appropriate NP size was found, we used this size of NP to evaluate the behavior of A-DEX release from the NPs. We carried out in vitro drug release in acetate buffer solution (ABS, pH = 4.5) containing 0.5% Tween 80 (w/v) using a dialysis method. Briefly, dialysis bags (MWCO = 3000 Da) containing 1 mL A-DEX NPs and A-DEX solution were separately submerged in 25 mL release medium with stirring at 100 r/min 37 °C. At designated time points (0.25, 0.5, 1, 2, 4, 8, 12, 24, 36, 48, and 72 h), 1 mL aliquots of the solution were withdrawn to determinate the released drug, and an equal volume of fresh buffer was added to keep the volume constant and to ensure sink conditions throughout the whole experiment. This was taken into account while calculating the cumulative drug release. These samples were filtered using 0.22-μm filter membranes, and A-DEX was determined by HPLC.
Cell experiments
Cellular uptake
For the qualitative experiment, the ability of the cells to internalize A-DEX solution and A-DEX NPs (10 μg/mL) was determined. In cellular uptake studies, cells were seeded in 6-well plates at a density of 1 × 104 cells per well and incubated for 24 h at 37 °C. The cells were incubated with A-DEX solution or A-DEX NPs for 0.25, 0.5, 1, or 2 h. After the incubation, cells were washed with ice-cold PBS three times and subjected to five freeze-thawing cycles following detachment from plates. The protein concentration of cells was determined using a BCA protein assay kit. Cellular uptake efficiency was evaluated by the uptake index (UI, ng/mg) [14], which was calculated by Eq. (1).
| 1 |
For energy depletion, the cellular uptake was performed at 4 or 37 °C or after pretreatment with NaN3 for 1 h. After that, cellular uptake efficiency was measured as mentioned above.
Mechanism of cellular uptake of A-DEX NPs
To explore the mechanism of A-DEX NP-mediated internalization, HBZY-1 cells were pre-incubated with the indicated inhibitors, including chlorpromazine (Chlo, 10 μg/mL), nystatin (10 μg/mL), and dimethyl amiloride (DMA, 10 μg/mL), for 1 h before adding A-DEX NPs (10 μg/mL) for another 1 h. The ensuing procedures were similar to those above.
Statistical analysis
All data are presented as the mean ± standard deviations (SD). Student’s t-test was used to assess the statistical significance of differences between two groups. A P < 0.05 was considered statistically significant, and a P < 0.01 was considered highly significant.
Results
Characterization of PEG-PLGA NPs
DLS results show that the average hydrodynamic diameters of our prepared PEG-PLGA NPs had narrow peaks (Fig. 2) [13]. TEM images of these NPs (70, 90, and 110 nm in diameter) also displayed a spherical shape and a smooth surface.
Fig. 2.

Size distribution of 70, 90, and 110 nm A-DEX NPs. Left: size distribution of A-DEX NPs detected by DLS. Right: TEM of A-DEX NPs. Scale bars: 100 nm
We monitored the NPs at various time points in vitro after preparation. We discovered that 70 nm NPs were the most unstable and became inhomogeneous at 2 h; the 110 nm NPs expanded at 8 h, and the solution gradually grew turbid. In comparison, the average size of 90 nm NPs remained unchanged over 12 h (Fig. 3).
Fig. 3.
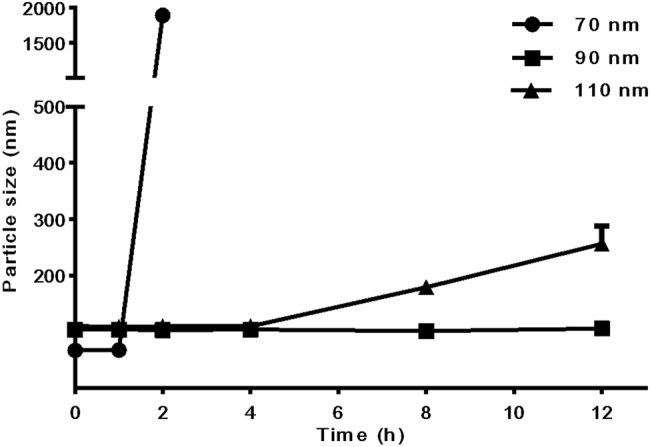
The variations in particle sizes of 70, 90, and 110 nm nanoparticles at 25 °C (mean ± SD)
Ninety nanometer PEG-PLGA NPs accumulate in rat kidneys
To find the most appropriate size for kidney targeting, DiD-NPs with sizes of 70, 90, and 110 nm were intravenously injected into rats separately, and IVIS was employed to visualize the distribution. Compared with free DiD, the fluorescence intensity of DiD-NPs increased in all imaged organs (Fig. 4a). The signal strengths of DiD-NPs with different sizes were significantly different in the kidney, while the majority of DiD signals were found in organs other than the kidney (Fig. 4b). PEG-PLGA NPs 90 nm in size showed much stronger kidney localization. The 90 nm DiD-NP-treated group also displayed a significantly higher DiD level than the free DiD group at all tested time points up to 4 h after administration (Fig. 4c). In summary, 90 nm was the optimal size for renal targeting by PEG-PLGA NPs.
Fig. 4.

Ex vivo imaging of DiD-NPs in the main organs and plasma. a Rats were intravenously injected with DiD solution or DiD-NPs of 70, 90, or 110 nm. Blood was collected and organs were harvested 1 h after the administration. b NP accumulation in kidneys. c Imaging of kidneys at different time points after injection of 90-nm DiD-NPs (15 min, 30 min, 1 h, 2 h, and 4 h)
NPs deposit within the glomerular mesangium
To determine whether i.v.-administered 90 nm NPs deposit in the mesangium, we counter-stained renal sections from rats that had received DiD-containing NPs with OX-7, a mesangial marker (Fig. 5). We observed co-localization of DiD and OX-7 signals 1 h after administration. These data suggest that our 90 nm NPs accumulated primarily in the mesangium after administration.
Fig. 5.

Immunofluorescence image of a rat glomerulus 1 h after administration of 90 nm DiD-NPs. Renal sections were co-stained for thymic antigen 1 (Thy1) mesangial marker with the OX-7 monoclonal antibody. Original magnification 400×. Scale bars: 20 μm
Biodistribution of 90 nm A-DEX NPs in rats
To gain a deeper understanding of the targeting effect of our NPs loaded with A-DEX, we further investigated their biodistribution in rats. Preliminary experiments found that both our A-DEX NPs and free A-DEX quickly degraded into dexamethasone (DEX) after injection into rats (data not shown). Therefore, DEX levels were used for the consequent biodistribution analyze.
At 1 h after administration, 90 nm A-DEX NPs showed biodistribution profiles different from those of A-DEX solution. The kidney was the main site of NP accumulation, and the kidney cortex had the highest NP level (Fig. 6). In the lung, spleen, and heart, the DEX level difference was non-significant. In the plasma and especially in the liver, the DEX level was actually reduced in the NP group. This significantly lower DEX level of the NP group in the liver may be attributed to the PEG chain, which might substantially reduce non-specific interactions with proteins through its hydrophilic and steric-repulsion effects, thus reducing opsonization and phagocytosed in the liver [15]. The AUC(0→t) of the 90 nm A-DEX NP-treated group in the kidney and kidney cortex was 1.27-fold and 1.37-fold higher than that of the A-DEX solution-treated group, respectively (Fig. 7), indicating that NPs with certain sizes could passively target the kidneys.
Fig. 6.

Biodistribution of 90 nm A-DEX NPs and A-DEX solution in rats at 1 h post-injection. The kidney and kidney cortex concentrations of DEX in the 90 nm A-DEX NP-treated group were, respectively, 1.30-fold and 1.45-fold higher than those in the A-DEX solution-treated group. Data represent the mean ± SD (n = 5). ##P < 0.01
Fig. 7.

Concentration–time curve of DEX in the kidney (a) and kidney cortex (b) after the intravenous injection of 90 nm A-DEX NPs or A-DEX solution in rats. The AUC (0→t) (μg/L*h) of 90 nm A-DEX NPs or A-DEX solution in the kidney was 1441.15 ± 65.41 or 1134.94 ± 56, respectively. The AUC (0→t) (μg/L*h) of 90-nm A-DEX NPs or A-DEX solution in the kidney cortex was 1567.40 ± 32.57 or 1140.99 ± 36.26, respectively. Data for each time point are expressed as the mean ± SD (n = 5). #P < 0.05
In conclusion, 90 nm A-DEX NPs can accumulate in the kidney and especially the kidney cortex after injection into rats. The distribution pattern of DEX in rats is similar to that of DiD, which shows the consistency of our method.
In vitro drug release
As dexamethasone acetate has an ester bond that is prone to degradation by hydrolyzation, we first studied the stability of dexamethasone acetate in PBS (pH = 7.4). The results revealed that 41.02% of the original drug was indeed degraded at 72 h (Fig. 8a). However, NPs in vivo will be phagocytosed by phagocytic cells and peripheral giant cells, where an acidic environment will be present [16]. In this regard, ABS (pH = 4.5) was selected as the in vitro release medium for modeling the release in vivo. Dexamethasone acetate was stable in ABS up to 72 h (Fig. 8a). Hence, ABS was used in subsequent investigations.
Fig. 8.

In vitro A-DEX release from 90 nm A-DEX NPs. a The stability of A-DEX in PBS (pH = 7.4) and ABS (pH = 4.5). b Cumulative release of A-DEX from A-DEX NPs and A-DEX solution. Data represent the mean ± SD (n = 3)
The release profile of 90 nm A-DEX NPs in ABS is shown in Fig. 8b. It can be seen that the release rate of A-DEX from prepared NPs was much slower than that from free A-DEX solution. The release behavior of A-DEX from the NPs exhibited a biphasic pattern, including an initial burst-release phase caused by fast A-DEX release from the surface of NPs, followed by a sustained release of the remaining drug. Only 32.09% of A-DEX was released from the 90 nm A-DEX NPs during the first 4 h, while 71.91% of A-DEX was released from A-DEX solution at the same time. These findings show that the 90 nm A-DEX NPs had a good sustained-release pattern.
Uptake pathway of the 90-nm A-DEX NPs
To function as an efficient drug or delivery vehicle, NPs generally must enter the cells effectively and release their payload into the cytoplasm. Cellular uptake experiments were performed using HBZY-1 rat mesangial cells. The results show that cellular uptake of 90 nm A-DEX NPs was significantly higher than that of free A-DEX at all time points measured. The uptake level increased rapidly with incubation time as well: it was 1.53, 1.47, 1.88, and 1.76 times that of the control at 15 min, 30 min, 1 h, and 2 h, respectively (Fig. 9a).
Fig. 9.

a Cellular uptake of 90 nm A-DEX NPs over time at 37 °C compared with that of A-DEX solution. #P < 0.05, ##P < 0.01. b Cellular uptake of A-DEX solution or 90-nm A-DEX NPs at 4 °C, at 37 °C, or in the presence of NaN3. The cellular uptake of 90-nm A-DEX NPs at 37 °C was set as 100%. #P < 0.05, ##P < 0.01. c Cellular uptake efficiency of 90 nm A-DEX NPs with inhibitors, including nystatin, chlorpromazine, and dimethyl amiloride. The cellular uptake of 90-nm A-DEX NPs without any treatment served as the control. Data represent the mean ± SD (n = 3). ##P < 0.01
Based on these results, we investigated the mechanisms of the excellent HBZY-1 cell delivery efficiency by 90 nm A-DEX NPs. First, we evaluated the cellular uptake of 90 nm A-DEX NPs at 4 °C or in the presence of metabolic inhibitors (NaN3) to determine whether the A-DEX NPs followed an energy-dependent or energy-independent pathway. Incubating HBZY-1 cells at 4 °C or after pretreatment with NaN3 resulted in a 56.71 or 65.66% reduction in the cellular uptake of A-DEX NPs, respectively, compared with the control (37 °C without NaN3), indicating that the uptake of 90 nm A-DEX NPs was both temperature-dependent and energy-dependent (Fig. 9b). Moreover, the uptake of A-DEX solution was not interfered with by temperature or NaN3, which suggests that the transport was passive.
Consequently, we explored the molecular mechanism of this active transport in more detail. A few endocytosis inhibitors were applied in uptake experiments, including nystatin, Chlo, and DMA. Nystatin is an inhibitor of cholesterol that disrupts caveola-mediated endocytosis [17]. Chlo inhibits clathrin-mediated endocytosis [18]. DMA blocks micropinocytosis [19]. The results show that both nystatin and DMA had relatively small effects on the uptake of our NPs, while Chlo reduced the uptake by approximately half (Fig. 9c). This implies that the 90 nm A-DEX NPs were mostly engulfed via clathrin-dependent endocytosis pathways.
Discussion
NPs are an emerging class of targeting drug carrier for tumor tissues due to the EPR effect [11]. The kidney possesses fenestrated endothelia that allow for passive accumulation of NPs and is hence an accessible site for NP delivery. Two different barriers in the kidney can select the nanoparticle size. One barrier has an exit cutoff size of 10 nm [3]. Hence, NPs with diameters larger than 10 nm will stay in the kidney. However, another barrier will block NPs larger than the endothelium pores (80–150 nm in diameter). In this study, we discovered that 90 nm NPs accumulated more in the kidneys after intravenous injection than did 70 and 110 nm NPs. The PEG-PLGA NPs with smaller sizes (70 nm) were less stable under the high-power manufacturing process (Fig. 3) and thus seemed to break into particles smaller than 10 nm more easily, which passed through the 10 nm barrier and exited the kidney. On the other hand, larger NPs (110 nm) might experience steric hindrance when permeating through the pores of the endothelium. Compared with these two, 90 nm NPs were more stable due to the milder preparation process and were thus less likely to break down and exit the kidney, but they could still pass through the pores of the endothelium. Therefore, the 90 nm NPs may come into contact with the mesangium, which directly adjoins the endothelium without contacting the GBM. Indeed, accumulation of 90 nm NPs in the mesangium was clearly shown by confocal microscopy (Fig. 5). The 90 nm A-DEX NPs displayed the same characteristics of distribution, particularly the enhanced accumulation in the kidney and kidney cortex (Fig. 6).
To function as an efficient drug or delivery vehicle, NPs generally must enter the cells effectively and release their payload into the cytoplasm. PLGA NPs are internalized rapidly, with NPs seen inside the vascular smooth muscle cells as early as 1 min after incubation [20]. Epifluorescence micrographs and flow cytometry have shown that rat mesangial cells internalized siRNA/CDP-NPs, but not free siRNA, after only 5 min of exposure [21]. Our data show that the uptake of 90 nm A-DEX NPs in HBZY-1 rat mesangial cells reached a high level at 15 min (24.92 ng/mg), indicating that HBZY-1 cells internalized 90 nm A-DEX NPs with high efficiency. The uptake of microparticles depends on the particles’ diameter, and the smaller the diameter is, the greater the uptake in cells [22, 23]. This may explain the fast uptake of 90 nm A-DEX NPs by HBZY-1 cells.
Guo et al. have demonstrated that albumin NPs with defined sizes were actively incorporated into mesangial cells via receptor-mediated endocytosis [24]. Inhibition of clathrin-coated pits (not of caveolae), which inhibit receptor-mediated endocytosis, has resulted in approximately 40% inhibition of PLGA NP uptake by HASMCs [25]. These findings are consistent with our result that chlorpromazine caused a significant drop in the uptake of 90 nm A-DEX NPs, indicating that clathrin was involved in the internalization of these NPs.
The PEG-PLGA NPs can be simply prepared by an emulsion/solvent evaporation method. PLGA itself is a biocompatible and biodegradable polymer and has been approved by the FDA for human use. PEG is also commonly utilized in medicine for various purposes, such as in laxatives and excipients. All these characteristics of our NPs make them advantageous for clinical application as drug delivery systems for the treatment of MsPGN.
Conclusion
Our work demonstrates that 90 nm PEG-PLGA NPs can effectively accumulate in the mesangium following intravenous administration into rats using IVIS and immunofluorescence microscopy. The 90 nm A-DEX NPs also had better stability at room temperature and exhibited a sustained release behavior. Furthermore, HBZY-1 rat mesangial cells could rapidly internalize the 90 nm A-DEX NPs in vitro.
Acknowledgements
The work was supported by the National Natural Science Foundation of China (Nos. 81473169 and 81130060).
Author contributions
LZ and SL designed the research; SL, Y-cZ, KP, and CL performed the research; LZ and Z-rZ contributed new reagents or analytic tools; SL analyzed the data and wrote the draft manuscript; LZ contributed to manuscript writing and proofreading.
Competing interests
The authors declare no competing interests.
References
- 1.Turner JE. Glomerulonephritis therapy: is there a role for green tea? Kidney Int. 2011;80:563–4. doi: 10.1038/ki.2011.173. [DOI] [PubMed] [Google Scholar]
- 2.Javaid B, Quigg RJ. Treatment of glomerulonephritis: will we ever have options other than steroids and cytotoxics? Kidney Int. 2005;67:1692–703. doi: 10.1111/j.1523-1755.2005.00266.x. [DOI] [PubMed] [Google Scholar]
- 3.Zuckerman JE, Davis ME. Targeting therapeutics to the glomerulus with nanoparticles. Adv Chronic Kidney Dis. 2013;20:500–7. doi: 10.1053/j.ackd.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 4.Lahdenkari AT, Lounatmaa K, Patrakka J, Holmberg C, Wartiovaara J, Kestilä M, et al. Podocytes are firmly attached to glomerular basement membrane in kidneys with heavy proteinuria. J Am Soc Nephrol. 2004;15:2611–8. doi: 10.1097/01.ASN.0000139478.03463.D9. [DOI] [PubMed] [Google Scholar]
- 5.Zuckerman JE, Choi CH, Han H, Davis ME. Polycation-siRNA nanoparticles can disassemble at the kidney glomerular basement membrane. Proc Natl Acad Sci USA. 2012;109:3137–42. doi: 10.1073/pnas.1200718109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shimizu H, Hori Y, Kaname S, Yamada K, Nishiyama N, Matsumoto S, et al. SiRNA-based therapy ameliorates glomerulonephritis. J Am Soc Nephrol. 2010;21:622–33. doi: 10.1681/ASN.2009030295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choi CH, Zuckerman JE, Webster P, Davis ME. Targeting kidney mesangium by nanoparticles of defined size. Proc Natl Acad Sci USA. 2011;108:6656–61. doi: 10.1073/pnas.1103573108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pollinger K, Hennig R, Bauer S, Breunig M, Tessmar J, Buschauer A, et al. Biodistribution of quantum dots in the kidney after intravenous injection. J Nanosci Nanotechnol. 2014;14:3313–9. doi: 10.1166/jnn.2014.8716. [DOI] [PubMed] [Google Scholar]
- 9.Campos IMF, Santos TM, Cunha GMF, Silva KMMN, Domingues RZ, Cunha AD, et al. Preparation and release characteristics of dexamethasone acetate loaded organochlorine-free poly (lactide-co-glycolide) nanoparticles. J Appl Polym Sci. 2014;131:41199. [Google Scholar]
- 10.Ameller T, Marsaud V, Legrand P, Gref R, Renoir JM. In vitro and in vivo biologic evaluation of long-circulating biodegradable drug carriers loaded with the pure antiestrogen. Int J Cancer. 2003;106:446–54. doi: 10.1002/ijc.11248. [DOI] [PubMed] [Google Scholar]
- 11.Guo J, Gao X, Su L, Xia H, Gu G, Pang Z, et al. Aptamer-functionalized PEG-PLGA nanoparticles for enhanced anti-glioma drug delivery. Biomaterials. 2011;32:8010–20. doi: 10.1016/j.biomaterials.2011.07.004. [DOI] [PubMed] [Google Scholar]
- 12.Sadat Tabatabaei Mirakabad F, Nejati-Koshki K, Akbarzadeh A, Yamchi MR, Milani M, Zarghami N, et al. PLGA-based nanoparticles as cancer drug delivery systems. Asian Pac J Cancer Prev. 2014;15:517–35. doi: 10.7314/APJCP.2014.15.2.517. [DOI] [PubMed] [Google Scholar]
- 13.Li S, Zeng YC, Zhang ZR. Preparation and characterization of dexamethasone acetate loaded PEG-PLGA nanoparticles. West China J Pharm Sci. 2017;32:124–6. [Google Scholar]
- 14.Zhang T, Zheng Y, Peng Q, Cao X, Gong T, Zhang ZR. A novel submicron emulsion system loaded with vincristine–oleic acid ion-pair complex with improved anticancer effect: in vitro and in vivo studies. Int J Nanomed. 2013;8:1185–96. doi: 10.2147/IJN.S41775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol Pharm. 2008;5:505–15. doi: 10.1021/mp800051m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Anderson JM, Shive MS. Biodegradation and biocompatibility of PLA and PLGA microspheres. Adv Drug Deliv Rev. 2012;64:72–82. doi: 10.1016/j.addr.2012.09.004. [DOI] [PubMed] [Google Scholar]
- 17.Nabi IR, Le PU. Caveolae/raft-dependent endocytosis. J Cell Biol. 2003;161:673–7. doi: 10.1083/jcb.200302028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mahmud A, Lavasanifar A. The effect of block copolymer structure on the internalization of polymeric micelles by human breast cancer cells. Colloids Surf B Biointerfaces. 2005;45:82–9. doi: 10.1016/j.colsurfb.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 19.Sun VZ, Li Z, Deming TJ, Kamei DT. Intracellular fates of cell-penetrating block copolypeptide vesicles. Biomacromolecules. 2011;12:10–3. doi: 10.1021/bm101036f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Panyam J, Labhasetwar V. Dynamics of endocytosis and exocytosis of poly (D, L-Lactide-co-Glycolide) nanoparticles in vascular smooth muscle cells. Pharm Res. 2003;20:212–20. doi: 10.1023/A:1022219003551. [DOI] [PubMed] [Google Scholar]
- 21.Zuckerman JE, Gale A, Wu P, Ma R, Davis ME. SiRNA delivery to the glomerular mesangium using polycationic cyclodextrin nanoparticles containing siRNA. Nucleic Acid Ther. 2015;25:53–64. doi: 10.1089/nat.2014.0505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Langston Suen WL, Chau Y. Size-dependent internalisation of folate-decorated nanoparticles via the pathways of clathrin and caveolae-mediated endocytosis in ARPE-19 cells. J Pharm Pharmacol. 2014;66:564–73. doi: 10.1111/jphp.12134. [DOI] [PubMed] [Google Scholar]
- 23.Qaddoumi MG, Ueda H, Yang J, Davda J, Labhasetwar V, Lee VHL. The characteristics and mechanisms of uptake of PLGA nanoparticles in rabbit conjunctival epithelial cell layers. Pharm Res. 2004;21:641–8. doi: 10.1023/B:PHAM.0000022411.47059.76. [DOI] [PubMed] [Google Scholar]
- 24.Guo L, Luo S, Du ZW, Zhou ML, Li PW, Fu Y, et al. Targeted delivery of celastrol to mesangial cells is effective against mesangioproliferative glomerulonephritis. Nat Commun. 2017;8:878. doi: 10.1038/s41467-017-00834-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Panyam J, Zhou WZ, Prabha S, Sahoo SK, Labhasetwar V. Rapid endo-lysosomal escape of poly (DL-lactide-co- glycolide) nanoparticles: implications for drug and gene delivery. FASEB J. 2002;16:1217–26. doi: 10.1096/fj.02-0088com. [DOI] [PubMed] [Google Scholar]