

Abstract
Berberine, berberrubine, thalifendine, demethyleneberberine, jatrorrhizine, and columbamine are six natural protoberberine alkaloid (PA) compounds that display extensive pharmacological properties and share the same protoberberine molecular skeleton with only slight substitution differences. The oral delivery of most PAs is hindered by their poor bioavailability, which is largely caused by P-glycoprotein (P-gp)-mediated drug efflux. Meanwhile, P-gp undergoes large-scale conformational changes (from an inward-facing to an outward-facing state) when transporting substrates, and these changes might strongly affect the P-gp-binding specificity. To confirm whether these six compounds are substrates of P-gp, to investigate the differences in efflux capacity caused by their trivial structural differences and to reveal the key to increasing their binding affinity to P-gp, we conducted a series of in vivo, in vitro, and in silico assays. Here, we first confirmed that all six compounds were substrates of P-gp by comparing the drug concentrations in wild-type and P-gp-knockout mice in vivo. The efflux capacity (net efflux) ranked as berberrubine > berberine > columbamine ~ jatrorrhizine > thalifendine > demethyleneberberine based on in vitro transport studies in Caco-2 monolayers. Using molecular dynamics simulation and molecular docking techniques, we determined the transport pathways of the six compounds and their binding affinities to P-gp. The results suggested that at the early binding stage, different hydrophobic and electrostatic interactions collectively differentiate the binding affinities of the compounds to P-gp, whereas electrostatic interactions are the main determinant at the late release stage. In addition to hydrophobic interactions, hydrogen bonds play an important role in discriminating the binding affinities.
Keywords: P-glycoprotein, protoberberine alkaloid, berberine, binding affinity, efflux
Introduction
Protoberberine alkaloids (PAs) are an important group of isoquinoline alkaloids that share the same protoberberine skeleton and are present in many plants, including Magnoliaceae, Ranunculaceae, Berberidaceae, and Menispermaceae [1]. Some PAs have demonstrated a wide spectrum of pharmacological effects and have been widely used in clinical practice. For instance, berberine (BBR) is currently among the most reported and studied PAs [2]. It displays broad-spectrum antibacterial [3], antidiabetic [4], antihyperlipidemic [5], and anticancer [6] activities and has been used as a nonprescription drug for gastroenteritis and diarrhea in clinical practice [7]. In recent decades, PAs have attracted increasing attention. New targets and relevant biological activities and mechanisms of action have been discovered and studied, and various PAs have been isolated and modified to improve their efficacy [8–10]. However, the suboptimal pharmacokinetic properties of certain PAs have limited further development. Among these, P-glycoprotein (P-gp)-mediated drug efflux is a key factor that causes poor bioavailability.
P-gp is a transmembrane transporter belonging to the ATP-binding cassette superfamily and is distributed widely at many physiological barriers, including the intestinal epithelium, capillary brain endothelium, liver hepatocytes, and kidney proximal tubules [11]. In general, P-gp is responsible for pumping cytotoxic compounds out of cells, thereby protecting the body from harmful xenobiotics [12, 13]. Meanwhile, in vivo and in vitro studies have indicated that P-gp also plays a significant role in drug absorption and disposition, reducing drug efficiency [14, 15].
Numerous in vivo and in vitro assays have been designed to profile the interaction of P-gp with potential substrates. The comparison of P-gp knockout (KO) mice with wild-type (WT) animals allows the contribution of intestinal P-gp to be quantitatively defined without the need for inhibitors, which often exhibit specificity and affinity problems in vivo [16, 17]. However, the amount of compound needed and the cost of KO animals limit the ability to assess all the novel compounds. The human epithelial colorectal adenocarcinoma cell (Caco-2) monolayer model is the most commonly and classically used in vitro method for assessing interactions of P-gp and substrates, but this assay also has certain disadvantages. It is expensive, laborious, and time consuming at the early discovery stage. Multiple in silico methods have been developed to rapidly and cost-effectively identify P-gp substrates or inhibitors. Based on the publication of the mouse P-gp crystal structure [18], docking of selective substrates into the binding cavity of P-gp confirmed the previously identified pharmacophore features of P-gp and revealed the interactions between atoms and functional groups in the compound structures and P-gp protein residues [19, 20]. With the development of molecular dynamic (MD) simulations, P-gp was found to undergo large-scale conformational changes (from an inward-facing to an outward-facing state) when transporting substrates, using energy from ATP hydrolysis [21–23]. These changes may greatly impact the P-gp-binding specificity. Therefore, treating P-gp transport as a flexible process in the simulation model is critical for evaluating the efflux capacity, which may be a critical reason why compounds with very similar structures can have different affinities for P-gp. However, to the best of our knowledge, no such studies have previously been performed.
This study examined the interactions of P-gp with six PAs: BBR, berberrubine (BER), thalifendine (THA), demethyleneberberine (DEM), jatrorrhizine (JAT), and columbamine (COL). These PAs share the same molecular skeleton and aromatic quaternary ammonium nitrogen and differ only slightly in the substitution of the A ring or the D ring (Scheme 1). We aim to investigate whether these six compounds are substrates of P-gp. As the last five compounds are major metabolites of BBR, it is feasible to investigate the contribution of P-gp to their pharmacokinetic behavior by administering BBR alone to P-gp-KO mice. In addition, as the five other PAs contribute to systemic pharmacological activities along with BBR [24–27], revealing the mechanism underlying the different efflux capacities they exhibit despite their highly similar structures is very important. We also hope to clarify the differences between these six PAs’ efflux capacities in vitro and to reveal the mechanism resulting in the different efflux capacities by comparing how these six highly similar compounds bind to P-gp during transport using MD simulation and molecular docking methods.
Scheme 1.

Chemical structures BBR, BER, COL, JAT, THA, and DEM
Materials and methods
Chemicals and reagents
BBR hydrochloride, BER, JAT hydrochloride, DEM, and COL standards were purchased from Chengdu Mansite Pharmaceutical Co., Ltd. (Chengdu, China). THA was kindly provided by Prof. Cheng-gang Huang (Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, China). The purity of all the chemicals was >98%. Ammonium acetate was purchased from Sigma-Aldrich (St. Louis, MO, USA). Fetal bovine serum (FBS), penicillin–streptomycin (PS), MEM nonessential amino acid (MEM-NEAA, 100×), Hanks’ balanced salt solution (HBSS, 1×, containing Ca2+), and N-2-hydroxyethylpiperazine-N-2-ethane sulfonic acid (HEPES, 100×) were purchased from Gibco Life Technologies (Carlsbad, CA, USA). Dulbecco’s modified Eagle’s medium/high glucose (DMEM-high glucose) was purchased from GE Healthcare Life Sciences (Logan, UT, USA). 2-Morpholinoethanesulfonic acid was purchased from TCI (Shanghai) Development Co., Ltd. (Shanghai, China). Acetonitrile and methanol were purchased from Merck kGaA (Darmstadt, Germany), and formic acid was obtained from Aladdin Company (Shanghai, China).
Animals and cells
Male P-gp-KO (mdr1a−/−) and WT C57 BL/6J (mdr1a+/+) mice (8 weeks old) were purchased from Nanjing Yinghe Biopharmaceuticals Co., Ltd. (Nanjing, China) and raised in a specific pathogen-free experimental animal room. All animals were housed under standard laboratory conditions (temperature of 25 ± 1 °C, humidity of 50 ± 10% and a 12 h light/12 h dark cycle) in the institutional animal facility with free access to food and water. All animal experiments were conducted according to the Guidelines for the Ethical Treatment of Laboratory Animals, and the experimental protocols were approved by the Institutional Animal Care and Use Committee at the Shanghai Institute of Materia Medica (Shanghai, China). Caco-2 cells were purchased from the Cell Culture Center of the Chinese Academy of Medical Sciences (Shanghai, China).
Animal experiment
Mice were fasted for 8–10 h prior to the experiment and then randomly divided into four groups. BBR was administered orally to WT (mdr1a+/+) and P-gp KO (mdr1a−/−) mice (n = 5/group/time point) at 150 mg/kg. The plasma and liver were then collected. The samples were frozen at −80 °C until liquid chromatography tandem mass spectrometry (LC-MS/MS) analysis. In addition, the tissue was homogenized in 3-fold excess volumes of normal saline before LC-MS/MS analysis.
Cell culture
Caco-2 cells were cultured at 37 °C in a humidified atmosphere of 5% CO2. The cells were maintained in DMEM-high glucose supplemented with 10% FBS, 1% PS, and 1% MEM-NEAA. In addition, routine passage was performed approximately every 3 days.
Studies of in vitro transport in Caco-2 monolayers
For the transport experiment, the cells were seeded on 12-well Transwell plates (1.12 cm2 area, 12 mm Transwell with 0.4 μm pore polycarbonate membrane; Product #3401, Corning Corporation, Corning, NY, USA) at a density of 1.5 × 105 cells/cm2. The culture medium was changed every 2 days. Permeability studies were conducted using monolayers cultured for 21 days. Transepithelial electrical resistance (TEER) was used to monitor the integrity and tight junction of the Caco-2 cell monolayer using a Millicell® ERS-2 Epithelial Volt-Ohm Meter (Millipore, Billerica, MA, USA). To simulate the pH environment of the intestinal mucosa, the pH of the transport medium was set according to a previous report and adjusted as necessary [28]. Monolayers with TEER values >500 Ω/cm2 were used for the experiments. Before the transepithelial transport studies, the cell monolayers were washed three times with 37 °C HBSS and equilibrated with transport medium (apical side: HBSS with 10 mM MES, pH 6.0; basolateral side: HBSS with 10 mM HEPES, pH 7.4) for 30 min at 37 °C. Transport medium containing BBR, BER, COL, JAT, THA, or DEM (10 μM for all) was added to either the apical or basolateral chamber (donor chamber), and blank apical/basolateral-side transport medium was added to the opposite chamber (receiver chamber). The total volume was 0.4 mL in the apical chamber and 1.2 mL in the basolateral chamber. Ninety microliter aliquots of the samples were taken separately from the receiver chambers at 30, 60, 90, and 120 min, and the same volume (90 μL) of blank transport medium was immediately added. Transport experiments were conducted in an incubator maintained at 37 °C with shaking at a speed of 50 rpm. The samples were frozen at −80 °C until LC-MS/MS analysis.
The apparent permeability coefficient (Papp) values across the cell monolayers were calculated according to the following equation:
where dQ/dt represents the rate of drug transport, A is the surface area of the cell monolayer (1.12 cm2), and C0 is the initial concentration of the drug in the donor chamber.
The efflux ratio (ER) and net efflux value were calculated according to the following equations:
LC-MS/MS analysis of biological samples
A sensitive LC-MS/MS (LCMS-8030 triple quadrupole system; Shimadzu, Kyoto, Japan) method was established to measure the BBR, BER, JAT, COL, THA, and DEM concentrations in plasma and in tissue homogenate. The mobile phase consisted of an organic phase (acetonitrile containing 0.2% formic acid) and an aqueous phase (ultrapure water containing 0.1% formic acid and 2.5 mM ammonium acetate), with an isocratic elution condition (85% aqueous phase) at a rate of 0.27 mL/min. The column temperature was maintained at 40 °C. The injection volume was 5 μL. The BBR, BER, JAT, COL, THA, and DEM concentrations were determined in positive electrospray ionization mode with a Waters LortecsTM C18 column (100 mm × 2.10 mm, 2.7 μm, Waters Corporation, Milford, MA, USA). The transitions selected for reaction monitoring were 336.10 m/z > 320.00 m/z for BBR, 322.20 m/z > 307.10 m/z for THA and BER, 338.20 m/z > 323.10 m/z for COL and JAT, 324.10 m/z > 308.10 m/z for DEM, and 356.00 m/z > 192.00 m/z for the internal standard (tetrahydropalmatine).
Samples were pretreated before LC-MS/MS as follows. First, 90 μL sample/control, 10 μL internal standard, and 300 μL acetonitrile were mixed and vortexed for 5 min and then centrifuged at 14,000 × g for 20 min at room temperature (RT). The supernatants were concentrated by vacuum concentrators (Thermo, Waltham, MA, USA), resuspended with 100 μL resuspension agent (water:acetonitrile = 7:3) and then centrifuged at 14,000 × g for 20 min at room temperature, and the supernatants were analyzed using the validated LC-MS/MS method.
Human P-gp modeling
An inward-facing model of human P-gp was built using a murine P-gp crystal structure (PDB code: 4M1M) as a template [29]. An outward-facing model was built based on the murine model reported by Pan et al. [30]. The resolutions of 4M1M and 2HYD are high, and these structures are often used to build P-gp models [31–35]. Sequence alignment and homology modeling were performed using Modeller (Ben Webb, Novato, CA, USA). The sequence identity between human and mouse P-gp was 88.7% after the removal of residues 1–30 at the N-terminal, residues 1275–1280 at the C-terminal and linker residues 631–684, which have no counterpart in the template sequence, indicating the reasonableness of building human P-gp structures using a template from mice. The resulting inward-facing and outward-facing human P-gp models are similar to the corresponding templates (root-mean-square deviation (RMSD)Cα < 0.4 Å) (Supplemental Fig. 4a). The two structures were minimized in Maestro and then evaluated using the MolProbity server (http://kinemage.biochem.duke.edu). The result showed that the constructed models were high quality (Supplemental Fig. 4b).
Normal mode analysis
The conformational transition pathway from the inward-facing to the outward-facing state of human P-gp was calculated using normal mode analysis (NMA) [36]. The constructed inward-facing model was set as the initial structure and the outward-facing model as the final target structure. Multiple iterations were run with these two structures as the end points, with the step size set to 10.0. Every intermediate structure calculated was minimized using 1000 MD simulation steps, and the optimized structure was then used in the next NMA iteration. One hundred conformations were selected from all transition conformations for the subsequent docking procedure.
Molecular docking
Molecular docking was performed using the Maestro software (Schrödinger, New York, NY, USA). The protein was prepared using the protein preparation wizard. Three-dimensional structures of BBR, BER, COL, JAT, THA, and DEM were downloaded from PubChem (https://www.ncbi.nlm.nih.gov/pccompound) and prepared using the Ligprep wizard (Schrödinger, NY, USA). The pKa values of COL, JAT, THA, and DEM were calculated using the Epik program (Schrödinger, NY, USA), and the pKa values of BER were obtained from the Human Metabolome database (see Supplemental Table 1). Therefore, at physiological pH 7.0 ± 2.0, only BER is partly deprotonated, resulting in zwitterionized and ionized mixed forms. The other five compounds (BBR, COL, JAT, THA, and DEM) all maintain their original protonation state. BBR, COL, JAT, THA, DEM, and ionized and zwitterionized BER were docked into P-gp conformations. The initial docking box center coordinate was set with reference to the center of mass of the ligand in a murine P-gp complex crystal structure (PDB code: 3G60). To obtain the whole transport pathway, the docking box center was shifted from 0 to 31.6 Å at intervals of ~0.4 Å along the Z-axis from the first to the last P-gp conformation. To constrain the docking poses at the settled Z coordinate, we set the inner box size to 30 Å × 30 Å × 2 Å and the outer box size to 40 Å × 40 Å × 20 Å. Then, molecular docking was performed using the Glide module with the self-propelled mode. To increase the ligand conformation variety generated, we used sampling enhanced four times at the conformer generation step. When selecting initial poses, we used expanded sampling by retaining 50,000 poses for the initial phase of docking and 4000 poses for energy minimization. Finally, the top 100 poses were outputted. When selecting the transporting intermediate docking poses, both the RMSD and binding affinity were considered. We assumed the RMSD between poses in neighboring protein conformations to be small. We first extracted the top 10 docking poses for each protein conformation and then sorted these poses according to the RMSD values with respect to the final pose. The docking pose with the lowest RMSD was selected as an intermediate conformation. The first starting ligand conformation was determined by the binding affinity.
Statistical analysis
Statistical analysis was performed using the GraphPad Prism 6 software (GraphPad Software Inc., La Jolla, CA, USA). The data are expressed as the mean ± SD. Differences between groups were analyzed using the unpaired Student’s t test. P values <0.05 were considered statistically significant (*P < 0.05, **P < 0.01, ***P < 0.001).
Results
BBR and five metabolite concentrations in the plasma and liver of WT and P-gp-KO mice
To determine whether BBR and the five other PAs are substrates of P-gp, we compared the concentration of each compound in the plasma and liver of WT (mdr1a+/+) and P-gp KO (mdr1a−/−) mice. As the last five PAs (BER, JAT, COL, THA, and DEM) are also major metabolites of BBR, it is possible to investigate the contribution of P-gp to their pharmacokinetic behavior by administering BBR alone to P-gp-KO mice. A sensitive LC-MS/MS method was used to identify and quantify BBR and its main metabolites (BER, JAT, COL, THA, and DEM) simultaneously after a single oral administration of 150 mg/kg BBR to WT and P-gp-KO mice. Both the plasma and liver concentrations of BBR, COL, JAT, THA, and DEM (Fig. 1) were significantly higher in P-gp-KO mice than in WT mice. The plasma concentrations at 2 h and liver concentrations at 6 h of BER increased without significance. The plasma concentrations of BER, BBR, COL, JAT, THA, and DEM in the P-gp KO group showed 3.9-, 4.3-, 9.2-, 15.8-, 30.1-, and 35.5-fold increases, respectively, compared with the values in the WT group after 6 h. The drug concentrations of BER, BBR, COL, JAT, THA, and DEM in the liver in the P-gp-KO group showed 1.1-, 3.6-, 4.1-, 4.3-, 4.6-, and 6.0-fold increases, respectively, compared with the WT group after 6 h.
Fig. 1.

The plasma and tissue concentrations of BBR and its metabolites in WT and P-gp-KO mice. Concentrations of BBR and its metabolites in the plasma (a) and liver (b) were detected at 2 and 6 h after the oral administration of 150 mg/kg BBR (n = 5/group/time point). Data are mean ± SD. Significance was analyzed by non-paired t test. P values <0.05 were considered statistically significant (*P < 0.05, **P < 0.01, ***P < 0.001)
In vitro permeability in Caco-2 monolayers
To compare the efflux capacity of the six compounds, the in vitro permeability across Caco-2 monolayers was measured for each compound. The Caco-2 cell monolayer model was used to determine the bidirectional transportation permeability (apical side to basolateral side and basolateral side to apical side, hereinafter referred to as AP-BL and BL-AP). The cumulative drug amounts in both directions were calculated (Fig. 2a, b). The Papp (AP-BL) values of the six compounds ranged from 0.08 × 10−6 to 3.65 × 10−6 cm/s (Fig. 2c). A drug with a Papp (AP-BL) < 5 × 10−6 cm/s is considered to exhibit poor penetration [37]. The Papp (BL-AP) values of the six compounds ranged from 0.31 × 10−6 to 70.6 × 10−6 cm/s (Fig. 2d). Therefore, all six tested compounds in this study showed poor permeability. Interestingly, BER exhibited much higher permeability than did the other compounds in both directions.
Fig. 2.

Transport of BBR and five other compounds in cultured Caco-2 cells. Caco-2 cell monolayers were incubated at 37 °C in HBSS, and test compounds were added to the apical or basolateral side. Four sequential samples were taken at different time points (30, 60, 90, and 120 min). The samples were withdrawn and the test compounds were determined by LC/MS/MS (n = 3). Time course for the AP-BL (a, b) and BL-AP (c, d) cumulative amounts and apparent permeability are shown. Data are mean ± SD
In addition, we calculated the ER (Papp (BL-AB)/Papp (AP-BL)) and net efflux (Papp (BL-AB) − Papp (AP-BL)) values, and the net efflux values of the six compounds across Caco-2 monolayers ranked as BER > BBR > COL ~ JAT > THA > DEM (Table 1).
Table 1.
The permeability characteristics of BBR and the five other PAs across the Caco-2 monolayer model in vitro (n = 3, three parallel wells)
| Papp (AP-BL) (×10−6 cm/s) | Papp (BL-AP) (×10−6 cm/s) | Net effluxa (×10−6 cm/s) | ERb | |
|---|---|---|---|---|
| BER (10 μM) | 3.64 ± 0.84 | 70.60 ± 9.70 | 66.69 ± 9.35 | 19.92 |
| BBR (10 μM) | 0.08 ± 0.04 | 2.63 ± 0.70 | 2.55 ± 0.67 | 33.54 |
| COL (10 μM) | 0.34 ± 0.02 | 1.94 ± 0.09 | 1.6 ± 0.07 | 5.68 |
| JAT (10 μM) | 0.50 ± 0.15 | 2.23 ± 0.28 | 1.73 ± 0.42 | 4.84 |
| THA (10 μM) | 0.89 ± 0.20 | 1.22 ± 0.07 | 0.35 ± 0.02 | 1.51 |
| DEM (10 μM) | 0.09 ± 0.04 | 0.31 ± 0.01 | 0.22 ± 0.05 | 3.71 |
aNet efflux = Papp (BL-AP) − Papp (AP-BL)
bER = Papp (BL-AP)/Papp (AP-BL)
Predicting the binding modes of BBR and metabolites along the translocation pathway
We modeled inward-facing and outward-facing human P-gp structures. Using NUMD [36] developed by our group, we obtained the whole transition pathway of P-gp from its inward-facing to outward-facing state. To simulate the translocation pathway of the six compounds, we docked each compound into intermediate P-gp conformations in which the docking box center was shifted from the binding site to an extracellular region along the Z-axis (Fig. 3b). By calculating the docking scores of each compound to the intermediate conformations, we obtained the binding affinities of the six compounds to P-gp in the translocation pathway. The binding affinities (docking scores) are also illustrated in Fig. 3a and Supplemental Fig. 2. One can clearly see in Supplemental Fig. 2 that ionized BER has a much lower binding affinity than does BBR, while the zwitterionized BER has much higher binding affinities than does BBR. The lower binding affinities of ionized BER indicate its larger efflux difficulty in being pumped by P-gp. Given that BER has a remarkably larger efflux capacity compared to BBR and the other compounds, it is reasonable to speculate that zwitterionized BER is the main efflux form. Therefore, we focus on the transport mechanism of zwitterionized BER through P-gp. As shown in Fig. 3a, DEM exhibited the highest binding affinity (lowest docking score) throughout the transport pathway, indicating that it has the greatest difficulty in being pumped by P-gp. The binding affinities for BBR, BER, COL, JAT, and THA in the releasing stage (22–32 Å) ranked as BER < BBR < JAT ~ COL < THA, indicating that the compounds ranked as BER > BBR > JAT ~ COL > THA in terms of P-gp pumping difficulty. These data agree well with the above experiments.
Fig. 3.

Predicting the binding of the six compounds to P-gp through the transport pathway. The complete transition pathway between the inward-facing and outward-facing conformations of human P-gp was modeled, and the six compounds were docked into a total of 100 intermediate P-gp conformations in which the docking box center was shifted from 0 to 31.6 Å at intervals of ~0.4 Å along the Z-axis; docking scores are averaged every Z ~ 1.5 Å (b). The complete transport pathway for all six compounds from the binding site to the extracellular region along with the corresponding binding affinities to P-gp. The binding affinities (docking scores) are illustrated in a
Binding modes of BBR and metabolites with P-gp in the binding stage
The six compounds bind to P-gp with highest affinity at Z ≈ 5 Å (Fig. 3a) when P-gp is in the inward-facing state (Fig. 4a, b). This result was consistent with the released murine complex crystal structure [18]. All six compounds primarily interact with P-gp through hydrophobic interactions (Fig. 4c, d). Compared with the other five compounds, DEM can form close contacts with more aromatic residues. Regarding electrostatic interactions, BBR, BER, COL, JAT, and THA can form only one or two hydrogen bonds with Y307, Y310, and N721 (Supplemental Figs. 3c–3g). In contrast, DEM can form six hydrogen bonds with Y307, Y310, and Q725 through two hydroxyl functional groups (Supplemental Fig. 3b). BBR, BER, COL, JAT, and THA form fewer hydrogen bonds than does DEM with P-gp (BBR: 1; BER: 1; COL: 3; JAT: 5; and THA: 2). Ambudkar et al. [38] reported that Y307, Y310, Q725, and N721 play critical roles in hydrogen bond formation, which is consistent with our findings. This indicates the high reliability of our docking experiment. In conclusion, the greater number of favorable closely packed hydrophobic interactions and stronger hydrogen bond network give DEM the highest binding affinity to P-gp in the early binding stage.
Fig. 4.

The binding modes of the six compounds interacting with P-gp at the lowest docking scores. All six compounds exhibit an inward-facing state binding mode and generally interact with P-gp through hydrophobic-driven binding modes. BBR, BER, COL, JAT, and THA binding to P-gp (a, c); DEM binding to P-gp (b, d). All residues within 4 Å of the compounds are shown in c and d
Binding modes of BBR and metabolites with P-gp in the release stage
The binding affinities of all six compounds to P-gp become distinguishable when Z > 17 Å. The greatest difference in binding affinity is observed at Z ~ 27 Å (Fig. 3a), with the compounds ranking BER < BBR < COL ~ JAT < THA < DEM. Obviously, the higher the binding affinity is, the more difficult the transportation of the compound. Therefore, the compounds rank as BER > BBR > COL ~ JAT > THA > DEM in terms of efflux capacity, which is consistent with the in vitro experiment results. As described in Scheme 1, the six compounds are highly similar in structure, with a common tetrahydroisoquinoline [2,1-b] isoquinoline core and one positive charge at the N. The position and number of hydroxyl groups on each compound make them different. For instance, the only difference between two pairs (BER vs. THA and COL vs. JAT) is the position of a hydroxyl group. The difference between three pairs (BBR vs. DEM, BBR vs. THA, and DEM vs. COL) is the number of hydroxyl groups. Intriguingly, these minor structural differences resulted in different efflux capacities.
To determine why such trivial structural differences among BBR and the five other PAs result in markedly different efflux capacities, we analyzed the docking poses at Z ~ 27 Å. The six compounds are bound to the extracellular part of P-gp (see the membrane border in gray color), which means that they have been transported out of the cell and reached the release stage (Fig. 5a). The docking poses of all six molecules are at nearly the same position, which indicates that the compounds have comparable hydrophobic interactions with F971, A80, F79, L975, and T76 (Fig. 5b, c). Therefore, it might be the individual differences in the electrostatic interactions with P-gp that determine the differences in their binding affinities, which are attributed to N84, D87, and E972 (Fig. 5b, c).
Fig. 5.
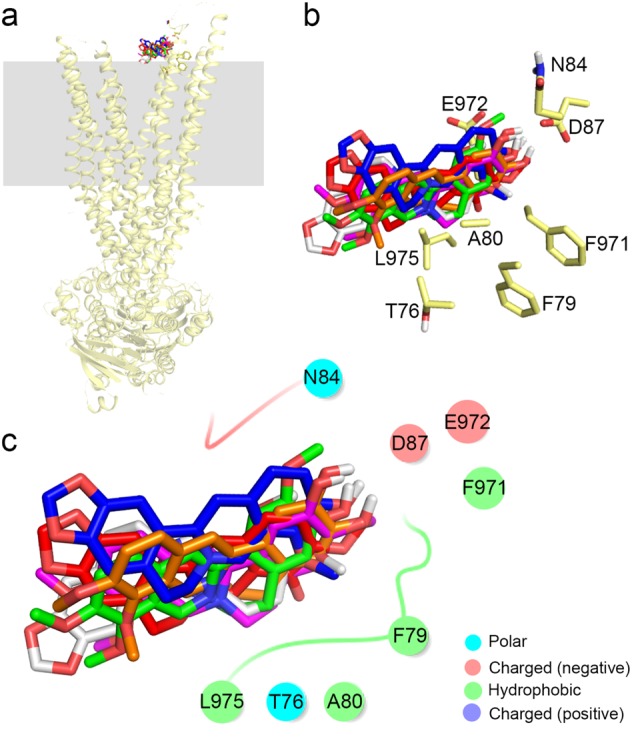
The binding docking results of the six compounds to P-gp in the release stage (Z ~ 27Å). Membranes are shown as gray squares, and the binding modes of the six compounds are shown (a). Amino acids within 4 Å of the compounds are shown as yellow sticks (b). Interactions of BBR, BER, COL, JAT, THA, and DEM with P-gp (c)
Then, we investigated how the hydroxyl group position influences the binding affinity of BER and THA or of COL and JAT. THA can form four hydrogen bonds with N84, D87, and E972, whereas BER cannot form any hydrogen bonds with P-gp in zwitterionic form (Fig. 6a). JAT and COL can form comparable hydrogen bonds (Fig. 6b). Next, we investigated the correlation between the hydroxyl group number and binding affinities by comparing BBR with DEM, BBR with THA, and DEM with COL. The compounds ranked as DEM > THA > COL > BBR in terms of the number of hydrogen bonds (DEM: 6; THA: 4; COL: 3; and BBR: 0) formed with P-gp (Fig. 6c–e).
Fig. 6.

Hydrogen bond networks of six compounds with P-gp in the release stage. The hydroxyl group position influences the binding affinity of BER and THA (a) and of COL and JAT (b). The correlation between hydroxyl group number and binding affinities by comparing BBR with THA (c), BBR with DEM (d), and DEM with COL (e). For clarity, the hydrogen bonds of DEM are shown in d but not in e
Based on the above result, the binding affinities are strongly correlated with the number of hydrogen bonds formed. Compounds with comparable hydrogen bond numbers appear to demonstrate comparable binding affinities, and compounds that form more hydrogen bonds demonstrate higher binding affinities than do counterparts that form fewer. We suggest that BBR and the five other PAs have comparable hydrophobic interactions with P-gp and that the electrostatic interaction (hydrogen bond number) is the main determinant of the different binding affinities at the release stage.
Discussion
The high levels of P-gp expressed in the intestine may directly limit oral drug absorption and increase variability in clinical practice [39]. Identifying the interactions of PAs with P-gp at the early stage of drug development could help to facilitate the drug screening process and predict potential drug–drug interaction risks.
In this study, both experimental assays (in vivo and in vitro) and in silico methods were employed to identify potential P-gp substrates. Mice lacking P-gp have been used extensively to investigate the role of P-gp in oral absorption [16]. P-gp is coded by mdr1a (or Abcb1a) and mdr1b (or Abcb1b) in rodents and by MDR1 (or ABCB1) in humans. The evidence suggests that mdr1a plays a dominant role in drug transport compared to mdr1b [40, 41]. In our study, the mRNA levels of both mdr1a and mdr1b were decreased in the livers of P-gp KO (mdr1a−/−) mice compared with those in WT mice (Supplemental Fig. 1). After oral administration of a 150 mg/kg dose of BBR, we compared the concentrations of BBR and five of its metabolites in WT and P-gp-KO mice. It was found that all six compounds’ plasma and liver concentrations were increased to different extents (Fig. 1) in the P-gp-KO mice. As the increases in the concentrations of the five other compounds were greater than the increase in BBR, it is reasonable to hypothesize that the absence of P-gp contributes substantially to this process. Of course, other factors, such as changes in cytochrome P450 and the compensating effect of other efflux transporters, could not be completely excluded.
It is difficult to quantitatively assess the P-gp efflux capacities of the six compounds with such a simple in vivo study. Therefore, a Caco-2 efflux in vitro experiment was conducted. Based on Papp (AP-BL) and Papp (BL-AP), the ER and net efflux value were calculated to assess the efflux capacity of each compound. In addition, we used the net efflux value as a reference parameter to assess the “strength” of the efflux capacities of the six compounds for the following reasons: (1) the ER can be used as a criterion to categorize compounds as substrates or non-substrates, but it cannot tell how “strong” the substrates are [42]; (2) the Caco-2 monolayer efflux assay can provide concordant results only for compounds with Papp values between 20 × 10−6 and 300 × 10−6 cm/s, while for drugs with low Papp (AP-BL), only Papp (BL-AP) can be used to assess substrate transport [42, 43]. The net efflux of BBR and the five other compounds across Caco-2 monolayers ranked as BER > BBR > COL ~ JAT > THA > DEM. Furthermore, the Caco-2 permeability is affected by both passive and active (uptake, efflux) transport [44]. The overall net permeability of a drug is the combination of passive diffusion and carrier-mediated transport (absorption and efflux) across the cell membrane [45]. Thus, both the ER and net efflux value maybe influenced by the passive membrane permeability, and we cannot rule out interference from this source.
In contrast to BBR, the other five PAs contain both iminium cations and hydroxyl groups, which results in multiple species in solution at different pH values, including protonated and deprotonated forms of each compound. This variety makes determination of the pKa values particularly important because at a pH value higher than the pKa value, these compounds could take on the characteristics of neutral molecules. The in silico-predicted pKa values demonstrated that COL, JAT, THA, and DEM have relatively high pKa values, and at physiological pH, they are present mainly as iminium cations, similar to BBR (Supplemental Table 1). In contrast, BER is deprotonated at pH 7.0 ± 2.0, and the pKa value of BER is 8.27; thus, BER is partly deprotonated and is present as a zwitterionized form. The hydrophobicity of the zwitterionized form is higher than that of the ionized form, which may result in increased passive permeability. This phenomenon may explain why BER exhibited much higher permeability than did the other compounds in both directions.
Our results clearly showed that while BBR and the five other PAs are highly similar in structure, they exhibit substantial differences in P-gp-related efflux capacities (Scheme 1 and Table 1). To investigate the underlying mechanism, we employed an in silico model to assess the transport pathway of the six compounds and their binding affinities to P-gp. The in silico model successfully ranked the compounds’ binding affinities consistently with the experimental results. The conformation corresponding to the highest binding affinity showed an inward-facing binding mode for all six compounds, which is consistent with the published murine complex crystal structure [18], and all six compounds showed a hydrophobic-driven binding mode. Compared to the other five compounds, DEM can form close contacts with more aromatic and hydrophobic amino acids and can form more hydrogen bonds with P-gp; thus, DEM displayed the highest binding affinity at the early binding stage. We found that the binding affinities to the extracellular part of P-gp show a strong correlation with the number of hydrogen bonds formed with P-gp: compounds with comparable hydrogen bond numbers exhibit comparable binding affinities, and compounds with more hydrogen bonds exhibit higher binding affinities than do their counterparts with fewer. That is, at the early binding stage, different hydrophobic and electrostatic interactions collectively differentiate the binding affinities to P-gp, while at the late release stage, the electrostatic interaction is the main determinant.
As shown in our study, the highest binding affinity at the binding stage facilitates binding, while the highest binding affinity at the release stage hinders dissociation from P-gp. In fact, “strong” P-gp substrates (competitive inhibitors) should be highly hydrophobic because they must first “dissolve” in the hydrophobic membrane medium according to the popular “hydrophobic vacuum model” [46]. Moreover, given the predominant distribution of hydrophobic residues in the binding pocket, researchers have generally thought that highly hydrophobic compounds would more easily bind to P-gp. However, Chufan et al. [38] noted the importance of hydrogen bond formation for an effective P-gp substrate [38]. After the mutation of residues that participate in hydrogen bond formation, he found that the effect of the third-generation inhibitor zosuquidar was reversed, resulting in a stimulatory effect on the ATPase activity of P-gp [38]. Similarly, this study indicated that DEM, with only one or two more hydroxyl groups than its counterparts, can form a much more complex hydrogen network with P-gp (Fig. 6d and Supplemental Fig. 3b) and thus displays a high binding affinity with P-gp and a low efflux capacity. Therefore, researchers should be sure to introduce hydrogen bond donors and acceptors to reduce the risk of producing P-gp substrates when designing PA analogs.
In the clinic, using existing P-gp inhibitors is not an ideal strategy to improve oral bioavailability [47, 48]. For example, concomitant use of the P-gp inhibitor cyclosporin A can suppress the immune system and cause medical complications [49]. Chemical modification is an alternative approach to overcoming the efflux of BBR by using P-gp as a target. The synthesized BBR analog pseudoberberine (IMB-Y53) has a low affinity to P-gp and exhibits improved bioavailability and a higher glucose-lowering efficacy compared to BBR [50, 51]. Moreover, some PAs, such as coptisine, have trivial structural differences from BBR but have been demonstrated to have very different binding affinities to P-gp [52]. It is crucial to clarify why such small structural differences can lead to substantial efflux capacity differences, which is essential for the design and optimization of the current PAs.
In conclusion, we proved that BBR and five other PAs were substrates of P-gp using in vivo experiments and identified their efflux capacity ranking, BER > BBR > COL ~ JAT > THA > DEM, by measuring the corresponding permeability across Caco-2 monolayers in vitro. The binding affinities of the six compounds to P-gp were successfully verified by an in silico model, and their transport pathways were further investigated. Our results suggested that hydrogen bonding plays an important role in the differences in the binding affinities of BBR and the five other PAs. At the early binding stage, different hydrophobic and electrostatic interactions collectively differentiate the binding affinities to P-gp, while at the late release stage, the electrostatic interaction is the main determinant. Our work highlights a new approach to reducing the P-gp efflux risk at the early drug discovery stages.
Electronic supplementary material
Acknowledgements
This work was partly supported by the National Natural Science Foundation of China (Grants 81573499 81872927) and Strategic Priority Research Program of the Chinese Academy of Sciences (Grant XDA16020205). We thank Prof. Cheng-gang Huang (Shanghai Institute of Materia Medica, Chinese Academy of Sciences) for providing the purified compound.
Author contributions
Y-tZ, Y-qY, X-tT, W-lZ, L-kG, and G-yP were responsible for the research design and wrote this manuscript; Y-tZ, Y-qY, X-xY, LW, and W-jW conducted the experiments; Y-qY and W-lZ contributed analytical tools; and Y-tZ and Y-qY performed the data analysis.
Competing interests
The authors declare no competing interests.
Contributor Information
Wei-liang Zhu, Email: wlzhu@mail.shcnc.ac.cn.
Li-kun Gong, Email: lkgong@cdser.simm.ac.cn.
Guo-yu Pan, Email: gypan@simm.ac.cn.
Electronic supplementary material
The online version of this article (10.1038/s41401-018-0183-7) contains supplementary material, which is available to authorized users.
References
- 1.Preininger V. Chemotaxonomy of papaveraceae and fumariaceae. In: Arnold B, editor. The alkaloids: chemistry and pharmacology. vol. 29. New York: Harcourt Brace Jovanovich; 1986. p. 1–98.
- 2.Kumar A, Ekavali, Chopra K, Mukherjee M, Pottabathini R, Dhull DK. Current knowledge and pharmacological profile of berberine: an update. Eur J Pharmacol. 2015;761:288–97. doi: 10.1016/j.ejphar.2015.05.068. [DOI] [PubMed] [Google Scholar]
- 3.Bandyopadhyay S, Patra PH, Mahanti A, Mondal DK, Dandapat P, Bandyopadhyay S, et al. Potential antibacterial activity of berberine against multi drug resistant enterovirulent Escherichia coli isolated from yaks (Poephagus grunniens) with haemorrhagic diarrhoea. Asian Pacific J Trop Med. 2013;6:315–9. [DOI] [PubMed]
- 4.Zhang Q, Xiao XH, Feng K, Wang T, Li WH, Yuan T, et al. Berberine moderates glucose and lipid metabolism through multipathway mechanism. Evid Based Complement Alternat Med. 2011; 1–10. [DOI] [PMC free article] [PubMed]
- 5.Chang XX, Yan HM, Xu Q, Xia MF, Bian H, Zhu TF, et al. The effects of berberine on hyperhomocysteinemia and hyperlipidemia in rats fed with a long-term high-fat diet. Lipids Health Dis. 2012;11:86. doi: 10.1186/1476-511X-11-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang LH, Liu LP, Shi Y, Cao HW, Chaturvedi R, Calcutt MW, et al. Berberine induces caspase-independent cell death in colon tumor cells through activation of apoptosis-inducing factor. PLoS ONE. 2012;7:e36418. doi: 10.1371/journal.pone.0036418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rabbani GH, Butler T, Knight J, Sanyal SC, Alam K. Randomized controlled trial of berberine sulfate therapy for diarrhea due to enterotoxigenic Escherichia coli and Vibrio cholerae. J Infect Dis. 1987;155:979–84. doi: 10.1093/infdis/155.5.979. [DOI] [PubMed] [Google Scholar]
- 8.Iranshahy M, Quinn RJ, Iranshahi M. Biologically active isoquinoline alkaloids with drug-like properties from the genus Corydalis. RSC Adv. 2014;4:15900–13. doi: 10.1039/C3RA47944G. [DOI] [Google Scholar]
- 9.Bhadra K, Kumar GS. Therapeutic potential of nucleic acid-binding isoquinoline alkaloids: binding aspects and implications for drug design. Med Res Rev. 2011;31:821–62. doi: 10.1002/med.20202. [DOI] [PubMed] [Google Scholar]
- 10.Grycova L, Dostal J, Marek R. Quaternary protoberberine alkaloids. Phytochemistry. 2007;68:150–75. doi: 10.1016/j.phytochem.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 11.Parcej D, Tampe R. ABC proteins in antigen translocation and viral inhibition. Nat Chem Biol. 2010;6:572–80. doi: 10.1038/nchembio.410. [DOI] [PubMed] [Google Scholar]
- 12.Thiebaut F, Tsuruo T, Hamada H, Gottesman MM, Pastan I, Willingham MC. Cellular-localization of the multidrug-resistance gene-product P-glycoprotein in normal human-tissues. Proc Natl Acad Sci USA. 1987;84:7735–8. doi: 10.1073/pnas.84.21.7735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Borst P, Schinkel AH. P-glycoprotein ABCB1: a major player in drug handling by mammals. J Clin Invest. 2013;123:4131–3. doi: 10.1172/JCI70430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu CS, Zheng YR, Zhang YF, Long XY. Research progress on berberine with a special focus on its oral bioavailability. Fitoterapia. 2016;109:274–82. doi: 10.1016/j.fitote.2016.02.001. [DOI] [PubMed] [Google Scholar]
- 15.Sun YH, He X, Yang XL, Dong CL, Zhang CF, Song ZJ, et al. Absorption characteristics of the total alkaloids from Mahonia bealei in an in situ single-pass intestinal perfusion assay. Chin J Nat Med. 2014;12:554–60. doi: 10.1016/S1875-5364(14)60085-6. [DOI] [PubMed] [Google Scholar]
- 16.Chen CP, Liu XR, Smith BJ. Utility of mdr1-gene deficient mice in assessing the impact of P-glycoprotein on pharmacokinetics and pharmacodynamics in drug discovery and development. Curr Drug Metab. 2003;4:272–91. doi: 10.2174/1389200033489415. [DOI] [PubMed] [Google Scholar]
- 17.Collett A, Tanianis-Hughes J, Hallifax D, Warhurst G. Predicting P-glycoprotein effects on oral absorption: correlation of transport in Caco-2 with drug pharmacokinetics in wild-type and mdr1a(−/−) mice in vivo. Pharm Res. 2004;21:819–26. doi: 10.1023/B:PHAM.0000026434.82855.69. [DOI] [PubMed] [Google Scholar]
- 18.Aller SG, Yu J, Ward A, Weng Y, Chittaboina S, Zhuo RP, et al. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science. 2009;323:1718–22. doi: 10.1126/science.1168750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pajeva IK, Globisch C, Wiese M. Combined pharmacophore modeling, docking, and 3D QSAR studies of ABCB1 and ABCC1 transporter inhibitors. ChemMedChem. 2009;4:1883–96. doi: 10.1002/cmdc.200900282. [DOI] [PubMed] [Google Scholar]
- 20.Dolghih E, Bryant C, Renslo AR, Jacobson MP. Predicting binding to p-glycoprotein by flexible receptor docking. PLoS Comput Biol. 2011;7:e1002083. doi: 10.1371/journal.pcbi.1002083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith PC, Karpowich N, Millen L, Moody JE, Rosen J, Thomas PJ, et al. ATP binding to the motor domain from an ABC transporter drives formation of a nucleotide sandwich dimer. Mol Cell. 2002;10:139–49. doi: 10.1016/S1097-2765(02)00576-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Verhalen B, Wilkens S. P-glycoprotein retains drug-stimulated ATPase activity upon covalent linkage of the two nucleotide binding domains at their C-terminal ends. J Biol Chem. 2011;286:10476–82. doi: 10.1074/jbc.M110.193151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Subramanian N, Condic-Jurkic K, O’Mara ML. Structural and dynamic perspectives on the promiscuous transport activity of P-glycoprotein. Neurochem Int. 2016;98:146–52. doi: 10.1016/j.neuint.2016.05.005. [DOI] [PubMed] [Google Scholar]
- 24.Qiu F, Zhu Z, Kang N, Piao S, Qin G, Yao X. Isolation and identification of urinary metabolites of berberine in rats and humans. Drug Metab Dispos. 2008;36:2159–65. doi: 10.1124/dmd.108.021659. [DOI] [PubMed] [Google Scholar]
- 25.Ma JY, Feng R, Tan XS, Ma C, Shou JW, Fu J, et al. Excretion of berberine and its metabolites in oral administration in rats. J Pharm Sci. 2013;102:4181–92. doi: 10.1002/jps.23718. [DOI] [PubMed] [Google Scholar]
- 26.Ma BL, Ma YM. Pharmacokinetic properties, potential herb-drug interactions and acute toxicity of oral Rhizoma coptidis alkaloids. Expert Opin Drug Metab Toxicol. 2013;9:51–61. doi: 10.1517/17425255.2012.722995. [DOI] [PubMed] [Google Scholar]
- 27.Li Y, Ren G, Wang YX, Kong WJ, Yang P, Wang YM, et al. Bioactivities of berberine metabolites after transformation through CYP450 isoenzymes. J Transl Med. 2011;9:62. doi: 10.1186/1479-5876-9-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang X, Qiu F, Jiang J, Gao C, Tan Y. Intestinal absorption mechanisms of berberine, palmatine, jateorhizine, and coptisine: involvement of P-glycoprotein. Xenobiotica. 2011;41:290–6. doi: 10.3109/00498254.2010.529180. [DOI] [PubMed] [Google Scholar]
- 29.Li J, Jaimes KF, Aller SG. Refined structures of mouse P-glycoprotein. Protein Sci. 2014;23:34–46. doi: 10.1002/pro.2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pan L, Aller SG. Equilibrated atomic models of outward-facing P-glycoprotein and effect of ATP binding on structural dynamics. Sci Rep. 2015;5:7880. doi: 10.1038/srep07880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li MJ, Nath A, Atkins WM. Differential coupling of binding, ATP hydrolysis, and transport of fluorescent probes with P-glycoprotein in lipid nanodiscs. Biochemistry. 2017;56:2506–17. doi: 10.1021/acs.biochem.6b01245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frank GA, Shukla S, Rao P, Borgnia MJ, Bartesaghi A, Merk A, et al. Cryo-EM analysis of the conformational landscape of human P-glycoprotein (ABCB1) during its catalytic cycle. Mol Pharmacol. 2016;90:35–41. doi: 10.1124/mol.116.104190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wise JG. Catalytic transitions in the human MDR1 P-glycoprotein drug binding sites. Biochemistry. 2012;51:5125–41. doi: 10.1021/bi300299z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ferreira RJ, Ferreira MJU, dos Santos DJVA. Insights on P-glycoprotein’s efflux mechanism obtained by molecular dynamics simulations. J Chem Theory Comput. 2012;8:1853–64. doi: 10.1021/ct300083m. [DOI] [PubMed] [Google Scholar]
- 35.McCormick JW, Vogel PD, Wise JG. Multiple drug transport pathways through human P-glycoprotein. Biochemistry. 2015;54:4374–90. doi: 10.1021/acs.biochem.5b00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang J, Shao Q, Xu Z, Liu Y, Yang Z, Cossins BP, et al. Exploring transition pathway and free-energy profile of large-scale protein conformational change by combining normal mode analysis and umbrella sampling molecular dynamics. J Phys Chem B. 2014;118:134–43. doi: 10.1021/jp4105129. [DOI] [PubMed] [Google Scholar]
- 37.Volpe DA, Faustino PJ, Ciavarella AB, Asafu-Adjaye EB, Ellison CD, Yu LX, et al. Classification of drug permeability with a Caco-2 cell monolayer assay. Clin Res Regul Aff. 2007;24:39–47. doi: 10.1080/10601330701273669. [DOI] [Google Scholar]
- 38.Chufan EE, Kapoor K, Ambudkar SV. Drug-protein hydrogen bonds govern the inhibition of the ATP hydrolysis of the multidrug transporter P-glycoprotein. Biochem Pharmacol. 2016;101:40–53. doi: 10.1016/j.bcp.2015.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin JH. Drug–drug interaction mediated by inhibition and induction of P-glycoprotein. Adv Drug Deliv Rev. 2003;55:53–81. doi: 10.1016/S0169-409X(02)00171-0. [DOI] [PubMed] [Google Scholar]
- 40.Zhang Y, Bachmeier C, Miller DW. In vitro and in vivo models for assessing drug efflux transporter activity. Adv Drug Deliv Rev. 2003;55:31–51. doi: 10.1016/S0169-409X(02)00170-9. [DOI] [PubMed] [Google Scholar]
- 41.Schinkel AH, Mayer U, Wagenaar E, Mol CA, van Deemter L, Smit JJ, et al. Normal viability and altered pharmacokinetics in mice lacking mdr1-type (drug-transporting) P-glycoproteins. Proc Natl Acad Sci USA. 1997;94:4028–33. doi: 10.1073/pnas.94.8.4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Volpe DA. Drug-permeability and transporter assays in Caco-2 and MDCK cell lines. Fut Med Chem. 2011;3:2063–77. doi: 10.4155/fmc.11.149. [DOI] [PubMed] [Google Scholar]
- 43.Polli JW, Wring SA, Humphreys JE, Huang LY, Morgan JB, Webster LO, et al. Rational use of in vitro P-glycoprotein assays in drug discovery. J Pharmacol Exp Ther. 2001;299:620–8. [PubMed] [Google Scholar]
- 44.Wang Q, Strab R, Kardos P, Ferguson C, Li J, Owen A, et al. Application and limitation of inhibitors in drug–transporter interactions studies. Int J Pharm. 2008;356:12–8. doi: 10.1016/j.ijpharm.2007.12.024. [DOI] [PubMed] [Google Scholar]
- 45.Lennernas H. Intestinal permeability and its relevance for absorption and elimination. Xenobiotica. 2007;37:1015–51. doi: 10.1080/00498250701704819. [DOI] [PubMed] [Google Scholar]
- 46.Ferreira RJ, Ferreira MJU, dos Santos DJVA. Do drugs have access to the P-glycoprotein drug-binding pocket through gates? J Chem Theory Comput. 2015;11:4525–9. doi: 10.1021/acs.jctc.5b00652. [DOI] [PubMed] [Google Scholar]
- 47.Bansal T, Akhtar N, Jaggi M, Khar RK, Talegaonkar S. Novel formulation approaches for optimising delivery of anticancer drugs based on P-glycoprotein modulation. Drug Discov Today. 2009;14:1067–74. doi: 10.1016/j.drudis.2009.07.010. [DOI] [PubMed] [Google Scholar]
- 48.Malingre MM, Beijnen JH, Rosing H, Koopman FJ, van Tellingen O, Duchin K, et al. A phase I and pharmacokinetic study of bi-daily dosing of oral paclitaxel in combination with cyclosporin A. Cancer Chemother Pharmacol. 2001;47:347–54. doi: 10.1007/s002800000226. [DOI] [PubMed] [Google Scholar]
- 49.Malingre MM, Richel DJ, Beijnen JH, Rosing H, Koopman FJ, Ten Bokkel Huinink WW, et al. Coadministration of cyclosporine strongly enhances the oral bioavailability of docetaxel. J Clin Oncol. 2001;19:1160–6. doi: 10.1200/JCO.2001.19.4.1160. [DOI] [PubMed] [Google Scholar]
- 50.Shan YQ, Ren G, Wang YX, Pang J, Zhao ZY, Yao J, et al. Berberine analogue IMB-Y53 improves glucose-lowering efficacy by averting cellular efflux especially P-glycoprotein efflux. Metabolism. 2013;62:446–56. doi: 10.1016/j.metabol.2012.09.009. [DOI] [PubMed] [Google Scholar]
- 51.Li YH, Yang P, Kong WJ, Wang YX, Hu CQ, Zuo ZY, et al. Berberine analogues as a novel class of the low-density-lipoprotein receptor up-regulators: synthesis, structure–activity relationships, and cholesterol-lowering efficacy. J Med Chem. 2009;52:492–501. doi: 10.1021/jm801157z. [DOI] [PubMed] [Google Scholar]
- 52.Cui HM, Zhang QY, Wang JL, Chen JL, Zhang YL, Tong XL. Poor permeability and absorption affect the activity of four alkaloids from Coptis. Mol Med Rep. 2015;12:7160–8. doi: 10.3892/mmr.2015.4288. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.