

Abstract
The persistence of latent HIV-1 reservoirs throughout combination antiretroviral therapy (cART) is a major barrier on the path to achieving a cure for AIDS. It has been shown that bromodomain and extra-terminal (BET) inhibitors could reactivate HIV-1 latency, but restrained from clinical application due to their toxicity and side effects. Thus, identifying a new type of BET inhibitor with high degrees of selectivity and safety is urgently needed. Apabetalone is a small-molecule selective BET inhibitor specific for second bromodomains, and has been evaluated in phase III clinical trials that enrolled patients with high-risk cardiovascular disorders, dyslipidemia, and low HDL cholesterol. In the current study, we examined the impact of apabetalone on HIV-1 latency. We showed that apabetalone (10−50 μmol/L) dose-dependently reactivated latent HIV-1 in 4 types of HIV-1 latency cells in vitro and in primary human CD4+ T cells ex vivo. In ACH2 cells, we further demonstrated that apabetalone activated latent HIV-1 through Tat-dependent P-TEFB pathway, i.e., dissociating bromodomain 4 (BDR4) from the HIV-1 promoter and recruiting Tat for stimulating HIV-1 elongation. Furthermore, we showed that apabetalone (10−30 μmol/L) caused dose-dependent cell cycle arrest at the G1/G0 phase in ACH2 cells, and thereby induced the preferential apoptosis of HIV-1 latent cells to promote the death of reactivated reservoir cells. Notably, cardiovascular diseases and low HDL cholesterol are known as the major side effects of cART, which should be prevented by apabetalone. In conclusion, apabetalone should be an ideal bifunctional latency-reversing agent for advancing HIV-1 eradication and reducing the side effects of BET inhibitors.
Keywords: latent HIV-1 reservoirs, latency reversing agent, BET inhibitors, apabetalone, P-TEFb, HIV eradication.
Introduction
Human immunodeficiency virus type 1 (HIV-1) is a viral antigen that causes incurable AIDS. Although combination antiretroviral therapy (cART) suppresses HIV-1 to undetectable levels and partially restores immune function in infected individuals [1], it produces serious side effects, such as lipid metabolism disorders and cardiovascular diseases [2]. Moreover, interruption of cART causes the virus to rapidly rebound to its pretreatment levels [3, 4]. The main cause of treatment failure is the existence of latent HIV-1 reservoirs [5–8]. Recent studies have explored a strategy named “shock and kill,” which would eradicate HIV-1 in latent HIV-1 reservoirs by activating HIV-1 transcription and viral antigen expression in the presence of cART [9–12]. When attempting to use this strategy, the first challenge is to find latency reversing agents (LRAs) to efficiently reactivate latent HIV-1. Several agents have been explored [13] and two types of LRAs have reached human testing. The histone deacetylase (HDAC) inhibitors romidepsin [14], vorinostat (SAHA) [15], and panobinostat [16, 17] transcriptionally reactivate latent HIV-1 but do so in a nonspecific manner [18, 19]. A second candidate, disulfiram, was proven effective as an LRA but failed to reduce the reservoir size [20, 21]. Therefore, the discovery of additional compounds and novel cellular targets will be required for an effective “shock and kill” strategy.
The pharmacological effects of bromodomain (BD) and extraterminal domain (BET) family members have recently received a great deal of attention as a possible therapeutic mechanism [22]. It is thought that targeting the binding site(s) of BET proteins to chromatin may be an effective means of regulating HIV-1 gene expression, particularly transcription elongation [23]. Several studies have reported that the BET inhibitors JQ1 and OTX015 could reactivate HIV-1 in different latency models [23–25]. However, both JQ1 and OTX015 are antitumor candidates, and their binding to both the first BD (BD1) and second BD (BD2) of BET proteins may increase their toxicity and side effects [26]. Recently, a novel BET inhibitor, apabetalone (RVX-208), has received much attention and is being developed by Resverlogix Corporation for the treatment of cardiovascular diseases and lipid metabolism disorders [27]. Apabetalone is specific for BET BDs and displays preferential binding to their BD2. Furthermore, it has high oral bioavailability and no reported side effects [28].
In this study, we examined the impact of apabetalone on HIV-1 latency. The results indicated that apabetalone could effectively reactivate latent HIV-1 by dissociating BDR4 from the HIV-1 promoter and recruiting Tat to stimulate HIV-1 elongation, and this effect was significantly enhanced when combined with prostratin or SAHA. Furthermore, we demonstrated that apabetalone produced cell cycle arrest at G1/G0 phase and thereby induced the preferential apoptosis of HIV-1 latent cells to promote the death of reactivated reservoir cells. Notably, low HDL cholesterol, lipid metabolism disorders, and cardiovascular diseases are all known side effects of cART and might be prevented by apabetalone. Therefore, apabetalone should be an ideal bifunctional LRA for advancing HIV eradication and reducing the side effects of cART.
Materials and methods
Cell culture
J-Lat A2 cells (harboring a single copy of latent HIV-1 long terminal repeat (LTR) and one copy of the green fluorescent protein (GFP) gene under HIV-1 LTR control), J-Lat 10.6 cells (containing a full-length integrated HIV-1 genome that expresses GFP upon activation), U1 cells (harboring two latent HIV-1 genomes with a defective Tat gene), and ACH2 cells (HIV-1 latent T-cell clone with one integrated proviral copy of Lymphaenopathy Associated Virus (LAV)) were cultured in RPMI 1640 medium with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin in a 37 °C incubator containing 5% CO2. All cell lines were obtained from the NIH AIDS Reagent Program. For reactivation of the HIV-1 LTR, cells were treated with prostratin (Sigma-Aldrich, St Louis, MO, USA), JQ1 (MCE, Vantaa, Finland), SAHA (vorinostat, MCE), or apabetalone (Selleck, Boston, MA) for 48 h. HIV-1 reactivation was quantified by measuring GFP expression with a flow cytometer. The data for J-Lat A2 cells and J-Lat 10.6 cells were analyzed using FlowJo Software, and the data for U1 cells and ACH2 cells were obtained using the HIV-1 p24 enzyme-linked immunosorbent assay (ELISA).
TZMbl cells used for transient transfection and luciferase assays were purchased from ATCC (Manassas, VA) and grown in Dulbecco’s modified Eagle’s medium supplemented with 10% FBS.
Isolation of CD4+ T cells from HIV-1-infected individuals
The HIV-infected patients were selected based on sustained plasma viral load suppression (plasma viral loads were < 20 copies/ml for > 12 months and their CD4 count was > 350 cells/ml) and their essential information is provided in Table S1. Human peripheral blood mononuclear cells (PBMCs) were isolated by Ficoll density gradient centrifugation using Histopaque-1077 (Sigma-Aldrich). Primary CD4+ T cells were isolated from populations of PBMCs with an EasySep kit (STEMCELL Technologies, Inc., Vancouver, BC, Canada) according to the manufacturer’s instructions and their purity was > 90%.
Cell viability assay
Cell viability was evaluated with a CCK-8 kit (Dojindo Molecular Technologies, Inc., Japan). Human PBMCs, J-Lat A2, J-Lat 10.6, ACH2, U1 cells (1 × 105 cells per well), or TZMbl cells (1 × 104 cells per well) were seeded into the wells of a 96-well plate and incubated with drugs for 48 h. Next, 10 μl of CCK-8 reagent was added to 100 μl of the cell culture mixture and incubated for an additional 4 h at 37 °C. Optical density was then recorded at a wavelength of 450 nm on a micro-plate reader (TECAN, Switzerland).
Detection of T-cell activation markers and HIV-1 receptors/co-receptors
Human PBMCs (1 × 106 cells per well) collected from healthy individuals were treated with drugs for 24 h or 72 h and then incubated with CD25-FITC, CD69-FITC, CD38-FITC, or HLA-DR-FITC antibodies (BD Biosciences, San Jose, CA, USA), respectively, at 4 °C for 30 min in the dark for purposes of surface staining. After staining, the cells were washed three times to eliminate fluorescence background interference and aliquots of 5 × 104 cells were collected for the analysis of T-cell activation markers via dual channels. Fluorescence values and dot plots were analyzed using FlowJo 7.6 software (Treestar, San Carlos, CA, USA).
Human PBMCs (1 × 106 cells per well) from healthy individuals were treated with drugs for 48 h and then incubated with either anti-CD4/anti-CCR5 antibodies or anti-CD4/anti-CXCR4 antibodies (BD Biosciences), respectively, at 4 °C for 30 min in the dark for purposes of surface staining. After staining, the cells were analyzed by flow cytometry.
ELISA for inflammatory cytokines
Supernatant fractions from PBMC cultures were collected at 24 h and 72 h after drug treatment and analyzed by ELISA assay according to the manufacturer’s protocols using the following: tumor necrosis factor (TNF)-α, interferon (IFN)-γ, interleukin (IL)-2, and IL-6 (eBioscience, San Diego, CA, USA).
Measurement of intracellular HIV-1 RNA transcripts
Isolated CD4+ T cells were either incubated with apabetalone or co-treated with LRAs for 48 h. Next, their total RNA was extracted using TRIZOL (Invitrogen, Carlsbad, CA, USA) and chloroform, and then precipitated with isopropanol. Reverse transcription of RNA to cDNA was performed according to a protocol provided with the PrimeScript RT Reagent Kit (TAKARA, Japan) and using gDNA Eraser (TAKARA) to remove genomic DNA. Quantitative PCR (qPCR) was performed using SYBR Select Master Mix (Applied Biosystems, Foster City, CA, USA) on the 7500 Real-Time PCR System (Applied Biosystems). The two-step procedure included a denaturation step (95 °C for 15 s) followed by annealing and extension (40 cycles of 60 °C for 60 s). The primers used for the HIV-1 LTR were as follows: forward (5′–3′) GCC TCC TAG CAT TTC GTC ACAT; reverse (5′–3′) GCT GCT TAT ATG TAG CAT CTG AGG. The 2−ΔΔCT method was used to analyze expression levels relative to the GAPDH gene.
Combination of apabetalone and anti-HIV drug luciferase assays
ACH2 cells (8 × 105 cells per well) were seeded into 96-well plates and then incubated with apabetalone (30 μM) and treated with anti-HIV-1 drugs, including Zidovudine (100 nM), Raltegravin (50 nM), Nevirapine (300 nM), and Plerisafor (100 nM), for 96 h at 37 °C. After centrifugation, cell debris was discarded and 100 μl supernatant was added into the 96-well polystyrene plates coated with TZMbl cells. After 48 h, TZMbl cells were lysed and luciferase activity was measured using the Dual-Luciferase Reporter Assay Kit (Promega, Madison, WI, USA) according to the manufacturer’s instructions.
Assessment of cART drugs’ antiviral activity in the presence or absence of apabetalone
The inhibitory activity of cART drugs against three different primary HIV-1 strains (HIV-1 IIIB (X4), Bal (R5), and 93BR020 (X4R5)) in the presence of preformed apabetalone was detected, respectively. Briefly, 1 × 105/ml TZMbl cells were seeded and incubated at 37 °C overnight. Apabetalone (30 μM) was incubated with Zidovudine, Raltegravin, Nevirapine, Maraviroc, or Plerisafor at graded concentrations, and the mixture was further coincubated with 2 ng of p24 of viruses at room temperature (RT) for 10 min before the addition of the mixture to TZMbl cells. At 3 h post infection, the culture supernatants were changed for fresh medium. At 72 h post infection, the luciferase activity was measured. The inhibition concentrations for 50% inhibition (IC50) values were calculated using Calcusyn software v. 40, kindly provided by Dr. T. C. Chou at Sloan-Kettering Cancer Center (New York, NY).
Transient transfection and luciferase assays
TZMbl cells were plated at 2 × 105 cells/well in 12-well culture plates 24 h before transfection and then transfected with either Tat or pcDNA 3.1 plasmids using Lipofectamine 3000 (Invitrogen) according to the manufacturer’s instructions. At 24 h post transfection, the cells were either mock-treated or treated with apabetalone. At 48 h post treatment, the cells were lysed and luciferase activity was measured using a Dual-Luciferase Reporter Assay Kit (Promega).
Protein extraction for western blot analysis
Following treatment, cells were lysed in RIPA lysis buffer (50 mM Tris HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.25% sodium deoxycholate, 0.1% SDS) and then incubated on ice for 10 min, after which they were centrifuged at 12,000 × g for 10 min at 4 °C. The supernatant fractions were collected for use as a whole protein extract. The nucleoproteins were extracted using NE-PER nuclear and cytoplasmic extraction reagents (Thermo Fisher Scientific, Carlsbad, CA, USA) according to the manufacturer’s protocol. The protein extract was quantified prior to being denatured by the addition of a loading buffer and then incubated at 100 °C for 10 min. The protein samples were either stored at − 80 °C or directly used for western blot analyses with the following antibodies: Tat (ab6539; Abcam), cyclin T1 (81464, CST), CDK9 (2316, CST), p-CDK9 (Thr186, sc-139604, Santa Cruz Biotechnology), Rpb1 CTD (2629, CST), P-Rpb1 CTD (Ser2, 13499, CST), p21waf1/Cip1 (2947, CST), CDK4 (12790, CST), CDK6 (13331, CST), cyclin D1 (2978, CST), Rb (9309, CST), p-Rb (Ser780, 8180, CST), p-Rb (Ser795, 9301, CST), and p-Rb (Ser807/811, 8516, CST).
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) assays were performed using kits (Millipore, Billerica, MA, USA) according to the manufacturer’s protocol and previously described procedures. Briefly, ACH2 cells (1 × 106 cells per well) were treated with apabetalone for 24 h, after which they were fixed in 4% formaldehyde, resuspended in lysis buffer, and sonicated to obtain DNA fragments of 500–1000 bp. The DNA fragments were incubated overnight at 4 °C with IgG, CDK9, BRD4, Tat, or Pol II CTD-Ser2P antibodies, and immune complexes were retrieved by incubation with Protein G agarose beads. After washing, the chromatin was eluted and reverse cross-linked overnight. qPCR was performed using SYBR Select Master Mix (Applied Biosystems) on the 7500 Real-Time PCR System (Applied Biosystems) using a standard two-step procedure (denaturation at 95 °C for 15 s, followed by 40 one-minute cycles of annealing/extension at 60 °C). The forward primer was 5′-AGACTGCTGACATCGAGCTTTCT-3′ and the reverse primer was 5′-GTGGGTTCCCTAGTTAGCCAGAG-3′. The results from fragments obtained after incubation with different antibodies were normalized against input DNA and are presented as fold-changes relative to a dimethyl sulfoxide control. The amplified DNA products (166 bp) were subjected to 2% agarose gel electrophoresis, and images were captured under the UV transilluminator (Alpha Innotech, CA, USA).
Cell cycle analysis
Cells were fixed with ice-cold 70% ethanol in phosphate-buffered saline (PBS) and then incubated with 50 μg/ml propidium iodide and 0.5 μg/ml RNase A at 37 °C for 0.5 h. The cells were then analyzed using a BD FACSCantoTM II flow cytometer (BD Biosciences). DNA histograms were generated and analyzed using FlowJo 7.6 software (Treestar, Inc., Ashland, OR, USA).
Annexin V labeling
After drug treatment, cells were collected and washed twice with PBS. Apoptosis was measured using a FITC Annexin V Apoptosis Detection Kit (BD Biosciences). The extent of apoptosis was quantified as the percentage of Annexin V-positive cells.
Quantitative analysis of synergy of LRA combinations
The Bliss independence model was adapted to calculate synergy when resveratrol was conventional LRAs. For drug x and drug y, faxy, P is the predicted fraction affected by a combination of drug x and drug y based on the experimentally observed fraction affected by drug x (ƒax) and drug y (ƒay) individually and is calculated from the equation ƒaxy, P = ƒax + ƒay − (ƒax)(ƒay). ƒaxy, O is the experimentally observed fraction affected by a combination of drug x and drug y. According to the Bliss model, ƒaxy, O was compared with ƒaxy, P with the following computation: Δƒaxy = ƒaxy, O − ƒaxy, P. If Δƒaxy > 0 showed statistical significance, the combination effect of two drugs exceeded that predicted by the Bliss model and the drug combination displayed synergy. If Δƒaxy < 0 showed statistical significance, the combination effect of two drugs was less than that predicted by the Bliss model and the drug combination displayed antagonism. In our analysis, ƒax = % GFP-positive cells after treatment with drug x − % GFP-positive cells of the untreated group.
Ethics statement
The patients’ peripheral blood was collected in the Eighth People’s Hospital of Guangzhou (Guangzhou, China) with written informed consent. The experiment was approved by the Ethical Committee of the Eighth People’s Hospital of Guangzhou and performed in accordance with relevant guidelines and regulations.
Statistical analysis
Experimental results are represented as the mean ± SD of at least three independent experiments. Statistical analyses of comparisons between groups were performed using one-way analysis of variance followed by Tukey’s t-test. All analyses were formed using GraphPad Prism 5.0 software (San Diego, CA, USA). P-values < 0.05 were considered statistically significant. P-values were defined as *P < 0.05 and **P < 0.01.
Results
Apabetalone reversed HIV-1 latency in vitro and ex vivo
The structures of apabetalone and JQ1 are shown in Fig. 1a,b. We initially assessed the ability of a biologically active of apabetalone to induce HIV-1 expression in J-Lat A2 and 10.6 cells, which contained a latent, transcriptionally competent HIV provirus that encodes a GFP gene as an indicator of reactivation. After treatment to J-Lat cells with apabetalone for 48 h, the percentages of GFP+ J-Lat A2 (Fig. 1c) and 10.6 cells (Fig. 1d) significantly increased as determined by flow cytometry. We further conducted the same studies using ACH2 cells and U1 cells, which were chronically infected with HIV-1. Results showed that apabetalone stimulated HIV-1 p24 expression in a dose-dependent manner in both ACH2 (Fig. 1e) and U1 cells (Fig. 1f) after 48 h of treatment.
Fig. 1.

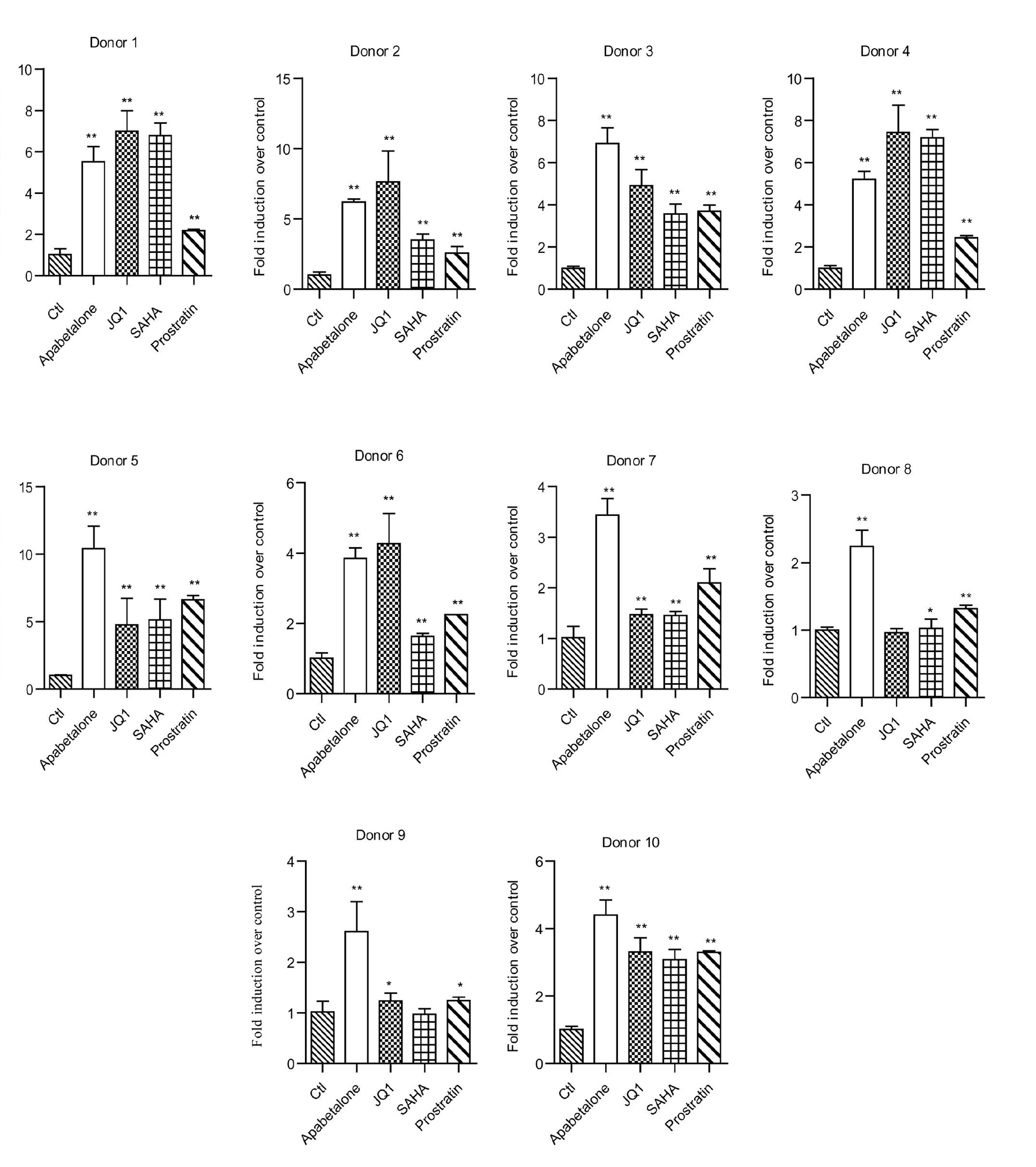
Apabetalone reversed HIV-1 latency in vitro and ex vivo. The chemical structure of a apabetalone and b JQ1. Apabetalone induced HIV-1 expression in c J-Lat A2 cells and d J-Lat 10.6 cells. Results are expressed as the percentage of GFP-positive cells within the entire population. Apabetalone promoted HIV-1 p24 antigen expression in e ACH2 cells and f U1 cells. g Apabetalone induced HIV-1 LTR expression in primary CD4+ T cells obtained from individuals receiving suppressive cART. All data are reported as the mean ± SD. The P-values are defined as *P < 0.05 and **P < 0.01 vs. control
We additionally investigated the ability of apabetalone to reactivate latent HIV-1 expression in isolated CD4+ T cells obtained from HIV-1-infected individuals receiving suppressive cART. Here, CD4+ T cells were incubated with apabetalone (30 μM), JQ1 (2 μM), SAHA (1 μM), or prostratin (1 μM) for 48 h, respectively. Then, the expression levels of HIV 5′-LTR transcription were analyzed by reverse transcription (RT)-qPCR. Apabetalone induced an increase in HIV-1 transcription in all ten donors, among which seven donors had a > 4-fold increase in intracellular HIV-1 mRNA levels compared with the negative control (Fig. 1g and Fig. S1). These results suggest that apabetalone is effective at reactivating transcription of HIV in latently infected cells in vitro and ex vivo.
Apabetalone displayed no toxicity and downregulated the HIV-1 receptor/co-receptors
The efficacy of an LRA depends on the balance between its specific activity and its safety. Therefore, we evaluated the cytotoxic effects of apabetalone on target cell lines, including human PBMCs, J-Lat A2, J-Lat 10.6, ACH2, U1, and TZMbl cells. After treatment with apabetalone for 48 h, CCK-8 assay was performed. Our experiment demonstrated that the CC50 of apabetalone up to 200 μM, which was ten times more than the CC50 of JQ1 (Table 1). The therapeutic index of apabetalone is > 8, indicating that it is safe and better than JQ1 when used at its reactivating concentration.
Table 1.
In vitro cytotoxicity of apabetalone and JQ1. Data were presented in means ± SD
| Cells | Apabetalone | JQ1 |
|---|---|---|
| CC50 (μM) | CC50 (μM) | |
| hPBMCs | > 500.00 | 37.53 ± 1.58 |
| J-Lat A2 | 248.57 ± 4.92 | 30.33 ± 1.66 |
| J-Lat 10.6 | 279.16 ± 6.32 | 26.79 ± 1.11 |
| ACH2 | 212.71 ± 7.24 | 18.76 ± 0.74 |
| U1 | 251.99 ± 5.78 | 21.61 ± 0.37 |
| TZMbl | 323.68 ± 3.46 | 31.44 ± 1.15 |
One of the major disadvantages of current therapeutic LRAs in clinical trials is the propensity to non-specifically activate bystander T cells. Here we further examined the expression of T-cell surface activation biomarkers following 24 and 72 h of stimulation with apabetalone in HIV-negative PBMCs. The results showed that apabetalone did not significantly affect the expression of T-cell biomarkers including CD69, CD25, CD38, and HLA-DR (Fig. 2a, b). However, significantly increased levels of T-cell activation biomarkers were observed after treatment with the positive control prostratin for both 24 h (Fig. 2a) and 72 h (Fig. 2b).
Fig. 2.

Apabetalone does not induce global T-cell activation and downregulated the HIV-1 receptor/co-receptor. Apabetalone did not induce the expression of T-cell activation markers after treatment for a 24 h or b 72 h in PBMCs from healthy HIV-negative donors. Data are reported as the mean ± SD. P-values are defined as *P < 0.05 and **P < 0.01 vs. control. c Apabetalone did not induce expression of pro-inflammatory cytokines in PBMCs from healthy HIV-negative donors. Data are reported as the mean ± SD. P-values are defied as *P < 0.05 and **P < 0.01 vs. control. d Apabetalone downregulated HIV receptor/co-receptor expression in PBMCs from healthy HIV-negative donors by FCM
It is important that any agent used for disrupting HIV latency does not exacerbate any unresolved chronic immune activation and inflammatory cytokine expression during HIV eradication interventions. We further measured pro-inflammatory cytokine expression following stimulation with apabetalone (30 μM) or prostratin (5 μM) for 24 or 72 h on HIV-negative PBMCs by ELISA. We found no significant increase in the expression of the TNF-α, IL-2, and IL-6 cytokines, and only a slight increase in IFN-γ expression (Fig. 2c), indicating that apabetalone did not inhibit the cytotoxic T lymphocyte (CTL) response of CD8+ T cells [29, 30].
Published data have identified that prostratin and JQ1 downregulate HIV receptor/co-receptor expression, which could protect cells against viral infection. Conversely, SAHA, which is known to induce latent HIV expression in HIV-infected individuals receiving cART, was reported to increase the susceptibility of naive CD4+ T cells to HIV acquisition. We sought to examine how apabetalone affected the expression of HIV receptors/co-receptors on the surface of HIV-negative PBMCs. The results showed that apabetalone significantly reduced the expression of the HIV co-receptors CCR5 and CXCR4 (Fig. 2d), suggesting that it might not pose a risk of increasing PBMCs susceptibility to HIV infection during its reactivation of HIV latency.
Apabetalone synergistically reactivated latent HIV-1 and could combine with cART drugs to inhibit viral infection
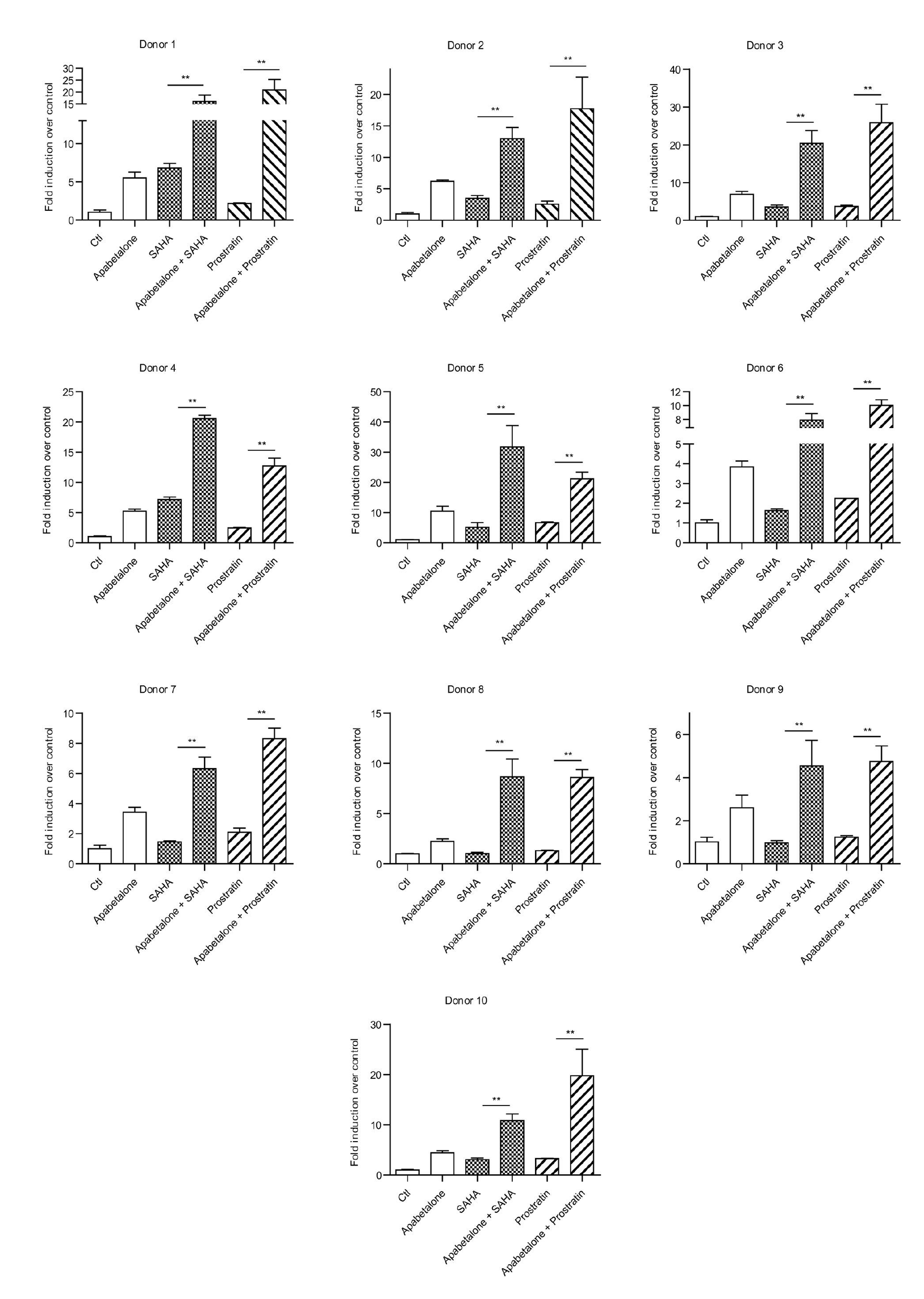
As several molecular pathways are involved in establishing and maintaining HIV-1 latency, our studies that examined the reactivation of latent HIV-1 were conducted using a protein kinase C agonist prostratin and a HDAC inhibitor SAHA in combination with apabetalone. J-Lat A2 cells were treated with SAHA (500 nM) or prostratin (500 nM) alone, or in combination with apabetalone (10 μM) for 48 h. A lower concentration was used in these assays, as apabetalone was found to be very potent in reactivating latent HIV-1 expression (Fig. 1) and the combined effects of apabetalone with other LRAs would be difficult to distinguish at higher concentrations. To assess whether the activity of the combined drugs met the criteria for drug synergy, we compared the experimentally observed effect of the combined agents to the effect predicted under the Bliss independence model described in the Materials and Methods. We observed that a combination of apabetalone and SAHA resulted in 28.80% of GFP+ cells, which was much higher than the predicted 14.17% of cells. The combination of apabetalone and prostratin resulted in 68.95% of GFP+ cells, which was much higher than the predicted 34.88% of cells (Fig. 3a). Similar results were observed in latent HIV-infected immune ACH2 cell lines. Although apabetalone (10 μM) alone induced an ~ 3.7-fold increase in latent HIV reactivation compared with controls, the addition of SAHA or prostratin increased HIV transcription > 10-fold and 83-fold, respectively (Fig. 3b). Those results indicated that apabetalone combined with SAHA or prostratin produced a synergistic effect on the reactivation of HIV-1 from latency in latent HIV-1 cell lines.
Fig. 3.

Apabetalone synergistically reactivated latent HIV-1 and could combine with cART drugs to inhibit viral infection. a Apabetalone combined with SAHA or prostratin had synergistic effects on the reactivation of latent HIV-1 in J-Lat A2 cells. *P < 0.05 and **P < 0.01 compared with LRA treatment alone. The Bliss independence model was utilized for calculating the synergy of LRA combinations. A dotted horizontal line signifies a purely additive effect (Δƒaxy = 0). Synergy is defined as Δƒaxy > 0, whereas Δƒaxy < 0 indicates antagonism. Statistical significance was determined using a one-tailed ratio t-test to compare the predicted and observed effects of drug combinations. b Apabetalone combined with SAHA or prostratin had synergistic effects on the reactivation of latent HIV-1 in ACH2 cells. *P < 0.05 and **P < 0.01 compared with LRA treatment alone. c Apabetalone combined with SAHA or prostratin had synergistic effects on the reactivation of latent HIV-1 in primary CD4+ T cells from individuals receiving suppressive cART. Data are reported as the mean ± SD from at least three independent experiments. P-values are defined as *P < 0.05 and **P < 0.01 compared with SAHA treatment alone. #P < 0.05 and ##P < 0.01 compared with prostratin treatment alone. d Combination of apabetalone with cART drugs to suppress infection of reactivated HIV-1. ACH2 cells (8 × 105) were treated with apabetalone in the presence or absence of cART drugs Zidovudine (100 nM), Raltegravir (50 nM), Nevirapine (300 nM), and Plerixafor (100 nM) for 96 h. The supernatants of ACH2 cells containing the residual reactivated HIV-1 were cultured with TZMbl cells for 48 h before testing residual HIV-1 infectivity using the luciferase assay. Data are reported as the mean ± SD from at least three independent experiments. P-values are defined as *P < 0.05 and **P < 0.01 compared with apabetalone treatment alone
The above results prompted us to examine whether a combination of SAHA or prostratin with apabetalone would significantly enhance the induction of latent HIV expression in CD4+ T cells obtained from HIV-1-infected individuals receiving suppressive cART. CD4+ T cells were treated with apabetalone (30 μM) alone or in combination with SAHA (500 nM) or prostratin (500 nM), and their HIV-1 transcription activity was measured by RT-qPCR. After 48 h of stimulation, the 5′-LTR assay indicated that the strong combined effect of apabetalone plus SAHA or prostratin induced a > 10-fold increase in HIV RNA transcription (Fig. 3c and Fig. S2). Thus, apabetalone given in combination with SAHA or prostratin exerted a synergic effect on the reactivation of HIV-1 transcription from latency in both HIV latency cell models and CD4+ T cells obtained from patients receiving suppressive cART.
To investigate whether apabetalone interfered with the effects of the antiretrovirals, we treated ACH2 cells with apabetalone and cART drugs including Zidovudine, Raltegravin, Nevirapine, and Plerisafor for 96 h. Then, the supernatant of ACH2 cells was added to TZMbl cells that contain an integrated HIV-1 LTR linked to the luciferase reporter gene in their genome to detect HIV-1 infection through luciferase assay. As shown in Fig. 3d, apabetalone antagonized HIV-1 latency efficiently at the concentration of 30 μM. However, the combination of apabetalone with cART drugs, a rapid decrease of HIV-1 expression could be evaluated by detecting the expression of luciferase reporter gene, which indicated that the virus reactivated by apabetalone could be inhibited by cART drugs.
Next, we wanted to know whether apabetalone would interfere with the anti-HIV activity of cART drugs. As shown in Table 2, although apabetalone could enhance HIV-1 infection, Zidovudine, Raltegravir, Nevirapine, Maraviroc, and Plerixafor still showed strong inhibitory activity against infection by all tested strains in the presence or absence of apabetalone. The IC50 values of cART drugs with or without apabetalone were similar across most HIV-1 strains. These data supported the notion that the combination of cART with apabetalone does not interfere with cART drugs’ antiretroviral activity, which suggested that the reactivation conducted under the cover of cART could lead to the selective killing of latently infected cells and the “purging” of the latent viral reservoir.
Table 2.
Inhibitory activity of cART drugs in the presence or absence of apabetalone on infection by HIV-1 clone strains
| Compounds | aInhibitory activity (mean ± SD) | |||||
|---|---|---|---|---|---|---|
| HIV-1NL4-3 (X4) | HIV-1SF162 (R5) | HIV-181A and NL4-3 (X4R5) | ||||
| EC50 (nM) | EC90 (nM) | EC50 (nM) | EC90 (nM) | EC50 (nM) | EC90 (nM) | |
| Zidovudine | 1.34 ± 0.17 | 14.80 ± 1.82 | 1.38 ± 0.13 | 8.05 ± 3.62 | 1.70 ± 0.35 | 23.40 ± 5.70 |
| Zidovudine + Apabetalone | 1.46 ± 0.20 | 26.92 ± 7.62 | 3.12 ± 0.11 | 34.02 ± 9.42 | 3.32 ± 0.64 | 35.74 ± 5.93 |
| Raltegravir | 0.18 ± 0.01 | 3.33 ± 0.76 | 0.09 ± 0.02 | 1.27 ± 0.56 | 0.13 ± 0.01 | 2.73 ± 0.32 |
| Raltegravir + Apabetalone | 0.15 ± 0.03 | 3.81 ± 0.43 | 0.02 ± 0.01 | 1.98 ± 0.18 | 0.07 ± 0.01 | 1.25 ± 0.44 |
| Nevirapine | 31.53 ± 11.43 | 179.00 ± 61.72 | 28.14 ± 6.35 | 157.34 ± 23.98 | 51.72 ± 11.52 | 214.38 ± 41.12 |
| Nevirapine + Aapbetalone | 23.77 ± 6.42 | 217.23 ± 64.57 | 35.71 ± 4.12 | 184.42 ± 35.91 | 45.07 ± 8.61 | 198.14 ± 27.33 |
| Maraviroc | / | / | 2.61 ± 0.46 | 12.80 ± 7.07 | 3.31 ± 0.84 | 41.88 ± 12.53 |
| Maraviroc + Apabetalone | / | / | 2.21 ± 0.18 | 12.25 ± 5.81 | 3.95 ± 0.45 | 48.21 ± 6.62 |
| Plerixafor | 10.38 ± 0.82 | 48.14 ± 8.77 | / | / | 18.14 ± 1.37 | 59.34 ± 8.39 |
| Plerxafor + Apabetalone | 9.22 ± 0.75 | 51.82 ± 4.97 | / | / | 20.38 ± 1.67 | 51.76 ± 5.44 |
Data were presented in means ± SD
Apabetalone activated latent HIV-1 by a Tat-dependent P-TEFb pathway
The HIV-1-encoded Tat protein has a key role in overcoming HIV-1 latency. Whether the reactivation of apabetalone was attributed to regulating the expression of Tat protein was unknown. Therefore, the expression of an integrated luciferase reporter gene driven by the HIV-1 LTR in the Hela-based TZMbl cells with or without Tat was detected first. The levels of Tat expression after transfection with 2 μg or 5 μg of Tat-plasmid are shown in Fig. 4a. Apabetalone (30 μM) alone induced LTR-driven luciferase expression by only 3.6-fold relative to the mock control. However, after transfection, the activation of HIV-1 LTR by apabetalone was dramatically increased up to 36.2-fold (2 μg Tat) and 49.4-fold (5 μg Tat), whereas Tat induced luciferase expression only by 8.3-fold (2 μg Tat) and 17.2-fold (5 μg Tat) (Fig. 4b).
Fig. 4.

Apabetalone stimulated latent HIV-1 via a Tat-dependent P-TEFb pathway. a The model of TZMbl cells transfected with Tat. HeLa-based TZMb1 cells were nucleofected with a plasmid expressing Tat ( + ) or a negative control plasmid ( − ) for 48 h; after which detected with Tat antibodies by western blotting. b Apabetalone predominantly stimulated Tat-dependent HIV-1 transcription. Whole cell extracts were assayed for luciferase activity. *P < 0.05 and **P < 0.01. Apabetalone increased cyclin T1 expression and induced CDK9 T-loop and RNAP II CTD phosphorylation in ACH2 cells in c dose-dependent and d time-dependent manners. e Apabetalone-induced BRD4 dissociation enabled Tat to recruit P-TEFb to the HIV promoter. The signals were normalized to those of the input. The error bars in all panels represent the mean ± SD. f The images were from agarose gel electrophorese. The input was made as 10% total amount and IgG was made as negative control
As transcription elongation from the HIV-1 promoter uniquely depends on P-TEFb, we wanted to know whether the effect of apabetalone on latent cells might influence P-TEFb activity. As shown in Fig. 4c,d, apabetalone treatment led to dose- and time-dependent increases in cyclin T1 expression, whereas CDK9 expression was not significantly changed. We further detected the CDK9 phosphorylation of Thr186. After treatment with apabetalone, CDK9 phosphorylation increased obviously. In addition, apabetalone treatment markedly enhanced the expression of the RNAP II C-terminal domain and its phosphorylation on Ser2, which in turn facilitates HIV-1 transcription elongation.
Next, we sought to determine whether apabetalone affected the occupancy of HIV-1 Tat, and the host cellular P-TEFb and RNAP II C-terminal domains of the HIV-1 promoter. Quantitative ChIP analyses were performed to detect the binding of these factors to a small region that overlapped with the HIV transcription starting site in ACH2 cells after treatment with apabetalone (30 μM). JQ1 (2 μM) served as a positive control. As expected, apabetalone induced a significant reduction (3.1-fold) in the level of BRD4 bound to the HIV-1 promoter when compared with the control. Moreover, it also significantly increased the levels of CDK9 and RNAP II CTD-Ser2P bound to the HIV-1 promoter by 3.4-fold and 2.6-fold, respectively (Fig. 4e). In addition, the ability of apabetalone to antagonize BRD4’s suppression of Tat-transactivation appeared to be achieved by removing BRD4 from the HIV-1 promoter, thereby allowing a 6.3-fold increase in the amount of Tat bound to the HIV-1 promoter (Fig. 4e). Corresponding to Fig. 4e, the PCR products were submitted to agarose electrophoresis and similar results were shown in Figure 4f.
Apabetalone preferentially induced apoptosis in HIV-1 latent cells
An ideal LRA should not only efficiently induce the activation of latent HIV-1 but also reduce the size of the HIV-1 latent reservoir. To explore whether apabetalone could induce apoptosis in HIV-1 latent cells, we used U937 cells and its subclone U1 cell line (a subclone of U937 that has been chronically infected with HIV-1). Intriguingly, although apabetalone (30 μM) failed to induce apoptosis in U937 cells, it induced significant levels of apoptosis among U1 cells in a dose-dependent manner after treatment for 72 h (Fig. 5a-c and Fig. S3B). In comparison, JQ1 (2 μM) induced apoptosis in both cell types and showed no selectivity. We also conducted the same experiment with CEM cells and their subclone ACH2 cells (CEM cells with one integrated proviral copy of LAV) and obtained similar results (Fig. 5d-f and Fig. S3C). These results are consistent with a preferential induction of apoptosis in apabetalone-treated HIV-expressing cells that leads to their elimination.
Fig. 5.

Apabetalone preferentially induced apoptosis in latent HIV-1 cells. a The percentage of apoptotic cells as determined by annexin-V staining of U937 cells or U1 cells (subclone of U937 that had been chronically infected with HIV-1) after 72 h of treatment with apabetalone. Summary of apoptotic effects of apabetalone in b U937 cells and c U1 cells are presented as histograms. The error bars in all panels represent the mean ± SD. d The percentage of apoptotic cells as determined by annexin-V staining of CEM cells or ACH2 cells (CEM cells with one integrated proviral copy of LAV) after 72 h of treatment with apabetalone. Summary of apoptotic effects of apabetalone in e CEM7 cells and f ACH2 cells are presented as histograms. The error bars in all panels represent the mean ± SD. **P < 0.01
Apabetalone induced apoptosis in HIV-1 latent cells by causing G1/G0 phase cell cycle arrest
To explore the mechanism by which apabetalone induces apoptosis in HIV-1 latent cells, we first performed a flow cytometric analysis to detect the effect of apabetalone on the ACH2 cell cycle. The results confirmed that apabetalone could induce G1/G0 cell cycle arrest in ACH2 cells in a dose-dependent manner. In particular, treatment with 30 μM apabetalone arrested 76.27% of the cells in their G1/G0 phase, and the percentages of cells in the S phase and G2 phase decreased accordingly (Fig. 6a).
Fig. 6.

Apabetalone specifically induced apoptosis in HIV-1 latent cells by causing p21waf1/Cip1-dependent G1/G0 phase cell cycle arrest. a Apabetalone induced G1/S phase arrest. Flow cytometry was used to determine the cell-cycle distribution of ACH2 cells treated with apabetalone for 48 h. The error bars in all panels represent the mean ± SD. **P < 0.01. b Apabetalone induced the expression of p21waf1/Cip1. c Apabetalone reduced the levels of cyclin D1 and CDK4/6 expression. d Apabetalone reduced the phosphorylation of Rb protein S780, S795, and S807/S811, and caused the accumulation of Rb protein
We next performed a western blot analysis to detect the expression of a cell cycle regulator, p21waf1/Cip1. As shown in Figure 6b, p21waf1/Cip1 expression was increased after 48 h of apabetalone (30 μM) treatment. Consistent with this finding, the expression levels of CDK4, CDK6, and cyclin D1, which are G1/S phase check point proteins, were significantly down-regulated (Fig. 6c). In addition, the phosphorylation levels of S780, S795, and S807/811 in the Rb protein, which are regulated by the cyclin–CDK complex, were also significantly reduced, resulting in the accumulation of Rb protein (Fig. 6d). In this manner, the cell cycle of treated cells was arrested in G1/G0 transition, leading to the apoptosis of HIV-1 latent-infected cells.
Discussion
BET proteins, which include four members in humans (BRD2, BRD3, BRD4, and the testis-specific BRDT), are transcriptional regulators that control the HIV-1 gene transcription elongation by binding acetylated histones [31–33]. BET proteins have two highly conserved BRD modules (BD1 and BD2), which show a high degree of sequence similarity but differ in their recognition of acetylated target sequences [28]. JQ1 is a first-generation pan-BET inhibitor and several studies have reported that JQ1 can reactivate HIV-1 in different latency models by inhibiting BRD2/4 [34–37]. However, JQ1 does not selectively bind the BD1 or BD2 domain of BET proteins, producing high degrees of cytotoxicity and side effects. Therefore, there is an urgent need to identify a new type of BET inhibitor that can effectively reactivate latent HIV-1 with high degrees of selectivity and safety.
Fortunately, sequence comparisons have revealed three residue positions in close proximity to the acetyl-lysine peptide binding site, which differs between the BD1 and BD2 domains [28]. The location of these residues suggests that these sequence variations should be explored for the purpose of developing inhibitors that specifically recognize one of the two BET BRDs [26]. Notably, apabetalone is a highly selective BET inhibitor that specifically targets the BD2 domain of BET proteins [28]. A microarray study showed that JQ1 strongly affects gene transcription with almost a 10-fold difference when compared with the BD2-specific inhibitor apabetalone. Apabetalone is being developed by Resverlogix Corporation for the treatment of cardiovascular diseases associated with atherosclerosis. In addition, apabetalone has recently entered clinical studies for the treatment of LDL dyslipidemia and recent phase III clinical data concerning the use apabetalone as an ApoA1 modulator have been encouraging [27, 38]. These disorders are all known as side effects of cART and might be prevented by apabetalone.
A recent study preliminarily showed that apabetalone could reactivate HIV-1 latency by increasing CDK9 Thr-186 phosphorylation [39]. However, our team has investigated the effects of apabetalone on latent HIV-1 and found that apabetalone could induce the preferential apoptosis of HIV-1 latent cells to promote the death of reactivated reservoir cells following viral reactivation, which makes apabetalone an ideal bifunctional LRA for HIV-1 eradication.
In the current study, the results indicated that apabetalone could significantly promote HIV-1 expression in various types of HIV-1 latency cells. Notably, the degrees of HIV-1 reactivation in the Tat-dependent latency cell models J-Lat A2 (Fig. 1c) and ACH2 (Fig. 1d) were significantly higher than the degrees of activation in the J-Lat 10.6 (Fig. 1e) and U1 cell models (Fig. 1f), suggesting that the effect of apabetalone on latent HIV-1 depends on Tat. Remarkably, we found that apabetalone induced the latent HIV-1 5’-LTR transcripts in primary CD4+ T cells from infected individuals receiving suppressive cART (Fig. 1g). Importantly, unlike other LRAs, such as Phorbol-12-myristate-13-acetate (PMA), prostratin, and bryostatin 1, which effectively reverse HIV-1 latency ex vivo but induce global T-cell activation and are too toxic for clinical use, apabetalone had no effect on T-cell activation (Fig. 2a,b) and pro-inflammatory cytokines (Fig. 2c), and showed no evidence of toxicity (Table 1). These traits may be related to data in previous reports that showed that apabetalone can act as an immunosuppressive agent and a chemokine inhibitor. All of these results support further investigations of apabetalone as a potential agent that can be incorporated into an HIV-1 curative strategy.
HIV-1 treatment strategies that employ combinational LRAs are widely assumed to be capable of efficiently reactivating latent HIV reservoirs [40, 41]. Prostratin and SAHA are conventional LRAs that work via different mechanisms. Apabetalone significantly promoted the reactivation effect of SAHA or prostratin in J-Lat A2 cells (Fig. 3a), ACH2 cells (Fig. 3b), and primary CD4+ T cells obtained from HIV-1-infected individuals receiving suppressive cART (Fig. 3c). The above results indicate that apabetalone is an efficacious and safe candidate for development as an HIV-1 LRA.
Notably, the role of cART drugs on viral clearance in the “shock and kill” strategy are crucial for preventing the spread of HIV-1 infection and achieving an HIV-1 cure [9, 42]. In this study, we combined apabetalone with different types of cART drugs with high antiviral activities including Zidovudine (nucleoside reverse transcriptase inhibitors (NRTI)), Raltegravir (HIV-1 integrase inhibitor), Nevirapine (non-NRTI), Maraviroc (CCR5-HIV-1 entry inhibitor), and Plerixafor (CXCR4-HIV-1 entry inhibitor). Our results showed that the infection of activated HIV-1 by apabetalone could significantly decrease after treatments of all tested cART drugs (Fig. 3d) and the combination of cART with apabetalone does not interfere with cART drugs’ antiretroviral activity (Table 2). This indicated that the combination of apabetalone with cART could protect uninfected target cells from becoming reinfected and is favorable to eliminating latent HIV-1 reservoirs.
Recent studies have reported that Tat has a critical role in JQ1 or OTX015-mediated reactivation of latent HIV-1 [43, 44] and this was also indicated by the reactivation activity conducted using apabetalone (Fig. 1c-f). To verify this hypothesis, we used TZMbl cells transfected with Tat to demonstrate that Tat has an important role in apabetalone-mediated HIV latent reactivation (Fig. 4a,b).
The promoter-proximal pausing of initiated RNAP II activity on integrated HIV-1 proviral DNA has long been recognized as a major rate-limiting step in viral gene expression [45]. To overcome this restriction, the viral protein Tat directly interacts with TAR and recruits P-TEFb, which is composed of CDK9 and cyclin T1, to the HIV-1 LTR. Enzymatically active CDK9, which requires the phosphorylation of Tr186 in its T-loop, phosphorylates the C-terminal domain of RNAP II and negative elongation factors, and thereby increases the rate of HIV-1 transcription [46, 47]. Results show that apabetalone-mediated reactivation involved increases in cyclin T1, CDK9 T-loop phosphorylation, and RNAP II CTD phosphorylation, all of which facilitated transcriptional elongation of the HIV-1 gene (Fig. 4c,d). The ChIP analysis showed that apabetalone can inhibit BRD4, but promote CDK9, Tat, and RNAP II CTD-Ser2P recruitment directly to the HIV-1 LTR (Fig. 4e,f). As apabetalone specifically binds to the BD2 domain of BRD2/BRD4, the role of BRD2 in apabetalone-mediated reactivation remains unknown. Further experiments are needed to explore the relationship between BRD2 and HIV-1 latency.
The reactivation of latent HIV-1 (“shock”) is not always concomitant with a reduction of the inducible latent reservoir (“kill”). For example, reactivation with SAHA failed to reduce the latent reservoir ex vivo unless CTLs were present and primed with antigen [48]. We demonstrated that apabetalone could induce a specific apoptosis of HIV latent cell lines (Fig. 5). Previous studies have shown that BET inhibitors can induce cell apoptosis by reducing c-Myc expression and inducing G1/G0 cell cycle arrest [49, 50]. p21waf1/Cip1 is a cyclin-dependent kinase inhibitor controlling G1/G0 cell-cycle progression through activation of cyclin D1-CDK4/6 complexes, regulates the expression and phosphorylation of the downstream Rb protein [51]. Results show that p21waf1/Cip1 expression was induced by apabetalone treatment (Fig. 6b), and this was followed by decreases in cyclin D1, CDK4, and CDK6 expression (Fig. 6c). Apabetalone impedes the function of the cyclinD/CDK complex by reducing cyclin D1 and CDK4/6 levels, and the phosphorylation of S780, S795, and S807/S811of Rb protein; in turn, this results in the accumulation of Rb proteins (Fig. 6d). Therefore, the cells become arrested at the G1/G0 checkpoint and enter apoptosis.
It is well known that resting CD4+ T cells harboring integrated proviruses would enter cell cycle progression for differentiation and proliferation after LRA stimulation [8]. Apabetalone could selectively reactivate the resting CD4+ T cells harboring integrated proviruses via a Tat-dependent pathway, induce them to enter the cell cycle and arrest them in the G1/G0 transition, leading to the apoptosis of HIV-1 latent-infected cells. It was notable that the pan-BET inhibitor JQ1 induced apoptosis in all the aforementioned cells. Previous studies have shown that JQ1 binds to both the BD1 and BD2 domains of BET proteins, whereas apabetalone only binds strongly to the BD2 domain [28]. In this way, apabetalone regulates far fewer genes than JQ1, which may explain the low toxicity and high selectivity of apabetalone. More possible scenarios can also account for these results. For example, a direct cytotoxic effect of HIV-1 may be responsible for this apoptosis. This would be in keeping with the fact that the latency model lacks the presence of natural killer or CTL cells that could otherwise eliminate reactivated cells [52]. Further investigations in this area are warranted.
In summary, we have provided strong evidence that the BET inhibitor apabetalone is a potent antagonist of HIV-1 latency. Apabetalone acts by activating P-TEFb via dissociating BDR4 from the HIV-1 promoter and recruiting Tat for stimulating HIV-1 elongation. Furthermore, apabetalone can down-regulate cyclin D1 expression, upregulate p21waf1/Cip1, and induce G1/G0 phase cell cycle arrest. It was particularly interesting that apabetalone induced the preferential apoptosis of HIV-1 latent cells and further promoted the death of reactivated reservoir cells. In conclusion, its low degree of cytotoxicity, coupled with its abilities to reactivate latent HIV-1 reservoirs, induce HIV-1 latent cell apoptosis, and reduce the side effects of cART, all make apabetalone worth investigating for development as a possible LRA for use in accelerating HIV-1 eradication.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers, and other organisations engaged with supporting research.
Electronic supplementary material
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank Dr. Shibo Jiang and Dr. Lu Lu at Fudan University, China, for generously providing HIV-1 latent cell models and reagents. This study was financially supported by grants from the Natural Science Foundation of China (81773787 to SL and 81673481 to LL) and National Science and Technology Major Project (2018ZX10301101 to SL).
Author contributions
XZ, LL, and SL conceived the project and wrote the manuscript. XZ, JL, and TL performed the experiments. HD and BX analyzed the data. XT provided the clinical samples. All authors reviewed the manuscript. LL and SL supervised the project and revised the manuscript.
Competing interests
The authors declare that the research was conducted in the absence of any commercial or finacial relationships that could be construed as a potential confict of interest.
Contributor Information
Lin Li, Email: li75lin@126.com.
Shu-wen Liu, Email: liusw@smu.edu.cn.
Electronic supplementary material
The online version of this article (10.1038/s41401-018-0027-5) contains supplementary material, which is available to authorized users.
References
- 1.Perelson AS, Essunger P, Cao Y, Vesanen M, Hurley A, Saksela K, et al. Decay characteristics of HIV-1-infected compartments during combination therapy. Nature. 1997;387:188–91. doi: 10.1038/387188a0. [DOI] [PubMed] [Google Scholar]
- 2.Vidal F, Gutierrez F, Gutierrez M, Olona M, Sanchez V, Mateo G, et al. Pharmacogenetics of adverse effects due to antiretroviral drugs. AIDS Rev. 2010;12:15–30. [PubMed] [Google Scholar]
- 3.Davey RT, Jr., Bhat N, Yoder C, Chun TW, Metcalf JA, Dewar R, et al. HIV-1 and T cell dynamics after interruption of highly active antiretroviral therapy (HAART) in patients with a history of sustained viral suppression. Proc Natl Acad Sci USA. 1999;96:15109–14. doi: 10.1073/pnas.96.26.15109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li TS, Tubiana R, Katlama C, Calvez V, Ait Mohand H, Autran B. Long-lasting recovery in CD4 T-cell function and viral-load reduction after highly active antiretroviral therapy in advanced HIV-1 disease. Lancet (Lond, Engl) 1998;351:1682–6. doi: 10.1016/S0140-6736(97)10291-4. [DOI] [PubMed] [Google Scholar]
- 5.Chun TW, Stuyver L, Mizell SB, Ehler LA, Mican JA, Baseler M, et al. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc Natl Acad Sci USA. 1997;94:13193–7. doi: 10.1073/pnas.94.24.13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Finzi D, Blankson J, Siliciano JD, Margolick JB, Chadwick K, Pierson T, et al. Latent infection of CD4 + T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat Med. 1999;5:512–7. doi: 10.1038/8394. [DOI] [PubMed] [Google Scholar]
- 7.Barouch DH, Deeks SG. Immunologic strategies for HIV-1 remission and eradication. Science. 2014;345:169–74. doi: 10.1126/science.1255512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hosmane NN, Kwon KJ, Bruner KM, Capoferri AA, Beg S. Proliferation of latently infected CD4 + T cells carrying replication-competent HIV-1: potential role in latent reservoir dynamics. J Exp Med. 2017;214:959–72. doi: 10.1084/jem.20170193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deeks SG. HIV: shock and kill. Nature. 2012;487:439–40. doi: 10.1038/487439a. [DOI] [PubMed] [Google Scholar]
- 10.Margolis DM, Garcia JV, Hazuda DJ, Haynes BF. Latency reversal and viral clearance to cure HIV-1. Science. 2016;353:aaf6517. doi: 10.1126/science.aaf6517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ruelas DS, Greene WC. An integrated overview of HIV-1 latency. Cell. 2013;155:519–29. doi: 10.1016/j.cell.2013.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu W, Li H, Wang Q. Advancements in developing strategies for sterilizing and functional HIV cures. Biomed Res Int. 2017;2017:6096134. doi: 10.1155/2017/6096134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang W, Sun Z, Hua C, Wang Q, Xu W, Deng Q, et al. Chidamide, a histone deacetylase inhibitor-based anticancer drug, effectively reactivates latent HIV-1 provirus. Microbes Infect. 2017. pii: S1286-4579(17)30178-8. doi: 10.1016/j.micinf.2017.10.003. [DOI] [PubMed]
- 14.Sogaard OS, Graversen ME, Leth S, Olesen R, Brinkmann CR, Nissen SK, et al. The depsipeptide romidepsin reverses HIV-1 latency in vivo. PLoS Pathog. 2015;11:e1005142. doi: 10.1371/journal.ppat.1005142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elliott JH, Wightman F, Solomon A, Ghneim K, Ahlers J, Cameron MJ, et al. Activation of HIV transcription with short-course vorinostat in HIV-infected patients on suppressive antiretroviral therapy. PLoS Pathog. 2014;10:e1004473. doi: 10.1371/journal.ppat.1004473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Olesen R, Vigano S, Rasmussen TA, Sogaard OS, Ouyang Z, Buzon M, et al. Innate immune activity correlates with CD4 T cell-associated HIV-1 DNA decline during latency-reversing treatment with panobinostat. J Virol. 2015;89:10176–89. doi: 10.1128/JVI.01484-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rasmussen TA, Tolstrup M, Brinkmann CR, Olesen R, Erikstrup C, Solomon A, et al. Panobinostat, a histone deacetylase inhibitor, for latent-virus reactivation in HIV-infected patients on suppressive antiretroviral therapy: a phase 1/2, single group, clinical trial. Lancet HIV. 2014;1:e13–21. doi: 10.1016/S2352-3018(14)70014-1. [DOI] [PubMed] [Google Scholar]
- 18.Gallo RC. Shock and kill with caution. Science. 2016;354:177–8. doi: 10.1126/science.aaf8094. [DOI] [PubMed] [Google Scholar]
- 19.Siliciano JD, Siliciano RF. Recent developments in the effort to cure HIV infection: going beyond N = 1. J Clin Invest. 2016;126:409–14. doi: 10.1172/JCI86047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Spivak AM, Andrade A, Eisele E, Hoh R, Bacchetti P, Bumpus NN, et al. A pilot study assessing the safety and latency-reversing activity of disulfiram in HIV-1-infected adults on antiretroviral therapy. Clin Infect Dis: Off Publ Infect Dis Soc Am. 2014;58:883–90. doi: 10.1093/cid/cit813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xing S, Bullen CK, Shroff NS, Shan L, Yang HC, Manucci JL, et al. Disulfiram reactivates latent HIV-1 in a Bcl-2-transduced primary CD4 + T cell model without inducing global T cell activation. J Virol. 2011;85:6060–4. doi: 10.1128/JVI.02033-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith SG, Zhou MM. Bromodomains as targets for therapeutic intervention. Drug Discov Today Technol. 2016;19:1–2. doi: 10.1016/j.ddtec.2016.09.002. [DOI] [PubMed] [Google Scholar]
- 23.Banerjee C, Archin N, Michaels D, Belkina AC, Denis GV, Bradner J, et al. BET bromodomain inhibition as a novel strategy for reactivation of HIV-1. J Leukoc Biol. 2012;92:1147–54. doi: 10.1189/jlb.0312165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li Z, Guo J, Wu Y, Zhou Q. The BET bromodomain inhibitor JQ1 activates HIV latency through antagonizing Brd4 inhibition of Tat-transactivation. Nucleic Acids Res. 2013;41:277–87. doi: 10.1093/nar/gks976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu P, Qu X, Shen Y, Jiang Z, Wang P, Zeng H, et al. The BET inhibitor OTX015 reactivates latent HIV-1 through P-TEFb. Sci Rep. 2016;6:24100. doi: 10.1038/srep24100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–73. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bailey D, Jahagirdar R, Gordon A, Hafiane A, Campbell S, Chatur S, et al. RVX-208: a small molecule that increases apolipoprotein A-I and high-density lipoprotein cholesterol in vitro and in vivo. J Am Coll Cardiol. 2010;55:2580–9. doi: 10.1016/j.jacc.2010.02.035. [DOI] [PubMed] [Google Scholar]
- 28.Picaud S, Wells C, Felletar I, Brotherton D, Martin S, Savitsky P, et al. RVX-208, an inhibitor of BET transcriptional regulators with selectivity for the second bromodomain. Proc Natl Acad Sci USA. 2013;110:19754–9. doi: 10.1073/pnas.1310658110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deng K, Pertea M, Rongvaux A, Wang L, Durand CM, Ghiaur G, et al. Broad CTL response is required to clear latent HIV-1 due to dominance of escape mutations. Nature. 2015;517:381–5. doi: 10.1038/nature14053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Walker-Sperling VE, Pohlmeyer CW, Tarwater PM, Blankson JN. The effect of latency reversal agents on primary CD8 + T cells: implications for shock and kill strategies for human immunodeficiency virus eradication. EBioMedicine. 2016;8:217–29. doi: 10.1016/j.ebiom.2016.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011;478:529–33. doi: 10.1038/nature10509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Denis GV. Bromodomain coactivators in cancer, obesity, type 2 diabetes, and inflammation. Discov Med. 2010;10:489–99. [PMC free article] [PubMed] [Google Scholar]
- 33.Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478:524–8. doi: 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bartholomeeusen K, Xiang Y, Fujinaga K, Peterlin BM. Bromodomain and extra-terminal (BET) bromodomain inhibition activate transcription via transient release of positive transcription elongation factor b (P-TEFb) from 7SK small nuclear ribonucleoprotein. J Biol Chem. 2012;287:36609–16. doi: 10.1074/jbc.M112.410746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhu J, Gaiha GD, John SP, Pertel T, Chin CR, Gao G, et al. Reactivation of latent HIV-1 by inhibition of BRD4. Cell Rep. 2012;2:807–16. doi: 10.1016/j.celrep.2012.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Belkina AC, Nikolajczyk BS, Denis GV. BET protein function is required for inflammation: Brd2 genetic disruption and BET inhibitor JQ1 impair mouse macrophage inflammatory responses. J Immunol (Baltim, Md: 1950) 2013;190:3670–8. doi: 10.4049/jimmunol.1202838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Karn J. A new BET on the control of HIV latency. Cell Cycle. 2013;12:545–6. doi: 10.4161/cc.23679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nicholls SJ, Gordon A, Johannson J, Ballantyne CM, Barter PJ, Brewer HB, et al. ApoA-I induction as a potential cardioprotective strategy: rationale for the SUSTAIN and ASSURE studies. Cardiovasc Drugs Ther. 2012;26:181–7. doi: 10.1007/s10557-012-6373-5. [DOI] [PubMed] [Google Scholar]
- 39.Lu P, Shen Y, Yang H, Wang Y, Jiang Z, Yang X, et al. BET inhibitors RVX-208 and PFI-1 reactivate HIV-1 from latency. Sci Rep. 2017;7:16646. doi: 10.1038/s41598-017-16816-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Burnett JC, Lim KI, Calafi A, Rossi JJ, Schaffer DV, Arkin AP. Combinatorial latency reactivation for HIV-1 subtypes and variants. J Virol. 2010;84:5958–74. doi: 10.1128/JVI.00161-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Laird GM, Bullen CK, Rosenbloom DI, Martin AR, Hill AL, Durand CM, et al. Ex vivo analysis identifies effective HIV-1 latency-reversing drug combinations. J Clin Invest. 2015;125:1901–12. doi: 10.1172/JCI80142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schwartz C, Bouchat S, Marban C, Gautier V, Van Lint C, Rohr O, et al. On the way to find a cure: Purging latent HIV-1 reservoirs. Biochem Pharmacol. 2017;146:10–22. doi: 10.1016/j.bcp.2017.07.001. [DOI] [PubMed] [Google Scholar]
- 43.Boehm D, Calvanese V, Dar RD, Xing S, Schroeder S, Martins L, et al. BET bromodomain-targeting compounds reactivate HIV from latency via a Tat-independent mechanism. Cell Cycle. 2013;12:452–62. doi: 10.4161/cc.23309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mousseau G, Kessing CF, Fromentin R, Trautmann L, Chomont N, Valente ST. The Tat inhibitor didehydro-cortistatin A prevents HIV-1 reactivation from latency. mBio. 2015;6:e00465. doi: 10.1128/mBio.00465-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cary DC, Fujinaga K, Peterlin BM. Molecular mechanisms of HIV latency. J Clin Invest. 2016;126:448–54. doi: 10.1172/JCI80565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Budhiraja S, Rice AP. Reactivation of latent HIV: do all roads go through P-TEFb? Future Virol. 2013;8:649–59. doi: 10.2217/fvl.13.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim YK, Mbonye U, Hokello J, Karn J. T-cell receptor signaling enhances transcriptional elongation from latent HIV proviruses by activating P-TEFb through an ERK-dependent pathway. J Mol Biol. 2011;410:896–916. doi: 10.1016/j.jmb.2011.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shan L, Deng K, Shroff NS, Durand CM, Rabi SA, Yang HC, et al. Stimulation of HIV-1-specific cytolytic T lymphocytes facilitates elimination of latent viral reservoir after virus reactivation. Immunity. 2012;36:491–501. doi: 10.1016/j.immuni.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kandela I, Jin HY, Owen K. Registered report: BET bromodomain inhibition as a therapeutic strategy to target c-Myc. eLife. 2015;4:e07072. doi: 10.7554/eLife.07072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Maruyama T, Farina A, Dey A, Cheong J, Bermudez VP, Tamura T, et al. A mammalian bromodomain protein, brd4, interacts with replication factor C and inhibits progression to S phase. Mol Cell Biol. 2002;22:6509–20. doi: 10.1128/MCB.22.18.6509-6520.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tan EP, Duncan FE, Slawson C. The sweet side of the cell cycle. Biochem Soc Trans. 2017;45:313–22. doi: 10.1042/BST20160145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brockman MA, Jones RB, Brumme ZL. Challenges and opportunities for T-cell-mediated strategies to eliminate HIV reservoirs. Front Immunol. 2015;6:506. doi: 10.3389/fimmu.2015.00506. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.