

Abstract
Chromosomal conformations, including promoter-enhancer loops, provide a critical regulatory layer for the transcriptional machinery. Therefore, schizophrenia, a common psychiatric disorder associated with broad changes in neuronal gene expression in prefrontal cortex and other brain regions implicated in psychosis, could be associated with alterations in higher order chromatin. Here, we review early studies on spatial genome organization in the schizophrenia postmortem brain and discuss how integrative approaches using cell culture and animal model systems could gain deeper insight into the potential roles of higher order chromatin for the neurobiology of and novel treatment avenues for common psychiatric disease.
Keywords: schizophrenia, epigenetics, chromatin, 3D genome, gene regulation, promoter-enhancer loops
INTRODUCTION TO THE EPIGENOMICS OF SCHIZOPHRENIA
Schizophrenia is a major psychiatric disorder often with onset in adolescence or young adulthood, with a wide range of symptoms ranging from delusions and hallucinations to severe social withdrawal and apathy, along with reduced lifespan primarily due to cardiovascular disease and suicide 1–3. Antipsychotic medications are widely prescribed and mostly target dopaminergic and serotonergic receptor systems4, 5 but often fail to deliver a satisfactory therapeutic response 6, 7. Cognitive symptoms in particular are severe, chronically disabling, and often persistent during the course of illness8. Regrettably, these symptoms are ineffectively treated with antipsychotic medication9. The genetic risk architecture of schizophrenia is exceedingly complex. For example, in a study involving 150,000 subjects, the Psychiatric Genomics Consortium identified altogether 108 haplotypes that by individual small effect contribute to the heritable risk for schizophrenia10. In addition, there are rare mutations and variants discovered by comprehensive sequencing of all protein coding genes (the ‘exome’, which comprises approximately 1% of total genome sequence)11, some of which may be causal to the disease etiology in some of the cases with schizophrenia12.
Importantly, there can be little doubt that dysregulation of neuronal gene expression in prefrontal cortex (PFC) and various other brain regions implicated in the neural circuitry of psychosis contributes to the pathophysiology of schizophrenia, broadly affecting excitatory and inhibitory neurotransmission, metabolism, myelination and immune signaling13–19. With the transcriptional process intimately connected to chromatin structure and function in human cells and model organisms alike20, 21, one would therefore expect that epigenomic markers associated with open (‘active’, ‘loose’) chromatin permissive for gene expression, versus repressed and silenced chromatin, will show significant alterations in brain tissue from subjects diagnosed with schizophrenia. Indeed, work conducted over the course of the last 15 years has provided first insights into epi-(Greek for ‘over’, ‘above’)-genomic aberrations encountered in brain tissue from subjects diagnosed with schizophrenia. Such types of epigenomic explorations in the postmortem brain initially focused on DNA methylation, one of the key epigenetic mechanisms involved in the regulation of gene expression22. Methylation at the cytosine C5 position, primarily in the context of cytosine-guanine (CpG) dinucleotides, when located in gene promoters often is implicated in gene repression by directly impeding the binding of transcription factors and by inducing repressive chromatin structure non-permissive to transcription23. Early studies, examining the DNA methylation status of candidate genes affected by dysregulated expression in brains of schizophrenia reported differential DNA methylation profiles in diseased cerebral cortex for key regulators of neuronal connectivity such as REELIN (RELN) 24, 25 and key transcription factors such as sex-determining region Y-box containing gene 10 (SOX10) 26, to mention just two examples. Recent genome-wide mapping of the DNA methylome 27, 28 reported DNA methylation changes for various genes implicated in excitatory or inhibitory neurotransmission, with one study exploring the DNA methylome in the prefrontal cortex of 191 subjects with schizophrenia in comparison to 335 controls collected across the lifespan identifying >2000 CpG sites with altered methylation levels in diseased tissue29. However, the functional implications of the overall extremely subtle methylation differences (on average, 1.3% difference between schizophrenia and control brains for significantly affected CpG sites) in the aforementioned study29 remain unclear.
Future studies, exploring the DNA methylome of specific brain cell populations in diseased vs. control brains30, 31 as opposed to the earlier studies which utilized tissue homogenate or correlational analyses between the brain’s DNA methylomes and structural or functional defects in neurons32, 33, may provide deeper insight into the role of epigenetic dysregulation affecting the brains of subjects with schizophrenia. Of note, some of the epigenomic determinants of chromatin structure and function in fetal and adult human brain, including histone methylation and acetylation markings associated with cis-regulatory sequences such as gene promoters, enhancers and repressors, show an impressive, up to 26-fold enrichment for single nucleotide polymorphisms associated with heritable risk for schizophrenia34. These effects were highly tissue-specific, because brain histone methylation landscapes showed no enrichment for polymorphisms associated with rheumatoid arthritis and other common disorders usually not affecting the central nervous system34. Therefore, given that 1) DNA methylation alterations have been reported in postmortem brain of subjects with schizophrenia, and 2) the significant association of histone modification landscapes from brain cells with the genetic risk architecture of the disease, it is very likely that ‘epigenomic dysregulation’—including alterations in the expression of specific genes or disruptions in the coordinated regulation of multiple transcriptional units—could be key drivers underlying cortical dysfunction in schizophrenia.
REGULATION OF CHROMOSOMAL CONFORMATIONS IN NEURODEVELOPMENT
As discussed in the Introduction, evidence arising both from candidate gene studies and genome-scale mappings of DNA methylation and histone modifications provided strong support for the hypothesis that alterations in chromatin structure and function could contribute to transcriptional dysregulation in brains of subject with schizophrenia. However, as discussed in a recent review35, comprehensive exploration of the epigenome in schizophrenia and other cognitive disease has to go far beyond DNA methylation, histone modifications and other regulators of gene expression that are typically ‘charted’ as epigenetic signals directly onto and above the ‘linear’ genome in two dimensions (Figure 2.1). Instead, it is increasingly recognized that the spatial configuration and packaging of interphase chromosomes, the equivalent of 250 cm of DNA from a single nucleus when unwound, involves myriads of non-randomly occurring chromosomal loop structures and DNA-DNA proximity conformations that bypass the linear genome across a wide range of genomic distance, from hundreds of base pairs (such as a short loop formation connecting a transcription start site and proximal gene promoter with a nearby enhancer) to many megabases of interspersed sequence (as in the case of some long-range contacts on the inactive X chromosome in female somatic cells). Proper regulation of these types of 3D genome structures are considered critical for the regulation of gene expression, maintenance of genome integrity and stability, control of growth and differentiation, among many other functions. Surprisingly, however, there is extremely little knowledge about the regulation of the three-dimensional (3D) genome in developing or mature brain, including potential alterations in neuropsychiatric disease, such as schizophrenia.
Figure 2.1:
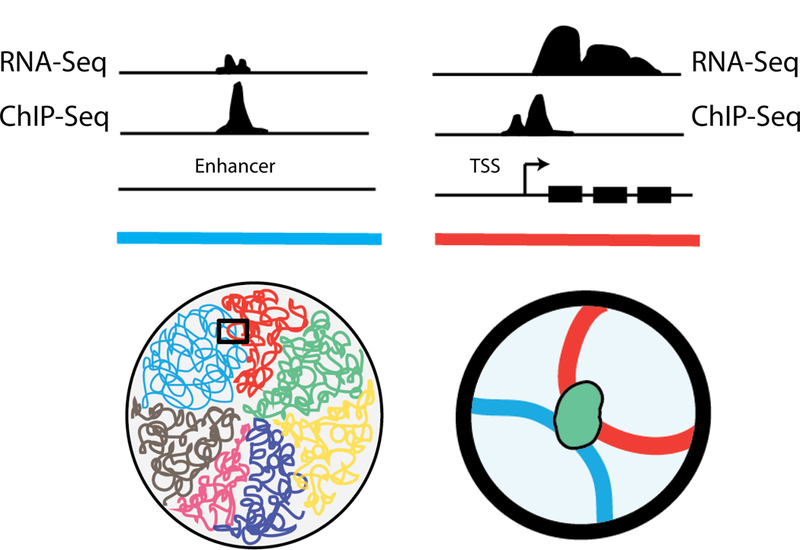
(Top) Conventional ChIP-Seq and RNA-seq tracks on the linear genome demonstrate enhancer and promoter features. (Bottom) However, these traditional approaches miss the fact that the two elements, although quite distal in the linear genome, are actually in close physical proximity through looping interactions.
Building blocks of the 3D genome
We will start our discussion of the 3D genome and its implications for schizophrenia research with a brief overview of its basic principles and organization. Nuclei, separated by a nuclear membrane from the cytoplasm, contain the genome packaged into chromatin fibers as nucleosomal arrays. Nucleosomes are comprised of 146bp DNA wrapped around a core histone octamer, and interconnected by linker DNA and linker histones. Chromatin can exist in different ‘states’, including ‘open’ (eu-) and condensed (hetero-) chromatin. These are differentially defined by three characteristics: (1) loose or dense nucleosomal packaging, (2) specific types of post-translational histone modifications, and (3) presence or absence of various chromatin regulatory proteins that either facilitate or repress transcription. For example, actively expressed genes in open chromatin show high levels of histone acetylation, with nucleosome-free intervals occupied by activator proteins (transcription factors) and the RNA polymerase II initiation complex (Figure 2.2). Superimposed upon this nucleosomal organization is the 3D conformation of chromatin fibers and entire chromosomes, often described in terms such as ‘loops’ or ‘globules’ and in toto referred to as the ‘3D genome’. These chromosomal conformations, to a certain extent, reflect the two alternative chromatin states mentioned above. For example, euchromatic and heterochromatic sequences tend to assemble into alternating regions of approximately ~5 megabases (Mb). These ‘compartments’, positioned along the same chromosome, could interact with compartments from different chromosomes36. Furthermore, A or B compartment designations are correlated with features such as DNA accessibility and transcriptional activity. Thus, A compartments are enriched for euchromatic regions with much higher overall levels of transcription, while B compartments primarily harbor inactive and heterochromatic sequences37 (Figure 2.2). Some of the largest clusters of heterochromatin are microscopically visible and enriched at the nuclear periphery and around nucleolar membranes38.
Figure 2.2:
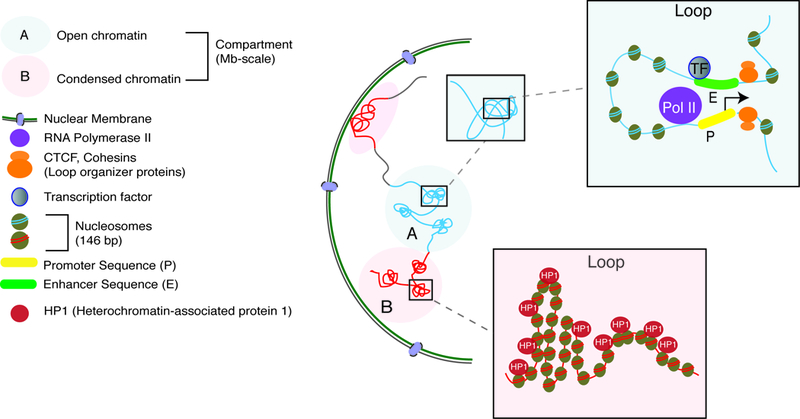
Within the nucleus, chromatin is organized into megabase-scale active or inactive compartments (blue and red circles, respectively), which contain many loops that are facilitative and allow promoter-enhancer contacts (top box) or are repressive and make heterochromatic the contained regions (bottom box). Loops are often scaffolded by key proteins, CTCF and cohesin (orange circles).
Studies exploring scaffolding and regulatory proteins with a critical role for the spatial conformations of the chromosomal materials have primarily focused on the cohesin complex and the CCCTC-binding factor/zinc finger protein CTCF. Cohesins are comprised of five core subunits SMC1, SMC3, RAD21/REC8, and STAG1–3 in humans39. In addition, accessory proteins load or release the complex onto chromosomes39. Cohesins were initially explored in the context of sister chromatid cohesion and segregation during cell division (mitosis) 39. However, these proteins maintain a heavy presence in the nuclear proteome of postmitotic cells including neurons40. Cohesins form ring-like structures, literally entrapping distant chromatin fibers into chromosomal loops39 (Figure 2.2). Cohesins are highly enriched at actively expressed genes in a tissue- and cell-type specific manner39. In contrast, CTCF, while dispensable for cohesin loading onto DNA, orchestrates cohesin enrichment at select binding sites40. As a result, chromosomal loops co-occupied by cohesins and CTCF at both ends often associate with broader stretches of regulatory domains, marking the co-regulated repression or expression of groups of genes in a cell-type specific manner41. The CTCF binding sites are thought to often (but not exclusively) be positioned in inward/convergent and, to a somewhat lesser degree, tandem orientation at the two contact sites of the loop37, 42. CTCF directionally recognizes binding sites via an 11 zinc finger array, while cohesin undergoes assembly from the CTCF’s C-terminal end, often resulting in higher order chromatin with loop-bound head-to-head CTCF configurations43.
Chromosomal scaffolding protein mutations and neuropsychiatric disease
Importantly, a rapidly increasing list of deleterious mutations in genes encoding scaffolding proteins for the 3D genome is linked to neuropsychiatric disease. These include neurodevelopmental disorders such as Cornelia de Lange Syndrome (CdLS) 39, 44, 45 and adult-onset progressive demyelination syndromes46. Neurodevelopmental disease phenotypes in CdLS include intellectual disability, psychosis and other psychiatric maladies. The underlying genetic defect includes microdeletions and copy number variations affecting core members of the cohesin complex including SMC1A and SMC3, and the accessory subunit NIPBL 44. The neurological manifestations could be due to 3D genome disorganization in brain cells and de-compaction of chromatin, albeit the precise molecular mechanisms remain to be elucidated47. In addition, genetic mutations in CTCF, as a key organizer for chromosomal loops, have been linked to monogenic causes of microcephaly and cognitive disorder48, 49. Consistent with these findings from clinical genetics, selective ablation of Ctcf in postnatal mouse brain causes behavioral alterations and dysregulated transcription of hundreds of neuronal transcripts50. It remains to be shown, however, whether the neurological manifestations of Cohesin gene mutations or CTCF are associated with widespread 3D genome alterations in brain. In addition to the aforementioned CTCF and cohesin complex, other examples of neurodevelopmental disease resulting from mutations in genes encoding 3D genome organizer proteins have been identified. These include Special AT-rich Sequence Binding Proteins 1 and 2 (SATB1, SATB2) that govern chromosomal territories extending across hundreds of kilobases51 and anchor chromatin fibers into the nuclear matrix52. Of note, SATB2 is essential for craniofacial development and proper differentiation of transcallosal cortical projection neurons53, 54. The gene has also been linked to some cases of Glass Syndrome (OMIM 612313) and mental retardation53, 55. The related protein, SATB1, is essential for connectivity and maturation of the GABAergic interneuron population in the cerebral cortex56, 57. These findings are of particular interest to the field, given that epigenomic dysregulation of GABAergic gene expression has been reported both for schizophrenia postmortem brain and in the animal and cell culture models33, 58–61.
LOOP DISRUPTIONS POTENTIALLY INVOLVED IN SCHIZOPHRENIA– EARLY FINDINGS
Promoter-enhancer loops and transcriptional regulation
Because, as discussed above, the molecular pathology of schizophrenia includes transcriptional dysregulation in cerebral cortex and other brain regions, chromosomal conformations associated with the regulation of gene expression are of particular interest to the field. To this end, promoter-enhancer loops represent one type of chromosomal interaction that is becoming increasingly understood. Promoters are often defined as cis-regulatory sequences within 1000 base pairs from the next gene transcription start site. In contrast, enhancers are a type of cis-regulatory sequence positioned >1kb from the nearest transcription start site62. Promoters (but not enhancers) typically include a core promoter as docking site for general transcription factors (TFIIA/B/D/E/F/H) and components of RNA polymerase II holoenzyme as part of the preinitiation complex62. These core promoters drive low levels of basal transcription. However, gene expression is heavily stimulated by ‘activators’ or transcription factors that bind, in sequence-specific fashion, at the site of promoters and enhancers62. The transcription factors bind to nucleosome-free intervals in open chromatin of promoters and enhancer sequences and recruit co-activator complexes, such as Mediator and CREB-binding protein (CPB)/p30062, to mention a few examples. Promoters in contrast to enhancers are often CpG rich62, which is of interest given that alterations in promoter CpG DNA methylation have been reported in the brains of schizophrenia patients24–27, 33. Importantly, enhancers, as distal regulatory elements, while critically important for transcriptional regulation are separated from their target gene often by many hundreds of base pairs, and in some cases many kilo- or even mega-bases of interspersed linear genome62. Various mechanisms have been proposed by which enhancer chromatin could regulate the expression of target genes from distant chromosomal locations. Such mechanisms include sliding along the chromosome to ‘track’ promoters63, or alternatively, a physical ‘bridge’ built via protein-protein interactions64. Presently, however, most studies implicate the promoter-enhancer (chromosomal) loop model which involves physical interaction, or at least spatial proximity, between the enhancer chromatin and the promoter target65.
Evidence for Loop Disruption in Brains of Subjects with Schizophrenia
Importantly, chromosome conformation capture (3C), a widely used DNA-DNA proximity assay on chromatin preparations treated by restriction digest followed by re-ligation, is applicable to postmortem tissue, with many chromosomal loop formations showing some level of preservation in brain tissue collected several hours after death66. One interesting and early example of 3C applied on human brain involved GAD167, encoding the 67Kda glutamic acid decarboyxlase GABA synthesis enzyme, a gene frequently showing dysregulated expression in multiple areas of the forebrain of subjects with schizophrenia 18, 68–86. One of the loop formations that were altered in a small pilot cohort of diseased prefrontal cortex67 also existed in neuronal cultures derived from induced pluripotent stem cells. Furthermore, this type of chromatin loop, which bypassed approximately 50kb of linear genome, was conserved in mouse and human brain67, which could indicate an important regulatory function for GAD1 expression across multiple mammalian lineages. In any case, according to the aforementioned study67, promoter-enhancer loops are potentially disrupted in diseased brain, thereby contributing to dysregulated gene expression. In the case of this 50kb GAD1 promoter-enhancer conformation, it has been suggested that the distal sequence (the enhancer) carries, via the loop, a cargo of transcription factors into close spatial proximity with the target gene promoter67(Figure 2.3A).
Figure 2.3:
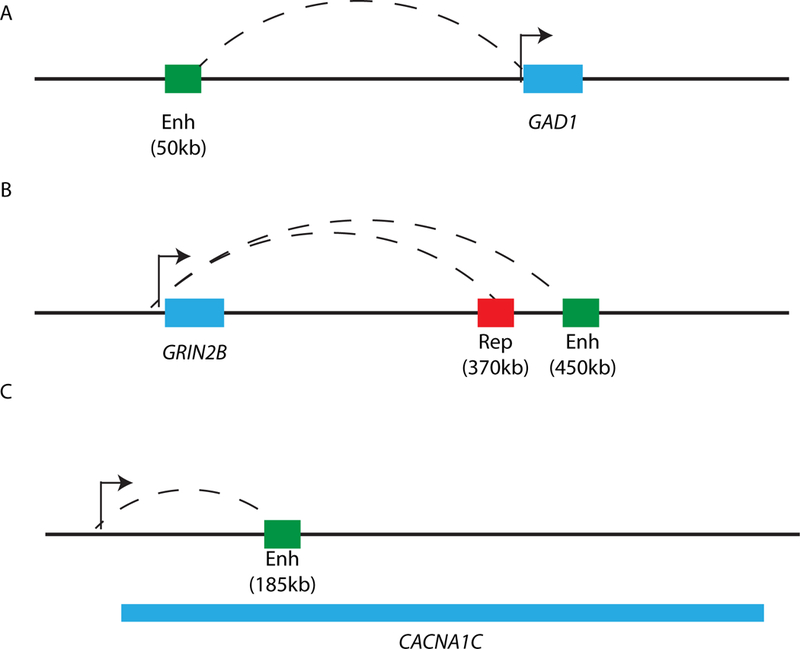
Depictions of distal promoter-enhancer loop interactions on the linear genome.
A) GAD1 (blue box) and its enhancer (green box) that lies 50kb upstream.
B) GRIN2B (blue box) and its enhancer (green box) that lies 450kb downstream and its repressor that lies 370kb downstream.
C) CACNA1 (introns and exons span the region demarcated by blue box) and its enhancer, which lies in an intronic sequence 185kb downstream from the promoter.
Another interesting example of higher order chromatin relevant for schizophrenia was recently described for the GRIN2B gene locus, encoding an NMDA glutamate receptor subunit87. Importantly, deleterious GRIN2B mutations rank prominently in exome sequencing studies capturing rare monogenic forms of neuropsychiatric disease, including intellectual disability, epilepsy, autism and psychosis spectrum disorders including schizophrenia88–92. Multiple loop-bound intronic and intergenic DNA sequences, up to 450kb downstream from the GRIN2B/Grin2b transcription start site (TSS), compete for access to the TSS87. These sequences are loaded with multiple transcription factors and, via chromosomal loop formations, are in physical proximity to the GRIN2B/Grin2b transcription start site87. However, in addition to such long-range promoter-enhancer loops, the GRIN2B/Grin2b promoter also interacts with intragenic repressive chromatin embedded in intron sequences87. Therefore, it was proposed that transcriptional regulation at the GRIN2B/Grin2b locus involves a dynamic and competitive interplay of multiple loop formations, each of which could engage with the GRIN2B promoter87(Figure 2.3B). However, this process is counterbalanced by promoter-bound higher order chromatin involving repressive intronic sequences 30 kb downstream from the transcription start site87, 93 (Figure 2.3B). Interestingly, multiple loop-bound sequences interacting with the GRIN2B promoter harbor single nucleotide polymorphisms (SNP) implicated with liability for working memory87, and schizophrenia and personality traits associated with schizophrenia87. Notably, one these risk-associated SNP alleles was associated with a promoter-enhancer loop formation and thought to convey decreased nucleoprotein binding and motif loss for the CCAAT/Enhancer Binding Protein CEBPB (C/EBPβ)87. CEBPB is a transcription factor implicated in consolidation of cortical and hippocampal learning and memory94–96. Because many enhancer elements are defined by sequential linear alignment of multiple transcription factors within short distances97, 98, additional activator proteins may synergistically cooperate with loop-bound CEBPB to regulate GRIN2B expression87. Taken together, these findings point to a complex and multilayered regulation of chromosomal conformations across at least one megabase of sequence surrounding the GRIN2B locus. Therefore, loop-bound DNA targeting the GRIN2B promoter and carrying disease-associated sequence polymorphisms, could either facilitate or repress expression, depending on the protein ‘cargo’, with multiple distal loop formations competing in a highly dynamic and activity-dependent manner for access to the GRIN2B promoter sequences87. Importantly, the DNA sequence variants affecting GRIN2B enhancer function, and working memory and liability for schizophrenia are located nearly half a megabase downstream from the GRIN2B transcription start site87. Therefore, these GRIN2B higher order chromatin studies provide the first example how chromosome conformation capture approaches could be harnessed to assign neurological function to risk-associated non-coding sequences that on the linear genome appear to be too far removed from gene promoters to impact expression.
Similarly, risk-associated sequences positioned in enhancer elements within the CACNA1C calcium channel gene body (a gene locus which ranks prominently in the polygenic risk maps of common psychiatric disease including schizophrenia and depression99) were recently identified as potent modulators of reporter gene activity34. Remarkably however, these CACNA1C intragenic enhancer sequences were, in human prefrontal cortex, physically bound to the gene transcription start site via a 185kb spanning chromosomal loop formation34, providing yet another example how the study of chromosomal conformations in human brain tissue could help to illuminate the role of non-coding DNA that, on the linear genome, is positioned far away from transcription start sites (Figure 2.3C).
However, there is evidence pointing to the importance of chromosomal conformations and the ‘3D genome’ for human cognition and schizophrenia far beyond the aforementioned candidate gene examples. For example, significant over-representation of enhancer sequences has been observed within the pool of polymorphisms and haplotypes associated with genetic risk for schizophrenia34, 100. Furthermore, postmortem studies using more conventional epigenomic assays, including genome-scale DNA methylation survey, have suggested that epigenetic dysregulation of enhancer sequences could contribute to the neurobiology of mood and psychosis spectrum disorders, including astrocyte dysfunction in depression101, and broad changes in neuronal gene expression in schizophrenia29, 102. Finally, recent genome-wide chromosomal conformation mapping in neural progenitor cells and fetal brain tissues reveals many additional examples of promoter-enhancer loops at the sites of schizophrenia relevant genes103.
(EPI)GENOMIC EDITING OF LOOP-BOUND REGULATORY SEQUENCES IN THE PRECLINICAL MODEL
In the paragraphs above, we provided specific examples of regulatory non-coding DNA that is associated with the genetic risk architecture of schizophrenia and via chromosomal loops physically associates with promoters and transcription start sites of important neuronal genes such as GAD1, GRIN2B and CACNA1C. One could argue that such types of promoter-enhancer loops provide unique opportunities for highly locus- and sequence-selective interventions, targeted against individual risk polymorphisms associated with schizophrenia, and aimed at conveying a therapeutic effect by affecting target gene expression. Thus, the field will soon be challenged with the task to ‘convert’ this molecular information into testable hypotheses aimed at gaining deeper insights into the neurobiology of schizophrenia. Perhaps more importantly, such newly gained information could be harnessed to develop novel therapies aimed at improving cognitive dysfunction. While there are already significant efforts to translate these evolving findings from schizophrenia genetics, genomics and epigenomics into drug discovery pipelines and clinical testing104, we predict that behavioral studies in mice and other small laboratory animals will serve as a critical preclinical intermediates towards this goal, in conjunction with molecular and cellular exploration of human cells in culture dishes105.
Importantly, the molecular toolboxes to test this approach, at least for the preclinical model, already exist. Specifically, genome editing strategies via RNA-guided nucleases, including the Clustered Regularly Interspaced Short Palindromic Repeat (CRISPR)-CRISPR-associated protein systems (CRISPR-Cas), introduced to the field only a few years ago106, have now been widely adopted in all areas of genomic medicine, including the neurosciences107. Mutations in and CRISPR-mediated disruption of enhancer sequences have been observed and accomplished for several neuropsychiatric risk genes, including the NMDA receptor gene GRIN2B 87 and the FOXG1 transcription factor103. Strikingly, mutations and manipulations in the enhancers have been linked with changes in the target gene expression even though the enhancer sequences lie distal to the target gene promoters on the linear genome. While the precise molecular mechanisms underlying these phenomena, such as allele-specific binding of specific transcription factors, often remain incompletely understood, it is noteworthy that such experiments have been conducted to date primarily in cell culture systems. Typically, transcriptional changes are explored after targeted mutations in the regulatory element, or by allele-specific comparisons of reporter gene expression activity. Therefore, the next phase of experiments should include in vivo genomic editing of risk-associated promoter and enhancer sequences, and then test for changes in cognition and behavior in the animal. In doing this, we must keep in mind that such genomic editing may carry drawbacks given that mutagenic interventions are likely to be irreversible. However, CRISPR-Cas and other RNA-guided nuclease systems can easily be converted into epigenomic editing tools (by using mutant protein with inactivated nuclease function, fused to a transcriptional activator such as VP64 or P300108, or a repressor such as KRAB109, even with multi-locus manipulation110. With the underlying DNA sequence left intact, it will be interesting to explore whether simultaneous epigenomic targeting of enhancer and promoter sequences within multiple risk haplotypes could offer a promising approach to effectively alter cognition and behavior. Just as in the aforementioned examples of genomic editing, proof-of-principle studies for epigenomic editing of schizophrenia risk loci by CRISPR-Cas-mediated loading of loop-bound sequences with artificial transcriptional activators and repressors already have been published. These experiments, performed on the clustered PROTOCADHERIN locus (regulating neuronal connectivity), have one caveat: they were conducted in cell culture and not in the animal111.
Obviously, preclinical assessment of cognitive and behavioral changes after genomic and epigenomic editing of psychiatric risk haplotypes is only feasible for genomic sites that show at least some degree of conservation between human and mouse (or other laboratory animals’) genomes. Such an ‘epigenomic conservation’ could include similarities in sequential arrangements of genes and transcriptional units at the locus of interest, conservation of histone modification landscapes, and similarities in chromosomal conformations including promoter-enhancer loops important for transcriptional regulation. While a more detailed investigation on the epigenomic conservation across species for genomic sites harboring schizophrenia risk haplotypes awaits further investigation, genome-scale112, 113 studies suggest that chromatin structure and function is conserved in human and mouse for a large number of regulatory non-coding sequences, even if these are not necessarily accompanied by DNA sequence conservation. This general observation also holds for brain chromatin, and there are striking similarities in locus-specific higher order chromatin landscapes in human and mouse for several of the aforementioned neuronal genes and loci, including GAD1, GRIN2B and the clustered Protocadherins67, 87, 111. With region-specific multiplex gene editing in adult mouse brain in vivo 114 now possible, we predict that the genomic and epigenomic editing of loop-bound regulatory DNA sequences will soon become an important avenue for preclinical schizophrenia research.
FUTURE DIRECTIONS
The application of 3D genome mapping technologies in the field of neuropsychiatry is still a relatively new endeavor. A few studies, as enumerated above, have delved into locus-specific dynamics in chromatin conformations and their potential impact in schizophrenia etiology. However, using agnostic, genome-wide approaches, we need to map the baseline 3D interaction landscape across various brain-relevant cell types, from neurons to microglia, to truly understand the nuanced ways in which disease risk variants impact loops in one or some but not in all cell types. Such an approach would not only elucidate any fundamental differences in overall genome topologies across cell types but would also allow us as a field to parse out cell-type-specific impacts of noncoding variants on promoter-enhancer loops and, as a result, variation in gene expression programs. Especially considering that various neuropsychiatric disorders are suspected to have neurodevelopmental origins, mapping chromatin states across differentiation from neural progenitor stage to other cell fates could prove fruitful.
With the advent of newer iterations in genome-wide as well as locus-targeted high throughput chromatin conformation mapping, paired with the trend in decreasing sequencing costs, the field is poised to uncover elements in the relatively underexplored epigenetic layer of three dimensional regulation. Moreover, innovations in CRISPR-Cas technologies, allowing for fine-tuned gene activation/inactivation, locus deletion, and multiplexing to target various disease-relevant loci at once will further enhance our abilities to tease out the functionality of newly discovered looping interactions, with the potential of uncovering novel therapeutic targets for complex neuropsychiatric illness.
ACKNOWLEDGMENT
This work is supported in part by 1F30MH113330–01 to P.R.
References
- 1.Hennekens CH, Hennekens AR, Hollar D, Casey DE. Schizophrenia and increased risks of cardiovascular disease. American heart journal 2005;150:1115–1121. [DOI] [PubMed] [Google Scholar]
- 2.Laursen TM, Munk-Olsen T, Vestergaard M. Life expectancy and cardiovascular mortality in persons with schizophrenia. Current opinion in psychiatry 2012;25:83–88. [DOI] [PubMed] [Google Scholar]
- 3.Saha S, Chant D, McGrath J. A systematic review of mortality in schizophrenia: is the differential mortality gap worsening over time? Archives of general psychiatry 2007;64:1123–1131. [DOI] [PubMed] [Google Scholar]
- 4.Taly A Novel approaches to drug design for the treatment of schizophrenia. Expert opinion on drug discovery 2013;8:1285–1296. [DOI] [PubMed] [Google Scholar]
- 5.Kim DH, Stahl SM. Antipsychotic drug development. Current topics in behavioral neurosciences 2010;4:123–139. [DOI] [PubMed] [Google Scholar]
- 6.Lieberman JA, Stroup TS, McEvoy JP, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. The New England journal of medicine 2005;353:1209–1223. [DOI] [PubMed] [Google Scholar]
- 7.Swartz MS, Perkins DO, Stroup TS, et al. Effects of antipsychotic medications on psychosocial functioning in patients with chronic schizophrenia: findings from the NIMH CATIE study. The American journal of psychiatry 2007;164:428–436. [DOI] [PubMed] [Google Scholar]
- 8.Ibrahim HM, Tamminga CA. Schizophrenia: treatment targets beyond monoamine systems. Annu Rev Pharmacol Toxicol 2011;51:189–209. [DOI] [PubMed] [Google Scholar]
- 9.Green MF. Impact of cognitive and social cognitive impairment on functional outcomes in patients with schizophrenia. J Clin Psychiatry 2016;77 Suppl 2:8–11. [DOI] [PubMed] [Google Scholar]
- 10.Schizophrenia Working Group of the Psychiatric Genomics C. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014;511:421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ruderfer DM, Hamamsy T, Lek M, et al. Patterns of genic intolerance of rare copy number variation in 59,898 human exomes. Nat Genet 2016;48:1107–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Genovese G, Fromer M, Stahl EA, et al. Increased burden of ultra-rare protein-altering variants among 4,877 individuals with schizophrenia. Nat Neurosci 2016;19:1433–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arion D, Corradi JP, Tang S, et al. Distinctive transcriptome alterations of prefrontal pyramidal neurons in schizophrenia and schizoaffective disorder. Mol Psychiatry 2015;20:1397–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao Z, Xu J, Chen J, et al. Transcriptome sequencing and genome-wide association analyses reveal lysosomal function and actin cytoskeleton remodeling in schizophrenia and bipolar disorder. Mol Psychiatry 2015;20:563–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Horvath S, Mirnics K. Schizophrenia as a disorder of molecular pathways. Biol Psychiatry 2015;77:22–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Middleton FA, Mirnics K, Pierri JN, Lewis DA, Levitt P. Gene expression profiling reveals alterations of specific metabolic pathways in schizophrenia. J Neurosci 2002;22:2718–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vawter MP, Shannon Weickert C, Ferran E, et al. Gene expression of metabolic enzymes and a protease inhibitor in the prefrontal cortex are decreased in schizophrenia. Neurochem Res 2004;29:1245–1255. [DOI] [PubMed] [Google Scholar]
- 18.Mirnics K, Middleton FA, Marquez A, Lewis DA, Levitt P. Molecular characterization of schizophrenia viewed by microarray analysis of gene expression in prefrontal cortex. Neuron 2000;28:53–67. [DOI] [PubMed] [Google Scholar]
- 19.Volk DW, Chitrapu A, Edelson JR, Roman KM, Moroco AE, Lewis DA. Molecular mechanisms and timing of cortical immune activation in schizophrenia. Am J Psychiatry 2015;172:1112–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brown JB, Celniker SE. Lessons from modENCODE. Annu Rev Genomics Hum Genet 2015;16:31–53. [DOI] [PubMed] [Google Scholar]
- 21.Lundberg SM, Tu WB, Raught B, Penn LZ, Hoffman MM, Lee SI. ChromNet: Learning the human chromatin network from all ENCODE ChIP-seq data. Genome Biol 2016;17:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends in biochemical sciences 2006;31:89–97. [DOI] [PubMed] [Google Scholar]
- 23.Bock C, Beerman I, Lien WH, et al. DNA methylation dynamics during in vivo differentiation of blood and skin stem cells. Mol Cell 2012;47:633–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abdolmaleky HM, Cheng KH, Russo A, et al. Hypermethylation of the reelin (RELN) promoter in the brain of schizophrenic patients: a preliminary report. American journal of medical genetics. Part B, Neuropsychiatric genetics : the official publication of the International Society of Psychiatric Genetics 2005;134B:60–66. [DOI] [PubMed] [Google Scholar]
- 25.Grayson DR, Jia X, Chen Y, et al. Reelin promoter hypermethylation in schizophrenia. Proc Natl Acad Sci U S A 2005;102:9341–9346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iwamoto K, Bundo M, Yamada K, et al. DNA methylation status of SOX10 correlates with its downregulation and oligodendrocyte dysfunction in schizophrenia. The Journal of Neuroscience 2005;25:5376–5381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mill J, Tang T, Kaminsky Z, et al. Epigenomic profiling reveals DNA-methylation changes associated with major psychosis. Am J Hum Genet 2008;82:696–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wockner LF, Noble EP, Lawford BR, et al. Genome-wide DNA methylation analysis of human brain tissue from schizophrenia patients. Translational psychiatry 2014;4:e339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jaffe AE, Gao Y, Deep-Soboslay A, et al. Mapping DNA methylation across development, genotype and schizophrenia in the human frontal cortex. Nat Neurosci 2016;19:40–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Siegmund KD, Connor CM, Campan M, et al. DNA methylation in the human cerebral cortex is dynamically regulated throughout the life span and involves differentiated neurons. PLoS One 2007;2:e895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang Y, Matevossian A, Huang HS, Straubhaar J, Akbarian S. Isolation of neuronal chromatin from brain tissue. BMC Neurosci 2008;9:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McKinney B, Ding Y, Lewis DA, Sweet RA. DNA methylation as a putative mechanism for reduced dendritic spine density in the superior temporal gyrus of subjects with schizophrenia. Transl Psychiatry 2017;7:e1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ruzicka WB, Subburaju S, Benes FM. Circuit- and Diagnosis-Specific DNA Methylation Changes at gamma-Aminobutyric Acid-Related Genes in Postmortem Human Hippocampus in Schizophrenia and Bipolar Disorder. JAMA Psychiatry 2015;72:541–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roussos P, Mitchell AC, Voloudakis G, et al. A role for noncoding variation in schizophrenia. Cell Rep 2014;9:1417–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rajarajan P, Gil SE, Brennand KJ, Akbarian S. Spatial genome organization and cognition. Nat Rev Neurosci 2016;17:681–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lieberman-Aiden E, van Berkum NL, Williams L, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science (New York, N.Y.) 2009;326:289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rao SS, Huntley MH, Durand NC, et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014;159:1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Padeken J, Heun P. Nucleolus and nuclear periphery: velcro for heterochromatin. Curr Opin Cell Biol 2014;28:54–60. [DOI] [PubMed] [Google Scholar]
- 39.Nasmyth K, Haering CH. Cohesin: its roles and mechanisms. Annu Rev Genet 2009;43:525–558. [DOI] [PubMed] [Google Scholar]
- 40.Wendt KS, Yoshida K, Itoh T, et al. Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature 2008;451:796–801. [DOI] [PubMed] [Google Scholar]
- 41.Dowen JM, Fan ZP, Hnisz D, et al. Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell 2014;159:374–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vietri Rudan M, Barrington C, Henderson S, et al. Comparative Hi-C reveals that CTCF underlies evolution of chromosomal domain architecture. Cell Rep 2015;10:1297–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guo Y, Xu Q, Canzio D, et al. CRISPR Inversion of CTCF Sites Alters Genome Topology and Enhancer/Promoter Function. Cell 2015;162:900–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Boyle MI, Jespersgaard C, Brondum-Nielsen K, Bisgaard AM, Tumer Z. Cornelia de Lange syndrome. Clin Genet 2015;88:1–12. [DOI] [PubMed] [Google Scholar]
- 45.Yan J, Zhang F, Brundage E, et al. Genomic duplication resulting in increased copy number of genes encoding the sister chromatid cohesion complex conveys clinical consequences distinct from Cornelia de Lange. J Med Genet 2009;46:626–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Finnsson J, Sundblom J, Dahl N, Melberg A, Raininko R. LMNB1-related autosomal-dominant leukodystrophy: Clinical and radiological course. Ann Neurol 2015;78:412–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nolen LD, Boyle S, Ansari M, Pritchard E, Bickmore WA. Regional chromatin decompaction in Cornelia de Lange syndrome associated with NIPBL disruption can be uncoupled from cohesin and CTCF. Hum Mol Genet 2013;22:4180–4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Watson LA, Wang X, Elbert A, Kernohan KD, Galjart N, Berube NG. Dual effect of CTCF loss on neuroprogenitor differentiation and survival. J Neurosci 2014;34:2860–2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gregor A, Oti M, Kouwenhoven EN, et al. De novo mutations in the genome organizer CTCF cause intellectual disability. Am J Hum Genet 2013;93:124–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hirayama T, Tarusawa E, Yoshimura Y, Galjart N, Yagi T. CTCF is required for neural development and stochastic expression of clustered Pcdh genes in neurons. Cell Rep 2012;2:345–357. [DOI] [PubMed] [Google Scholar]
- 51.Cai S, Lee CC, Kohwi-Shigematsu T. SATB1 packages densely looped, transcriptionally active chromatin for coordinated expression of cytokine genes. Nat Genet 2006;38:1278–1288. [DOI] [PubMed] [Google Scholar]
- 52.Britanova O, Akopov S, Lukyanov S, Gruss P, Tarabykin V. Novel transcription factor Satb2 interacts with matrix attachment region DNA elements in a tissue-specific manner and demonstrates cell-type-dependent expression in the developing mouse CNS. Eur J Neurosci 2005;21:658–668. [DOI] [PubMed] [Google Scholar]
- 53.Docker D, Schubach M, Menzel M, et al. Further delineation of the SATB2 phenotype. Eur J Hum Genet 2014;22:1034–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Alcamo EA, Chirivella L, Dautzenberg M, et al. Satb2 regulates callosal projection neuron identity in the developing cerebral cortex. Neuron 2008;57:364–377. [DOI] [PubMed] [Google Scholar]
- 55.Leoyklang P, Suphapeetiporn K, Siriwan P, et al. Heterozygous nonsense mutation SATB2 associated with cleft palate, osteoporosis, and cognitive defects. Hum Mutat 2007;28:732–738. [DOI] [PubMed] [Google Scholar]
- 56.Close J, Xu H, De Marco Garcia N, et al. Satb1 is an activity-modulated transcription factor required for the terminal differentiation and connectivity of medial ganglionic eminence-derived cortical interneurons. J Neurosci 2012;32:17690–17705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Denaxa M, Kalaitzidou M, Garefalaki A, et al. Maturation-promoting activity of SATB1 in MGE-derived cortical interneurons. Cell Rep 2012;2:1351–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Labouesse MA, Dong E, Grayson DR, Guidotti A, Meyer U. Maternal immune activation induces GAD1 and GAD2 promoter remodeling in the offspring prefrontal cortex. Epigenetics 2015;10:1143–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Guidotti A, Ruzicka W, Grayson DR, et al. S-adenosyl methionine and DNA methyltransferase-1 mRNA overexpression in psychosis. Neuroreport 2007;18:57–60. [DOI] [PubMed] [Google Scholar]
- 60.Kundakovic M, Chen Y, Guidotti A, Grayson DR. The reelin and GAD67 promoters are activated by epigenetic drugs that facilitate the disruption of local repressor complexes. Mol Pharmacol 2009;75:342–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Costa E, Grayson DR, Mitchell CP, Tremolizzo L, Veldic M, Guidotti A. GABAergic cortical neuron chromatin as a putative target to treat schizophrenia vulnerability. Crit Rev Neurobiol 2003;15:121–142. [DOI] [PubMed] [Google Scholar]
- 62.Vernimmen D, Bickmore WA. The Hierarchy of Transcriptional Activation: From Enhancer to Promoter. Trends Genet 2015;31:696–708. [DOI] [PubMed] [Google Scholar]
- 63.Hatzis P, Talianidis I. Dynamics of enhancer-promoter communication during differentiation-induced gene activation. Mol Cell 2002;10:1467–1477. [DOI] [PubMed] [Google Scholar]
- 64.Mueller-Storm HP, Sogo JM, Schaffner W. An enhancer stimulates transcription in trans when attached to the promoter via a protein bridge. Cell 1989;58:767–777. [DOI] [PubMed] [Google Scholar]
- 65.Gorkin DU, Leung D, Ren B. The 3D genome in transcriptional regulation and pluripotency. Cell Stem Cell 2014;14:762–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mitchell AC, Bharadwaj R, Whittle C, et al. The genome in three dimensions: a new frontier in human brain research. Biol Psychiatry 2014;75:961–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bharadwaj R, Jiang Y, Mao W, et al. Conserved chromosome 2q31 conformations are associated with transcriptional regulation of GAD1 GABA synthesis enzyme and altered in prefrontal cortex of subjects with schizophrenia. J Neurosci 2013;33:11839–11851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Volk DW, Austin MC, Pierri JN, Sampson AR, Lewis DA. Decreased glutamic acid decarboxylase67 messenger RNA expression in a subset of prefrontal cortical gamma-aminobutyric acid neurons in subjects with schizophrenia. Archives of general psychiatry 2000;57:237–245. [DOI] [PubMed] [Google Scholar]
- 69.Impagnatiello F, Guidotti AR, Pesold C, et al. A decrease of reelin expression as a putative vulnerability factor in schizophrenia. Proceedings of the National Academy of Sciences of the United States of America 1998;95:15718–15723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Akbarian S, Kim JJ, Potkin SG, et al. Gene expression for glutamic acid decarboxylase is reduced without loss of neurons in prefrontal cortex of schizophrenics. Archives of general psychiatry 1995;52:258–266. [DOI] [PubMed] [Google Scholar]
- 71.Guidotti A, Auta J, Davis JM, et al. Decrease in reelin and glutamic acid decarboxylase67 (GAD67) expression in schizophrenia and bipolar disorder: a postmortem brain study. Archives of general psychiatry 2000;57:1061–1069. [DOI] [PubMed] [Google Scholar]
- 72.Hashimoto T, Volk DW, Eggan SM, et al. Gene expression deficits in a subclass of GABA neurons in the prefrontal cortex of subjects with schizophrenia. The Journal of neuroscience : the official journal of the Society for Neuroscience 2003;23:6315–6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Veldic M, Guidotti A, Maloku E, Davis JM, Costa E. In psychosis, cortical interneurons overexpress DNA-methyltransferase 1. Proceedings of the National Academy of Sciences of the United States of America 2005;102:2152–2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Torrey EF, Barci BM, Webster MJ, Bartko JJ, Meador-Woodruff JH, Knable MB. Neurochemical markers for schizophrenia, bipolar disorder, and major depression in postmortem brains. Biol Psychiatry 2005;57:252–260. [DOI] [PubMed] [Google Scholar]
- 75.Veldic M, Kadriu B, Maloku E, et al. Epigenetic mechanisms expressed in basal ganglia GABAergic neurons differentiate schizophrenia from bipolar disorder. Schizophrenia research 2007;91:51–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Benes FM, Lim B, Matzilevich D, Walsh JP, Subburaju S, Minns M. Regulation of the GABA cell phenotype in hippocampus of schizophrenics and bipolars. Proceedings of the National Academy of Sciences of the United States of America 2007;104:10164–10169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Thompson Ray M, Weickert CS, Wyatt E, Webster MJ. Decreased BDNF, trkB-TK+ and GAD67 mRNA expression in the hippocampus of individuals with schizophrenia and mood disorders. J Psychiatry Neurosci 2011;36:195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Curley AA, Arion D, Volk DW, et al. Cortical deficits of glutamic acid decarboxylase 67 expression in schizophrenia: clinical, protein, and cell type-specific features. The American journal of psychiatry 2011;168:921–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sheng G, Demers M, Subburaju S, Benes FM. Differences in the circuitry-based association of copy numbers and gene expression between the hippocampi of patients with schizophrenia and the hippocampi of patients with bipolar disorder. Archives of general psychiatry 2012;69:550–561. [DOI] [PubMed] [Google Scholar]
- 80.Volk DW, Matsubara T, Li S, et al. Deficits in transcriptional regulators of cortical parvalbumin neurons in schizophrenia. Am J Psychiatry 2012;169:1082–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gilabert-Juan J, Varea E, Guirado R, Blasco-Ibanez JM, Crespo C, Nacher J. Alterations in the expression of PSA-NCAM and synaptic proteins in the dorsolateral prefrontal cortex of psychiatric disorder patients. Neurosci Lett 2012;530:97–102. [DOI] [PubMed] [Google Scholar]
- 82.Hashimoto T, Bazmi HH, Mirnics K, Wu Q, Sampson AR, Lewis DA. Conserved regional patterns of GABA-related transcript expression in the neocortex of subjects with schizophrenia. The American journal of psychiatry 2008;165:479–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hashimoto T, Arion D, Unger T, et al. Alterations in GABA-related transcriptome in the dorsolateral prefrontal cortex of subjects with schizophrenia. Molecular psychiatry 2008;13:147–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Huang HS, Matevossian A, Whittle C, et al. Prefrontal dysfunction in schizophrenia involves mixed-lineage leukemia 1-regulated histone methylation at GABAergic gene promoters. The Journal of neuroscience : the official journal of the Society for Neuroscience 2007;27:11254–11262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bullock WM, Cardon K, Bustillo J, Roberts RC, Perrone-Bizzozero NI. Altered expression of genes involved in GABAergic transmission and neuromodulation of granule cell activity in the cerebellum of schizophrenia patients. The American journal of psychiatry 2008;165:1594–1603. [DOI] [PubMed] [Google Scholar]
- 86.Fatemi SH, Stary JM, Earle JA, Araghi-Niknam M, Eagan E. GABAergic dysfunction in schizophrenia and mood disorders as reflected by decreased levels of glutamic acid decarboxylase 65 and 67 kDa and Reelin proteins in cerebellum. Schizophrenia research 2005;72:109–122. [DOI] [PubMed] [Google Scholar]
- 87.Bharadwaj R, Peter CJ, Jiang Y, et al. Conserved higher-order chromatin regulates NMDA receptor gene expression and cognition. Neuron 2014;84:997–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Talkowski ME, Rosenfeld JA, Blumenthal I, et al. Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell 2012;149:525–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Epi KC, Epilepsy Phenome/Genome P, Allen AS, et al. De novo mutations in epileptic encephalopathies. Nature 2013;501:217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hamdan FF, Srour M, Capo-Chichi JM, et al. De novo mutations in moderate or severe intellectual disability. PLoS Genet 2014;10:e1004772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.O’Roak BJ, Deriziotis P, Lee C, et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet 2011;43:585–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hu C, Chen W, Myers SJ, Yuan H, Traynelis SF. Human GRIN2B variants in neurodevelopmental disorders. J Pharmacol Sci 2016;132:115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jiang Y, Jakovcevski M, Bharadwaj R, et al. Setdb1 histone methyltransferase regulates mood-related behaviors and expression of the NMDA receptor subunit NR2B. J Neurosci 2010;30:7152–7167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Taubenfeld SM, Milekic MH, Monti B, Alberini CM. The consolidation of new but not reactivated memory requires hippocampal C/EBPbeta. Nat Neurosci 2001;4:813–818. [DOI] [PubMed] [Google Scholar]
- 95.Taubenfeld SM, Wiig KA, Monti B, Dolan B, Pollonini G, Alberini CM. Fornix-dependent induction of hippocampal CCAAT enhancer-binding protein [beta] and [delta] Co-localizes with phosphorylated cAMP response element-binding protein and accompanies long-term memory consolidation. J Neurosci 2001;21:84–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Merhav M, Kuulmann-Vander S, Elkobi A, Jacobson-Pick S, Karni A, Rosenblum K. Behavioral interference and C/EBPbeta expression in the insular-cortex reveal a prolonged time period for taste memory consolidation. Learn Mem 2006;13:571–574. [DOI] [PubMed] [Google Scholar]
- 97.Dickel DE, Visel A, Pennacchio LA. Functional anatomy of distant-acting mammalian enhancers. Philos Trans R Soc Lond B Biol Sci 2013;368:20120359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Stampfel G, Kazmar T, Frank O, Wienerroither S, Reiter F, Stark A. Transcriptional regulators form diverse groups with context-dependent regulatory functions. Nature 2015;528:147–151. [DOI] [PubMed] [Google Scholar]
- 99.Cross-Disorder Group of the Psychiatric Genomics C. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet 2013;381:1371–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gusev A, Lee SH, Trynka G, et al. Partitioning heritability of regulatory and cell-type-specific variants across 11 common diseases. Am J Hum Genet 2014;95:535–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nagy C, Suderman M, Yang J, et al. Astrocytic abnormalities and global DNA methylation patterns in depression and suicide. Mol Psychiatry 2015;20:320–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Roussos P, Giakoumaki SG, Zouraraki C, et al. The Relationship of Common Risk Variants and Polygenic Risk for Schizophrenia to Sensorimotor Gating. Biol Psychiatry 2015. [DOI] [PubMed]
- 103.Won H, de la Torre-Ubieta L, Stein JL, et al. Chromosome conformation elucidates regulatory relationships in developing human brain. Nature 2016;538:523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Breen G, Li Q, Roth BL, et al. Translating genome-wide association findings into new therapeutics for psychiatry. Nat Neurosci 2016;19:1392–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Brennand KJ, Landek-Salgado MA, Sawa A. Modeling heterogeneous patients with a clinical diagnosis of schizophrenia with induced pluripotent stem cells. Biol Psychiatry 2014;75:936–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014;346:1258096. [DOI] [PubMed] [Google Scholar]
- 107.Heidenreich M, Zhang F. Applications of CRISPR-Cas systems in neuroscience. Nat Rev Neurosci 2016;17:36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hilton IB, D’Ippolito AM, Vockley CM, et al. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat Biotechnol 2015;33:510–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Thakore PI, D’Ippolito AM, Song L, et al. Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat Methods 2015;12:1143–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Stricker SH, Koferle A, Beck S. From profiles to function in epigenomics. Nat Rev Genet 2017;18:51–66. [DOI] [PubMed] [Google Scholar]
- 111.Jiang Y, Loh YE, Rajarajan P, et al. The methyltransferase SETDB1 regulates a large neuron-specific topological chromatin domain. Nat Genet 2017;49:1239–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Xiao S, Xie D, Cao X, et al. Comparative epigenomic annotation of regulatory DNA. Cell 2012;149:1381–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Dincer A, Gavin DP, Xu K, et al. Deciphering H3K4me3 broad domains associated with gene-regulatory networks and conserved epigenomic landscapes in the human brain. Transl Psychiatry 2015;5:e679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zetsche B, Heidenreich M, Mohanraju P, et al. Multiplex gene editing by CRISPR-Cpf1 using a single crRNA array. Nat Biotechnol 2017;35:31–34. [DOI] [PMC free article] [PubMed] [Google Scholar]