

Abstract
Chronic liver diseases that inevitably lead to hepatic fibrosis, cirrhosis and/or hepatocellular carcinoma have become a major cause of illness and death worldwide. Among them, cholangiopathies or cholestatic liver diseases comprise a large group of conditions in which injury is primarily focused on the biliary system. These include congenital diseases (such as biliary atresia and cystic fibrosis), acquired diseases (such as primary sclerosing cholangitis and primary biliary cirrhosis), and those that arise from secondary damage to the biliary tree from obstruction, cholangitis or ischaemia. These conditions are associated with a specific pattern of chronic liver injury centered on damaged bile ducts that drive the development of peribiliary fibrosis and, ultimately, biliary cirrhosis and liver failure. For most, there is no established medical therapy and, hence, these diseases remain one of the most important indications for liver transplantation. As a result, there is a major need to develop new therapies that can prevent the development of chronic biliary injury and fibrosis. This mini-review briefly discusses the pathophysiology of liver fibrosis and its progression to cirrhosis. We make a special emphasis on biliary fibrosis and current therapeutic options, such as angiotensin converting enzyme-2 (known as ACE2) over-expression in the diseased liver as a novel potential therapy to treat this condition.
Keywords: Chronic liver disease, Biliary fibrosis, Current therapies for biliary fibrosis, Angiotensin converting enzyme-2, Gene therapy
Core tip: This mini-review focuses on the pathophysiology of chronic liver fibrosis, with a special emphasis on biliary fibrosis. We also attempted to provide information on current clinically available therapeutic options for biliary fibrosis and other potential therapeutic options that are in the preclinical stage of development, and discuss their advantages and disadvantages. In particular, work from the author’s laboratory described in this review indicates that liver-specific over-expression of angiotensin converting enzyme-2 (known as ACE2) of the alternate renin angiotensin system dramatically reduces biliary fibrosis in mouse models of biliary disease. This suggests that ACE2 gene therapy has the potential to treat patients with chronic biliary fibrosis.
INTRODUCTION
The prevalence of chronic liver diseases is rising worldwide, and approximately 1.7 million deaths are reported annually[1,2]. The aetiology of chronic liver diseases is multifactorial, and evidence from the literature indicates that these causative agents vary according to geographical location[3]. Major causes are chronic viral infections (e.g., hepatitis B and C), excessive alcohol consumption, non-alcoholic fatty liver disease (NAFLD), inherited diseases (e.g., Wilson’s disease, biliary fibrosis) and primary sclerosing cholangitis (PSC), side effects of medications, toxic chemicals and idiopathic or cryptogenic causes[3,4]. Regardless of the aetiology, the events associated with pathogenesis and fibrogenic progression of chronic liver injury appear to share common intracellular pathways.
Hepatic fibrosis is the result of the wound-healing response of the liver to repeated injury. As a result, the balance between parenchymal cell regeneration and the wound healing response is shifted towards the wound healing response with impaired regenerative pathways over time, and hepatocytes are substituted with abundant extracellular matrix (ECM), eventually leading to accumulation of excess fibrotic scar tissue[5]. Cirrhosis is the end result of chronic liver diseases in which much of the hepatic parenchymal tissue is replaced by fibrous tissue, altering the liver function and distorting liver architecture with septae and nodule formation. This leads to alterations in blood flow with collateral formation, which ultimately results in cirrhosis and liver failure[4,6]. There are no established medical therapies for cirrhosis, and the ultimate therapy for this condition is liver transplantation, which is limited by the lack of donor livers and carries the risk of post-transplantation complications[4]. Thus, there remains a major need to identify potentially modifiable factors that exacerbate liver injury and fibrosis, and to develop therapies that can prevent or slow liver scarring.
Liver injuries are categorized into three major groups: cell-indiscriminate, cholestasis and hepatocyte-associated injuries. Mechanical trauma, ischemia and liver resection lead to cell-indiscriminate, whilst either mechanical or autoimmune bile duct injuries cause cholestasis. The major types of hepatocyte-associated injuries are either direct injuries (alcohol, drugs and hepatotropic infectious viruses, such as hepatitis B and C) or immune-mediated[2,7].
As injury persists, regardless of the initial cause, liver tissue responds by depositing ECM[8], which is known as the wound healing response. In addition, ECM synthesis is considered an effort of liver tissue to localize the injury by encapsulating the area of injury. Even though it is as an essential part of the wound healing process, the condition progresses to “liver fibrosis” once it is deregulated, which becomes an inefficient attempt at liver tissue remodelling[9,10]. Thus, liver fibrosis is mainly characterized by the excessive accumulation of ECM in the liver parenchyma that replaces functional hepatic tissue[11].
Interestingly, the microenvironment in the liver is an organized multidirectional interaction complex (cell-matrix-cell), which delivers the molecular signals crucial for normal liver homeostasis. In this process, each cell type in the liver, including hepatocytes, hepatic stellate cells (HSCs), Kupffer cells (KCs) and liver sinusoidal epithelial cells (LSECs), have their own roles to play while talking to each other, a process referred to as cellular “crosstalk”[12].
Activated HSCs are the main cell type that is responsible for ECM synthesis in the injured liver. In addition, they exert contractile and pro-inflammatory properties. During liver injury, HSC activation proceeds as a result of two major intercellular crosstalk pathways[12,13], which include capillarization[14,15] of LSECs and apoptosis of hepatocytes[12,16]. It has been shown that KCs are also involved in cellular crosstalk during the process of fibrosis[12]. KCs are liver-resident macrophages that engulf apoptotic bodies arising from the apoptotic hepatocytes[12,17] and become activated. The activated KCs begin to express death ligands, such as Fas, TNF-α and TNF-related apoptosis-inducing ligand, that induce hepatocyte apoptosis in a feed-forward manner[17]. The activated KCs also release cytokines and reactive oxygen species through which they trigger the activation of HSCs in a paracrine manner[18].
Other than HSCs, there are myofibroblasts that are predominantly located around the portal tracts, particularly in cholestatic liver injuries. These myofibroblasts are derived from either bone marrow or small portal vessels as a response to cholestasis, and proliferate around biliary tracts[19]. In addition, the periportal myofibroblast cell population has been postulated to also derive from activated cholangiocytes[20]. These myofibroblast are also considered to play a role in collagen synthesis and perform a similar role to HSCs[19,21].
There is evidence that Mast cells are also involved in liver fibrosis as a response to injury (Figure 1). A Mast cell is a white blood cell in the circulation that contains histamine and heparin granules. It has been shown that Mast cell infiltration is evident during liver fibrosis in several rat models, including the bile duct-ligated model[22,23]. The infiltration of Mast cells into the liver during the progression of biliary fibrosis has also been described in multiple drug resistant gene 2 knockout (Mdr2-KO) mice, a mouse model of progressive biliary fibrosis. The presence of Mast cells increases local levels of histamine, which is a pro-fibrogenic and proliferative factor. It induces intrahepatic bile duct masses and ductular proliferation during fibrogenesis[24]. Transforming growth factor-beta 1 (TGF-β1), released by Mast cells, is a key pro-fibrotic cytokine that subsequently activates quiescent HSCs that produce ECM, leading to fibrosis[22,25,26]. In addition, Mast cells have the ability to induce the production of ECM components by overproduction of basement membrane, which induces fibroblast attachment, spreading and proliferation[22,27].
Figure 1.
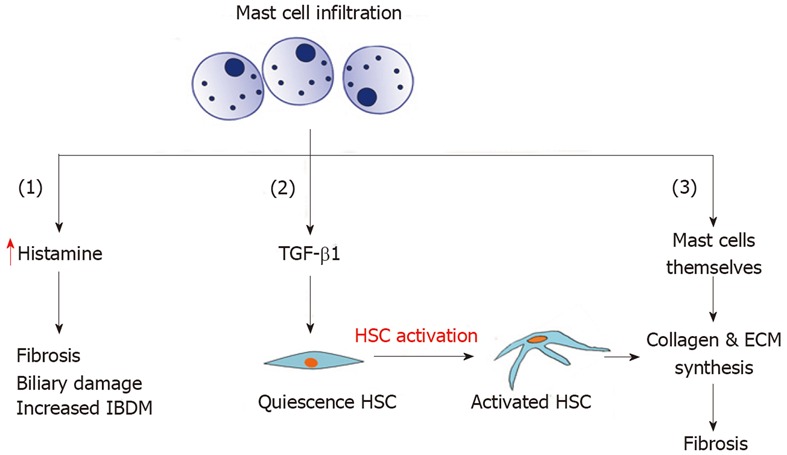
Mast cell infiltration and its role in biliary fibrosis. HSC: Hepatic stellate cell; ECM: Extracellular matrix; IBDM: Intrahepatic bile duct mass; TGF-β1: Transforming growth factor-beta 1.
Cirrhosis is the end stage of liver fibrosis, and characterized by abnormal continuation of fibrogenesis and distortion of hepatic vasculature by neo-angiogenesis, a process involved in new sinusoid formation. In advanced stages of fibrogenesis, there is collective ECM synthesis from activated HSCs, myofibroblasts derived from bone marrow, portal fibroblasts and Mast cells that are closely associated with neo-angiogenesis and capillarization[10]. Cirrhosis is histologically characterised by vascularised fibrotic septa that link portal tracts and central veins, forming clusters of hepatocyte islands surrounded by fibrotic septa[28]. Thus, cirrhotic liver is characterized by diffuse fibrosis, regenerative nodules, altered lobular architecture and the establishment of intrahepatic vascular shunts between afferent vessels and efferent hepatic veins of the liver[10,29]. Some of the major clinical consequences of these distortions are the loss of liver function, development of portal hypertension (PHT), variceal bleeding and ascites, which can lead to renal failure and hepatic encephalopathy[28].
BILIARY DISEASES
Biliary diseases or cholangiopathies are a group of chronic liver diseases characterized by cholestasis and progressive biliary fibrosis that can lead to end stage liver failure. There are numerous aetiologies for these diseases. Two of the common cholangiopathies are immune disorders, primary biliary cholangitis/primary biliary cirrhosis (PBC)[30] and PSC. Infectious agents of bacterial, viral or fungal origin, vascular or ischemic causes (such as post-liver transplantation), hepatic artery stenosis, drugs/toxin and genetical abnormalities (such as cystic fibrosis) are also causes of cholangiopathies. There are also idiopathic cholangiopathies, including biliary atresia and idiopathic ductopenia. Many cholangiopathies, including PBC and drug-induced cholangiopathies, primarily affect small bile ducts. In contrast, diseases like PSC and cholangiocarcinoma affect both intra and extrahepatic large bile ducts[31]. Once bile flow is impaired, bile accumulates in the liver, causing primary damage to the biliary epithelium and eventually the liver parenchyma. A majority of cholangiopathies has similar features, including peri-portal inflammations that lead to liver fibrosis/cirrhosis. Given their progressive nature, most cholangiopathies cause substantial morbidity and mortality in patients and, thus, they are a major indication for liver transplantation[31,32].
PATHOGENESIS OF CHOLESTASIS AND BILIARY FIBROSIS
Cholestasis is defined as a decrease in bile flow due to impaired secretion by hepatocytes or obstruction of bile flow. Obstruction of bile flow can occur due to intrahepatic or extrahepatic causes. Whilst intrahepatic bile duct obstruction and alterations in bile secretion by hepatocytes are considered as intrahepatic causes, obstruction in the extrahepatic bile duct is referred to as an extrahepatic cause of cholestasis[33]. Once bile flow is impaired, increased accumulation of bile within hepatocytes causes primary damage to biliary epithelium and eventually the liver parenchyma[34]. Many cholangiopathies, including PBC and drug-induced cholangiopathies, primarily affect small bile ducts. In contract, diseases like PSC and cholangiocarcinoma affect both intra and extrahepatic large bile ducts[31,35].
In chronic cholestatic liver injury, two major pathways are responsible for repairing the damaged cells and maintaining biliary homeostasis. The first is the proliferation of existing cholangiocytes (Figure 2A) of both small and large injured bile ducts, leading to the subsequent expansion of existing bile ducts. The second pathway is via activation of hepatic progenitor cells (HPCs) or oval cells[36,37], which differentiate into cholangiocytes, leading to the formation of new bile ducts, a condition referred to as “ductular reaction”[38] (Figure 2B). These newly formed ductules will eventually form a tubular network that restores the ductal mass in an attempt to prevent further liver injury and the leakage of bile acids into the liver parenchyma. In order to sustain newly formed tubules, a fibro-vascular stromal area is developed as a result of extensive cross-talk between hepatocytes, HSCs, LSECs and KCs[39]. On the other hand, ductular reaction is accompanied by continuous inflammatory signals resulting from key signalling molecules, such as TGF-β1, TNF-α and vascular endothelial growth factor, which then lead to liver fibrosis and later cirrhosis[36,40,41]. In late-stage cholangiopathies, ductopenia can occur that predominates over proliferation, leading to a state of vanishing bile ducts. The apoptosis rate of cholangiocytes becomes higher than that of the proliferation rate, and subsequently the cholangiocyte number is reduced, contributing to progressive portal fibrosis as seen in advanced cholangiopathies[31,42].
Figure 2.
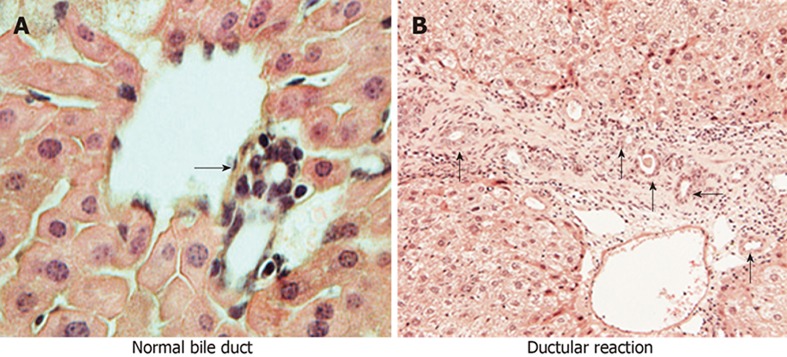
A bile duct consists of cholangiocytes in normal liver (A) and ductular reaction with reactive ductular cells in biliary diseases (B) (arrows indicate bile ducts).
CURRENT TREATMENT OPTIONS FOR CHOLANGIOPATHIES
PBC and PSC are considered to be the most common cholangiopathies in humans. Both conditions lead to end stage liver failure, an indication for liver transplantation[43,44]. There has been a decrease in the number of liver transplantations for PBC in the United States and Europe after the clinical use of ursodeoxycholic acid (UDCA) in PBC patients[45]. Although it is the only Food and Drug Administration (FDA)-approved medical treatment for PBC, it has not been proven as a therapy for any other cholangiopathies[32,46]. PSC is the second most common cholangiopathy with no specific medical therapy, and current evidence shows that there is no reduction in the number of PSC patients listed for liver transplantation[47]. This indicates that there is no effective medical therapy to prevent PSC patient progression to cirrhosis[43,46]. Moreover, recurrence of PSC after liver transplantation emphasises the critical need for an effective medical therapy to treat this condition[48].
The development of antifibrotic therapies holds promise in the treatment of liver fibrosis, including biliary diseases, irrespective of the cause of disease. They can be used to either prevent the formation of excessive ECM by inhibiting the activation of myofibroblastic cell populations or stimulate ECM degradation. However, the lack of availability of an effective antifibrotic therapy with minimal or no side effects is the main hurdle. As a result, liver transplantation has inevitably become the only option for patients with biliary fibrosis. An increased incidence of chronic liver disease, lack of donor organs, post-transplant complications and high cost associated with liver transplantation make the current situation worse. Therefore, there is a major need to develop and formulate specific, effective, safe and inexpensive medical treatments.
An exciting potential target to develop antifibrotic therapies is the local renin angiotensin system (RAS). In normal physiology, the RAS plays a pivotal role in blood pressure regulation and sodium and water homeostasis, as well as tissue remodelling after injury. It is now well-established that the RAS consists of two arms called the “classical arm” and the “alternate arm”, which play counter-balancing roles. There is substantial evidence that angiotensin II (Ang II) is a main mediator in hepatic fibrosis, and circulating Ang II levels are elevated in patients with cirrhosis[49]. It has also been shown that the local RAS is also activated in the liver as a response to injury. Studies published by our laboratory and others have shown that once activated, there is increased expression of components of the classical RAS, including hepatic angiotensin converting enzyme (ACE) and Ang II type 1 receptor (AT1-R)[26,49]. Moreover, increased expression of classical RAS components is localized to the areas of active fibrogenesis, confirming that local RAS plays a pivotal role during hepatic fibrogenesis[22,50,51]. Consequently, attempts have been made to inhibit either the production of Ang II by ACE inhibitors (ACEi) or AT1-R activation by angiotensin receptor blockers (ARBs) in cirrhotic patients. This implies that ACEi and ARBs can be considered as potential pharmacological agents to block the effects of classical RAS to inhibit liver fibrosis[52]. Unfortunately, a major setback with this approach is that they produce off-target systemic side effects, including systemic hypotension and reduced renal perfusion.
Work from our laboratory has demonstrated that the alternate RAS, comprising ACE2 and the antifibrotic peptide angiotensin-(1-7) [Ang-(1-7)], is also activated in liver injury[22,53]. The alternate RAS is expected to counter the deleterious effects produced by activated classical RAS. In experimental cholestasis induced by bile duct ligation (BDL) in rats, the components of the classical RAS (including angiotensinogen, ACE and AT1-R) are upregulated at 1 wk post-BDL. However, the expression of components of the alternate RAS, such as ACE2, Ang-(1-7) and putative Ang-(1-7) receptor Mas (Mas-R), are delayed until the third week post-BDL. Upon activation, however, the expression of the alternate RAS parallels the changes of the classical RAS[53]. This, in turn, results in elevated circulating Ang-(1-7) levels[53]. These findings were corroborated with elevated levels of circulating Ang-(1-7) in patients with liver disease, confirming the activation of the alternate RAS during chronic liver injury[49,54].
Although inhibition of the components of the classical RAS has been extensively investigated in animal models of liver disease, there were only a few studies carried out to investigate the role of the alternate RAS in liver disease. Emerging evidence suggests that the alternate RAS is an attractive target for drug intervention in biliary fibrosis. One possible way of achieving a therapeutic outcome in biliary fibrosis would be to increase the level of antifibrotic peptide Ang-(1-7), the effector peptide of the alternate RAS, which opposes many of the deleterious effects of Ang II. Animal studies performed using BDL rats and cultured rat HSCs have confirmed that Ang-(1-7) peptide has the ability to reduce collagen secretion, leading to a profound improvement in hepatic fibrosis[49]. Moreover, the same study showed that the non-peptide Mas-R agonist AVE0991 produced a significant decrease in α-SMA protein content and collagen production in rat HSCs. The findings that these effects were inhibited by Mas-R antagonist D-Ala7-Ang-(1-7) (A779) suggest that the antifibrotic effects of Ang-(1-7) are mediated via Mas-R[49]. Moreover, an oral formulation of Ang-(1-7) has recently been developed where the peptide is encapsulated with oligosaccharide hydroxypropyl-cyclodextrin (HPβCD) to protect it from degradation by enzymes in the digestive system. This study showed that this oral Ang-(1-7) formulation was cardioprotective in rats with myocardial infarction[55].
Published work from the author’s laboratory, however, suggested that the best way to achieve a therapeutic outcome in liver fibrosis is to target ACE2 of the alternate RAS. This is because enhanced expression and activity of liver ACE2 would be expected to provide dual benefits by increasing the degradation of profibrotic peptide Ang II with simultaneous generation of antifibrotic peptide Ang-(1-7). The evidence comes from animal studies showing that recombinant human ACE2 (rhACE2) is beneficial for the prevention of hypertension in cardiovascular disease[56] and the improvement of kidney function in diabetic nephropathy[57]. Recombinant hACE2 was shown to be well-tolerated by a group of healthy human volunteers in a phase 1 clinical trial without exerting any unwanted cardiovascular side effects[58]. However, randomized clinical trials with an adequate number of healthy individuals and patients assigned to receive rhACE2 treatment are yet to be undertaken. There is one study that reported therapeutic effects of rACE2 in experimental liver fibrosis, in which liver injury was induced by BDL or carbon tetrachloride (CCl4) intoxication[59]. This study demonstrated that rACE2 reduced hepatic fibrosis in two animal models of liver disease[59]. Additionally, ACE2 gene knockout mice had elevated α-SMA protein and collagen content in the liver of CCl4-induced cirrhotic animals compared with those of wild-type controls[59]. These findings suggest that ACE2 of the alternate RAS is a potential target for liver fibrosis.
A major disadvantage of systemic therapy is that the treatment will inevitably produce off-target effects, which in many cases are undesirable. Thus, there are several disadvantages with systemic administration of rACE2. This includes daily injections of ACE2[59], a procedure that is invasive in a clinical setting and an expensive approach[52]. Increased circulating ACE2 is highly likely to produce off-target effects, including an effect on blood pressure. To circumvent this problem, an ideal approach would be to increase tissue- or organ-specific ACE2 levels. Thus, organ-specific increased ACE2 activity would not only produce long-term organ-specific benefits, but would also minimize unwanted off-target effects.
ACE2 OVER-EXPRESSION IN THE LIVER
Viral vectors are effective and safe vehicles to introduce a transgene into specific tissues or organs. Of the viral vectors that have been used to date to increase the delivery of genes, adeno-associated viral (AAV) vectors appears to be the most safe and effective, and are widely used in Phase I-II clinical trials. The AAV vector has been shown to be efficient in the delivery of a transgene, and provides many advantages over other candidate viral vectors that include replicative defectiveness, non-pathogenicity, minimal immunogenicity and broad tissue tropism in both animal models and humans. The AAV system has become a popular tool for gene delivery with its ability to maintain long-term gene and protein expression following a single injection of the vector. This type of gene delivery system has been widely tested for inherited metabolic diseases[60]. It is significant that, for the first time, the FDA has approved a pioneering gene therapy protocol using an AAV vector for a rare form of childhood blindness in 2017, the first such treatment cleared in the United States for an inherited disease. Moreover, gene therapy using the AAV vector was approved in 2012 by the European Commission for the treatment of patients with lipoprotein lipase deficiency (LPLD)[61]. However, because LPL deficiency is an extremely rare genetic disorder in human and the treatment is expensive, UniQure, the company that produced AAV vector to treat LPLD, has not renewed its EU license in 2017.
In line with this, our group has developed a safe and effective therapeutic approach using a pseudotyped AAV vector, which uses the AAV2 genome and liver-specific AAV8 capsid (AAV2/8) to deliver murine ACE2 (AAV2/8-mACE2). This showed that a single intraperitoneal injection of rAAV2/8-mACE2 produces sustained elevation of liver ACE2 expression for up to 6 mo. The treatment was administered to three short-term mouse models with liver disease[62], which included liver disease induced by BDL (2-wk model), CCl4 (8-wk model) and a methionine and choline deficient diet (8-wk model), representing cholestatic biliary fibrosis alcoholic liver fibrosis and NAFLD, respectively (Figure 3). AAV2/8-mACE2 therapy markedly reduced hepatic fibrosis in all three models. More importantly, they further demonstrated that, in addition to sustained expression of liver ACE2 for up to 6 mo, ACE2 over-expression was absent in other major organs such as heart, lungs, brain, intestines and kidneys. Increased liver ACE2 expression and activity was accompanied by increased hepatic Ang-(1-7) levels with a concomitant decrease in hepatic Ang II levels[62].
Figure 3.
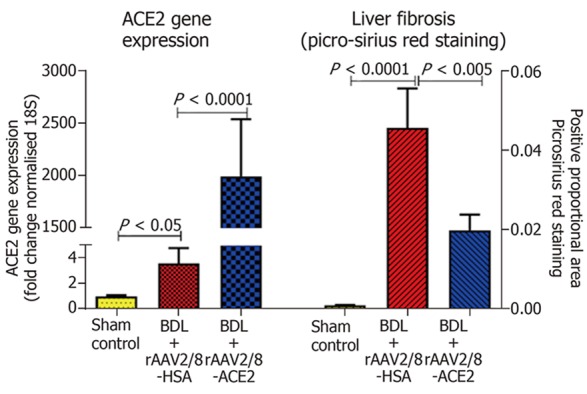
Hepatic ACE2 gene expression and fibrosis in a short-term model of biliary fibrosis with rAAV2/8-ACE2 therapy. ACE2 gene expression was significantly increased in ACE2-treated mice with biliary fibrosis compared to BDL mice injected with a control human serum albumin vector (rAAV2/8-HSA). rAAV2/8-ACE2 gene therapy markedly reduced the liver fibrosis in BDL mice compared to mice injected with rAAV2/8-HSA.
We have now confirmed the effectiveness of this treatment strategy in Mdr2-KO mice, a long-term animal model with progressive hepato-biliary fibrosis. This model, which has been widely used for studies that investigated pathophysiology of biliary fibrosis, produces lesions that resemble those of human PSC[63-65]. Gene therapy using the AAV2/8-mACE2 vector was very effective in Mdr2-KO mice, showing 50% and 80% reduction in liver fibrosis in both established and advanced liver disease, respectively (Table 1).
Table 1.
mACE2-rAAV2/8 therapy increased hepatic ACE2 expression, resulting in a marked reduction in biliary fibrosis in a long-term model of chronic biliary fibrosis (Mdr2-KO mice)
| Stage of the disease | Hepatic ACE2 expression, fold | Liver fibrosis reduction, % |
| Early: 3-6 mo | 60 | 50% |
| Advanced: 7-9 mo | 160 | 80% |
SUMMARY
In clinical practice, although UDCA is the standard treatment for PBC, reports indicate that approximately 35%-40% of PBC patients do not achieve optimum responses to UDCA[30]. On the other hand, PSC among other cholangiopathies is a significant biliary disease, and studies in patients with PSC showed that whilst standard doses of UDCA are not effective, higher doses produce serious adverse events[66,67]. Thus, the lack of an effective pharmacotherapy for biliary diseases is often associated with the condition progressing to biliary cirrhosis, and bears the risk of developing into HCC or cholangiocarcinoma. Therefore, liver transplantation is considered as the only treatment option for patients with chronic cholangiopathies, such as end-stage PSC and PBC. However, the shortage of donor livers creates a large, unmet need to develop effective therapies for these conditions.
ACE2 gene therapy is a potential strategy to treat human biliary fibrosis by delivering ACE2 using human liver-specific novel vectors with high transduction efficiency[68]. Therefore, it is important to select an AAV vector specific for human hepatocytes with enhanced transduction efficiency[68]. Recent studies have shown that novel AAV vectors, such as AAV-LK03, AAV3B and AAVrh10, which have been identified by AAV DNA re-shuffling, transduce human primary hepatocytes at higher efficiency[68,69]. Since the FDA and EU have now endorsed human gene therapy, novel approaches of gene therapy research that employ human liver-specific AAV vectors will lead to the formulation of therapeutic gene therapy applications for human biliary fibrosis.
Footnotes
Conflict-of-interest statement: None.
Manuscript source: Invited manuscript
Peer-review started: August 9, 2018
First decision: October 19, 2018
Article in press: December 11, 2018
Specialty type: Medicine, research and experimental
Country of origin: Canada
Peer-review report classification
Grade A (Excellent): A
Grade B (Very good): B
Grade C (Good): C, C
Grade D (Fair): 0
Grade E (Poor): 0
P- Reviewer: Morini S, Tao R, Tsoulfas G, Zhu YL S- Editor: Dou Y L- Editor: Filipodia E- Editor: Wu YXJ
Contributor Information
Indu G Rajapaksha, Department of Medicine, The University of Melbourne, Melbourne, VIC 3084, Australia.
Peter W Angus, Department of Gastroenterology and Hepatology, Austin Health, Melbourne, VIC 3084, Australia.
Chandana B Herath, Department of Medicine, The University of Melbourne, Melbourne, VIC 3084, Australia. cherath@unimelb.edu.au.
References
- 1.World Health Organization. Global health estimates 2014 summary tables: deaths by cause, age and sex, 2000-2012 Geneva, Switzerland, 2014 [Google Scholar]
- 2.Tu T, Calabro SR, Lee A, Maczurek AE, Budzinska MA, Warner FJ, McLennan SV, Shackel NA. Hepatocytes in liver injury: Victim, bystander, or accomplice in progressive fibrosis? J Gastroenterol Hepatol. 2015;30:1696–1704. doi: 10.1111/jgh.13065. [DOI] [PubMed] [Google Scholar]
- 3.Zhou WC, Zhang QB, Qiao L. Pathogenesis of liver cirrhosis. World J Gastroenterol. 2014;20:7312–7324. doi: 10.3748/wjg.v20.i23.7312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schuppan D, Afdhal NH. Liver cirrhosis. Lancet. 2008;371:838–851. doi: 10.1016/S0140-6736(08)60383-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Benyon RC, Iredale JP. Is liver fibrosis reversible? Gut. 2000;46:443–446. doi: 10.1136/gut.46.4.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655–1669. doi: 10.1053/j.gastro.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Perz JF, Armstrong GL, Farrington LA, Hutin YJ, Bell BP. The contributions of hepatitis B virus and hepatitis C virus infections to cirrhosis and primary liver cancer worldwide. J Hepatol. 2006;45:529–538. doi: 10.1016/j.jhep.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 8.Minton K. Extracellular matrix: Preconditioning the ECM for fibrosis. Nat Rev Mol Cell Biol. 2014;15:766–767. doi: 10.1038/nrm3906. [DOI] [PubMed] [Google Scholar]
- 9.Albanis E, Friedman SL. Hepatic Fibrosis: Pathogenesis and Principles of Therapy. Clin Liver Dis. 2001;5:315–334. doi: 10.1016/s1089-3261(05)70168-9. [DOI] [PubMed] [Google Scholar]
- 10.Pinzani M, Rosselli M, Zuckermann M. Liver cirrhosis. Best Pract Res Clin Gastroenterol. 2011;25:281–290. doi: 10.1016/j.bpg.2011.02.009. [DOI] [PubMed] [Google Scholar]
- 11.Liang S, Kisseleva T, Brenner DA. The Role of NADPH Oxidases (NOXs) in Liver Fibrosis and the Activation of Myofibroblasts. Front Physiol. 2016;7:17. doi: 10.3389/fphys.2016.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marrone G, Shah VH, Gracia-Sancho J. Sinusoidal communication in liver fibrosis and regeneration. J Hepatol. 2016;65:608–617. doi: 10.1016/j.jhep.2016.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol. 2017;14:397–411. doi: 10.1038/nrgastro.2017.38. [DOI] [PubMed] [Google Scholar]
- 14.DeLeve LD. Liver sinusoidal endothelial cells in hepatic fibrosis. Hepatology. 2015;61:1740–1746. doi: 10.1002/hep.27376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ana Claudia Maretti-Mira XW, Lei Wang, Laurie D. DeLeve. 1667 Role of incomplete stem cell maturation in hepatic fibrosis. AASLD. 2016;64:825A. [Google Scholar]
- 16.Jiang JX, Mikami K, Venugopal S, Li Y, Török NJ. Apoptotic body engulfment by hepatic stellate cells promotes their survival by the JAK/STAT and Akt/NF-kappaB-dependent pathways. J Hepatol. 2009;51:139–148. doi: 10.1016/j.jhep.2009.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Canbay A, Feldstein AE, Higuchi H, Werneburg N, Grambihler A, Bronk SF, Gores GJ. Kupffer Cell Engulfment of Apoptotic Bodies Stimulates Death Ligand and Cytokine Expression. Hepatology. 2003;38:1188–1198. doi: 10.1053/jhep.2003.50472. [DOI] [PubMed] [Google Scholar]
- 18.Boyer TD, Wright TL, Manns MP. Zakim and Boyer’s Hepatology. Elsevier Inc. 2006 [Google Scholar]
- 19.Bataller R, Brenner DA. Liver fibrosis. The Journal of Clinical Investigation. 2005:115; 209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao YL, Zhu RT, Sun YL. Epithelial-mesenchymal transition in liver fibrosis. Biomedical Reports. 2019;4:269–274. doi: 10.3892/br.2016.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kinnman N, Housset C. Peribiliary myofibroblasts in biliary type liver fibrosis. Frontiers in bioscience: a journal and virtual library; 2002. pp. 7; d496–503. [DOI] [PubMed] [Google Scholar]
- 22.Paizis G, Cooper ME, Schembri JM, Tikellis C, Burrell LM, Angus PW. Up-regulation of components of the renin-angiotensin system in the bile duct-ligated rat liver. Gastroenterology. 2002;123:1667–1676. doi: 10.1053/gast.2002.36561. [DOI] [PubMed] [Google Scholar]
- 23.Rioux KP, Sharkey KA, Wallace JL Swain MG. Hepatic mucosal mast cell hyperplasia in rats with secondary biliary cirrhosis. Hepatology. 1996;23:888–895. doi: 10.1002/hep.510230433. [DOI] [PubMed] [Google Scholar]
- 24.Jennifer Demieville LH, Lindsey Kennedy, Verinica Jarido, Heather L. Francis. 181 Knockout of the HDC/histamine axis and reduction of mast cell number/function rescues Mdr2-KO mice from PSC-related biliary proliferation and fibrosis. AASLD. 2016;64:96A. [Google Scholar]
- 25.Grizzi F, Di Caro G, Laghi L, Hermonat P, Mazzola P, Nguyen DD, Radhi S, Figueroa JA, Cobos E, Annoni G, et al. Mast cells and the liver aging process. Immun Ageing. 2013;10:9. doi: 10.1186/1742-4933-10-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paizis G, Gilbert RE, Cooper ME, Murthi P, Schembri JM, Wu LL, Rumble JR, Kelly DJ, Tikellis C, Cox A, et al. Effect of angiotensin II type 1 receptor blockade on experimental hepatic fibrogenesis. J Hepatol. 2001;35:376–385. doi: 10.1016/s0168-8278(01)00146-5. [DOI] [PubMed] [Google Scholar]
- 27.Thompson HL, Burbelo PD, Gabriel G, Yamada Y, Metcalfe DD. Murine mast cells synthesize basement membrane components. A potential role in early fibrosis. J Clin Invest. 1991;87:619–623. doi: 10.1172/JCI115038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schuppan D. Liver fibrosis: Common mechanisms and antifibrotic therapies. Clin Res Hepatol Gastroenterol. 2015;39:S51–S59. doi: 10.1016/j.clinre.2015.05.005. [DOI] [PubMed] [Google Scholar]
- 29.Fernández M, Semela D, Bruix J, Colle I, Pinzani M, Bosch J. Angiogenesis in liver disease. J Hepatol. 2009;50:604–620. doi: 10.1016/j.jhep.2008.12.011. [DOI] [PubMed] [Google Scholar]
- 30.de Vries E, Beuers U. Management of cholestatic disease in 2017. Liver Int. 2017;37 Suppl 1:123–129. doi: 10.1111/liv.13306. [DOI] [PubMed] [Google Scholar]
- 31.Lazaridis KN, Strazzabosco M, LaRusso NF. The cholangiopathies: Disorders of biliary epithelia. Gastroenterology. 2004;127:1565–1577. doi: 10.1053/j.gastro.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 32.Lazaridis KN, LaRusso NF. The Cholangiopathies. Mayo Clin Proc. 2015;90:791–800. doi: 10.1016/j.mayocp.2015.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kumar D, Tandon RK. Use of ursodeoxycholic acid in liver diseases. J Gastroen Hepatol (Australia) 2001;16:3–14. doi: 10.1046/j.1440-1746.2001.02376.x. [DOI] [PubMed] [Google Scholar]
- 34.Pinzani M, Luong TV. Pathogenesis of biliary fibrosis. Biochim Biophys Acta Mol Basis Dis. 2018;1864:1279–1283. doi: 10.1016/j.bbadis.2017.07.026. [DOI] [PubMed] [Google Scholar]
- 35.Chung BK, Karlsen TH, Folseraas T. Cholangiocytes in the pathogenesis of primary sclerosing cholangitis and development of cholangiocarcinoma. Biochim Biophys Acta Mol Basis Dis. 2018;1864:1390–1400. doi: 10.1016/j.bbadis.2017.08.020. [DOI] [PubMed] [Google Scholar]
- 36.Strazzabosco M, Fabris L. Development of the bile ducts: essentials for the clinical hepatologist. J Hepatol. 2012;56:1159–1170. doi: 10.1016/j.jhep.2011.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Williams MJ, Clouston AD, Forbes SJ. Links between hepatic fibrosis, ductular reaction, and progenitor cell expansion. Gastroenterology. 2014;146:349–356. doi: 10.1053/j.gastro.2013.11.034. [DOI] [PubMed] [Google Scholar]
- 38.O’Hara SP, Tabibian JH, Splinter PL, Larusso NF. The dynamic biliary epithelia: Molecules, pathways, and disease. J Hepatol. 2013;58:575–582. doi: 10.1016/j.jhep.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yoo KS, Lim WT, Choi HS. Biology of Cholangiocytes: From Bench to Bedside. Gut Liver. 2016;10:687–698. doi: 10.5009/gnl16033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hirschfield GM, Heathcote EJ, Gershwin ME. Pathogenesis of cholestatic liver disease and therapeutic approaches. Gastroenterology. 2010;139:1481–1496. doi: 10.1053/j.gastro.2010.09.004. [DOI] [PubMed] [Google Scholar]
- 41.Pérez Fernández T, López Serrano P, Tomás E, Gutiérrez ML, Lledó JL, Cacho G, Santander C, Fernández Rodríguez CM. Diagnostic and therapeutic approach to cholestatic liver disease. Rev Esp Enferm Dig. 2004;96:60–73. doi: 10.4321/s1130-01082004000100008. [DOI] [PubMed] [Google Scholar]
- 42.Alpini G, McGill JM, LaRusso NF. The pathobiology of biliary epithelia. Hepatology. 2002;35:1256–1268. doi: 10.1053/jhep.2002.33541. [DOI] [PubMed] [Google Scholar]
- 43.Blum HE. Chronic cholestatic liver diseases. J Gastroen Hepatol. 2002;17:S399–S402. doi: 10.1046/j.1440-1746.17.s3.34.x. [DOI] [PubMed] [Google Scholar]
- 44.Lazaridis KN, LaRusso NF. Primary Sclerosing Cholangitis. N Engl J Med. 2016;375:1161–1170. doi: 10.1056/NEJMra1506330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carbone M, Neuberger J. Liver transplantation in PBC and PSC: indications and disease recurrence. Clin Res Hepatol Gastroenterol. 2011;35:446–454. doi: 10.1016/j.clinre.2011.02.007. [DOI] [PubMed] [Google Scholar]
- 46.Genda T, Ichida T. Ohira H. Autoimmune Liver Diseases: Perspectives from Japan. Springer Japan; 2014. Liver Transplantation for Primary Biliary Cirrhosis; pp. 287–300. [Google Scholar]
- 47.Lindor KD, Kowdley KV, Harrison ME; American College of Gastroenterology. ACG Clinical Guideline: Primary Sclerosing Cholangitis. Am J Gastroenterol. 2015;110:646–59; quiz 660. doi: 10.1038/ajg.2015.112. [DOI] [PubMed] [Google Scholar]
- 48.Tabibian JH, Lindor KD. Primary sclerosing cholangitis: a review and update on therapeutic developments. Expert Rev Gastroenterol Hepatol. 2013;7:103–114. doi: 10.1586/egh.12.80. [DOI] [PubMed] [Google Scholar]
- 49.Lubel JS, Herath CB, Tchongue J, Grace J, Jia Z, Spencer K, Casley D, Crowley P, Sievert W, Burrell LM, et al. Angiotensin-(1-7), an alternative metabolite of the renin-angiotensin system, is up-regulated in human liver disease and has antifibrotic activity in the bile-duct-ligated rat. Clin Sci (Lond) 2009;117:375–386. doi: 10.1042/CS20080647. [DOI] [PubMed] [Google Scholar]
- 50.Grace JA, Herath CB, Mak KY, Burrell LM, Angus PW. Update on new aspects of the renin-angiotensin system in liver disease: clinical implications and new therapeutic options. Clin Sci (Lond) 2012;123:225–239. doi: 10.1042/CS20120030. [DOI] [PubMed] [Google Scholar]
- 51.Paul M, Poyan Mehr A, Kreutz R. Physiology of Local Renin-Angiotensin Systems. Physiol Rev. 2006;86:747. doi: 10.1152/physrev.00036.2005. [DOI] [PubMed] [Google Scholar]
- 52.Herath CB, Mak KY, Angus PW. Unger T, Steckelings UM, Souza dos Santos RA. The Protective Arm of the Renin Angiotensin System (RAS): Functional Aspects and Therapeutic Implications; 2015. Role of the Alternate RAS in Liver Disease and the GI Tract; pp. 239–247. [Google Scholar]
- 53.Herath CB, Warner FJ, Lubel JS, Dean RG, Jia Z, Lew RA, Smith AI, Burrell LM, Angus PW. Upregulation of hepatic angiotensin-converting enzyme 2 (ACE2) and angiotensin-(1-7) levels in experimental biliary fibrosis. J Hepatol. 2007;47:387–395. doi: 10.1016/j.jhep.2007.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Paizis G, Tikellis C, Cooper ME, Schembri JM, Lew RA, Smith AI, Shaw T, Warner FJ, Zuilli A, Burrell LM, et al. Chronic liver injury in rats and humans upregulates the novel enzyme angiotensin converting enzyme 2. Gut. 2005;54:1790–1796. doi: 10.1136/gut.2004.062398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Marques FD, Ferreira AJ, Sinisterra RD, Jacoby BA, Sousa FB, Caliari MV, Silva GA, Melo MB, Nadu AP, Souza LE, et al. An oral formulation of angiotensin-(1-7) produces cardioprotective effects in infarcted and isoproterenol-treated rats. Hypertension. 2011;57:477–483. doi: 10.1161/HYPERTENSIONAHA.110.167346. [DOI] [PubMed] [Google Scholar]
- 56.Wysocki J, Ye M, Rodriguez E, González-Pacheco FR, Barrios C, Evora K, Schuster M, Loibner H, Brosnihan KB, Ferrario CM, et al. Targeting the degradation of angiotensin II with recombinant angiotensin-converting enzyme 2: prevention of angiotensin II-dependent hypertension. Hypertension. 2010;55:90–98. doi: 10.1161/HYPERTENSIONAHA.109.138420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Oudit GY, Liu GC, Zhong J, Basu R, Chow FL, Zhou J, Loibner H, Janzek E, Schuster M, Penninger JM, et al. Human recombinant ACE2 reduces the progression of diabetic nephropathy. Diabetes. 2010;59:529–538. doi: 10.2337/db09-1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Haschke M, Schuster M, Poglitsch M, Loibner H, Salzberg M, Bruggisser M, Penninger J, Krähenbühl S. Pharmacokinetics and pharmacodynamics of recombinant human angiotensin-converting enzyme 2 in healthy human subjects. Clin Pharmacokinet. 2013;52:783–792. doi: 10.1007/s40262-013-0072-7. [DOI] [PubMed] [Google Scholar]
- 59.Osterreicher CH, Taura K, De Minicis S, Seki E, Penz-Osterreicher M, Kodama Y, Kluwe J, Schuster M, Oudit GY, Penninger JM, et al. Angiotensin-converting-enzyme 2 inhibits liver fibrosis in mice. Hepatology. 2009;50:929–938. doi: 10.1002/hep.23104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Alexander IE, Cunningham SC, Logan GJ, Christodoulou J. Potential of AAV vectors in the treatment of metabolic disease. Gene Ther. 2008;15:831–839. doi: 10.1038/gt.2008.64. [DOI] [PubMed] [Google Scholar]
- 61.Ferreira V, Petry H, Salmon F. Immune Responses to AAV-Vectors, the Glybera Example from Bench to Bedside. Front Immunol. 2014;5:82. doi: 10.3389/fimmu.2014.00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mak KY, Chin R, Cunningham SC, Habib MR, Torresi J, Sharland AF, Alexander IE, Angus PW, Herath CB. ACE2 Therapy Using Adeno-associated Viral Vector Inhibits Liver Fibrosis in Mice. Mol Ther. 2015;23:1434–1443. doi: 10.1038/mt.2015.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fickert P, Fuchsbichler A, Wagner M, Zollner G, Kaser A, Tilg H, Krause R, Lammert F, Langner C, Zatloukal K, et al. Regurgitation of bile acids from leaky bile ducts causes sclerosing cholangitis in Mdr2 (Abcb4) knockout mice. Gastroenterology. 2004;127:261–274. doi: 10.1053/j.gastro.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 64.Fickert P, Zollner G, Fuchsbichler A, Stumptner C, Weiglein AH, Lammert F, Marschall HU, Tsybrovskyy O, Zatloukal K, Denk H, et al. Ursodeoxycholic acid aggravates bile infarcts in bile duct ligated and Mdr2 knockout mice via disruption of cholangioles. Gastroenterology. 2002;123:1238–1251. doi: 10.1053/gast.2002.35948. [DOI] [PubMed] [Google Scholar]
- 65.Van Nieuwkerk CM, Elferink RP, Groen AK, Ottenhoff R, Tytgat GN, Dingemans KP, Van Den Bergh Weerman MA, Offerhaus GJ. Effects of Ursodeoxycholate and cholate feeding on liver disease in FVB mice with a disrupted mdr2 P-glycoprotein gene. Gastroenterology. 1996;111:165–171. doi: 10.1053/gast.1996.v111.pm8698195. [DOI] [PubMed] [Google Scholar]
- 66.Lindor KD. Ursodiol for Primary Sclerosing Cholangitis. New Engl J Med. 1997;336:691–695. doi: 10.1056/NEJM199703063361003. [DOI] [PubMed] [Google Scholar]
- 67.Lindor KD, Kowdley KV, Luketic VA, Harrison ME, McCashland T, Befeler AS, Harnois D, Jorgensen R, Petz J, Keach J, et al. High-dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis. Hepatology. 2009;50:808–814. doi: 10.1002/hep.23082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lisowski L, Dane AP, Chu K, Zhang Y, Cunningham SC, Wilson EM, Nygaard S, Grompe M, Alexander IE, Kay MA. Selection and evaluation of clinically relevant AAV variants in a xenograft liver model. Nature. 2014;506:382–386. doi: 10.1038/nature12875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang L, Bell P, Somanathan S, Wang Q, He Z, Yu H, McMenamin D, Goode T, Calcedo R, Wilson JM. Comparative Study of Liver Gene Transfer With AAV Vectors Based on Natural and Engineered AAV Capsids. Mol Ther. 2015;23:1877–1887. doi: 10.1038/mt.2015.179. [DOI] [PMC free article] [PubMed] [Google Scholar]