

Abstract
Autism spectrum disorder (ASD) is a heterogeneous group of neurobehavioral disorders characterized by the two core domains of behavioral deficits, including sociability deficits and stereotyped repetitive behaviors. It is not clear whether the core symptoms of ASD are produced by dysfunction of the overall neural network of the brain or that of a limited brain region. Recent studies reported that excessive glutamatergic or dopaminergic inputs in the dorsal striatum induced sociability deficits and repetitive behaviors. These findings suggest that the dorsal striatum plays a crucial role in autistic-like behaviors. The present study addresses whether functional deficits of well-known ASD-related genes in the dorsal striatum also produce ASD core symptoms. This study also examines whether these behavioral changes can be modulated by rebalancing glutamate and/or dopamine receptor activity in the dorsal striatum. First, we found that the siRNA-mediated inhibition of Shank3, Nlgn3, Fmr1, Mecp2, or Tsc1 in the dorsal striatum produced mild to severe behavioral changes in sociability, cognition, and/or repetitive behaviors. The knockdown effects of Mecp2 and Tsc1 on behavioral changes were the most prominent. Next, we demonstrated that behavioral changes induced by striatal inhibition of MeCP2 and TSC1 were rescued by D-cycloserine (an NMDA agonist), fenobam (an mGluR5 antagonist), SCH23390 (a D1 antagonist), and/or ecopipam (a D1 partial antagonist), pharmacological drugs that are known to regulate ASD-like symptoms in animal models. Collectively, these results suggest that the dorsal striatum is a critical brain region that, when dysfunctional, produces the core symptoms of ASD.
Keywords: Autism spectrum disorder, dorsal striatum, MeCP2, TSC1
Graphical Abstract
INTRODUCTION
Autism spectrum disorder (ASD) is a heterogeneous group of neurobehavioral disorders that is characterized by sociability deficits and restricted stereotypies [1]. Recent studies have identified a number of genes associated with ASD. Statistical modeling based on published results predicts that 1,000~1,500 genes may be associated with ASD [2,3]. The complexity of the functional profiles of known ASD genes makes it difficult to define shared mechanisms in ASD. It is also unclear whether ASD core symptoms are produced by dysfunction of the brain's overall neural network or by that of a specific brain region.
A growing body of evidence indicates that functional changes in the dorsal striatum promote sociability deficits and repetitive behaviors [4,5,6]. The dorsal striatum receives excitatory glutamatergic inputs from cortical and subcortical regions including the prefrontal cortex and thalamus [7], and dopaminergic inputs from the substantia nigra [8]. Adenylyl cyclase 5 (AC5) is preferentially expressed in the dorsal striatum, where it functions as an essential mediator of D1 and D2 dopamine receptors [9]. Recently, we reported that AC5 KO or D2 KO mice exhibit typical autistic-like behaviors. Similarly, siRNA-mediated inhibition of AC5 or D2 dopamine receptors locally in the dorsal striatum also produces the autistic-like phenotypes displayed by AC5 KO or D2 KO mice [5,6,10]. Furthermore, siRNA-mediated inhibition of mGluR3, GluN2B, or GluA1 in the dorsal striatum also produces sociability deficits and repetitive behaviors [5]. The gene knockdown effects were experimentally independent events in those experiments, whereas all produced similar autistic-like behaviors in these studies. The genes are related to the glutamate or dopamine receptor systems that act in the dorsal striatum [5,6,10]. The results of these studies raise the question of whether striatal inhibition of well-known ASD-related genes would produce autistic-like behaviors, although they are not related directly to glutamate or dopamine receptor signaling pathways.
Of the well-characterized ASD-related genes in previous studies, SHANK (SH3 and multiple ankyrin repeat domains), NLGN (neuroligin), and NRXN (neurexin) [11,12,13,14] are key components in synapse formation, MeCP2 (methyl CpG binding protein 2) [15] and FMR1 (fragile X mental retardation 1) play a role in transcription/post-transcriptional processes, and TSC1 and TSC2 (tuberous sclerosis proteins 1 and 2) [16] are cytosolic factors that regulate the mTOR signaling pathway.
In this study, we investigated whether striatal inhibition of well-known ASD-related genes could produce autistic-like behaviors. We found that siRNA-mediated suppression of Shank3, Nlgn3, Fmr1, Mecp2, or Tsc1 in the dorsal striatum induced mild to severe behavioral changes in autistic behaviors.
MATERIALS AND METHODS
Animals
Seven-week-old C57BL/6 male mice were purchased from Daehan BioLink (Eumsung, Chungbuk, Republic of Korea). Upon arrival, mice were housed in pairs in standard plastic cages and were fed lab chow and water ad libitum. The animal room was maintained at a temperature of 22~23℃ with 50~60% humidity and with a normal light-dark cycle (light on at 7:00 a.m.). It was a specific-pathogen-free environment. All animals were handled in accordance with the Guidelines of Animal Care at Ewha Womans University through permission of EWU-IACUC (No. 16-020).
Real-time PCR analysis
Real-time PCR analysis was performed as described previously [5,6]. Total RNA was purified from striatal tissues using TRI reagent (Sigma-Aldrich, St. Louis, MO, USA), and treated with DAase I to avoid genomic contamination. Real-time PCR was performed with iQTM SYBR Green Supermix (Bio-Rad Laboratories, Foster City, CA, USA) using the CFX 96 Real-Time PCR System Detector (Bio-Rad Laboratories; Foster City, CA, USA). The following primer sets were used: 5′- ACGAAGTGCCTGCGTCTGGAC-3′ and 5′-CTCTTGCCAACCATTCTCATCAGTG-3′ for Shank3; 5′- TGGAATGGGGACCAGGATGC-3′ and 5′-CAGTACTGTTGAACCCTGCGGC-3′ for Nlgn3; 5′-GCGTAGATGGGCTGCAAC-3′ and 5′-CCTGTGCCATCTTGCCTACT-3′ for Fmr1; 5′ -TGCTTTCAGTCATTTGGCTATA-3′ and 5′ TGTCGTATTCACCTTCAGTT-3′ for Mecp2; 5′-GAGGTAGAGTCACTGGAGGC-3′ and 5′-GAACTGGGAAGTGAGCCAAC-3′ for Tsc1; 5′-GCTGCCATCTGTTTTACGG-3′ and 5′-TGACTGGTGCCTGATGAACT-3′ for Gapdh; and 5′-GCTGCCATCTGTTTTACGG-3′ and 5′-TGACTGGTGCCTGATGAACT-3′ for L32.
Stereotaxic siRNA injection
Stereotaxic injection of siRNA was performed as described previously [5,6]. Briefly, mice were anesthetized using an injection of a mixture of ketamine hydrochloride and xylazine hydrochloride (the ratio of 2.5: 1) at a dose of 2.5 µl/g of body weight. The siRNA was injected at a volume of 1.5 µL (18 ng of siRNA) into each side of the dorsal striatum (AP, +1.0; ML, ±1.5; DV, −3.6 mm) using a stereotaxic apparatus (Stoelting Company, Wood Dale, IL, USA). Behavioral tests were examined 48 h after siRNA injection based on the known knockdown profile of target transcripts and behavioral modification effects from previous studies [17,18]. The following siRNAs were used: control-siRNA (SN-1012), Shank3-siRNA (1424750, NM_021423.2), Nlgn3-siRNA (1392209, NM_172932.1), Fmr1-siRNA (1358762, NM_008031.2), Mecp2-siRNA (1385135, NM_010788.2), and Tsc1-siRNA (1441941, NM_022887.2). These siRNAs were purchased from Bioneer Co. (Deajun, Korea). The FAM-labeled RISK-independent siRNA transfection control siGLO Green (D-001630-01-05) was purchased from Dharmacon Inc. (Chicago, IL, USA).
Drug administration
SCH23390, ecopipam, and D-cycloserine (DSC) were administered intraperitoneally (i.p.) 30 min prior to the behavioral tests. Fenobam was administered 1 h prior to the behavioral tests. The drug doses used were as follows: SCH23390 (0.02 mg/kg), ecopipam (0.02 mg/kg), DCS (20 mg/kg), and fenobam (30 mg/kg). These doses were chosen based on the dose tests in this study and in previous studies [5,6].
Behavioral assessments
Behavioral assessments were carried out using a computerized video tracking system (Panlab SMART, Harvard Apparatus, Barcelona, Spain) as described previously [5,6]. The behavior testing room was illuminated by 20 lux in the three-chamber assays, U-field assays, open field test, and repetitive behavior test. In contrast, it was illuminated by 250 lux in the marble burying test. White noise (65 dB) was used to mask background noise in the behavior testing room. Test equipment was frequently cleaned using 70% ethanol.
Sociability and social preference tests
The sociability test and social novelty preference test were carried out as described previously [5,6,10]. The U-field two-choice field was also used to test for social preference as described previously [5,6,19]. For the social novelty preference test, a subject mouse was allowed to freely explore the U-shaped two-choice field (45×45 cm2) for 10 min (habituation). While the subject mouse was returned to its home cage for two min, a familiar mouse (a cage mate) and an unfamiliar mouse (a new social target) were separately loaded into wire cages (12 cm in diameter) and placed at the corner of each closed square of the U-shaped arena. The subject mouse was placed in the center of the U-shaped field and allowed to freely explore the two-choice field for 10 min. During this time, the time spent and trajectory in each field were recorded.
Cage mates and strangers were prepared as described previously [5,6]. Mice were randomly housed in pairs two to three days prior to behavioral tests. The cage mate conditions were maintained until the behavioral tests were started. One cage mate was used as the subject mouse, while the other was used as the familiar target.
The sociability test using three chambers has been previously described [5,6]. Briefly, a subject mouse was allowed to freely explore the three chambers (22×32 cm2 each) for 10 min. After this habituation, a circular wire cage (12 cm in diameter) containing a social target (stranger; a naïve C57BL/6, same sex, same age) was placed in one side chamber, and while an empty wire cage was placed in the other side chamber. The subject mouse was allowed to freely explore both chambers for 10 min. Similarly, the time spent and the trajectory between chambers were recorded during this test. This test step was regarded as the sociability test.
Novel object preference test
The novel object preference test was performed as described previously [5,6]. Mice were presented with two identical objects (100-ml-glass-flasks containing cage bedding at a 3-cm-depth) placed 20 cm apart in the open field (30 cm×45 cm) and were familiarized with the objects for 10 min (habituation session). While the subject mouse was returned to its home cage for 2 min, one familiar object was exchanged for a novel object (a wooden block; 3.5 cm×3.5 cm×7 cm). After 2 min, the mice were placed in the center of the open-field arena, and allowed to freely explore the open-field. The time spent investigating an object by directing its nose within 2 cm of the object and/or sniffing was recorded for 5 min. The familiar object and novel object were used randomly and counterbalanced.
Open field test
The open field test was conducted as described previously [20]. In brief, mice were allowed to freely explore the open field (45×45 cm2) for 1 hour, and the moving distance was recorded.
Repetitive behavior assessments
Repetitive behavior assessments were carried out as described previously [5,6]. In brief, mice were placed individually in a home cage with new bedding. The times spent performing repetitive behaviors including grooming and digging were measured from video recordings of the 10 minute test session.
The marble burying test was carried out as described previously [5,6]. Mice were placed individually in a home cage that was filled with moderately fine wood chip bedding (JRS 3-4, Rosenberg, Germany). The bedding was layered up to 5 cm from the cage floor. Twelve marbles (diameter: 1.5 cm) were placed uniformly throughout the cage. Mice were allowed to freely explore the cage for 30 min. The number of buried marbles was recorded every five minutes. A marble was considered “burying” when <25% of it was visible.
Statistical analysis
Two-sample comparisons were performed using Student's t-test. Multiple comparisons were performed by one-way and two-way ANOVA or two-way repeated measures ANOVA followed using a post hoc test using Graphpad Prism 6 (San Diego, CA, USA). All data are presented as mean±SEM or box plot diagrams. Statistical significance was defined by the 5% level.
RESULTS
The siRNA-mediated inhibition of Shank3, Nlgn3, Fmr1, Mecp2, or Tsc1 in the dorsal striatum produced mild to severe autistic-like behaviors
To determine whether functional deficits of well-known ASD-related genes in the dorsal striatum would produce autistic-like behaviors, Shank3-siRNA, Nlgn3-siRNA, Fmr1-siRNA, Mecp2-siRNA, or Tsc1-siRNA was injected bilaterally in the dorsal striatum of normal mice. Two days later, when the siRNA-mediated knockdown effects are fully effective [10,21,22] (Fig. 1B, I), standard behavioral tests were used to assess the mice (Fig. 1A).
Fig. 1. siRNA-mediated suppression of Shank3, Nlgn3, Fmr1, Mecp2 or Tsc1 in the dorsal striatum produced mild to severe autistic-like phenotypes. (A) Experimental design of siRNA injection into the dorsal striatum and following behavioral tests. A diagram showing siRNA injection sites in the dorsal striatum (AP, +1.0; ML, ±1.5; DV, −3.6 mm). Representative photomicrograph showing the dorsal striatum injected with fluorescence-tagged siRNA-control. Brain sections were prepared 2 days after siRNA injection. dST: dorsal striatum; cc: corpus callosum. (B) Real-time PCR data showing the level of siRNA-induced knockdown of Shank3, Nlgn3, Fmr1, Mecp2 and Tsc1 in HT22 cells. Cells were harvested 24 h after siRNA transfection. (C, D) Grooming (C) and digging (D) behaviors of mice with the knockdown of Shank3, Nlgn3, Fmr1, Mecp2 or Tsc1 in the dorsal striatum. (E, F) Experimental design of the social novelty preference test using U-field two-choice field (E). The exploration time of mice with the knockdown of Shank3, Nlgn3, Fmr1, Mecp2 or Tsc1 in the dorsal striatum (F). (G, H) Experimental design of novel object preference (G), and sniffing time against the object of mice with the knockdown of Shank3, Nlgn3, Fmr1, Mecp2 or Tsc1 in the dorsal striatum (H). n=6–8 animals. (I) Real-time PCR data showing the siRNA-induced knockdown of Mecp2 and Tsc1 in the dorsal striatum of WT mice (I). n=3 animals, and 3 PCR repeats for each siRNA. (J, K) Experimental design of the three chamber test (J). Social interaction time between stranger vs. empty cage for mice with the knockdown of Mecp2 and Tsc1 in the dorsal striatum (K). (L, M) Locomotion in the open field test (L), and marble burying (M) behaviors of mice with the knockdown of Mecp2 and Tsc1 in the dorsal striatum. ** in Figure 1M denotes the difference between Con-siRNA and Tsc1-siRNA at p<0.01 at time points marked by arrowheads. n=6–9 animals. Data are presented as mean±SEM. * and ** denote the differences between the indicated groups at p<0.05 and p<0.01, respectively (Student's t-test, two-way ANOVA, two-way repeated measures ANOVA and Holm-Sidak post hoc test).

Mice injected with Nlgn3-siRNA, Mecp2-siRNA, or Tsc1-siRNA showed increased grooming behavior compared to those that received control-siRNA injection. Mice injected with Nlgn3-siRNA or Shank3-siRNA exhibited reduced digging behavior or only a tendency of reduction in digging compared to those with control-siRNA injection. However, mice injected with Fmr1-siRNA injection displayed no significant change in grooming or digging behavior (Fig. 1C, D). We recently demonstrated that the sociability test using a U-shaped two-choice field is useful to determine whether an animal is sociable with a novel social target in the presence of a familiar cage-mate [5,6]. This test involves presenting the subject mouse with a social novelty (a non-mate) and a socially familiar target (a cage-mate) in the U-shaped two-choice field. In this U-shaped two-choice field test, mice injected with Fmr1-siRNA, Mecp2-siRNA, or Tsc1-siRNA in the dorsal striatum exhibited decreased time interacting with an unfamiliar target over a cage-mate compared to that of mice injected with the control-siRNA. In contrast, mice injected with Shank3-siRNA or Nlgn3-siRNA did not show significant changes in this test (Fig. 1E, F). In the novel object preference test, mice injected with Shank3-siRNA, Nlgn3-siRNA, Fmr1-siRNA, Mecp2-siRNA, or Tsc1-siRNA spent less time sniffing a novel object over a familiar one compared to that of mice injected with the control-siRNA (Fig. 1G, H).
Overall, these results indicate that injection of Shank3-siRNA, Nlgn3-siRNA, Fmr1-siRNA, Mecp2-siRNA, or Tsc1-siRNA in the dorsal striatum produced mild to severe repetitive behavior, and deficits in sociability or novel object preference. Of these, the knockdown of Mecp2 or Tsc1 produced the most robust changes in sociability and grooming behaviors.
Next, we examined the behavioral features of mice with striatal knockdown of Mecp2 or Tsc1 in more detail. In the sociability test using a three chamber apparatus, mice with Mecp2-siRNA injection displayed preference of a social target over an empty object. However, mice with Mecp2-siRNA injection tended to have less interaction with a social target compared to that of mice injected with the control-siRNA (siRNA×social target interaction; F(2, 33)=6.433, p=0.0044, two-way ANOVA). Mice injected with Tsc1-siRNA also showed decreased social interaction with a social target (Fig. 1I–K).
Mice with Tsc1 knockdown showed increased locomotion in the open field and reduced marble burying behavior compared to their respective controls with control-siRNA injection. Mice with Mecp2 knockdown tended to have increased locomotion (p=0.1876) and reduced marble burying (Fig. 1L, M) (siRNA×time interaction; F (6, 84)=3.694, p=0.0026; the siRNA main effects; F (1, 14)=3.556, p=0.0803, repeated two-way ANOVA and following Sidak post-hoc test).
Collectively, these results suggest that local inhibition of Shank3, Nlgn3, Fmr1, Mecp2, or Tsc1 in the dorsal striatum produces mild to severe behavioral changes with regard to repetitive behavior, sociability, or novel object preference, although there were some differences in phenotypic severity across these genetic changes and behavioral tests. The siRNA-mediated knockdown of Mecp2 or Tsc1 in the dorsal striatum resulted in ~50% reduction in Mecp2 or Tsc1 transcript levels compared to that of the controls, as assessed by RT-PCR (Fig. 1H). Despite this modest effect, mice with siRNA-mediated inhibition of Mecp2 or Tsc1 in the dorsal striatum exhibited autistic-like phenotypes.
Autistic-like phenotypes induced by striatal inhibition of MeCP2 or TSC1 were rescued by rebalancing glutamate or dopaminergic activity
Recent pharmacological studies combined with siRNA-mediated functional analyses [5,6] demonstrated that the autistic-like phenotypes of AC5 KO mice or D2 KO mice were modulated by D-cycloserine (an NMDA agonist), fenobam (an mGluR5 antagonist), SCH23390 (a D1 antagonist) and/or ecopipam (a D1/5 antagonist). We examined whether the autistic-like phenotypes induced by inhibition of MeCP2 or TSC1 in the dorsal striatum could be modulated by pharmacologically targeting the dorsal striatum.
Mice with siRNA-mediated inhibition of Mecp2 in the dorsal stratum exhibited increased grooming behavior compared to that of controls. This increased grooming was suppressed by D-cycloserine (an NMDA agonist), fenobam (an mGluR5 antagonist), or ecopipam (a D1 partial antagonist), but not with SCH23390 (a D1 antagonist) (Fig. 2A, B). The behavioral deficits in the sociable novelty preference of mice with Mecp2 knockdown were reversed by D-cycloserine, SCH23390, or ecopipam, but not by fenobam (Fig. 2D, E). The impairment in novel object preference of mice with Mecp2 inhibition was rescued by D-cycloserine, fenobam, or ecopipam, but not by SCH23390 (Fig. 2G, H).
Fig. 2. Autistic-like phenotypes induced by the striatal inhibition of MeCP2 or TSC1 were rescued by rebalancing glutamate or dopaminergic activity. (A) Experimental design of siRNA injection into the dorsal striatum and following behavioral tests. A diagram showing the siRNA injection sites in the dorsal striatum (AP, +1.0; ML, ±1.5; DV, −3.6 mm). (B, C) Grooming behaviors of Mecp2-knockdown (B) or Tsc1-knockdown (C) mice after treatment with D-cycloserine (DCS), fenobam, SCH23390, or ecopipam. (D~F) Experimental design of the social novelty preference test (D). Social novelty preference test of Mecp2-knockdown (E) or Tsc1-knockdown (F) mice after treatment with D-cycloserine (DCS), fenobam, SCH23390, or ecopipam. (G~I) Experimental design of the novel object preference test (G). Novel object preference test of Mecp2-knockdown (H) or Tsc1-knockdown (I) mice after treatment with D-cycloserine (DCS), fenobam, SCH23390, or ecopipam. Mecp2-siRNA, n=6–8 animals; Tsc1-siRNA, n=6–8 animals. Data are presented as mean±SEM. * and ** denote the differences between the indicated groups at p<0.05 and p<0.01, respectively (One-way ANOVA, two-way ANOVA, and Holm-Sidak post hoc test).

Mice with Tsc1 knockdown in the dorsal stratum exhibited increased grooming behavior compared to that of controls. However, this enhanced grooming could not be significantly reversed by D-cycloserine, fenobam, SCH23390, or ecopipam (Fig. 2A, C). In contrast, the social novelty preference deficits of mice with Tsc1 knockdown was rescued by D-cycloserine, fenobam, SCH23390 or ecopipam (Fig. 2D, F). The bluntness in novel object preference of mice with Tsc1 knockdown was also rescued by D-cycloserine, SCH23390 or ecopipam, but not by fenobam (Fig. 2G, I).
Overall, these results suggest that autistic-like phenotypes induced by siRNA-mediated inhibition Mecp2 or Tsc1 can be modified by modulation of the glutamate or dopamine receptors. However, the specific mechanisms involved in each of these pharmacological effects on behavioral outputs appear to be complex.
DISCUSSION
Striatal dysfunction of ASD-related genes produced sociability deficits and repetitive behaviors
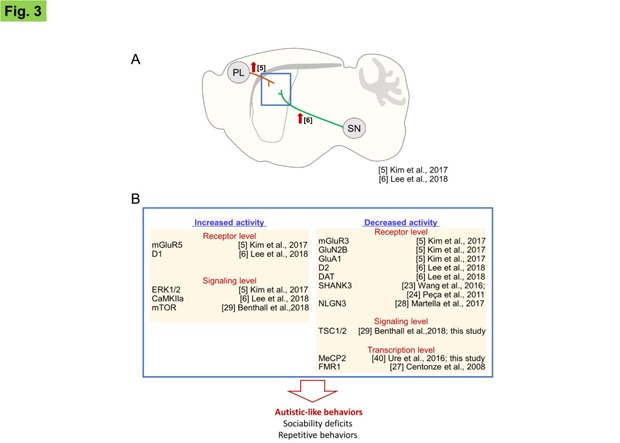
In this study, we investigated the neuroanatomical correlates of ASD core symptoms. Our results and evidence from the literatures support the hypothesis that striatal dysfunction can induce impairments in the two core domains of ASD behavioral symptoms, including sociability deficits and repetitive behaviors. First, as demonstrated here, local inhibition of MeCP2 and TSC1 in the dorsal striatum, and although to a much lesser extent, that of SHANK3, NLGN3, and FMR1, produced ASD-related behaviors. These findings are based on the results of the typical behavioral tests used in preclinical studies. Second, optogenetic stimulation of cortico-striatal glutamatergic neurons in wildtype mice produced sociability deficits (Fig. 3A; ref [5]). Thus, increased glutamatergic inputs to the dorsal striatum, which might induce the activation of various types of glutamatergic receptors, were pro-autistic [5]. Partly consistent with this finding, AC5 KO mice [5], D2 KO mice [6], or Shank3 KO mice (Shank3Δe4-22−/−) [23] that exhibited autistic-like behaviors had an increased activity of mGluR5 in the dorsal striatum, whereas inhibition of mGluR5 in the dorsal striatum antagonized their autistic phenotypes [5,6,23]. In contrast, siRNA-mediated knockdown of GluN2B, GluA1 or mGluR3 in the dorsal striatum in normal mice also produced autistic-like behaviors (Fig. 3B; ref [5]). These somewhat complex and contradictory findings raise the possibility that both enhancement of corticostriatal glutamatergic inputs and imbalance in the activities of specific glutamate receptors in the dorsal striatum could be pro-autistic, although detailed mechanisms should be elaborated in the future. Third, mice with increased dopamine release in the dorsal striatum via siRNA-mediated suppression of dopamine transporter in synaptic terminals of substantia nigra neurons or optogenetic stimulation of the nigro-striatal circuitry showed sociability deficits and repetitive behaviors (Fig. 3A). These behavioral changes were blocked by D1 receptor antagonists [6]. Moreover, the siRNA-mediated knockdown of D2 dopamine receptors or overactivation of D1 dopamine receptors in the dorsal striatum induced autistic-like behaviors (Fig. 3B; ref [6]). Fourth, SHANK3 was preferentially expressed in the dorsal striatum. Mice with a genetic deletion of Shank3 showed reduced excitability of striatal neurons and autistic-like behaviors (Fig. 3B; ref [24]). Autistic-like behaviors could also be generated by the transgenic and viral vector-mediated disruption of cholinergic interneurons and parvalbumin-positive interneurons in the dorsal striatum [25] or conditional knockout of diacylglycerol lipase α (DGLα; a synthetic enzyme for endocannabinoid 2-arachidonoylglycerol) in D1-expressing medium spiny neurons [26]. Fmr1 −/− mice show increased GABA release in striatal neurons (Fig. 3B; ref [27]). Mice with the R451C-neuroligin3 mutation exhibit autistic-like behaviors and impaired long-term synaptic depression at corticostriatal glutamatergic synapses (Fig. 3B; ref [28]). Collectively, these results suggest that dysfunction of the dorsal striatum is the main source of the core ASD symptoms. However, the results of this study do not necessarily exclude important contributions from other brain regions, such as the prefrontal cortex and hippocampus, in the full expression of ASD-related symptoms. Many ASD-related phenotypes, whether they are neurologic or not, require immediate clinical attention.
Fig. 3. The striatal dysfunction hypothesis of autistic-like behaviors. (A) Optogenetic stimulation of the glutamatergic inputs from the prefrontal cortex (PL) to the dorsal striatum (dST) suppressed social interaction with a social novelty and novel object preference [5]. Similarly, optogenetic stimulation of the dopaminergic inputs from the substantia nigra (SN) to the dorsal striatum reduced social interaction [6]. (B) A list of the genes whose dysfunction in the dorsal striatum produced autistic-like behaviors. Molecular, genetic, and pharmacological data suggest that increased activity of mGluR5, D1, ERK1/2, CaMKIIα, or mTOR or decreased activity of mGluR3, GluN2B, GluA1, SHANK3, NLGN3, D2, DAT, TSC1/2, MeCP2, or FMR1 promoted pro-autistic behaviors. References are indicated.

As demonstrated here, siRNA-mediated inhibition of Mecp2 or Tsc1 in the dorsal striatum produced sociability deficits and increased grooming behavior. The siRNA-mediated inhibition of Shank3, Nlgn3, or Fmr1 produced only weak or specific ASD-related phenotypes. The following possibilities must be considered to appropriately interpret these results. First, although sociability and stereotyped repetitive behaviors are complex, they can be manipulated using the siRNA-mediated knockdown technique [5,6,21,22]. Therefore, this method is powerful and has contributed to dissecting the role of a specific gene in regulating behaviors. However, this method also has limitations. The siRNA-mediated inhibition of a specific gene is transient and usually incomplete. Therefore, it is possible that siRNA-mediated knockdown effects at the injected site are not strong enough to produce behavioral changes. siRNA methods are prone to potential off-target effects. Complementary methods, such as pharmacological or genetic knockout methods, might be required in parallel to overcome siRNA off-target effects. It is also possible that there is a compensatory mechanism. SHANK and NLGN include multiple families and isoforms. Each group of proteins or isoforms exhibits a different expression pattern depending on brain region. We also do not exclude the possibility that SHANK3, NLGN3, and FMR1 play an important role in a specific development stage, and then, their developmental dysfunction may contribute to developmental defects and autistic symptoms. The disruption in the balance between excitatory and inhibitory activities in the dorsal striatum was reported in various ASD models, including mutant mice for Fmr1, Shank3, Met receptor and Cntnap2 [4]. Furthermore, the synaptic transmission was impaired in MSNs of mice with the mutation of Shank3, Nlgn3, Fmr1, Mecp2 and Tsc1 [24,27,28,29,30].
Local inhibition of MeCP2 or TSC1 in the dorsal striatum produced sociability deficits and repetitive behaviors
In this study, we demonstrated that siRNA-mediated inhibition of Mecp2 or Tsc1 in the dorsal striatum produced sociability deficits and repetitive behaviors. MeCP2 is a global transcription regulator that interacts with HDACs, Sin3A and CREB [31,32]. Mutations in MECP2 cause Rett syndrome [33], which is characterized by microcephaly, seizures, autism, language problems, and stereotypic hand movements [34]. Rett syndrome is not listed in the ASD section of the DSM 5, but it does include autistic phenotypes. In addition, MECP2 is involved in regulation of ASD-related genes [3,35]. Tuberous sclerosis complex (TSC) 1 and 2 are cytosolic factors that negatively regulate mammalian target of rapamycin (mTOR), via a small G protein, called Ras homologue enriched in the brain (Rheb) [36]. Mutations in TSC1/2 are well-known genetic causes of ASD [16,37]. Mice with Tsc1 or Tsc2 heterozygous mutation have deficits in learning and memory [38], and social interaction [39]. Tsc1 KO mice show increased cortico-striatal excitatory synaptic transmission into D1-expressing medium spiny neurons [29]. Regarding those results, it might be possible that siRNA-mediated knockdown of Mecp2 or Tsc1 induces autistic-like phenotypes through the alternation in excitability of striatal neurons.
Mutations in MeCP2 and TSC1 disrupt the mTOR pathway in the opposite direction and change GABA-mediated inhibitory activity [35]. Mecp2 conditional knockout mice have reduced striatal GABAergic activity [40]. Mecp2-null male mice (Mecp2-/y) have decreased mTOR signaling in the cerebral cortex and hippocampus [41]. In contrast, Tsc1 or 2 mutations result in increased mTOR signaling, which interferes with cell proliferation and neural development [36], whereas rapamycin (an mTOR inhibitor) treatment alleviates the cognitive and social deficits of Tsc1 or Tsc2 heterozygous mice [39]. These results raise the question of whether anti-autistic effects of rapamycin were produced through the inhibition of the mTOR signaling pathway or other unknown targets. A simple possibility is that anti-autistic effects of rapamycin could be produced through the mechanism that is not directly related to the mTOR signaling. However, considering the results of the present study that siRNA-mediated inhibition of Tsc1/2 or Mecp2 in the dorsal striatum produced autistic-like behaviors, it is also necessary to investigate whether inhibition of Tsc1/2 or Mecp2 in the dorsal striatum also change the mTOR pathway in the opposite direction.
Mice with Mecp2-siRNA or Tsc1-siRNA injection in the dorsal striatum exhibited increased grooming, but decreased marble burying behavior (Fig. 2). Thus, mice with Mecp2-siRNA or Tsc1-siRNA injection exhibited complex phenotypes in the view of the repetitive behaviors. The marble burying test is commonly used to measure not only repetitive behavior, but also anxiety in rodents. In addition, rodents display burying behavior in response to an aversive stimulus, and this behavior is referred to “defensive burying” [42]. Thus, marble burying behavior could present multiple behavioral aspects. Furthermore, Mecp2 and Tsc1/2 mutant mice display increased anxiety [15,43]. Therefore, it will require further analysis what the changes in marble burying behavior displayed by mice with Mecp2-siRNA or Tsc1-siRNA injection mean.
Autistic-like phenotypes can be rescued by rebalancing glutamate or dopamine receptor activity
We recently demonstrated that autistic-like phenotypes displayed by AC5 KO mice or D2 KO mice were rescued by pharmacological inhibition of mGluR5 with fenobam and of D1 receptors with SCH23390/ecopipam (SCH39166) or pharmacological activation of GluN with D-cycloserine [5,6]. Although functional inhibition of MeCP2 or TSC1 is not directly related to the genetic deletion of AC5 or D2 receptor, autistic-like behaviors induced by striatal inhibition of MeCP2 or TSC1 (Fig. 2) were also rescued by treatment with D-cycloserine, fenobam, SCH23390 and/or ecopipam (Fig. 2). These results raise the possibility that autistic-like phenotypes induced by functional deficits in the dorsal striatum could be reversed by modulation of the glutamate or dopamine receptor systems. It will be worth to examine whether similar mechanisms work for other cases of ASD animal models.
Concerning the pharmacological effects of SCH23390 and ecopipam, the impairment in novel object preference in mice with MeCP2 inhibition was rescued by ecopipam, but not by SCH23390 (Fig. 2G, H). Thus, there seems to be a subtle difference in pharmacological effects of SCH23390 and ecopipam. SCH23390 is a halobenzazepine that acts as a selective antagonist of D1 and D5 (Kis=0.2 and 0.3 nM, respectively) [44], and also show high affinity at the serotonin receptor subtype 5-HT2C (Ki, 9.3 nM) [45], and 5-HT1C. However, the doses required to induce a similar response in vivo are greater than 10-fold higher than those required to induce a D1-mediated response [44]. Ecopipam (SCH 39166) is a synthetic benzazepine derivative that acts as a selective antagonist of D1 and D5. Ecopipam has reduced affinity at 5-HT2 to compared that of SCH23390 [46]. Therefore, it might be possible that the subtle difference in pharmacological profiles of SCH23390 and ecopipam produces slightly differential behavioral responses.
ACKNOWLEDGEMENTS
This research was supported by a grant (HI15C1834) from the Ministry of Health and Welfare, Republic of Korea.
References
- 1.American Psychiatric Association. Diagnostic and statistical manual of mental disorders. Washington, D.C: American Psychiatric Association; 2013. [Google Scholar]
- 2.Krumm N, O'Roak BJ, Shendure J, Eichler EE. A de novo convergence of autism genetics and molecular neuroscience. Trends Neurosci. 2014;37:95–105. doi: 10.1016/j.tins.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ziats MN, Rennert OM. The evolving diagnostic and genetic landscapes of autism spectrum disorder. Front Genet. 2016;7:65. doi: 10.3389/fgene.2016.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fuccillo MV. Striatal circuits as a common node for autism pathophysiology. Front Neurosci. 2016;10:27. doi: 10.3389/fnins.2016.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim H, Lee Y, Park JY, Kim JE, Kim TK, Choi J, Lee JE, Lee EH, Kim D, Kim KS, Han PL. Loss of adenylyl cyclase type-5 in the dorsal striatum produces autistic-like behaviors. Mol Neurobiol. 2017;54:7994–8008. doi: 10.1007/s12035-016-0256-x. [DOI] [PubMed] [Google Scholar]
- 6.Lee Y, Kim H, Kim JE, Park JY, Choi J, Lee JE, Lee EH, Han PL. Excessive D1 dopamine receptor activation in the dorsal striatum promotes autistic-like behaviors. Mol Neurobiol. 2018;55:5658–5671. doi: 10.1007/s12035-017-0770-5. [DOI] [PubMed] [Google Scholar]
- 7.Huerta-Ocampo I, Mena-Segovia J, Bolam JP. Convergence of cortical and thalamic input to direct and indirect pathway medium spiny neurons in the striatum. Brain Struct Funct. 2014;219:1787–1800. doi: 10.1007/s00429-013-0601-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roeper J. Dissecting the diversity of midbrain dopamine neurons. Trends Neurosci. 2013;36:336–342. doi: 10.1016/j.tins.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 9.Lee KW, Hong JH, Choi IY, Che Y, Lee JK, Yang SD, Song CW, Kang HS, Lee JH, Noh JS, Shin HS, Han PL. Impaired D2 dopamine receptor function in mice lacking type 5 adenylyl cyclase. J Neurosci. 2002;22:7931–7940. doi: 10.1523/JNEUROSCI.22-18-07931.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim H, Lee Y, Kim JE, Han PL. Reversal of an unconditioned behavioral preference for specific food pellets by intervention of whisker sensory inputs. Exp Neurobiol. 2016;25:79–85. doi: 10.5607/en.2016.25.2.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huguet G, Ey E, Bourgeron T. The genetic landscapes of autism spectrum disorders. Annu Rev Genomics Hum Genet. 2013;14:191–213. doi: 10.1146/annurev-genom-091212-153431. [DOI] [PubMed] [Google Scholar]
- 12.O'Roak BJ, Vives L, Fu W, Egertson JD, Stanaway IB, Phelps IG, Carvill G, Kumar A, Lee C, Ankenman K, Munson J, Hiatt JB, Turner EH, Levy R, O'Day DR, Krumm N, Coe BP, Martin BK, Borenstein E, Nickerson DA, Mefford HC, Doherty D, Akey JM, Bernier R, Eichler EE, Shendure J. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science. 2012;338:1619–1622. doi: 10.1126/science.1227764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.O'Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, Levy R, Ko A, Lee C, Smith JD, Turner EH, Stanaway IB, Vernot B, Malig M, Baker C, Reilly B, Akey JM, Borenstein E, Rieder MJ, Nickerson DA, Bernier R, Shendure J, Eichler EE. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485:246–250. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Toro R, Konyukh M, Delorme R, Leblond C, Chaste P, Fauchereau F, Coleman M, Leboyer M, Gillberg C, Bourgeron T. Key role for gene dosage and synaptic homeostasis in autism spectrum disorders. Trends Genet. 2010;26:363–372. doi: 10.1016/j.tig.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 15.Chahrour M, Zoghbi HY. The story of Rett syndrome: from clinic to neurobiology. Neuron. 2007;56:422–437. doi: 10.1016/j.neuron.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 16.Smalley SL. Autism and tuberous sclerosis. J Autism Dev Disord. 1998;28:407–414. doi: 10.1023/a:1026052421693. [DOI] [PubMed] [Google Scholar]
- 17.Kim KS, Kim H, Park SK, Han PL. The dorsal striatum expressing adenylyl cyclase-5 controls behavioral sensitivity of the righting reflex to high-dose ethanol. Brain Res. 2012;1489:27–36. doi: 10.1016/j.brainres.2012.10.016. [DOI] [PubMed] [Google Scholar]
- 18.Kim H, Kim TK, Kim JE, Park JY, Lee Y, Kang M, Kim KS, Han PL. Adenylyl cyclase-5 in the dorsal striatum function as a molecular switch for the generation of behavioral preferences for cue-directed food choices. Mol Brain. 2014;7:77. doi: 10.1186/s13041-014-0077-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park JY, Kim TK, Choi J, Lee JE, Kim H, Lee EH, Han PL. Implementation of a two-dimensional behavior matrix to distinguish individuals with differential depression states in a rodent model of depression. Exp Neurobiol. 2014;23:215–223. doi: 10.5607/en.2014.23.3.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim KS, Lee KW, Baek IS, Lim CM, Krishnan V, Lee JK, Nestler EJ, Han PL. Adenylyl cyclase-5 activity in the nucleus accumbens regulates anxiety-related behavior. J Neurochem. 2008;107:105–115. doi: 10.1111/j.1471-4159.2008.05592.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bortolozzi A, Castañé A, Semakova J, Santana N, Alvarado G, Cortés R, Ferrés-Coy A, Fernández G, Carmona MC, Toth M, Perales JC, Montefeltro A, Artigas F. Selective siRNA-mediated suppression of 5-HT1A autoreceptors evokes strong anti-depressant-like effects. Mol Psychiatry. 2012;17:612–623. doi: 10.1038/mp.2011.92. [DOI] [PubMed] [Google Scholar]
- 22.Mehta SL, Kim T, Vemuganti R. Long noncoding RNA FosDT promotes ischemic brain injury by interacting with REST-associated chromatin-modifying proteins. J Neurosci. 2015;35:16443–16449. doi: 10.1523/JNEUROSCI.2943-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang X, Bey AL, Katz BM, Badea A, Kim N, David LK, Duffney LJ, Kumar S, Mague SD, Hulbert SW, Dutta N, Hayrapetyan V, Yu C, Gaidis E, Zhao S, Ding JD, Xu Q, Chung L, Rodriguiz RM, Wang F, Weinberg RJ, Wetsel WC, Dzirasa K, Yin H, Jiang YH. Altered mGluR5-Homer scaffolds and corticostriatal connectivity in a Shank3 complete knockout model of autism. Nat Commun. 2016;7:11459. doi: 10.1038/ncomms11459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peça J, Feliciano C, Ting JT, Wang W, Wells MF, Venkatraman TN, Lascola CD, Fu Z, Feng G. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature. 2011;472:437–442. doi: 10.1038/nature09965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rapanelli M, Frick LR, Xu M, Groman SM, Jindachomthong K, Tamamaki N, Tanahira C, Taylor JR, Pittenger C. Targeted interneuron depletion in the dorsal striatum produces autism-like behavioral abnormalities in male but not female mice. Biol Psychiatry. 2017;82:194–203. doi: 10.1016/j.biopsych.2017.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shonesy BC, Parrish WP, Haddad HK, Stephenson JR, Báldi R, Bluett RJ, Marks CR, Centanni SW, Folkes OM, Spiess K, Augustin SM, Mackie K, Lovinger DM, Winder DG, Patel S, Colbran RJ. Role of striatal direct pathway 2-arachidonoylglycerol signaling in sociability and repetitive behavior. Biol Psychiatry. 2018;84:304–315. doi: 10.1016/j.biopsych.2017.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Centonze D, Rossi S, Mercaldo V, Napoli I, Ciotti MT, De Chiara V, Musella A, Prosperetti C, Calabresi P, Bernardi G, Bagni C. Abnormal striatal GABA transmission in the mouse model for the fragile X syndrome. Biol Psychiatry. 2008;63:963–973. doi: 10.1016/j.biopsych.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 28.Martella G, Meringolo M, Trobiani L, De Jaco A, Pisani A, Bonsi P. The neurobiological bases of autism spectrum disorders: the R451C-neuroligin 3 mutation hampers the expression of long-term synaptic depression in the dorsal striatum. Eur J Neurosci. 2018;47:701–708. doi: 10.1111/ejn.13705. [DOI] [PubMed] [Google Scholar]
- 29.Benthall KN, Ong SL, Bateup HS. Corticostriatal transmission is selectively enhanced in striatonigral neurons with postnatal loss of Tsc1. Cell Reports. 2018;23:3197–3208. doi: 10.1016/j.celrep.2018.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chao HT, Chen H, Samaco RC, Xue M, Chahrour M, Yoo J, Neul JL, Gong S, Lu HC, Heintz N, Ekker M, Rubenstein JL, Noebels JL, Rosenmund C, Zoghbi HY. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature. 2010;468:263–269. doi: 10.1038/nature09582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jones PL, Veenstra GJ, Wade PA, Vermaak D, Kass SU, Landsberger N, Strouboulis J, Wolffe AP. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet. 1998;19:187–191. doi: 10.1038/561. [DOI] [PubMed] [Google Scholar]
- 32.Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, Zoghbi HY. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320:1224–1229. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 34.Amir RE, Zoghbi HY. Rett syndrome: methyl-CpG-binding protein 2 mutations and phenotype-genotype correlations. Am J Med Genet. 2000;97:147–152. doi: 10.1002/1096-8628(200022)97:2<147::aid-ajmg6>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 35.Winden KD, Ebrahimi-Fakhari D, Sahin M. Abnormal mTOR activation in autism. Annu Rev Neurosci. 2018;41:1–23. doi: 10.1146/annurev-neuro-080317-061747. [DOI] [PubMed] [Google Scholar]
- 36.Huang J, Manning BD. The TSC1-TSC2 complex: a molecular switchboard controlling cell growth. Biochem J. 2008;412:179–190. doi: 10.1042/BJ20080281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wiznitzer M. Autism and tuberous sclerosis. J Child Neurol. 2004;19:675–679. doi: 10.1177/08830738040190090701. [DOI] [PubMed] [Google Scholar]
- 38.Goorden SM, van Woerden GM, van der Weerd L, Cheadle JP, Elgersma Y. Cognitive deficits in Tsc1+/− mice in the absence of cerebral lesions and seizures. Ann Neurol. 2007;62:648–655. doi: 10.1002/ana.21317. [DOI] [PubMed] [Google Scholar]
- 39.Sato A, Kasai S, Kobayashi T, Takamatsu Y, Hino O, Ikeda K, Mizuguchi M. Rapamycin reverses impaired social interaction in mouse models of tuberous sclerosis complex. Nat Commun. 2012;3:1292. doi: 10.1038/ncomms2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ure K, Lu H, Wang W, Ito-Ishida A, Wu Z, He LJ, Sztainberg Y, Chen W, Tang J, Zoghbi HY. Restoration of Mecp2 expression in GABAergic neurons is sufficient to rescue multiple disease features in a mouse model of Rett syndrome. Elife. 2016;5:e14198. doi: 10.7554/eLife.14198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ricciardi S, Boggio EM, Grosso S, Lonetti G, Forlani G, Stefanelli G, Calcagno E, Morello N, Landsberger N, Biffo S, Pizzorusso T, Giustetto M, Broccoli V. Reduced AKT/mTOR signaling and protein synthesis dysregulation in a Rett syndrome animal model. Hum Mol Genet. 2011;20:1182–1196. doi: 10.1093/hmg/ddq563. [DOI] [PubMed] [Google Scholar]
- 42.De Boer SF, Koolhaas JM. Defensive burying in rodents: ethology, neurobiology and psychopharmacology. Eur J Pharmacol. 2003;463:145–161. doi: 10.1016/s0014-2999(03)01278-0. [DOI] [PubMed] [Google Scholar]
- 43.Ehninger D, Silva AJ. Increased levels of anxiety-related behaviors in a Tsc2 dominant negative transgenic mouse model of tuberous sclerosis. Behav Genet. 2011;41:357–363. doi: 10.1007/s10519-010-9398-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bourne JA. SCH 23390: the first selective dopamine D1-like receptor antagonist. CNS Drug Rev. 2001;7:399–414. doi: 10.1111/j.1527-3458.2001.tb00207.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Millan MJ, Newman-Tancredi A, Quentric Y, Cussac D. The “selective” dopamine D1 receptor antagonist, SCH23390, is a potent and high efficacy agonist at cloned human serotonin2C receptors. Psychopharmacology (Berl) 2001;156:58–56. doi: 10.1007/s002130100742. [DOI] [PubMed] [Google Scholar]
- 46.Barnett A, McQuade RD, Tedford C. Highlights of D1 dopamine receptor antagonist research. Neurochem Int. 1992;20(Suppl):119S–122S. doi: 10.1016/0197-0186(92)90223-e. [DOI] [PubMed] [Google Scholar]