

Abstract
The microscope is the quintessential tool for discovery in cell biology. From its earliest incarnation as a tool to make the unseen visible, microscopes have been at the center of most revolutionizing developments in cell biology, histology and pathology. Major quantum leaps in imaging involved the dramatic improvements in resolution to see increasingly smaller structures, methods to visualize specific molecules inside of cells and tissues, and the ability to peer into living cells to study dynamics of molecules and cellular structures. The latest revolution in microscopy is Deep Imaging—the ability to look at very large numbers of samples by high-throughput microscopy at high spatial and temporal resolution. This approach is rooted in the development of fully automated high-resolution microscopes and the application of advanced computational image analysis and mining methods. Deep Imaging is enabling two novel, powerful approaches in cell biology: the ability to image thousands of samples with high optical precision allows every discernible morphological pattern to be used as a read-out in large-scale imaging-based screens, particularly in conjunction with RNAi-based screening technology; in addition, the capacity to capture large numbers of images, combined with advanced computational image analysis methods, has also opened the door to detect and analyze very rare cellular events. These two applications of Deep Imaging are revolutionizing cell biology.
Keywords: High-throughput imaging, Deep Imaging, High-content screening
Introduction
It can be argued that the microscope has been the principal catalyst of modern cell and molecular biology. The realization that organisms are made up of the individual cells seen through the early microscopes in the late nineteenth century sparked the quest to first understand the nature of organisms and led to the revolutionary discovery of cells as the building blocks of all living things (Mazzarello 1999). But as we now know, that was only the beginning. Improvements in optics and lens manufacturing allowed, by the early twentieth century, to begin to see the wealth of subcellular structures, particularly chromosomes during mitosis and the large organelles such as mitochondria and the Golgi complex. The application of electron microscopy in the 1950s to biological specimens allowed for the first time the visualization of details of cellular morphology and opened up the doors to a detailed characterization of the inner workings of the cell. The development in the 1970s of methods to localize molecules by indirect immunofluorescence and immune-electron microscopy elevated the study of cells to a new level and permitted for the first time the probing of molecular processes inside of cells, rather than just the morphological description of cellular structures (Lazarides and Weber 1974; Roth et al. 1978). The following decades saw improvements in resolution first by the invention of confocal microscopy and then by using elegant tricks to overcome the optical resolution limit of light microscopes, leading the development of the now popular super-resolution imaging modalities (Cremer and Cremer 1978; Patterson et al. 2010). These innovations in imaging technology were complemented by the ground-breaking discovery of genetically encoded fluorescent proteins which allowed the study of protein localization and their dynamics in living cells (Chalfie et al. 1994). The sum total of these advances is our current ability to observe, track and quantitate individual molecules inside of living cells, and increasingly in intact organisms, with remarkably high sensitivity and accuracy.
Despite all these major contributions, microscopy has traditionally been a limited tool for unbiased discovery for several reasons. Most microscopy methods require the use of antibodies or the expression of fluorescently tagged proteins. As a consequence, most strategies are restricted to candidate approaches in which the behavior of a known molecule upon a specific perturbation, such as a change in growth condition or drug treatment, is assessed (Simpson et al. 2001). However, it has been difficult to use imaging to identify novel, unexpected players in a given cellular process, as for example is possible in unbiased large-scale screening approaches. Another limitation of traditional imaging approaches is their single cell nature. While the inherent property of analyzing individual cells offers the advantage of assessing the degree of heterogeneity in a population of cells and of discerning differences amongst subpopulations of cells, the limited throughput imposed by the need to observe cells one by one makes it very difficult to capture rare events by traditional imaging methods. This is a particularly consequential drawback considering that many biologically relevant events occur very infrequently, as for example the formation of tumorigenic chromosome translocations or the eviction of a cell with metastatic potential from the primary tumor. Recent progress in microscopy automation has overcome these obstacles. Large numbers of cells can now be imaged routinely without loss of image quality. These high-throughput imaging approaches are, once again, revolutionizing shaping how we do cell and molecular biology.
High-throughput imaging: seeing more, seeing deeper
As the name implies, high-throughput imaging (HTI) describes the ability to analyze a large number of samples (Conrad and Gerlich 2010). In HTI, cells are typically plated in 96-, 384- or 1,536-well plates and all manipulations are performed in a fully, or at least partially, automated pipeline, including automated cell seeding and robotic sample manipulations such as dispensing of small molecules or siRNA solutions, washes and staining procedures (Fig. 1). Due to the inherent large sample numbers, image acquisition is performed in a fully automated manner and images are analyzed using image analysis software rather than examined by visual inspection as is done in traditional microscopy. The scale of HTI experiments varies considerable between various applications, but high-end imaging systems are able to acquire several hundred thousands of images per day. Given the very large primary datasets, an integral part of HTI is the use of automated image analysis approaches to detect distribution patterns of detected proteins or of morphological features (Fig. 1). Ironically, despite being based on high-end imaging capacities, in these approaches, the user relies heavily on computerized image analysis and quantitative analysis parameters, such as signal intensity or quantitation of distribution patterns, and only visually inspects images for quality control. However, an added benefit of the mandatory use of image analysis methods in HTI is the highly quantitative nature of the resulting data, overcoming another weakness of traditional imaging approaches, which are often based on anecdotal observations or qualitative analysis. The quantitative data also allow careful statistical analysis of any potential differences among samples, resulting in a very sensitive tool to probe cellular organization. In addition, user bias is reduced by the highly quantitative nature of analysis and the large sample size. A key aspect of HTI is the fact that the ability to image very large numbers of samples does not come with a trade-off in image quality and resolution (Fig. 1). HTI approaches have been implemented in a wide range of imaging modalities including confocal, spinning-disk and FRET-based microscopy methods (Conrad and Gerlich 2010).
Fig. 1.
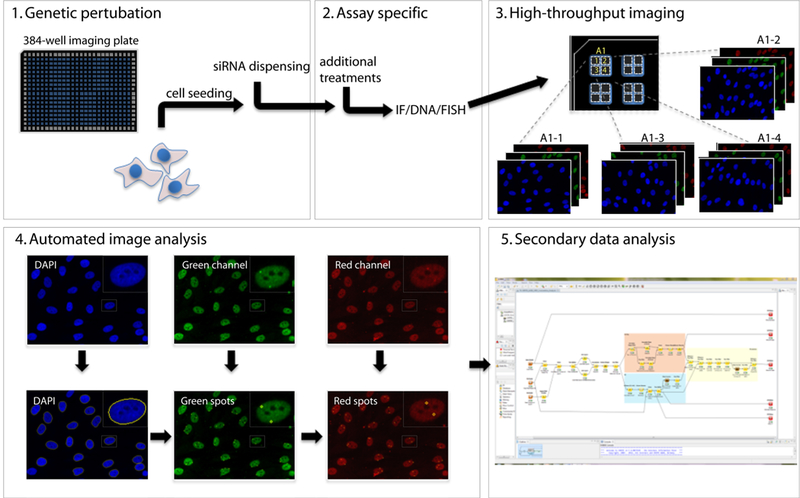
Imaging-based screening. The workflow of an imaging-based RNAi screens typically involves the plating of cells in multi-well plates, and their automated manipulation including addition of siRNA reagents before a specific biological assay is performed. Detection of signal may occur via immunocytochemical methods, by use of genetically encoded markers, or analysis of morphological features. Samples are imaged in a fully automated high-throughput fashion and dedicated image analysis algorithms used to detect and quantitate signals. Secondary data analysis identifies hits based on statistical analysis
The seemingly trivial advance of being able to image very large numbers of samples has overcome several central limitations of imaging and has opened the door to two major applications of imaging: the routine use of imaging in screening assays and the observation of rare biological events.
Optical screens: imaging as a powerful tool for unbiased discovery
It can be argued that any localization pattern or morphological feature ever seen through a microscope can in principle be used as the basis for a cellular assay to ask what cellular factors are involved in establishing or maintaining the observed pattern. For example, what cellular factors determine the distribution of mitochondria in cells or what are the proteins that govern the organization of genes and chromosomes inside the cell nucleus? These types of questions have now become amenable to unbiased discovery approaches due to the confluence of HTI and the emergence of RNA interference methods. In HTI-RNAi approaches, cells are typically plated in 384-wells, transfected with siRNA oligonucleotides or infected with virus expressing shRNAs, to knock-down individual gene products in each well. After appropriate incubation, cells are imaged and the particular pattern of interest imaged in every assay well using HTI; siRNAs, which affect the assay pattern are identified as positive hits (Conrad and Gerlich 2010; Pepperkok and Ellenberg 2006).
Examples of such imaging-based RNAi screens now abound. The mother of such screen was the systematic identification of factors that affect mitosis (Neumann et al. 2010). Using a whole-genome RNAi library, Neumann et al. monitored the effect of individually silenced 21,000 protein-coding human genes on mitosis in HeLa cells by automated high-throughput time-lapse microscopy. Then, based on automated image analysis algorithms of several millions of timelapse movies, several classes of mitotic defects were defined, including delayed entry into mitosis, formation of aberrant daughter nuclei and failure to undergo cytokinesis. This approach yielded several hundred novel putative mitosis factors (Neumann et al. 2010). The fact that more than half of them were novel and not previously implicated in mitosis powerfully underlines the potential of imaging-based screens for unbiased discovery. Another impressive example of a genome-wide screen is the recent identification of factors which mediate the translocation of the parkin protein to damaged mitochondria (Hasson et al. 2013). Parkin is a component of an E3 ubiquitin complex involved in protein degradation, and mutations in parkin are known to cause familial Parkinson’s disease (Shen and Cookson 2004). The protein translocates into mitochondria upon mitochondrial damage and the screen was designed to discover novel factors that mediate this event. The screen identified several components of the parkin translocation and accumulation mechanisms including TOM7 as an importer, the heat-shock protein HSPA1L and BAG4 as antagonistic regulator of import and SIAH3 as an inhibitor of parkin accumulation (Hasson et al. 2013). The screen was based on the detection by HTI of GFP-labeled parkin inside of mitochondria, which were labeled by using a red fluorescent marker, overcoming the limited throughput of biochemical assays to detect parkin in mitochondria. This screen identified not only individual factors involved in parkin mitochondrial translocation, but outlined several regulatory steps of the mitophagy pathway (Hasson et al. 2013).
While siRNA screens such these can obviously be performed at a genome-wide scale, focused screens using smaller siRNA libraries representing a particular class of gene, such as kinases, chromatin proteins or protein degradation factors, for example, are increasingly popular. Examples of such focused screens are the identification human protein quality control factors using a library of 1,591 siRNA genes involved in the ubiquitin-proteasome system (UPS) (Pegoraro et al. 2012) or the discovery of novel regulators of synaptogenesis in a screen of 597 lentivirally expressed shRNAs targeting synapse-related genes (Nieland et al. 2014). Such focused screens are attractive as they are logistically less complex, require lower throughput and are thus faster to execute. In addition, custom-designed focused libraries targeting a particular pathway of interest can also be used to systematically probe the role of each pathway component in a given process.
Numerous variations on the theme of HTI-based screens exist. One powerful approach is the use of genetically manipulatable organisms, such as yeast, Drosophila or C. elegans, rather than RNA interference methods to eliminate individual genes. An impressive example of this approach is the identification of factors involved in various stages of yeast spindle assembly using a collection of yeast deletion strains each lacking a single gene (Vizeacoumar et al. 2010). Another modification of HTI approaches is the use of overexpression libraries rather than knockdown libraries. In these strategies, libraries of fluorescently tagged proteins are expressed and their localization classified after HTI to identify novel proteins with particularly localization patterns. Examples of such screens are the mapping of the localization of several hundred proteins of the secretory pathway (Starkuviene et al. 2004) or the identification of several hundred proteins localization to various nuclear subcompartments such as the nucleolus, Cajal bodies, PML bodies or nuclear splicing speckles (Fong et al. 2013). Given the versatility and power of imaging-based screens, combined with the increasing ease of their execution due to the availability of custom-made siRNA or overexpression libraries and of sophisticated turn-key imaging systems and imagine analysis tools, it is safe to predict that imaging-based high-throughput screens will increasingly become a standard discovery tool in cell biology.
Deep imaging: chromosome translocations as a case study
Many important cellular events, particularly some with catastrophic consequences, occur with low frequency and are rare; capturing these events and studying them in a cell population is thus a formidable challenge. In addition to enabling screening approaches, the ability to image large numbers of samples by HTI has also made it possible to capture and analyze rare cellular events. Having high-throughput imaging tools in hand, the strategy to visualize rare events is simple—imaging a large number of cells over long periods of time, often using time-lapse microscopy, and then mine the data to locate the rare events of interest. A prime case study to illustrate the power of such an approach, referred to as “Deep Imaging” in analogy to large-scale sequencing methods, is the recent visualization and analysis of the formation of chromosome translocations (Roukos et al. 2013).
Chromosome translocations are recombination events between broken chromosomes that create aberrant fusions with key roles in tumorigenesis (Mitelman et al. 2007). During a translocation, a part of a chromosome breaks off and reattaches to another chromosome. Translocations are found in most tumors and in some cases have been shown to lead to cancer by either the formation of fusion genes expressing chimeric proteins with oncogenic potential or by disrupting genes or their regulatory elements causing gene misregulation (Roukos and Misteli 2014). Because of their detrimental nature, and because cells have developed sophisticated DNA damage repair mechanisms, translocations are exceedingly rare. As a consequence, it has been impossible to observe them in intact cells and the mechanistic studies of their formation have been limited to population studies, which do not fully reflect what happens in an individual cell.
The application of a deep imaging approach, combined with a custom-tailored system to induce DNA breaks, has recently provided first insights into the cell biology of translocations (Roukos et al. 2013) (Fig. 2). To visualize the formation of chromosome translocations, a cell-based system was engineered in NIH3T3 fibroblasts to mimic the events that are required for a translocation to form. In this system, DNA double-strand breaks (DSBs) are induced in distinct chromosomes in a controlled fashion by the integration of the restriction sequence for the yeast endonuclease IScel, leading to the formation of DSBs when the endonuclease is expressed experimentally (Roukos et al. 2014). The endonuclease restriction sites are integrated adjacent to DNA arrays of the Lac and Tet operator repeats, which mark the chromosome ends and can be used to visualize and track in space and time the location of the induced breaks by virtue of binding of specific fluorescently tagged DNA binding proteins (LacR, TetR; Fig. 2). Using this system, the fate of individual chromosome breaks can be followed in thousands of cells for long periods of time by high-throughput timelapse microscopy. Among all the cells imaged, the few in which a translocation is formed are extracted computationally based on the persistent superimposition of Lac and Tet arrays (Fig. 2). The image datasets associated with these cells are then used for further analysis to ask questions about the genesis of the translocation.
Fig. 2.
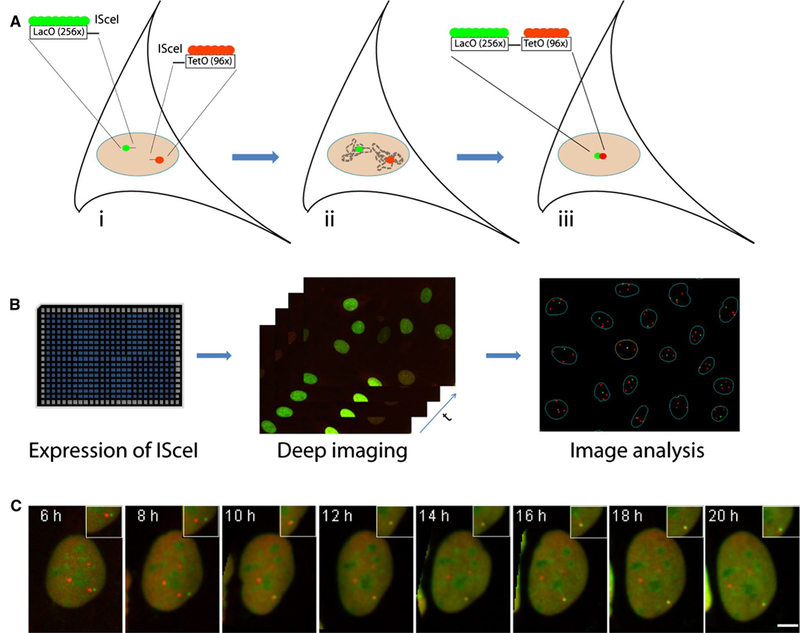
Observation of a rare cellular event by deep imaging. a Design of a cell-based system to capture the formation of chromosome translocations in living cells. (i) Double-strand breaks (DSBs) are induced at engineered ISceI restriction sites integrated on distinct chromosomes adjacent to the LacO and TetO array sequences, which can be visualized through the LacO/TetO operator/repressor system (red, green, respectively). (ii) The fluorescently marked chromosome ends are tracked in space and in time by timelapse deep imaging and (iii) the rare joining of broken chromosomes in the form of a translocation is captured by the persistent superimposition of the two fluorescently labeled chromosome ends as identified by automated image analysis. b workflow of a deep imaging experiment. Cells are plated in multi-well plates (96, 384 wells), thousands of cells imaged in a fully automated fashion over time and features of interest (LacO/TetO spots) automatically extracted using image analysis software. Imaging data are then mined for analysis. c visualization of a chromosome translocation by timelapse microscopy. Cells were transfected with the ISceI endonuclease, and 6 h later, timelapse microscopy was performed to follow DSBs marked by the LacO (green) and TetO (red) for up to 20 h. Broken chromosome ends are seen to congregate and ultimately fuse as they translocate. Maximal projected image sequences are shown. Scale bar 5 μm
Using this system, it became possible for the first time to directly track the fate of DSBs and to fully describe the events leading to a translocation in an individual living cell. These studies showed that DSBs in mammalian cells undergo a nondirectional, locally restricted, saltatory motion, which is indistinguishable from the motion of intact chromosome loci (Roukos et al. 2013). Some of the broken chromosome ends pair with other DSBs and undergo transient cycles of pairing and dissociation, and while many of these pairs eventually separate, some DSBs persistently synapse, making them susceptible for illegitimate joining. These studies also provided unique insights into the cell biology of the occurrence of reciprocal translocations, in which pieces of two chromosomes breaking off and switch places, because it became apparent in these tracking studies that the two intrachromosomal ends generated by the DSB move together to the area of synapsis with the other chromosome partners, indicating that the illegitimate joining takes place after the chromosomal partners are placed in proximity (Roukos et al. 2013). Therefore, the probability of a mutual chromosome exchange of both chromosome ends to reciprocal translocations is higher than if chromosome ends were separated before their synapsis.
A key question in the field has long been when in the cell cycle translocations occur. Using deep imaging in combination with a novel tool to assess the cell cycle status of individual cells by imaging of its DNA content (Roukos et al. 2013), it was found that the percentage of cells with synapsed DSBs remains constant in the various cell cycle phases, suggesting that the observed variability in mobility previously observed at various points in the cell cycle (Heun et al. 2001; Krawczyk et al. 2012; Walter et al. 2003) does not affect the probability of DSB pairing (Roukos et al. 2013). These findings are in line with the emerging notion that nonhomologous end joining, a repair pathway which is active in all stages of the cell cycle, is the major repair pathway in the formation of translocations (Mao et al. 2008; Symington and Gautier 2011; Weinstock et al. 2006).
Deep imaging also laid to rest another key question in the field, which was whether the nonrandom organization of genes and chromosomes in the 3D space of the nucleus contributes to determining which chromosome translocates with which other chromosome (Misteli 2007). As the physical association of DSBs within the nuclear space is a fundamental step in the formation of chromosome translocations, it had long been proposed that the spatial arrangement of the broken chromosomes relative to each other contributes to the propensity to form translocations. This hypothesis has been extensively assessed over the last decade by correlative cytogenetic and population-based genome-wide mapping approaches, and the emerging evidence strongly suggested spatial genome organization as a driver of determining translocation frequency (Hakim et al. 2012; Meaburn et al. 2007; Roix et al. 2003; Zhang et al. 2012). To directly test this model, it was, however, necessary to identify, in an individual cell, the spatial origin of DSBs involved in translocations and follow them until they form a translocation. Using deep imaging, translocating DSBs were tracked by high-throughput timelapse microscopy and image analysis was used to assess their relative position several hours prior to their synapsis. This analysis revealed that more than 80 % of translocating breaks originated from locations within 2.5 μm of each other suggesting that translocations are formed predominantly, albeit not exclusively, by proximal DSBs and that the spatial arrangement of chromosomes in the nucleus significantly contributes to translocation partner choice (Roukos et al. 2013).
Deep imaging methods can also be used to probe cellular mechanisms. In the case of translocation formation, it is known that the formation of a chromosome translocation requires the involvement of the DNA repair machinery to join, albeit incorrectly, the chromosome ends. By using high-throughput imaging and automated image analysis to measure the rate of synapsed DSBs in the presence of inhibitors or after siRNA-mediated knockdown of key players of the DNA damage response (DDR), it was revealed that inhibition of upstream components of the DNA damage response pathway decreased the percentage of cells with paired DSBs and translocations and in this way identified specific factors involved in determining translocation frequency (Roukos et al. 2013). Taken together, these observations are a powerful demonstration of the types of questions Deep Imaging can answer.
Conclusions
High-throughput imaging methods are allowing researchers to do two things that were previously very difficult: to perform large-scale and highly sensitive imaging-based screens and to observe and mechanistically analyze rare biological events. While screening approaches can be applied to just about any area of cell biology, automated methods to detect rare events or characterize the behavior of subpopulations of cells by tracing them individually will be particularly impactful in fields characterized by cell- to-cell heterogeneity in the population, such as stem cell biology. High-throughput imaging is a powerful new tool in biology, and its emergence extends the long tradition of imaging as a driver of innovation in biomedicine.
Acknowledgments
We thank Gianluca Pegoraro and Sigal Shachar for Fig. 1. Work in the Misteli laboratory is supported by the Intramural Research Program of the National Institutes of Health (NIH), NCI, Center for Cancer Research.
Contributor Information
Vassilis Roukos, National Cancer Institute, NIH, Bethesda, MD 20892, USA.
Tom Misteli, National Cancer Institute, NIH, Bethesda, MD 20892, USA.
References
- Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC (1994) Green fluorescent protein as a marker for gene expression. Science (New York, NY) 263:802–805 [DOI] [PubMed] [Google Scholar]
- Conrad C, Gerlich DW (2010) Automated microscopy for high-content RNAi screening. J Cell Biol 188:453–461. doi: 10.1083/jcb.200910105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremer C, Cremer T (1978) Considerations on a laser-scanning- microscope with high resolution and depth of field. Microsc Acta 81:31–44 [PubMed] [Google Scholar]
- Fong KW et al. (2013) Whole-genome screening identifies proteins localized to distinct nuclear bodies. J Cell Biol 203:149–164. doi: 10.1083/jcb.201303145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakim O et al. (2012) DNA damage defines sites of recurrent chromosomal translocations in B lymphocytes. Nature 484:69–74. doi: 10.1038/nature10909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasson SA et al. (2013) High-content genome-wide RNAi screens identify regulators of parkin upstream of mitophagy. Nature 504:291–295. doi: 10.1038/nature12748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heun P, Laroche T, Shimada K, Furrer P, Gasser SM (2001) Chromosome dynamics in the yeast interphase nucleus. Science (New York, NY) 294:2181–2186. doi: 10.1126/science.1065366 [DOI] [PubMed] [Google Scholar]
- Krawczyk PM et al. (2012) Chromatin mobility is increased at sites of DNA double-strand breaks. J Cell Sci. doi: 10.1242/jcs.089847 [DOI] [PubMed] [Google Scholar]
- Lazarides E, Weber K (1974) Actin antibody: the specific visualization of actin filaments in non-muscle cells. Proc Natl Acad Sci USA 71:2268–2272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Z, Bozzella M, Seluanov A, Gorbunova V (2008) DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle 7:2902–2906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzarello P (1999) A unifying concept: the history of cell theory. Nat Cell Biol 1:E13–E15. doi: 10.1038/8964 [DOI] [PubMed] [Google Scholar]
- Meaburn KJ, Misteli T, Soutoglou E (2007) Spatial genome organization in the formation of chromosomal translocations. Semin Cancer Biol 17:80–90. doi: 10.1016/j.semcancer.2006.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misteli T (2007) Beyond the sequence: cellular organization of genome function. Cell 128:787–800. doi: 10.1016/j.cell.2007.01.028 [DOI] [PubMed] [Google Scholar]
- Mitelman F, Johansson B, Mertens F (2007) The impact of translocations and gene fusions on cancer causation. Nat Rev Cancer 7:233–245. doi: 10.1038/nrc2091 [DOI] [PubMed] [Google Scholar]
- Neumann B et al. (2010) Phenotypic profiling of the human genome by time-lapse microscopy reveals cell division genes. Nature 464:721–727. doi: 10.1038/nature08869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieland TJ et al. (2014) High content image analysis identifies novel regulators of synaptogenesis in a high-throughput RNAi screen of primary neurons. PloS One 9:e91744. doi: 10.1371/journal.pone.0091744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson G, Davidson M, Manley S, Lippincott-Schwartz J (2010) Superresolution imaging using single-molecule localization. Annu Rev Phys Chem 61:345–367. doi: 10.1146/annurev.physchem.012809.103444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pegoraro G, Voss TC, Martin SE, Tuzmen P, Guha R, Misteli T (2012) Identification of mammalian protein quality control factors by high-throughput cellular imaging. PloS One 7:e31684. doi: 10.1371/journal.pone.0031684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepperkok R, Ellenberg J (2006) High-throughput fluorescence microscopy for systems biology. Nat Rev Mol Cell Biol 7:690–696. doi: 10.1038/nrm1979 [DOI] [PubMed] [Google Scholar]
- Roix JJ, McQueen PG, Munson PJ, Parada LA, Misteli T (2003) Spatial proximity of translocation-prone gene loci in human lymphomas. Nat Genet 34:287–291. doi: 10.1038/ng1177 [DOI] [PubMed] [Google Scholar]
- Roth J, Bendayan M, Orci L (1978) Ultrastructural localization of intracellular antigens by the use of protein A-gold complex. J Histochem Cytochem 26:1074–1081 [DOI] [PubMed] [Google Scholar]
- Roukos V, Misteli T (2014) The biogenesis of chromosome translocations. Nat Cell Biol 16:293–300. doi: 10.1038/ncb2941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roukos V, Voss TC, Schmidt CK, Lee S, Wangsa D, Misteli T (2013) Spatial dynamics of chromosome translocations in living cells. Science (New York, NY) 341:660–664. doi: 10.1126/science.1237150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roukos V, Burgess RC, Misteli T (2014) Generation of cell-based systems to visualize chromosome damage and translocations in living cells. Nat Protoc (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Cookson MR (2004) Mitochondria and dopamine: new insights into recessive parkinsonism. Neuron 43:301–304. doi: 10.1016/j.neuron.2004.07.012 [DOI] [PubMed] [Google Scholar]
- Simpson JC, Neubrand VE, Wiemann S, Pepperkok R (2001) Illuminating the human genome. Histochem Cell Biol 115:23–29 [DOI] [PubMed] [Google Scholar]
- Starkuviene V, Liebel U, Simpson JC, Erfle H, Poustka A, Wiemann S, Pepperkok R (2004) High-content screening microscopy identifies novel proteins with a putative role in secretory membrane traffic. Genome Res 14:1948–1956. doi: 10.1101/gr.2658304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symington LS, Gautier J (2011) Double-strand break end resection and repair pathway choice. Annu Rev Genet 45:247–271. doi: 10.1146/annurev-genet-110410-132435 [DOI] [PubMed] [Google Scholar]
- Vizeacoumar FJ et al. (2010) Integrating high-throughput genetic interaction mapping and high-content screening to explore yeast spindle morphogenesis. J Cell Biol 188:69–81. doi: 10.1083/jcb.200909013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter J, Schermelleh L, Cremer M, Tashiro S, Cremer T (2003) Chromosome order in HeLa cells changes during mitosis and early G1, but is stably maintained during subsequent interphase stages. J Cell Biol 160:685–697. doi: 10.1083/jcb.200211103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstock DM, Richardson CA, Elliott B, Jasin M (2006) Modeling oncogenic translocations: distinct roles for double-strand break repair pathways in translocation formation in mammalian cells. DNA Repair 5:1065–1074. doi: 10.1016/j.dnarep.2006.05.028 [DOI] [PubMed] [Google Scholar]
- Zhang Y et al. (2012) Spatial organization of the mouse genome and its role in recurrent chromosomal translocations. Cell 148:908–921. doi: 10.1016/j.cell.2012.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]