

Abstract
Tryptophan (Trp) catabolizing enzymes play an important and complex role in the development of cancer. Significant evidence implicates them in a range of inflammatory and immunosuppressive activities. Whereas inhibitors of indoleamine 2,3-dioxygenase-1 (IDO1) have been reported and analyzed in the clinic, fewer inhibitors have been described for tryptophan dioxygenase (TDO) and indoleamine 2,3-dioxygenase-2 (IDO2) which also have been implicated more recently in cancer, inflammation and immune control. Consequently the development of dual or pan inhibitors of these Trp catabolizing enzymes may represent a therapeutically important area of research. This is the first report to describe the development of dual and pan inhibitors of IDO1, TDO and IDO2.
Graphical Abstract
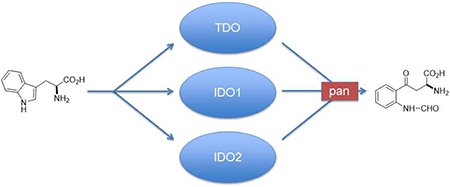
Introduction
In mammals and other vertebrates, three enzymes, IDO1, IDO2 and TDO, catalyze the rate-limiting first step of tryptophan catabolism down the kynurenine pathway which accounts for ≥95% of tryptophan catabolism1, 2. Whereas TDO is a predominantly hepatic enzyme functioning to maintain tryptophan homeostasis, IDO1 is induced by inflammation, particularly by IFNγ, and can be found in lymphoid tissues and organs, along mucosal surfaces, and in the placenta and epididymis3. IDO2 has been less thoroughly investigated than the other two enzymes, but the available data indicate it has a more restricted expression than IDO13. In cancer patients, IDO1 expression has been observed at varying levels and frequencies in most tumor types while TDO is strongly expressed in hepatocarcinoma, but also weakly in many other tumor types3Extensive preclinical evidence indicates that IDO1 inhibitors can relieve T cell suppression and enhance cancer therapeutic responses4, 5, including when combined with cytotoxic chemotherapy, radiotherapy or immune checkpoint therapy6–9. Nevertheless, the failure of the IDO1-selective inhibitor epaccadostat to provide a benefit in combination with the immune checkpoint agent pembrolizumab in a recent phase III clinical trial10 in melanoma patients highlights the continuing challenges to exploit the role IDO1 has in regulating the immune system.
While this trial has raised many questions11, attendant with its failure has been the growing recognition of the complex role and importance of the related dioxygenases, TDO and IDO24, 5, which, like IDO1, also exert root functions in cancer inflammatory programming. It has become clear that IDO1 drives both neovascularization along with immunosuppression in tumors.12, 13 TDO promotes immunosuppression, but also other processes that promote metastatic progression3, 14–17. IDO2 drives inflammation in autoimmune and cancer settings including pancreas cancer18–20. Combo or pan IDO1/TDO/IDO2 inhibitors will potentially offer metabolic adjuvants to illuminate key questions concerning the peculiar nature of chronic inflammatory states that sustain cancer21. As metabolic adjuvants, IDO1-selective inhibitors do not plumb the full potential of blunting Trp catabolism. Emerging evidence indicates that TDO and IDO2 are jointly activated with IDO1 in many tumors3, 14–17, 22, arguing for independent as well as overlapping functions. Whereas IDO1 modulates T-cell inflamed states, IDO2 influences B-cell inflamed states critical for autoimmunity20 as well as certain cancers like pancreatic cancer23, 24. IDO2 targeting may pose an additional benefit in mitigating autoimmune side-effects of immune checkpoint therapy based on evidence of a role in autoimmune inflammation20, 25. Lastly, resistance to IDO1 blockade in preclinical models is associated with upregulated Trp catabolism down the kynurenine pathway by TDO or IDO212. In the wake of these discoveries, the next logical step would be the development of dual or broad spectrum inhibitors of tryptophan catabolism to target IDO1, IDO2 and TDO.
Although a large number of IDO1 inhibitors have been communicated26, 27, there are significantly fewer TDO22, 28–32 and IDO2 inhibitors33–35 described in the literature. To the best of our knowledge, there is only one report of dual inhibition in the literature31, although a collection of patents have appeared36–39. Our recent report of monoaryl hydroxylamine activity as an impressive, structurally simple IDO1 inhibitor40 prompted us to explore this structural class of compounds further for combinatorial activity against IDO/TDO enzymes. Cognizant of the potential therapeutic benefits that might accrue from joint inhibition of IDO2 and TDO, we therefore screened more broadly for the effects of diaryl hydroxylamines on IDO2 and TDO as well as IDO1, to identify pan or selective inhibitors.
Based on literature defining two major pockets termed A and B in the active site of IDO126, 41–43. The expansion of our previously reported monoaryl hydroxylamines to diaryl derivatives was conceived initially as an effort to exploit both pockets in IDO1 to achieve more potent and selective inhibition. In addition, IDO1’s substrate profile has always suggested greater promiscuity relative to TDO44, 45 and the reported flexibility of the enzyme41 likely contributes to the ability of IDO1 to accommodate a range of structures. We were cognizant of these challenges and opportunities as we explored a new structural derivative of the monoaryl hydroxylamines. Herein we report our results with regards to these studies.
Results and Discussion
Synthesis of diaryl inhibitors.
The synthesis of the diaryl inhibitors followed the pattern of the previously reported monoaryl hydroxylamines40 in using the Mitsunobu reaction of an alcohol with N-hydroxyphthalimide to create the critical hydroxylamine functional group. The alcohol precursors of the Mitsunobu reaction were usually synthesized by adding phenyllithium or phenyl Grignard to the appropriate aldehyde (Scheme 1). Details for the synthesis of all alcohol precursors to the Mitsunobu reaction are shown in the Supplementary Data appendix. The Mitsunobu sequence shown in Scheme 2 illustrates the transformation of the alcohol substrates to O-alkylhydroxylamines.
Scheme 1.

Synthesis of Diaryl Alcohols with One Ring Substituted
Scheme 2.

Mitsunobu Reaction to Convert Alcohols to O-Alkyl Hydroxylamines
Enzyme inhibition data analysis.
Analysis of the diaryl compounds with isolated enzyme assays for IDO1, IDO2 and TDO activity allowed a preliminary ranking of the derivatives (Table 1). These results illustrated an array of activity and selectivity in this class of dioxygenase inhibitors. In all cases, compounds were tested as racemic mixtures, so it is possible that one enantiomer is the preferred and more potent stereoisomer. IDO1, which is the primary focus of most therapeutic work, was most potently inhibited by compounds with bromo and chloro substituents on the aromatic rings (compounds 1–7) with a potency in the range of 1–4 μM. This was also consistent with the results from our studies with the monoaryl hydroxylamines40. Methoxy or hydroxy substituted derivatives (20, 23, 25) were noticeably less potent (23 μM, 85% inhibition and 18% inhibition, respectively). The preference for bromo and chloro substitution on IDO1 inhibitors has also been found with the most potent compounds that have entered the clinic4.
Table 1.
Enzyme Inhibition Data for Diaryl Hydroxylamines with Ring Substitutiona
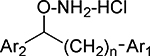 | ||||
|---|---|---|---|---|
| Compound no. | Structurea | IDO1 (μM or % inhibition)b | IDO2 (μM or % inhibition)b | TDO (μM or % inhibition)b |
| 1 | 3-bromo | 1 μM | 77.6% | 92.6% |
| 2 | 3,5-dichloro | 2 μM | 69.8% | 83.4% |
| 3 | 3,4-dichloro | 2 μM | 32 μM | 4 μM |
| 4 | 4,4’-dichloro | 3 μM | 69.3% | 2 μM |
| 5 | 3 μM | 84.3% | 75.5% | |
| 6 | 2,4-dichloro | 4 μM | 73% | 6 μM |
| 7 | 3,5-difluoro | 4 μM | 83% | 92.6% |
| 8 | 4-chloro | 5 μM | 74.3% | 5 μM |
| 9 | 3 -fluoro | 5 μM | 82% | 6 μM |
| 10 | 2-fluoro-3 -trifluoromethyl | 6 μM | 81.1% | 93.2% |
| 11 | 7 μM | 78.1% | 93.1% | |
| 12 | 2,4-difluoro | 7 μM | 46% | 90.3% |
| 13 | 2-fluoro-5 -trifluoromethyl | 8 μM | 73.8% | 73.4% |
| 14 | 2,3-dichloro | 8 μM | 72.4% | 9 μM |
| 15 | 4,4’-difluoro | 9 μM | 73.6% | 9 μM |
| 16 | 2-fluoro | 12 μM | 77.8% | 87.2% |
| 17 | 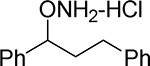 |
16 μM | 76.0% | 89.9% |
| 18 | 2-bromo | 17 μM | 81.0% | 90.4% |
| 19 | 2-chloro | 18 μM | 25 μM | 89.8% |
| 20 | 2-methoxy | 23 μM | 80.0% | 80.3% |
| 21 | 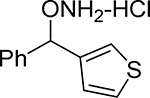 |
83% | 76.5% | 93.3% |
| 22 | 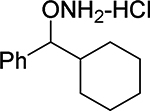 |
76% | 88.6% | 43.6% |
| 23 | 2-methoxy-4-methyl-4’chloro | 85% | 73.8% | 66% |
| 24 | 3,5 -bis(trifuoromethyl) | 56% | 53.3% | 50.8% |
| 25 | 4-hydroxy | 18.3% | 72.3% | 48.7% |
| 26 | 3 -trifluoromethyl | 18% | 36.1% | 12 μM |
In all structures, n=0 unless otherwise shown.
The inhibition activity of each compound as listed was first tested with 100 μΜ inhibitor (except 24 and 25, which were tested with 200 μM inhibitor), IC50 values were then determined for those exhibiting >95% inhibition activity.
In addition to the potency enhancing effects of halogenated derivatives towards IDO1 inhibition, we found two modifications that often led to IDO1 selective inhibitors: 2-fluoro substitution of one aryl ring (10, 12, 13, 16) and diaryl derivatives with a longer chain of atoms between the two aryl rings (5, 11, 17). In the ortho-fluoro substitution cases, the two most potent examples also have a trifluoromethyl group on the same ring and therefore the highly electron deficient aryl ring may be critical for IDO1 selectivity. With respect to the diaryls with greater distance between the two aryl rings, their selectivity is likely the result of the extended second aryl group better occupying the B pocket of IDO1. Inhibitors that occupy both the A and B pockets of IDO1 often have greater potency42, 43, although, to the best of our knowledge, our discovery is the first to illustrate selectivity from an extended binding inhibitor. This could be critical to the development of more selective inhibitors of the dioxygenase family.
New pan, dual and selective dioxygenase inhibitors were also identified (Figure 1). For example, compound 3 with the 3,4-dichloro aryl ring substitution, was a pan inhibitor (IDO1/IDO2/TDO potency: 2, 32, 4 μM, respectively). Several of the chlorinated and fluorinated derivatives demonstrated dual IDO1/TDO inhibition, including 4 (4,4’-dichloro), 6 (2,4-dichloro), 8 (4-chloro), 9 (3-fluoro), 14 (2,3-dichloro), and 15 (4,4’-difluoro). All six had IDO1 and TDO IC50 values in the single digit micromolar range. One dual IDO1/IDO2 inhibitor was identified in the 2-chloro substituted diaryl derivative, compound 19, which had IC50 values of 18 and 25 μM for IDO1 and IDO2, respectively. There was one diaryl compound (26) tested that was a selective TDO inhibitor (12 μM). Twelve of the other compounds were IDO1 selective: 1, 2, 5, 7, 10–13, 16–18, and 20. As noted earlier, seven of these derivatives fit into one of two categories: (1) 2-fluoro substitution (10, 12, 13, 16) or (2) extended distance between the two aromatic rings (5, 11, 17). Four of the IDO1 selective inhibitors had 3,5-dihalo (2 and 7) or 2-position substitution (18 and 20). The most potent compound, 1, fit none of these patterns as it had a 3-bromo substitution, thereby demonstrating the empirical nature of much of this analysis.
Figure 1.

Venn diagram of selectivities seen with diaryl hydroxylamines in enzyme inhibition assay.
Cell-based data analysis.
The first point that was noticeable in the cell-based IC50 data (Table 2) was that almost all inhibitors demonstrated more potent inhibition than in the isolated enzyme assay. This is quite common with IDO1 screens46, 47 and seems reasonable to expect for the similarly challenging isolated enzyme assays for TDO and IDO2. In particular, the IDO1 enzyme assay uses a surrogate reduction system (typically methylene blue and ascorbic acid) which is not the native reduction operation, to maintain the IDO1 in its catalytically active ferrous state; for although there is no redox cycling in the catalytic reaction, IDO1 is quite prone to oxidation to the ferric heme in the presence of oxygen. Also, as with the isolated enzyme assays, all compounds were tested as racemic mixtures.
Table 2.
Inhibition Data (IC50 values) for Cell-based Assays of Diaryl Hydroxylamines with Ring Substitutiona
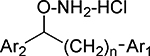 | ||||
|---|---|---|---|---|
| Compound no. | Structurea | IDO1 (μM)b | IDO2 (μM) | TDO (μM) |
| Epacadostat (INCB24360) |  |
0.07 | 18.9 | 25.2 |
| PF-06840003 |  |
0.21 | 327 | 378 |
| 1 | 3 -bromo | 0.590 | 11.8 | 7.15 |
| 2 | 3,5-dichloro | 0.870 | 8.66 | 5.70 |
| 3 | 3,4-dichloro | 1.91 | 19.5 | 8.11 |
| 4 | 4,4’-dichloro | 1.09 | 12.5 | 4.53 |
| 5 | 1.43 | 36.1 | 123 | |
| 6 | 2,4-dichloro | 1.99 | 7.68 | 4.82 |
| 7 | 3,5-difluoro | 0.357 | 8.75 | 3.48 |
| 8 | 4-chloro | 7.15 | 27.8 | 23.7 |
| 9 | 3-fluoro | 0.939 | 9.99 | 6.21 |
| 10 | 2-fluoro-3 -trifluoromethyl | 0.933 | 16.8 | 5.62 |
| 11 | 6.74 | 20.5 | 58.7 | |
| 12 | 2,4-difluoro | 1.21 | 18.9 | 6.37 |
| 13 | 2 -fluoro-5 -trifluoromethyl | 1.09 | 8.94 | 9.02 |
| 14 | 2,3-dichloro | 1.87 | 8.95 | 4.54 |
| 15 | 4,4’-difluoro | 1.68 | 12.1 | 5.15 |
| 16 | 2-fluoro | 1.26 | 30.5 | 3.92 |
| 17 | 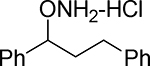 |
14.7 | 19.3 | 49.7 |
| 18 | 2-bromo | 1.46 | 12.3 | 22.5 |
| 19 | 2-chloro | 2.48 | 10.7 | 21.8 |
| 20 | 2-methoxy | 1.11 | 16.5 | 48.8 |
| 21 | 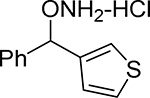 |
3.36 | 49.9 | 4.41 |
| 22 | 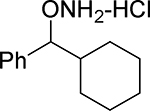 |
21.0 | 40.2 | 2770 |
| 23 | 2 -methoxy −4 -methyl −4’chloro | 1.76 | 8.77 | 19.1 |
| 24 | 3,5-bis(trifuoromethyl) | 3.35 | 14.5 | 77.4 |
| 25 | 4-hydroxy | 2.89 | 12.6 | 29.8 |
| 26 | 3 -trifluoromethyl | 1.73 | 24.7 | 13.6 |
In all structures,n=0 unless otherwise shown.
The IDO1 assay used HeLa cells, the IDO2 assay used mouse Trex cells and the TDO assay used Trex cells. Data is from a single data point except for the most potent compounds, which were done in triplicate and the results averaged.
The cell-based and isolated enzyme screens generally demonstrated similar relative potency for IDO1, IDO2 and TDO in most cases, although some differences in selectivity profiles were observed. For example, the same bromo and dichloro substituted derivatives (1–4) that were potent IDO1 inhibitors in the isolated enzyme assay were also potent IDO1 inhibitors in the cell-based assays (0.6–2 μM). However, the cell-based data for these compounds exhibited more significant potency with IDO2 and TDO as well. Indeed, most of the diaryl compounds demonstrated pan inhibition with high nanomolar or low micromolar inhibition of all three tryptophan catabolizing enzymes (Figure 2). In all these cases, the potency for IDO1 was the strongest, albeit in some examples only marginally so or within experimental error. For comparison and as proof of the significant pan inhibitor potency of the diaryl hydroxylamines, two clinical IDO1 inhibitor candidates, epacadostat (INCB24360) and PF-068400003, were tested and are included in Table 2 and the Venn Diagram Figure 2. Although INCB24360 shows pan inhibition at roughly the same level as many of the more IDO1 selective diaryl inhibitors, the structural simplicity of the diaryl hydroxylamine inhibitors and their ease of synthesis (3 steps versus 14 steps for epacadostat) makes them an important discovery and an exciting new lead compound series.
Figure 2.

Venn diagram of selectivities seen with diaryl hydroxylamines in cell-based assay.
As with the enzyme assays, the halogenated derivatives (fluoro, chloro and bromo) were the most potent and, within these examples, the dihalogenated versions (2–4, 6, 7, 12, 14, 15) demonstrated stronger pan inhibition than the monohalogenated analogs (5, 8, 11, 16, 18, 19), which had at least one inhibition constant above 20 μM. The meta monofluorinated 9 demonstrated IDO1 selective inhibition, along with three ortho monosubstituted derivatives 18–20, which had roughly five- to ten-fold greater potency with IDO1 (~1 μM) than either IDO2 (~10 μM) or TDO (>20 μM). The power of ring halogenation was also witnessed in our previous report40 on monoaryl O-alkylhydoxylamine derivatives as IDO1 inhibitors.
As with the isolated enzyme assay, the extended binding inhibitor 5 with the greater distance between the two aryl rings demonstrated more than 20-fold selectivity for IDO1 over IDO2 and TDO. However, the other extended binding inhibitors, 11 and 17, were less potent and less selective, illustrating that compound 5 has the better distance between the two aryl rings to generate selective and maximum IDO1 inhibition. Interestingly, there were two compounds, 16 and 21, which showed dual IDO1/TDO inhibitor activity with roughly 10-fold greater potency for IDO1 and TDO (~1–5 μM) over IDO2 (~30–50 μM). Notably, the loss of the second aryl ring, as in compound 22, resulted in significant attenuation in overall inhibition activity and for TDO in particular. This example serves as an important illustration of the requisite diaryl character in these inhibitors. Surprisingly, the four least potent compounds in the isolated enzyme assay (23–26) showed single micromolar potency towards IDO1 in the cell-based assay and retained low micromolar inhibition of IDO2 and TDO. Again, this is likely attributed to differences in the isolated enzyme assay procedure versus the cell-based assay.
Compound associated cytotoxicity could be a possible complicating factor for interpreting the cell-based enzyme inhibition data and a potential concern with regard to safety and tolerability. Therefore, a viability screen with HeLa cells was undertaken which demonstrated roughly ≥80% viability up to 100 μM for all compounds except for 1, 12 and 24 (See Supplementary Data appendix, Figure S3). For these three compounds, the cell viability dropped to approximately 60% around 100 μM. The overall lack of cytotoxicity apparent at the effective inhibitory dose range for these compounds suggests that general cytotoxicity is not likely to be an issue for advancing the diaryl hydroxylamine series. It was clear that the diaryl hydroxylamines, like the monoaryl hydroxylamines, have little to suggest that they would be problematic. Assessment of impact of human serum protein binding indicated a substantial reduction in inhibitory activity for one of the compounds, 6, while the inhibitory activity of 2 was relatively unaffected (see Supplementary Data appendix, Figure S4). This analysis indicates that it should be possible to develop diaryl hydroxylamines inhibitors that are not unduly compromised by serum protein binding.
Conclusions
Several important discoveries were made in the studies reported here. First, diaryl hydroxylamines demonstrated strong potency in isolated enzyme assays and were even stronger in cell-based assays. This reaffirms a common pattern for IDO1 inhibition studies and alerts researchers in the field that isolated enzyme studies should be interpreted with caution. This is understandable because the isolated enzyme assay for these Trp catabolizing enzymes is not an accurate replication of the actual cell environment. Second, the value of halogenation (fluoro, chloro and bromo) to potency in all three enzymes is a critical recognition, echoing the most potent IDO1 inhibitors that have entered the clinic. Third, as seen with 5, extended binding inhibitors do offer selectivity for IDO1 over the two other dioxygenase enzyme targets. Most notably, this study illustrates a rare example of potent pan and IDO1/TDO selective inhibitors. Although IDO1 inhibitors have been developed and reported for over a decade26, the development of pan or variably selective inhibitors is still in its infancy. Indeed, given the recent failure of the IDO1-selective inhibitor epacadostat in the ECHO-301 Phase 3 trial in melanoma, it seems likely that dual or pan inhibitors may be important to the development of effective therapeutic inhibitors of tryptophan catabolism11. The illustration in this report that these relatively simple inhibitors have significant potency and unique selectivity is an important development in the field that should benefit other researchers. Future studies will seek to further advance these understandings in the creation of more therapeutically relevant structures.
Experimental Section
General Synthetic Chemistry Experimental Parameters
All reactants and reagents were commercially available and were used without further purification unless otherwise indicated. Anhydrous CH2Cl2 was obtained by distillation from calcium hydride under nitrogen. Anhydrous THF was freshly distilled from Na and benzophenone. All reactions were carried out under an inert atmosphere of argon or nitrogen unless otherwise indicated. Concentrated refers to the removal of solvent with a rotary evaporator at normal water aspirator pressure followed by further evacuation with a direct-drive rotary vane vacuum pump. Thin layer chromatography was performed using silica gel 60 Å precoated glass or aluminum backed plates (0.25 mm thickness) with fluorescent indicator, which were cut. Developed TLC plates were visualized with UV light (254 nm), iodine, or ninhydrin. Flash column chromatography was conducted with the indicated solvent system using normal phase silica gel 60 Å, 230–400 mesh. Yields refer to chromatographically and spectroscopically pure (>95%) compounds except as otherwise indicated. Melting points were determined using an open capillary and are uncorrected. 1H NMR spectra were recorded at 400 MHz. Chemical shifts are reported in δ values (ppm) relative to an internal reference (0.05% v/v) of tetramethylsilane (TMS) for 1H NMR. Peak splitting patterns in the 1H NMR are reported as follows: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br,broad. 13C NMR experiments were conducted with the attached proton test (APT) pulse sequence. 13C multiplicities are reported as δu (up) for methyl and methine, and δd (down) for methylene and quaternary carbons. HPLC was conducted on an Agilent 1100 with an Ascentis Express C-18 column (100 × 4.6 mm, 2.7 μm) and a mobile phase of 80:20 MeCN:H2O. GC analyses were performed on the free hydroxylamine with an EI-MS detector fitted with a 30 m x 0.25 mm column filled with crosslinked 5% PH ME siloxane (0.25 μm film thickness); gas pressure 7.63 psi He. Analysis of samples involved heating from 70 to 250°C (10°C/min) and finally holding at 250°C for 7 min. All compounds were found to be >95% purity by elemental analysis or GC as indicated.
General Synthesis of O-Alkyl Hydroxylamines.
To a solution of alcohol (1 mmol) in freshly distilled THF (5 ml) was added triphenylphosphine (1.1 mmol) and N-hydroxylphthalimide (1.1 mmol). After the solution was cooled to 0°C diisopropylazodicarboxylate (1.1 mmol) was added dropwise. The solution was allowed to warm to room temperature over 3 hours. Reaction progress was monitored by TLC (1:1 heptanes:ethyl acetate). Hydrazine monohydrate (1.1 mmol) was then added and the solution was allowed to stir for 30 min. Water was added to dissolve the white precipitate. The aqueous layer was then extracted with ethyl acetate 3 times, washed with brine, and dried over Na2SO4. Removal of the solvent under reduced pressure afforded a yellow oil that was purified by flash chromatography (1:1 heptanes/ethyl acetate with 0.1% triethylamine).
HCl salt formation:
The O-alkyl hydroxylamine was dissolved in a minimal amount of diethyl ether. HCl in anhydrous diethyl ether (1.0 M, 1 eq) was added slowly. The white precipitate was filtered and washed with diethyl ether and dried under high vacuum.
O-(3-Bromo-α-phenylbenzyl)hydroxylamine hydrochloride (1).
Synthesized from 3-bromo-α-phenyl-benzenemethanol according to the general procedure to afford 1 as white crystals in 66% yield. Mp=159–162°C. 1H NMR (CD3OD) δ 7.57–7.60 (m, 2H, ArH), 7.33–7.52 (m, 7H, ArH), 6.11 (s, 1H, ArCH). 13C NMR (CD3OD) δu 132.0, 130.5, 130.0, 129.3, 128.9, 127.3, 126.0, 87.0; δd 139.8, 136.5, 122.5. Anal. calcd. for C13H13ClBrNO C=49.63, H=4.17, N=4.45.Found: C=49.63%, H =4.48%, N =4.38%.
O-(3,5-Dichloro-α-phenylbenzyl)hydroxylamine hydrochloride (2).
Synthesized from 3,5-dichloro-α-phenyl-benzenemethanolaccording to the general procedure to afford 2 as white crystals in 88%yield. Mp= 172–177 °C. 1H NMR (CD3OD) δ 7.38–7.44 (m, 4H, ArH), 7.24(d, 2H, ArH, J=8.6 Hz), 6.84–6.87 (d, 2H, ArH, J=6.68 Hz), 6.01 (s, 1H, ArCH). 13C NMR (CD3OD) δu 131.7, 131.2, 130.9, 129.5, 127.8, 88.5; δd 143.6, 138.4, 137.6. Anal. calcd. for C13H12Cl3NO: C=51.26; H=3.97; N=4.60. Found: C=51.54%, H = 3.69%, N =4.57%.
O-(3,4-Dichlorophenylbenzyl)hydroxylamine hydrochloride (3).
Synthesized from respective alcohol according to the general procedure to afford 3 as white crystals in 67% yield. Mp=155–156°C.1H NMR (CD3OD) δ 7.61 (d, 2H, ArH, J=8.0Hz),δ 7.50–7.44 (m, 4H, ArH),δ 7.36–7.34 (d, 1H, ArH, J=8.0Hz), δ 6.18 (s, 1H, ArCH(ONH2)). 13C NMR (CD3OD) δu 130.8, 129.4, 129.1, 129.0, 127.2, 126.9, 86.4; δd 138.0, 136.2, 132.8, 132.6. Anal. calcd. for C13H12Cl3NO:C=51.26; H=3.97; N=4.60, Found: C=51.26%, H = 3.65%, N =4.41%.
O-[Bis(4-chlorophenyl)methyl]-hydroxylamine hydrochloride monohydrate (4).
Synthesized from bis-4-chlorophenyl methanol according to the general procedure to afford 4 as white crystals in 49% yield. Mp=201–202°C. 1H NMR (CD3OD) δ 7.46–7.48 (d, 2H, ArH, J=8.40 Hz), 7.39–7.41 (d, 2H, ArH, J=8.48 Hz), 6.08 (s, 1H, ArCH). 13C NMR (CD3OD) δu 128.9, 86.3; δd 135.6, 135.0. Anal. calcd. for C13H14Cl2NO-0.5 HCl C=48.36, H=3.91, N=4.34. Found: C=48.52%, H =3.87%, N =4.19%.
5-(Aminooxy)-5-(3-chlorophenyl)-N-phenylpentanamide hydrochlorie (5).
Synthesized from the respective alcohol according to the general procedure with the following modifications: A resin-bound triphenylphosphine was used and the intermediate N-hydroxy-phthalimide intermediate was purified by column chromatography and then used immediately in the next hydrazinolysis step. The product was obtained as a white solid in 62% yield. Mp=182–184°C. 1H NMR (d6-DMSO) δ 10.97 (br s, 3 H, NH3), 10.05 (s, 1 H, NH-C(=O)), 7.6 (d, 1 H, ArH, J=8.0 Hz), 7.47 (d, 3 H, ArH, J=4.0 Hz), 7.39–7.37 (m, 1 H, ArH), 7.27 (t, 2 H, ArH, J=8.0 Hz), 7.01 (t, 1 H, ArH, J=8.0 Hz), 5.19 (t, 1 H, NO-CH, J=6.4 Hz), 2.37 (t, 2 H, O=CCH2, J=4.0 Hz), 1.971.88 (m, 1 H, CH2), 1.78–1.60 (m, 2 H, CH2), 1.58–1.47 (m, 1 H, CH2). 13C NMR (d6-DMSO) δu 131.1, 129.3, 129.1, 127.6, 126.4, 123.4, 119.5, 85.0; δd 171.3, 141.0, 139.8, 133.8, 36.1, 34.8, 21.6. GC tR = 22.528 min.
O-(2,4-Dichlorophenylbenzyl)hydroxylamine hydrochloride (6).
Synthesized from 2,4-dichlorophenylbenzhydrol according to the general procedure to afford 6 as white crystals in 76% yield. Mp=170–171.5°C. 1H NMR (CD3OD) δ 7.56–7.57 (m, 2H, ArH),7.47–7.51 (m, 6H, ArH) 6.46 (s, 1H, ArCH). 13C NMR (CD3OD) δu 129.6, 128.9, 128.8, 127.8, 127.7, 84.0; δd 135.4, 135.0, 134.0, 133.7. Anal. calcd. for C13H12Cl3NO-0.25 HCl: C=49.77, H=3.94, N=4.47. Found: C=49.66%, H =4.17%, N =4.40%.
O-(3,5-Diflouro-α-phenylbenzyl)hydroxylamine hydrochloride (7).
Synthesized from 3,5-diflouro-α-phenyl benzenemethanol according to the general procedure to afford 7 as white crystals in 89% yield. Mp=177–181°C.1H NMR (CD3OD) δ 7.65–7.68 (m, 3H, ArH) 7.45–7.48 (m, 1H, ArH),7.36–7.44 (m, 4H, ArH), 6.52 (s, 1H, ArCH). 13C NMR (CD3OD) δu 129.5, 128.9, 127.3, 110.0 (q, 2JC-F=19, 8 Hz), 103.9 (t, 2JC-F=25 Hz) 86.4; δd 163.3 (dd, 1JC-F=250 Hz, 3JC-F=13 Hz), 141.7 (d, JC-F=8 Hz), 136.1. GC tR = 10.764 min.
O-(4-Chloro-α-phenylbenzyl)hydroxylamine hydrochloride (8).
Synthesized from 4-chloro-α-phenyl-benzenemethanol according to the general procedure to afford 8 as white crystals in 54% yield. Mp-171–175°C.1H NMR (CD3OD) δ 7.38–7.44 (m, 4H, ArH), 7.24 (d, 2H, ArH, J=8.6 Hz), 6.85 (t, 1H, ArH, J=6.7 Hz), 6.01 (s, 1H, ArCH2). 13C NMR (CD3OD) δu 129.2, 128.8, 128.4, 127.2, 87.2; δd 136.6, 136.0, 134.9. Anal. calcd. for C13H13Cl2NO: C=57.80; H=4.85; N=5.18. Found: C=58.02%, H = 4.58%, N =5.14%.
O-(3-Flouro-α-phenylbenzyl)hydroxylamine hydrochloride (9).
Synthesized from 3-flouro-α-phenyl-benzenemethanol according to the general procedure to afford 9 as white crystals in 77% yield. Mp=169–170°C.1H NMR (CD3OD) δ 7.43–7.44 (m, 6H, ArH) 7.24–7.26 (m, 1H, ArH),7.14–7.20 (m, 2H, ArH), 6.15 (s, 1H, ArCH). 13C NMR (CD3OD) δu 130.6 (d, 3JC-F=8 Hz), 129.2, 128.8, 127.3, 123.0 (d, 3JC-F=3 Hz), 115.6 (d, 2JC-F=21 Hz), 113.8 (d, 2JC-F=23 Hz), 87.0; δd 163.0 (d, 1JC-F=245 Hz), 139.9 (d, 3JC-F=7 Hz), 136.6. Anal. calcd. for C13H13ClFNO C=61.55; H=5.17; N=5.52. Found: C=61.21%, H = 4.79%, N =5.48%.
O-(2-Fluoro-3-trifluoromethyl-α-phenylbenzyl)hydroxylamine hydrochloride (10).
Synthesized from 2-fluoro-4-trifluoromethyl)-α-phenyl benzenemethanol according to the general procedure to afford 10 as white crystals in 61% yield. Mp=157–158°C. 1H NMR (CD3OD) δ 7.38–7.44 (m, 4H, ArH), 7.24 (d, 2H, ArH, J=8.6), 6.85 (t, 1H, ArH, J=6.7), 6.01 (s,1H, ArCH(ONH2)). 13C NMR (CD3OD) δu 132.1 (d, 3JC-F=3 Hz), 129.6, 129.0, 127.9 (m), 127.2, 124.9 (d, 3JC-F=5 Hz), 81.1 (d, 3JC-F=3 Hz); δd 157.2 (d, 1JC-F=250 Hz), 135.2, 126.7 (t, 2JC-F=13 Hz), 122.5 (d, 1JC-F=270 Hz), 118.4 (q, 2JC-F=33, 12 Hz). Anal. calcd. for C14H12ClF4NO: C=52.27, H=3.76, N=4.35. Found: C=52.39%, H =3.49%, N =4.29%.
O-(3-(3-chlorophenyl)-1-phenylpropyl)hydroxylamine hydrochloride (11)
Synthesized from the respective alcohol according to the general procedure to afford 11. Mp=140.5–141.5°C. 1H NMR (CD3OD) δ 7.44–7.54 (m, 5 H, ArH), 7.28 (t, 1 H, ArH, J=8.0 Hz), 7.22 (s, 2 H, ArH), 7.13 (d, 1 H, ArH, J=8.0 Hz), 4.98 (t, 1 H, ArCH(ONH2), J=6.9 Hz), 2.59–2.76 (m, 2 H, ArCH2), 2.27–2.37 (m, 1 H, ArCH2CH2), 2.05–2.15 (m, 1 H, ArCH2CH2). 13C NMR (CD3OD) δu 129.7, 129.6, 129.0, 128.1, 127.2, 126.5, 126.0, 86.5; δd 143.0, 136.6, 134.0, 36.6, 30.8. GC tR =14.656 min.
O-(2,4-Difluorophenylbenzyl)hydroxylamine hydrochloride (12).
Synthesized from respective alcohol according to the general procedure to afford 12 as white crystals in 62% yield. Mp=167–168°C. 1H NMR (CD3OD) δ 7.46–7.43 (overlapping signal, 5H, ArH), δ 7.11–7.05 (overlapping signal, 2H, ArH), δ 6.38 (s, 2H, ArCH(ONH2)). 13C NMR (CD3OD) δu 129.6 (d, 3JC-F=4 Hz), 129.5 (d, 3JC-F=4 Hz), 129.3, 128.8, 127.1, 111.7 (q, 2JC-F=22 Hz, 3JC-F=4 Hz), 104.0 (t, 2JC-F=25 Hz), 81.4 (d, 3JC-F=3 Hz); δd 163.6 (d, 1JC-F=249 Hz, 3JC-F=13 Hz), 160.8 (d, 1JC-F=249 Hz, 3JC-F=12 Hz), 135.6, 120.9 (q, 2JC-F=13 Hz, 4JC-F=4 Hz). GC tR = 10.835 min.
O-((2-Fluoro-5-trifluoromethyl-α-phenylbenzyl)hydroxylamine hydrochloride (13).
Synthesized from the respective alcohol according to the general procedure to afford 13 as white crystals in 91% yield. Mp=157–158.5°C. 1H NMR (CD3OD) δ 7.78–7.82 (m, 2H, ArH ), 7.40–7.51(m, 6H, ArH),6.45(1H, ArCH(ONH2)). 13C NMR (CD3OD) δu 129.6, 129.0, 128.5, 127.3, 125.0 (t, 3JC-F=4 Hz), 116.9 (d, 2JC-F=23 Hz), 81.3; δd 162.1 (d, 1JC-F=254 Hz), 135.1, 129.6, 126.3 (d, 2JC-F=14 Hz). GC tR = 10.494 min.
O-(2,3-Dichloro-α-phenylbenzyl)hydroxylamine hydrochloride (14).
Synthesized from 2,3-dichloro-α-phenyl-benzenemethanol according to the general procedure to afford 14 as white crystals in 63%yield. Mp=170–175°C. 1H NMR (CD3OD) δ 7.64 (dd, 1H, ArH, J=2.1, 8.0 Hz), 7.60 (dd, 1H, ArH, J=1.4 Hz, 7.8 Hz), 7.42–7.80 (m, 6H, ArH), 6.53 (s,1H, ArCH). 13C NMR (CD3OD) δu 130.8, 129.5, 128.8, 128.2, 128.0, 125.9, 84.8; δd 137.2, 135.0, 133.6, 131.1. Anal. calcd. for C13H12Cl3NO: C=51.26; H=3.97; N=4.60. Found: C=51.27%, H = 3.90%, N =4.52%.
O-[Bis(4-fluorophenyl)methyl]-hydroxylamine hydrochloride (15).
Synthesized from bis-4-fluorophenyl methanol according to the general procedure to afford 15 as white crystals in 58% yield. Mp=164–165.5°C. 1H NMR (CD3OD) δ 7.44–7.46 (m, 4H, ArH), 7.18–7.23 (m, 4H, ArH), 6.12 (s,1H, ArCH). 13C NMR (CD3OD) δu 129.5 (d, 3JC-F=8 Hz), 115.5 (d, 2JC-F=22 Hz), 86.4; δd 163.2 (d, 1JC-F=246 Hz), 133.0 (d, 4JC-F=3 Hz). Anal. calcd. for C13H12ClF2NO C=57.47, H=4.45; N=5.16. Found: C=57.29%, H = 4.51%, N =5.16%.
O-(2-Fluoro-α-phenylbenzyl)hydroxylamine hydrochloride (16).
Synthesized from 2-fluoro-α-phenyl benzenemethanol according to the general procedure to afford 16 as white crystals in 85%yield. Mp=184–185°C. 1H NMR (CD3OD) δ 7.42–7.50 (m, 7H, ArH), 7.27–7.31 (t, 1H, ArH, J=6.60 Hz), 7.18–7.23 (t, 1H, ArH, J=8.64 Hz), 6.44 (s, 1H, ArCH). 13C NMR (CD3OD) δu 131.0 (d, 3JC-F=8 Hz), 129.2, 128.8, 127.8 (d, 4JC-F=3 Hz), 127.2, 124.6 (d, 3JC-F=4 Hz), 115.6 (d, 2JCF=22 Hz), 81.8 (d, 3JC-F=4 Hz); δd 160.4 (d, 1JC-F=246 Hz), 135.9, 124.5 (d, 2JC-F=13 Hz). Anal. Calcd. for C13H13ClFNO C=61.83; H=5.17; N=5.52. Found: C=61.83%, H = 4.89%, N =5.48%.
O-(1,3-diphenylpropyl)hydroxylamine hydrochloride (17).
Synthesized from 1,3-diphenylpropan-1-ol according to the general procedure to afford 17 in 40% yield. Mp=156–159°C. 1H NMR (CD3OD) δ 7.45–7.54 (m, 5 H, ArH), 7.30 (t, 2 H, ArH, J=7.9 Hz), 7.20 (t, 2 H, ArH, J=6.2 Hz), 4.97 (t, 1 H, ArCH(ONH2), J=6.9 Hz), 2.60–2.74 (m, 2 H, ArCH2), 2.28–2.37 (m, 1 H, ArCH2CH2), 2.05–2.14 (m, 1 H, ArCH2CH2). 13C NMR (CD3OD) δu 129.5, 129.0, 128.2, 128.0, 127.2, 126.8, 125.9, 86.9; δd 140.5, 136.8, 37.0, 31.1. GC tR = 12.784 min.
O-(2-Bromo-α-phenylbenzyl)hydroxylamine hydrochloride (18).
Synthesized from 2-bromo-α-phenyl-benzenemethanol according to the general procedure to afford 18 as white crystals in 34% yield. Mp=166–167°C. 1H NMR (CD3OD) δ 7.71 (dd, 1H, ArH, J=8.0, 1.6 Hz) 7.61 (dd, 1H, ArH, J=8.0, 1.6 Hz), 77.53 (dt, 1H, ArH, J=8.0, 1.6 Hz), 7.40–7.48 (m, 5H, ArH), 7.36 (dt, 1H, J=8.0, 1.6 Hz), 6.47 (s, 1H, ArCH). 13C NMR (CD3OD) δu 135.4, 130.6, 129.3, 129.0, 128.7, 128.0, 127.9, 127.2, 86.6; δd 136.2, 135.4, 123.1. Anal. calcd. for C13H13ClBrNO: C=49.63; H=4.17; N=4.45. Found: C=49.78%, H =4.38%, N =4.63%.
O-(2-Chloro-α-phenylbenzyl)-hydroxylamine hydrochloride (19).
Synthesized from 3-chloro-α-phenyl-benzenepropanol according to the general procedure to afford 19 as white crystals in 84%yield. Mp=169–174°C.1H NMR (CD3OD) δ 7.50 (d, 1H, ArH, J=4.0 Hz), 7.41–7.48 (m, 8H, ArH), 6.51 (s, 1H, ArCH). 13C NMR (CD3OD) δu 130.3, 129.9, 129.3, 128.9, 128.7, 127.9, 127.6, 127.5, 84.5; δd 135.5, 134.7, 133.2. GC tR = 13.022 min.
O-(2-Methoxy-α-phenylbenzyl)hydroxylamine hydrochloride (20).
Synthesized from 2-methoxy-α-phenyl-benzenemethanol according to the general procedure to afford 20 as white crystals in 89%yield. Mp=134–134.5°C. 1H NMR (CD3OD) δ 7.35–7.46 (m, 6H, ArH), 6.98–7.01 (m, 3H, ArH), 6.08 (s, 1H, ArCH), 3.81 (s, 3H, ArOCH3). 13C NMR (CD3OD) δu 129.9, 129.0, 128.6, 127.2, 119.3, 114.2, 112.9, 87.8, 54.4; δd 160.2, 138.4, 137.0. Anal. calcd. for C14H16ClNO2: C=63.28, H=6.07, N=5.27. Found: C=63.44%, H = 6.23%, N =5.31%.
O-(phenyl(thiophen-3-yl)methyl)hydroxylamine hydrochloride (21).
Synthesized from 2-methoxy-α-phenyl-benzenemethanol according to the general procedure to afford 21 as white crystals in 61% yield. Mp=138–139.5°C. 1H NMR (CD3OD) δ 7.41–7.53 (m, 7 H, ArH), 7.14 (dd, 1 H, ArH, J=5.0, 1.0 Hz), 6.27 (s, 1 H, ArCH(ONH2)). 13C NMR (CD3OD) δu 129.3, 128.7, 128.6, 127.2, 127.0, 126.1, 125.2, 84.2; δd 138.1, 136.8. GC tR =10.857 min.
O-(cyclohexyl(phenyl)methyl)hydroxylamine hydrochloride (22).
Synthesized from cyclohexyl(phenyl)methanol according to the general procedure to afford 22 in 87% yield. Mp=193–196.5°C. 1H NMR (CD3OD) δ 7.38–7.52 (m, 5 H, ArH), 4.70 (d, 1 H, ArCH(ONH2), J=8.2 Hz), 2.14 (d, 1 H, CHCHONH2, J=6.4 Hz), 1.76–1.85 (m, 2 H, CH2), 1.65–1.68 (m, 2 H, CH2), 1.12–1.37 (m, 5 H, CH2), 0.9–1.03 (m, 1 H, CH2). 13C NMR (CD3OD) δu 129.3, 128.8, 127.6, 91.7, 42.7 δd 135.9, 29.2, 28.5, 25,8, 25.4, 25.2. GC tR =10.232 min.
O-[4-Chloro-α-(2’-methyl,4’-methoxyphenyl)benzenemethyl]hydroxylamine hydrochloride (23).
Synthesized from the respective alcohol according to the general procedure to afford 23 as white crystals in 72% yield. Mp=139.5–140.5°C. 1H NMR (CD3OD) δ 7.36–7.43 (m, 4H, ArH), 7.19–7.21 (d, 1H, ArH J=7.76 Hz), 6.88–6.93 (m, 2H, ArH), 6.43(s, 1H, ArCHONH2), 3.87 (s, 3H, ArOCH3), 2.39 (s, 3H, ArCH3). 13C NMR (CD3OD) δu 129.2, 128.5, 126.7, 121.3, 111.8, 81.6, 54.7, 20.3; δd 157.3, 141.2, 136.0, 134.4, 121.6. Anal. calcd. for C15H17Cl2NO2: C=57.34, H=5.45, N=4.46. Found: C=57.60%, H = 5.52%, N =4.36%.
O-(3,5-Bis-trifluoromethyl-α-phenyl-benzyl)hydroxylamine hydrochloride (24).
Synthesized from 3,5-bis-trifluoromethyl-α-phenyl benzenemethanol according to the general procedure to afford 24 as white crystals in 88%yield. Mp=158–160°C.1H NMR (CD3OD) δ 7.90 (m, 3H, ArH), 7.38–7.56 (m, 5H, ArH), 6.31(s, 1H, ArCH). 13C NMR (CD3OD) δu 129.8, 129.2, 127.5, 122.5 (m), 86.0; δd 141.0, 135.7, 132.0 (q, 2JC-F=33 Hz), 123.2 (q, 1JC-F=270 Hz). Anal. calcd. for C15H12ClF6NO: C= 48.47, H=3.25, 3.77. Found: C=48.90%, H =2.98%, N =3.76%.
O-(4-Hydroxy-α-phenyl)-benzyl hydroxylamine hydrochloride.(25)
Synthesized from the 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-α-phenyl-benzenemethanol (25s) according to the general procedure to afford O-[4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-α-phenyl-benzyl]hydroxylamine. To a solution of the protected phenol (0.3–3.5mmol) in anhydrous tetrahydrofuran (5–25 mL) was added tetrabutylammonium fluoride (1 M, 1.0 equiv per TBDMS), and the pale yellow solution was stirred for 45 min. The mixture was poured into water and extracted with ethyl acetate. Removal of the solvent in vacuo from the organic phase afforded a tan oil that was subjected to gravity column chromatography (9:1 hexane/ethyl acetate). Formation of the HCl salt followed the general procedure to afford 25 as white crystals in 57% yield. Mp=168.5–169.5°C. 1H NMR (CD3OD) δ 7.38–7.46 (m, 5H, ArH), 7.24 (d, 2H, ArH, J=8.6 Hz), 6.86 (d, 2H, ArH, J=8.6 Hz), 6.01 (s, 1H, ArCH). ). 13C NMR (CD3OD) δu 129.8, 128.8, 127.7, 115.6, 69.1; δd 158.5, 134.1, 123.9. Anal. calcd. for C13H14ClNO2-H2O: C=57.89; H=5.98; N=5.19. Found: C=58.03% H=6.01% N= 5.20%.
O-(3-Trifluoromethyl-α-phenylbenzyl)hydroxylamine hydrochloride (26).
Synthesized from phenyl(3-(trifluoromethyl)phenyl)methanol according to the general procedure to afford 26 as white crystals in 77%yield. Mp=199–199.5°C. 1H NMR (CD3OD) δ 7.61 (d, 1H, ArH, J=2.3 Hz), 7.58 (s, 1H, ArH), 7.50 (dd, 1H, ArH, J=8.6, 2.2 Hz), 6.47 (s, 1H, ArCH). ). 13C NMR (CD3OD) δu 132.8, 129.6, 129.3, 128.8, 128.7, 127.6, 126.2 (q, 3JC-F=6 Hz), 83.4; δd 136.1, 135.0, 128.3 (d, 2JC-F=30 Hz), 124.2 (q, 1JC-F=272 Hz). Anal. calcd. for C14H13ClF3NO: C=55.37; H=4.31; N=4.61. Found: C=55.52%, H = 4.58%, N =5.04%.
Enzyme-based IDO1, IDO2 and TDO inhibition assays.
The steady-state activity of each enzyme was measured in the absence or presence of an inhibitor at 20 °C with a protocol reported previously48, 49 (with small modifications). For IDO1, each reaction was initiated by mixing 50 μM L-tryptophan with 100 nM enzyme in 50 mM Tris buffer (pH 7.4) that contained 200 nM catalase, 12 μM methylene blue, 20 mM ascorbate and 100 μM inhibitor (unless otherwise indicated). The same protocol was used for IDO2 except that 4 mM L-tryptophan and 1 μM enzyme were used; in addition pyocyanin (39 μM) instead of methylene blue was used as an electron mediator. For TDO, the experiment was initiated by mixing 100 μM L-tryptophan with 250 nM enzyme in 50 mM Tris buffer (pH 7.4) that contained 100 μM ascorbate and 100 μM inhibitor (unless otherwise indicated). The inhibitor stocks were prepared in dimethyl sulfoxide (DMSO), the final DMSO concentration for all reactions was kept at 3.5% (v/v). The initial linear velocities of the reactions, v, were obtained by monitoring the formation of the product N-formyl kynurenine at 321 nm (ℇ = 3750 M−1cm−1) as a function of time with a UV2400 spectrophotometer (Shimadzu Scientific Instruments, Inc.) with a spectral slit width of 2 nm. The % inhibition for each inhibitor was calculated based on (1-v/vo )x100, where vo was obtained in the absence of any inhibitor. For each inhibitor that exhibited ≥95% inhibition activity for IDO1 or TDO, or ≥85% inhibition activity for IDO2, the IC50 value was determined by measuring the inhibition activity as a function of inhibitor concentration. It is noted that the threshold for IDO2 was set lower (85%) for the initial screening due to the generally lower inhibition activity of the inhibitors. All the data were analyzed with Origin 6.1 software (Microcal Software, Inc., MA).
Cell-based IDO1, IDO2 and TDO inhibition assays.
Compounds were evaluated for inhibitory activity against human IDO1 expressed endogenously in HeLa cells and human TDO and mouse IDO2 expressed exogenously in T-Rex cells. HeLa cells were seeded in a 96 well plate at a density of 10,000 cells per well in 100 μl DMEM + 10% fetal bovine serum + 1% penicillin-streptomycin with L-tryptophan being adjusted to 100 μM and IDO1 expression was induced by the addition of IFNγ to a final concentration of 100 ng/mL. T-Rex cells containing a tet-regulated human TDO were seeded in a 96-well plate at a density of 10,000 cells per well in 100 μL of DMEM + 10% FBS + 1% penicillin-streptomycin with L-tryptophan adjusted to 100 μM and TDO expression was induced by the addition of 100 μL of media containing 2 ng/mL doxycycline with adjusted L-tryptophan. T-Rex cells containing a tet-regulated mouse IDO2 cDNA were seeded in a 96-well plate at a density of 10,000 cells per well in 100 μL of DMEM + 10% FBS + 1% penicillin-streptomycin and IDO2 expression was induced by the addition of 100 μL of media containing 2 ng/mL doxycycline + 8 mM 5-aminolevulinic acid + 20 μM hemin + 4 mM L-tryptophan. The IDO1 and TDO induction was allowed to proceed for 24 hours, the IDO2 was induced for 48 hours. After induction, the media was discarded, the wells rinsed once, and serial dilutions of compound in 200 μL of appropriate media with the final concentration of tryptophan adjusted to 100 μM for IDO1 and TDO and to 2mM for IDO2. Following incubation at 37 °C for an additional 20 hrs, the assay was stopped by the addition of 50 μL of 50% (w/v) TCA to each well, and the cells were fixed by incubating for 1 hr at 4 °C.
Assessment of IDO1, IDO2 and TDO activity
Compound IC50 values were assessed from single point dilution series with the most potent compounds subsequently retested two or more times and the results reported as averages. Following the TCA fixation step, the supernatants were transferred to a round-bottomed 96-well plate and incubated at 65 °C for 15 min. The plates were then centrifuged at 1250 × g for 10 min, and 100 μL of clarified supernatant was transferred to a new flat-bottomed 96-well plate and mixed at equal volume with 2% (w/v) p-dimethylaminobenzaldehyde in acetic acid. The yellow reaction was measured at 490 nm using a Synergy HT microtiter plate reader (Bio-Tek, Winooski, VT). Graphs of inhibition curves with IC50 values were generated using Prism v.5.0 (GraphPad Software, Inc.).
Supplementary Material
HIGHLIGHTS.
Extension of monoaryl hydroxylamines to diaryl hydroxylamines illuminates activities as potent pan and dual inhibitors of the tryptophan catabolizing enzymes, IDO1, IDO2 and TDO.
This capability is unique given the lack of previous reports of dual and pan inhibitors for these enzymes, which jointly regulate the kynurenine pathway of tryptophan catabolism to modulate immune, inflammatory and neurological functions in the body.
Aryl halide substitution generated the most potent derivatives studied.
Acknowledgements
Financial support for this work was provided by NIH grants R01 CA109542 (GCP, AJM and WPM) and R01 GM086482 (SRY). AJM and GCP acknowledge additional support from the Lankenau Medical Center Foundation and Main Line Health. GCP holds the Havens Chair for Biomedical Research at the Lankenau Institute for Medical Research. WPM acknowledges additional support from NIH grant GM087291 and Bryn Mawr College. A generous award (CHE-0958996) from the National Science Foundation enabling acquisition of the 400 MHz NMR spectrometer used in these studies is gratefully acknowledged
Footnotes
Supplementary Data
Full experimental details for the synthesis of alcohols that were not commercially available are described. Copies of 1H and 13C NMR spectra for all tested hydroxylamine compounds are also available. Raw IC50 data for top inhibitors in the enzyme and cell-based inhibition assays are shown. Toxicity screen with HeLa cells for diaryl hydroxylamines, epacadostat and PF-0684003 are available. In addition, an assay assessing the impact of human serum protein binding on the apparent IC50 values is also shown. This material is available at …
Conflict of Interest Statement
AJM, WPM and GCP declare interests as inventors of IDO technology licensed to New Link Genetics Corporation, a biopharmaceutical company involved in the clinical development of IDO inhibitors, as described in U.S. Patents Nos. 7705022, 7714139, 8008281, 8058416, 8383613, 8389568, 8436151, 8476454 and 8586636. WPM and GCP are also former scientific advisors and GCP a former grants recipient of New Link Genetics. GCP is currently a scientific advisor for Kyn Therapeutics, which is developing IDO/TDO/AhR small molecule antagonists for cancer treatment. AJM is a scientific advisor and grant recipient of I-O Biotech AG, which is developing IDO vaccines for cancer treatment.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ball HJ, Jusof FF, Bakmiwewa SM, Hunt NH, Yuasa HJ. Tryptophan-Catabolizing Enzymes – Party of Three. Frontiers in Immunology. 2014;5: 485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Badawy AAB. Kynurenine Pathway of Tryptophan Metabolism: Regulatory and Functional Aspects. International Journal of Tryptophan Research : IJTR. 2017;10: 1178646917691938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van Baren N, Van den Eynde BJ. Tumoral Immune Resistance Mediated by Enzymes That Degrade Tryptophan. Cancer Immunol Res. 2015;3(9): 978–985. [DOI] [PubMed] [Google Scholar]
- 4.Prendergast GC, Malachowski WJ, DuHadaway J, Muller AJ. Discovery of IDO1 inhibitors: from bench to bedside. Cancer Res. 2017;in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Prendergast GC, Malachowski WJ, Mondal A, Scherle P, Muller AJ. Chapter Four - Indoleamine 2,3-Dioxygenase and Its Therapeutic Inhibition in Cancer In: Galluzzi L, ed. International Review of Cell and Molecular Biology. Vol 336 Academic Press; 2018:175–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muller AJ, DuHadaway JB, Donover PS, Sutanto-Ward E, Prendergast GC. Inhibition of indoleamine 2,3-dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nat Med. 2005;11. [DOI] [PubMed] [Google Scholar]
- 7.Li M, Bolduc AR, Hoda MN, et al. The indoleamine 2,3-dioxygenase pathway controls complement-dependent enhancement of chemo-radiation therapy against murine glioblastoma. Journal for ImmunoTherapy of Cancer. 2014;2(1): 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Monjazeb AM, Kent MS, Grossenbacher SK, et al. Blocking Indolamine-2,3-Dioxygenase Rebound Immune Suppression Boosts Antitumor Effects of Radio-Immunotherapy in Murine Models and Spontaneous Canine Malignancies. Clin Cancer Res. 2016;22(17): 4328–4340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Holmgaard RB, Zamarin D, Munn DH, Wolchok JD, Allison JP. Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. J Exp Med. 2013;210(7): 1389–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Long GV, Dummer R, Hamid O, et al. Epacadostat (E) Plus Pembrolizumab (P) Versus Pembrolizumab Alone in Patients (pts) with Unresectable or Metastatic Melanoma: Results of the Phase 3 ECHO-301/KEYNOTE-252 Study. 2018 American Society of Clinical Oncology Annual Meeting 36 (suppl; abstract 108). Chicago, IL: American Society of Clinical Oncology; 2018. [Google Scholar]
- 11.Muller AJ, Manfredi M, Zakharia Y, Prendergast GC IDO inhibitors for cancer treatment: lessons from ECHO-301 (invited review). Seminars in Immunopathology. 2018: under review. [DOI] [PubMed] [Google Scholar]
- 12.Smith C, Chang MY, Parker KH, et al. IDO Is a nodal pathogenic driver of lung cancer and metastasis development. Cancer Discov. 2012;2(8): 722–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mondal A, Smith C, DuHadaway JB, et al. IDO1 is an Integral Mediator of Inflammatory Neovascularization. EBioMedicine. 2016;14: 74–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Opitz CA, Litzenburger UM, Sahm F, et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature. 2011;478(7368): 197–203. [DOI] [PubMed] [Google Scholar]
- 15.Puccetti P, Fallarino F, Italiano A, et al. Accumulation of an endogenous tryptophan-derived metabolite in colorectal and breast cancers. PLoS One. 2015;10(4): e0122046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ochs K, Ott M, Rauschenbach KJ, et al. Tryptophan-2,3-dioxygenase is regulated by prostaglandin E2 in malignant glioma via a positive signaling loop involving prostaglandin E receptor-4. J Neurochem. 2015. [DOI] [PubMed] [Google Scholar]
- 17.D’Amato NC, Rogers TJ, Gordon MA, et al. A TDO2-AhR signaling axis facilitates anoikis resistance and metastasis in triple-negative breast cancer. Cancer Res. 2015;75(21): 4651–4664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Witkiewicz AK, Costantino CL, Metz R, et al. Genotyping and expression analysis of IDO2 in human pancreatic cancer: a novel, active target. J Am Coll Surg. 2009;208(5): 781–787; discussion 787–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Metz R, Smith C, Duhadaway JB, et al. IDO2 is critical for IDO1-mediated T cell regulation and exerts a non-redundant function in inflammation. Int Immunol. 2014;26: 357–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prendergast GC, Metz R, Muller AJ, Merlo LM, Mandik-Nayak L. IDO2 in Immunomodulation and Autoimmune Disease. Front Immunol. 2014;5: 585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peek RM, Mohla S, DuBois RN. Inflammation in the genesis and perpetuation of cancer: summary and recommendations from a National Cancer Institute-sponsored meeting. Cancer Res. 2005;65: 8583–8586. [DOI] [PubMed] [Google Scholar]
- 22.Pilotte L, Larrieu P, Stroobant V, et al. Reversal of tumoral immune resistance by inhibition of tryptophan 2,3-dioxygenase. Proc Natl Acad Sci U S A. 2012;109(7): 2497–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gunderson AJ, Coussens LM. B cells and their mediators as targets for therapy in solid tumors. Exp Cell Res. 2013;319(11): 1644–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Affara NI, Ruffell B, Medler TR, et al. B cells regulate macrophage phenotype and response to chemotherapy in squamous carcinomas. Cancer Cell. 2014;25(6): 809–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Merlo LM, DuHadaway JB, Grabler S, Prendergast GC, Muller AJ, Mandik-Nayak L. IDO2 Modulates T Cell-Dependent Autoimmune Responses through a B Cell-Intrinsic Mechanism. J Immunol. 2016;196(11): 4487–4497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rohrig UF, Majjigapu SR, Vogel P, Zoete V, Michielin O. Challenges in the Discovery of Indoleamine 2,3-Dioxygenase 1 (IDO1) Inhibitors. J Med Chem. 2015;58(24): 9421–9437. [DOI] [PubMed] [Google Scholar]
- 27.Qian S, Zhang M, Chen Q, He Y, Wang W, Wang Z. IDO as a drug target for cancer immunotherapy: recent developments in IDO inhibitors discovery. RSC Advances. 2016;6(9): 7575–7581. [Google Scholar]
- 28.Wu J-S, Lin S-Y, Liao F-Y, et al. Identification of Substituted Naphthotriazolediones as Novel Tryptophan 2,3-Dioxygenase (TDO) Inhibitors through Structure-Based Virtual Screening. Journal of Medicinal Chemistry. 2015;58(19): 7807–7819. [DOI] [PubMed] [Google Scholar]
- 29.Pantouris G, Loudon-Griffiths J, Mowat CG. Insights into the mechanism of inhibition of tryptophan 2,3-dioxygenase by isatin derivatives. Journal of Enzyme Inhibition and Medicinal Chemistry. 2016;31(sup1): 70–78. [DOI] [PubMed] [Google Scholar]
- 30.Salter M, Hazelwood R, Pogson CI, Iyer R, Madge DJ. The effects of a novel and selective inhibitor of tryptophan 2,3-dioxygenase on tryptophan and serotonin metabolism in the rat. Biochemical Pharmacology. 1995;49(10): 1435–1442. [DOI] [PubMed] [Google Scholar]
- 31.Pantouris G, Mowat CG. Antitumour agents as inhibitors of tryptophan 2,3-dioxygenase. Biochemical and Biophysical Research Communications. 2014;443(1): 28–31. [DOI] [PubMed] [Google Scholar]
- 32.Dolušić E, Larrieu P, Moineaux L, et al. Tryptophan 2,3-Dioxygenase (TDO) Inhibitors. 3-(2-(Pyridyl)ethenyl)indoles as Potential Anticancer Immunomodulators. Journal of Medicinal Chemistry. 2011;54(15): 5320–5334. [DOI] [PubMed] [Google Scholar]
- 33.Bakmiwewa SM, Fatokun AA, Tran A, Payne RJ, Hunt NH, Ball HJ. Identification of selective inhibitors of indoleamine 2,3-dioxygenase 2. Bioorganic & Medicinal Chemistry Letters. 2012;22(24): 7641–7646. [DOI] [PubMed] [Google Scholar]
- 34.Li J, Li Y, Yang D, et al. Establishment of a human indoleamine 2, 3-dioxygenase 2 (hIDO2) bioassay system and discovery of tryptanthrin derivatives as potent hIDO2 inhibitors. European Journal of Medicinal Chemistry. 2016;123: 171–179. [DOI] [PubMed] [Google Scholar]
- 35.Röhrig UF, Majjigapu SR, Caldelari D, et al. 1,2,3-Triazoles as inhibitors of indoleamine 2,3-dioxygenase 2 (IDO2). Bioorganic & Medicinal Chemistry Letters. 2016;26(17): 4330–4333. [DOI] [PubMed] [Google Scholar]
- 36.Cowley P, Wise A. Pharmaceutical Compounds.
- 37.Wang Z, Guo W, Zhu J, Hu X. Preparation of highly-efficient IDO/TDO dual inhibitor with nitrogen-containing heterocyclic helical structure.
- 38.Wang H, Guo Y, Ren B, Wang Z, Zhang G, Zhou C. Preparation of 5 or 8-substituted imidazo[1,5-a]pyridines as selective inhibitors of indoleamine and/ortryptophan 2,3-dioxygenases.
- 39.Bingham M, Armitage SE, Pesnot T. Preparation of imidazo-pyrrolo-pyridinyl derivatives as IDO and TDO inhibitors for the treatment of IDO - and TDO - mediated diseases.
- 40.Malachowski WP, Winters M, DuHadaway JB, et al. O-alkylhydroxylamines as rationally-designed mechanism-based inhibitors of indoleamine 2,3-dioxygenase-1. Eur J Med Chem. 2016;108: 564–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sugimoto H, Oda S-i, Otsuki T, Hino T, Yoshida T, Shiro Y Crystal structure of human indoleamine 2,3-dioxygenase: Catalytic mechanism of O2 incorporation by a heme-containing dioxygenase. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(8): 2611–2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lewis-Ballester A, Pham KN, Batabyal D, et al. Structural Insights into Substrate and Inhibitor Binding Sites in Human Indoleamine 2,3-Dioxygenase 1. Nature Communications. 2017: accepted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tojo S, Kohno T, Tanaka T, et al. Crystal Structures and Structure–Activity Relationships of Imidazothiazole Derivatives as IDO1 Inhibitors. ACS Medicinal Chemistry Letters. 2014;5(10): 1119–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chauhan N, Basran J, Rafice Sara A, et al. How is the distal pocket of a heme protein optimized for binding of tryptophan? The FEBS Journal. 2012;279(24): 4501–4509. [DOI] [PubMed] [Google Scholar]
- 45.Forouhar F, Anderson JLR, Mowat CG, et al. Molecular insights into substrate recognition and catalysis by tryptophan 2,3-dioxygenase. Proceedings of the National Academy of Sciences. 2007;104(2): 473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yue EW, Douty B, Wayland B, et al. Discovery of Potent Competitive Inhibitors of Indoleamine 2,3-Dioxygenase with in Vivo Pharmacodynamic Activity and Efficacy in a Mouse Melanoma Model. Journal of Medicinal Chemistry. 2009;52(23): 7364–7367. [DOI] [PubMed] [Google Scholar]
- 47.Yang S, Li X, Hu F, et al. Discovery of Tryptanthrin Derivatives as Potent Inhibitors of Indoleamine 2,3-Dioxygenase with Therapeutic Activity in Lewis Lung Cancer (LLC) Tumor-Bearing Mice. Journal of Medicinal Chemistry. 2013;56(21): 8321–8331. [DOI] [PubMed] [Google Scholar]
- 48.Batabyal D, Yeh SR. Human tryptophan dioxygenase: a comparison to indoleamine 2,3-dioxygenase. Journal of the American Chemical Society. 2007;129(50): 15690–15701. [DOI] [PubMed] [Google Scholar]
- 49.Lu C, Lin Y, Yeh SR. Inhibitory substrate binding site of human indoleamine 2,3-dioxygenase. J Am Chem Soc. 2009;131(36): 12866–12867. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.