

Abstract
Prostaglandin E2 (PGE2) via its Gαs-coupled EP2 receptor protects cerebral cortical neurons from excitotoxic and anoxic injury, though EP2 receptor activation can also cause secondary neurotoxicity in chronic inflammation. We performed a high-throughput screen of a library of 292 000 small molecules and identified several compounds that have a 2-piperidinyl phenyl benzamide or trisubstituted pyrimidine core as positive modulators for human EP2 receptor. The most active compounds increased the potency of PGE2 on EP2 receptor 4−5-fold at 20 μM without altering efficacy, indicative of an allosteric mechanism. These compounds did not augment the activity of the other Gαs-coupled PGE2 receptor subtype EP4 and showed neuroprotection against N-methyl-D-aspartate (NMDA)-induced excitotoxicity. These newly developed compounds represent second-generation allosteric potentiators for EP2 receptor and shed light on a promising neuroprotective strategy. They should prove valuable as molecular tools to achieve a better understanding of the dichotomous action of brain EP2 receptor activation.
Keywords: Allosteric potentiator; cAMP; cyclooxygenase, excitotoxicity; GPCR; high-throughput screening; neuroinflammation; neuronal injury; neuroprotection; NMDA; PGE2; TR-FRET
Graphical abstract
INTRODUCTION
Cyclooxygenase-2 (COX-2), the inducible form of COX, is dynamically regulated by synaptic activities,1 and is rapidly upregulated in the brain after cerebral ischemia2,3 and other neurological insults.4–12 As a major enzymatic product of COX2 in the cerebral cortex, prostaglandin E2 (PGE2) can activate four G protein-coupled receptors (GPCRs) named EP1 through EP4, that together regulate a myriad of physiological and pathological signaling events. For instance, the EP1 receptor is coupled to Gαq that mediates the mobilization of cytosolic Ca2+, resulting in the activation of protein kinase C (PKC); EP2 and EP4 are associated with Gαs that activates adenylyl cyclase, leading to cAMP-dependent signaling; EP3 is mainly coupled to Gαi and its activation downregulates cytosolic cAMP.13,14 Following excitotoxic and anoxic injury, upregulated PGE2 signaling via EP2 receptor can protect cerebral neurons from neurotoxicity in a cAMP-dependent manner.2,3,15,16 Moreover, activation of the EP2 receptor is protective against hemin-mediated toxicity in mouse primary cortical neurons also in a cAMP-dependent manner,17 and protects middle-aged brains from intracerebral hemorrhage injury with small hematomas,18 but not young brains with large lesion size.19 These findings support a neuroprotective strategy via selectively enhancing EP2 receptor activation before or during injury.
On the other hand, EP2 receptor activation can also promote chronic inflammatory processes within the brain that exacerbate neurotoxicity and neuronal death in models of neurodegenerative diseases, such as Alzheimer’s disease,20 Parkinson’s disease,21 and amyotrophic lateral sclerosis.22 The secondary neurotoxic action of EP2 receptor is associated with brain innate immunity, particularly microglia-mediated neuroinflammation.23,24 Indeed, in animal models of status epilepticus delayed pharmacological inhibition of EP2 receptor is anti-inflammatory, neuroprotective and maintains the bloodbrain barrier integrity.6,9,25–27 Moreover, intracerebroventricular administration of the EP2 selective agonist butaprost immediately after status epilepticus provides moderate protection of hippocampal neurons.4 These seemingly contradicting results suggest a dichotomy of action of EP2 receptor in the central nervous system (CNS), depending on its cell typespecific (neuronal vs glial) and/or model-specific (acute vs chronic) downstream targets.5,12–14,28 Developing new selective small-molecule modulators provides valuable pharmacological tools aiming at a better understanding of the dual and opposing consequences of EP2 receptor activation in different disease states.14
Over the past decade, remarkable advances in the development of allosteric modulators for GPCRs have led to encouraging therapeutic strategies for a variety of CNS conditions, including neurodegenerative diseases and psychiatric disorders.29 While orthosteric ligands including agonists and antagonists have traditionally been pursued to target GPCRs, allosteric regulators provide multiple mechanistic advantages owing to their ability to distinguish among closely associated receptor subtypes and to provide temporal and spatial context-dependence.29–33 We previously reported a series of thiophene-carboxylate compounds as positive allosteric modulators that are selective for EP2 receptor over EP4 and β2-adrenergic receptors.34 In the present study, we report two additional groups of small-molecule compounds with distinct chemical structures that can augment EP2 receptor activity only in the presence of its natural agonist PGE2. Moreover, we study the selectivity, potency, toxicity, and neuroprotection of these compounds and provide insight into the correlations between their chemical structures and biological activities.
RESULTS AND DISCUSSION
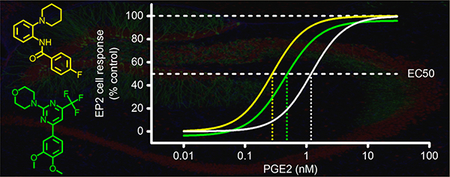
We previously reported high-throughput screening (HTS) of a library of 292 000 small molecules, in which a cell-based time resolved fluorescence energy transfer (TR-FRET) cAMP assay was used to monitor the activity of PGE2 receptor EP2.34 The HTS and a follow-up systematic medicinal chemistry campaign led to the discovery of a series of novel positive allosteric modulators for EP2 receptor with a chemical scaffold of thiophene-carboxylate. From the same screening, we have also identified other two groups of small molecules that markedly enhanced the response of rat C6 glioma (C6G) cells stably expressing human EP2 receptor to an EC15 of PGE2 (∼0.3 nM) (Figure 1). These compounds did not show a similar effect on C6G cells expressing other Gαs-coupled receptors, such as EP4 and β2-adrenergic receptors. In addition, none of these newly identified compounds affected the parent C6G cells or the TRFRET cAMP assay itself. These counter-assay data indicated that the new groups of compounds increased EP2-mediated cAMP production of the cells but not through directly acting on the downstream adenylyl cyclase (Figure 1). The results from primary and secondary screens together suggest that these new primary hits are EP2-selective allosteric potentiators.
Figure 1.

Identification of small-molecule compounds as EP2 allosteric potentiators by high-throughput screening (HTS). The HTS strategy consisted of a primary screen and a panel of secondary assays.34 In the primary screen, library compounds were tested at 20 μM in rat C6 glioma (C6G) cells stably expressing EP2 receptor for their potentiation of the cell response to an EC15 of PGE2 (∼0.3 nM). The EP2 receptor activity was assessed by measuring cellular cAMP levels using a cell-based time-resolved fluorescence energy transfer (TR-FRET) assay. Compounds with at least 50% potentiation in the primary screen were subject to secondary screens, which contained a series of counter-tests aiming to eliminate false-positive hits that might also act on other Gαs-coupled receptors, such as EP4 and β2-adrenergic receptors, or their downstream cAMP production components. These counter-assays also excluded compounds that interfered with the TR-FRET assay itself. Compounds that survived all screens were grouped into structural clusters, among which are 2-piperidinyl phenyl benzamide and trisubstituted pyrimidine.
The first group of compounds, typified by CID890517 and CID2340325, possesses a 2-piperidinyl phenyl moiety coupled to a benzamide moiety; the second group of compounds (CID2992168 and CID891729) is characterized by a trisubstituted pyrimidine core attached to a morpholine ring (Figure 1 and Table 1). For confirmation, we purchased and/or resynthesized these four hit compounds (Scheme 1), and tested response to 0.3 nM PGE2 (∼EC15). Cellular cAMP level was them in C6G-EP2 cells for their potentiation of the cell measured by TR-FRET assay as a surrogate for the cell response. Indeed, all four hit compounds increased the PGE2induced EP2 receptor activity in a concentration-dependent manner, and 2-piperidinyl phenyl benzamide compounds in general showed higher activities than trisubstituted pyrimidines. In contrast, the C6G-EP2 cells had no response to compounds (up to 40 μM) in the absence of PGE2 (Figure 2A), suggesting an allosteric mechanism of EP2 receptor activation.
Table 1.
Chemical Structure and Activity of EP2 Positive Allosteric Modulators
| Compound | Chemical structure | Potentiation on EP2 receptora |
| 2-piperidinyl phenyl benzamides | ||
| CID890517 (Synthesized as TG6–270) | 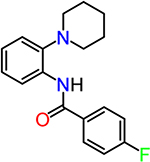 |
4.3 |
| CID2340325 |  |
3.3 |
| CID953673 (TG6–133-1) |  |
2.5 |
| CID1254488 | 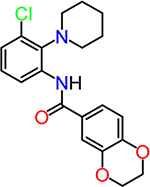 |
2.5 |
| TG6-127-1 | 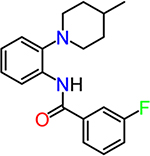 |
1.4 |
| TG6-127-2 | 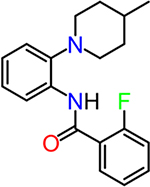 |
1.2 |
| TG6-264 | 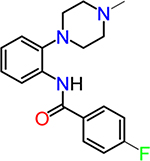 |
1.0 |
| TG6-268 | 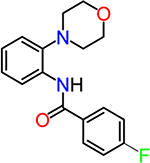 |
1.0 |
| Compound | Chemical structure | Potentiation on EP2 receptora |
| Trisubstituted pyrimidines | ||
| CID2992168 | 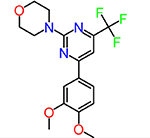 |
2.7 |
| CID891729 | 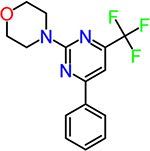 |
2.4 |
| CID852313 | 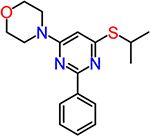 |
2.4 |
| CID852308 | 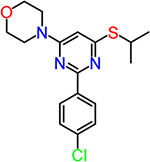 |
2.4 |
| CID3239428 | 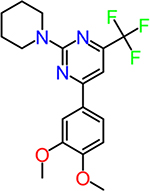 |
2.2 |
| CID630454 | 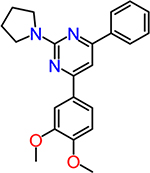 |
1.7 |
| CID2992722 | 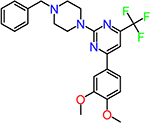 |
1.5 |
| CID2987851 | 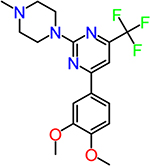 |
1.1 |
Potentiation on EP2 receptor: the mean fold shift to the left of the PGE2 EC50 in the presence of 20 μM tested compound (n = 3−6).
Scheme 1a.

aReagents and conditions: 3, para-fluorophenacyl chloride; 4, meta-fluorophenacyl chloride; 5, ortho-fluorophenacyl chloride; (i) DMAP, DCM, RT, overnight. Yield 65−75%.
Figure 2.

Selective allosteric potentiation of EP2 receptor activation. (A) C6G-EP2 cells were treated by tested compounds with increasing concentrations (0.3−40 μM) and 5 min later by an EC15 of PGE2 (∼0.3 nM) for 40 min. The EP2 receptor activity was assessed by measuring the cellular cAMP production using the cell-based TR-FRET assay. Data were scaled between responses to 0.3 nM PGE2 (= 0%) and a saturating concentration of PGE2 (= 100%). Note that all tested compounds increased PGE2-induced EP2 receptor activity in a concentration-dependent manner, but the cells did not response to compounds in the absence of PGE2. (B) C6G-EP2 cells were treated by tested compounds (20 μM) and 5 min later by increasing concentrations of PGE2 (0.01−30 nM). The PGE2 concentration−response curves were generated and EC50 values were calculated using OriginPro. Note that PGE2 induced EP2 receptor activation in a concentration-dependent manner; treatment with tested compounds shifted the PGE2 concentration−response curve to the left. Control, EC50 = 1.19 nM; CID890517, EC50 = 0.28 nM; CID2340325, EC50 = 0.36 nM; CID2992168, EC50 = 0.44 nM; CID891729, EC50 = 0.50 nM. (C) C6G cells stably expressing EP4 receptor (C6G-EP4) were treated with tested compounds (20 μM) and 5 min later by increasing concentrations of PGE2 (0.003−10 nM). The EP4 receptor activity was assessed using the cell-based TR-FRET cAMP assay. Note that PGE2 induced EP4 receptor activation in a concentration-dependent manner; treatment with compounds had no leftward-shift effect on the PGE2 concentration−response curve. Control, EC50 = 0.02 nM; CID890517, EC50 = 0.02 nM; CID2340325, EC50 = 0.08 nM; CID2992168, EC50 = 0.03 nM; CID891729, EC50 = 0.02 nM. Data are shown as mean ± SEM (n = 3).
The primary screening assay was designed to identify compounds that can augment the response of EP2 receptor to a low concentration of PGE2. Therefore, the hit compounds could enhance EP2 activity by increasing either the efficacy or the potency of PGE2. To further determine their mechanism of action on EP2 receptor, we treated the C6G-EP2 cells with compounds for 5 min and then with increasing concentrations of PGE2 for 40 min. The cell response (i.e., cAMP level) to PGE2 stimulation was assessed by the TR-FRET cAMP assay. We showed that PGE2 induced EP2 cell response in a concentration-dependent manner with an EC50 of 1.19 nM. Pretreatment with these compounds did not enhance the maximum response of PGE2 for activating EP2 receptor, arguing against an effect of tested compounds on efficacy. Instead, they produced a marked leftward-shift in the PGE2 concentration−response curve, suggesting that these compounds enhanced potency of PGE2 (Figure 2B). In line, 2piperidinyl phenyl benzamide compounds on average showed higher EP2 potentiation than trisubstituted pyrimidines, reflected by their larger fold shift of PGE2 EC50 (Figure 2B and Table 1).
Among the four PGE2 receptor subtypes, Gαs-coupled EP2 and EP4 are the most closely related, as they share a common endogenous ligand PGE2 and much of the downstream cAMPdependent and independent signaling pathways.14,27,35 PGE2 signaling through EP4 receptor has also been shown to exert anti-inflammatory effects in brain innate immunity,36 as well as neuronal and vascular protection in animal models of cerebral ischemia and hypoxic-ischemic encephalopathy.37,38 Therefore, it is important for these allosteric modulators to be able to distinguish between the two closely associated PGE2 receptor subtypes. To study their effect on EP4 receptor, we treated C6G cells that express human EP4 (C6G-EP4) with these newly identified compounds for 5 min and then with PGE2 for 40 min. The TR-FRET cAMP assay result revealed that PGE2 induced EP4 cell response in a concentration-dependent manner with an EC50 of 0.02 nM and treatment with these compounds was unable to potentiate the effect of PGE2 on EP4 receptor at 20 μM (Figure 2C). On the contrary, 2-piperidinyl phenyl benzamide compound CID2340325 inhibited EP4 receptor, as evidenced by a 4-fold rightward-shift of the PGE2 concentration−response curve in the presence of 20 μM CID2340325 (Figure 2C).
We next examined the cytotoxicity of these EP2 allosteric compounds in C6G parent cells using the CellTiter-Glo luminescent cell viability assay, which measures the cellular ATP level that is commonly used to indicate the cell viability. The cells were incubated with tested compounds or doxorubicin as the positive control for 48 h before the measurement for viability. The CellTiter-Glo assay revealed that all tested compounds had low cytotoxicity, demonstrated by their high CC50 values (>130 μM for 2-piperidinyl phenyl benzamides; >500 μM for trisubstituted pyrimidines), whereas the positive control treatment with doxorubicin showed significant toxicity with an anticipated low CC50 of 0.52 μM in the same assay (Figure 3A).
Figure 3.

Cytotoxicity and neuroprotection of EP2 positive allosteric modulators. (A) C6G cells were incubated with tested compounds for 48 h and cell viability was measured by CellTiter-Glo luminescent assay. Doxorubicin treatment was used as the positive control. The concentrationdependent cytotoxicity curves were generated and CC50 values were calculated using OriginPro. Doxorubicin, CC50 = 0.52 μM; CID890517, CC50 = 154 μM; CID2340325, CC50 = 135 μM; CID2992168, CC50 = 807 μM; CID891729, CC50 = 575 μM. Data are shown as mean ± SEM (n = 4). (B) Rat primary hippocampal neurons (DIV14) were pretreated with EP2 allosteric potentiators (20 μM) for 30 min, followed by overnight incubation with NMDA (30 μM) and glycine (10 μM). Excitotoxic damage to the cultured neurons was assessed by measuring the fraction of lactate dehydrogenase (LDH) released into the culture medium. Note that NMDA-induced neuronal damage was partially to fully prevented by the pretreatment with tested compounds (*P < 0.05; ***P < 0.001, one-way ANOVA and posthoc Bonferroni test for multiple comparisons with selected pairs indicated). Data are shown as mean + SEM (n = 4−8).
EP2 receptor activation has been reported to prevent injury of neurons from excitotoxicity induced by N-methyl-D-aspartic acid (NMDA) and cerebral ischemia both in vitro and in vivo.2,3,15 We previously also showed that thiophene-carboxylate EP2 allosteric potentiators provided considerable neuroprotection in NMDA-treated rat primary neurons.34 Thus, we next examined the effect of these new EP2 allosteric potentiators on NMDA-induced neuronal excitotoxicity. Rat hippocampal neurons (DIV14) were incubated with tested compounds for 30 min, followed by 30 μM NMDA treatment overnight in the continued presence of the tested compounds. Lactate dehydrogenase (LDH) release into the culture medium was used to indicate neuronal injury. NMDA treatment caused a nearly 3-fold increase of LDH release in the cultured hippocampal neurons (P < 0.001, Figure 3B), suggesting a substantial neuronal damage induced by NMDA treatment. However, the NMDA-triggered neuronal excitotoxicity was partially to largely prevented by coincubation with these EP2 positive allosteric modulators (P < 0.05 for CID2992168; P < 0.001 for CID890517, CID2340325 and CID891729, Figure 3B). Compared to our previously reported lead EP2 potentiator of thiophene-carboxylates,34 trisubstituted pyrimidine hit CID891729 showed a higher reduction of NMDA-induced neuronal injury (63% vs 45%) when tested at the same concentration (20 μM) (Figure 3B).
To obtain information on structure−activity relationship (SAR), we purchased and/or synthesized analogs based on the 2-piperidine phenyl benzamide hit compounds CID890517 [4-fluoro-N-(2-piperidin-1-ylphenyl)benzamide] and CID2340325 [2-methyl-N-(2-piperidin-1-ylphenyl)benzamide] (Scheme 1), and the trisubstituted pyrimidine core of compounds CID2992168[4-[4-(3, 4-dimethoxyphenyl)-6 (trifluoromethyl)pyrimidin-2-yl]morpholine] and CID891729 [4-[4-phenyl-6-(trifluoromethyl)pyrimidin-2-yl]morpholine]. All compounds were evaluated for potentiation on EP2 receptor by calculating the fold shift of PGE2 concentrationdepend curve (Table 1). It appeared that, for the 2-piperidinyl phenyl benzamides, para-fluorobenzamide is important for EP2 potentiation, as moving fluorine from para to meta or ortho position (as shown by TG6–127-1 and TG6–127-2) decreased the fold shift of PGE2 EC50. Interestingly, incorporation of a dioxane ring connecting para to meta positions of the benzene ring and addition of a chlorine atom next to 2-piperidine (e.g., CID1254488) maintained compound activity. To further extend SAR around these compounds, we tested with piperazine and morpholine moieties (TG6–264 and TG6–268, respectively) in the place of piperidine at 2-position (TG6270). Disappointingly, we did not observe any EP2 potentiation by these derivatives, reinforcing the notion that the piperidine moiety is essential for the potentiation activity within this class. For the trisubstituted pyrimidine analogues (Table 1), the morpholine ring maintains high EP2 potentiation activity compared with piperidine, pyrrolidine and piperazine rings (cf. CID2992168 vs CID3239428, CID630454, CID2992722, and CID2987851). Two other closely related isopropylthio pyrimidine derivatives (CID852313 and CID852308) also displayed EP2 potentiation activity similar to the HTS hit CID891729. Additional SAR studies are necessary to understand the full scope of these two scaffolds regarding their potency and efficacy of EP2 receptor potentiation.
Finally, eight key characteristics in adsorption, distribution, metabolism, and excretion (ADME) of these hits and analogs were estimated by QikProp software (Schrödinger Release 2017–3: QikProp, Schrödinger, LLC, New York, NY, 2017). The predicted ADME properties of all active EP2 allosteric modulators fall into the projected range of 95% of known drugs (Table 2). Moreover, the prediction by QikProp also suggests that these newly developed EP2 compounds should be able to cross the brain-blood barrier and are anticipated to have positive CNS activity (Table 2).
Table 2.
ADME Properties (Absorption, Distribution, Metabolism, Excretion) of EP2 Allosteric Potentiators Predicted by QikPropa
| MW |
QPlogPo/w |
QPlogS |
QPPCaco |
QPPMDCK |
QPlogBB |
CNS |
RO3 |
RO5 |
|
|---|---|---|---|---|---|---|---|---|---|
| range or recommended valuesb | |||||||||
| compd | <500 | −2.0 to 65. | −6.5 to 0.5 | <25 poor/>500 great | <25 poor/>500 great | −3.0 to 1.2 | −2 to + 2 | 0 best/3 poor | 0 best/≥4 poor |
| CID890517 (TG6–270) | 298.359 | 2.277 | −2.807 | 6823.583 | 6570.798 | 0.529 | 2 | 0 | 0 |
| CID2340325 | 294.396 | 2.821 | −2.917 | 8918.539 | 5266.293 | 0.526 | 2 | 0 | 0 |
| CID953673 (TG6–133-1) | 312.386 | 4.611 | −5.784 | 3847.153 | 3837.694 | 0.144 | 1 | 1 | 0 |
| CID1254488 | 372.85 | 2.653 | −4.054 | 6823.844 | 6405.129 | 0.503 | 2 | 0 | 1 |
| TG6–127-1 | 312.386 | 4.602 | −5.753 | 4095.364 | 4106.024 | 0.172 | 1 | 1 | 0 |
| TG6–127-2 | 312.386 | 4.676 | −5.868 | 4099.12 | 3657.917 | 0.145 | 1 | 1 | 0 |
| TG6–264 | 313.374 | 3.146 | −3.807 | 823.019 | 801.774 | 0.473 | 2 | 0 | 0 |
| TG6–268 | 300.332 | 3.326 | −4.323 | 3851.291 | 3842.16 | 0.169 | 1 | 0 | 0 |
| CID2992168 | 369.343 | 2.391 | −4.04 | 7665.817 | 10 000 | 0.632 | 2 | 0 | 1 |
| CID891729 | 309.29 | 3.149 | −4.384 | 9130.118 | 10 000 | 0.708 | 2 | 0 | 0 |
| CID852313 | 315.432 | 2.619 | −3.602 | 9425.404 | 7241.735 | 0.593 | 2 | 0 | 0 |
| CID852308 | 349.877 | 2.854 | −3.761 | 9016.764 | 10 000 | 0.728 | 2 | 0 | 0 |
| CID3239428 | 367.37 | 2.442 | −4.106 | 7665.817 | 10 000 | 0.632 | 2 | 0 | 1 |
| CID630454 | 361.443 | 2.654 | −5.022 | 7713.944 | 4501.848 | 0.441 | 0 | 0 | 1 |
| CID2992722 | 458.482 | 3.033 | −5.651 | 7663.333 | 10 000 | 0.613 | 2 | 0 | 1 |
| CID2987851 | 382.385 | 1.597 | −3.518 | 7663.333 | 10 000 | 0.627 | 2 | 0 | 1 |
MW = Molecular weight. QPlogPo/w = Predicted octanol/water partition coefficient. QPlogS = Predicted aqueous solubility, logS. S in mol/L is the concentration of the solute in a saturated solution that is in equilibrium with the crystalline solid. QPPCaco = Predicted apparent Caco-2 cell permeability in nm/s. Caco-2 cells are considered a model for the gut-blood barrier. QPPMDCK = Predicted apparent MDCK cell permeability in nm/s. MDCK cells are considered a mimic for the blood-brain barrier. QPlogBB = Predicted brain/blood partition coefficient. CNS = Predicted central nervous system (CNS) activity on a −2 (inactive) to +2 (active) scale. RO3 = Predicated Jorgensen rule of three, an indication of oral availability. RO5 = Predicted Lipinski rule of five, an indication of drug-likeness.
Range is for 95% of known drugs.
CONCLUSIONS
We report a series of small-molecule compounds with a 2piperidinyl phenyl benzamide or trisubstituted pyrimidine core as positive allosteric modulators for the EP2 receptor of prostaglandin PGE2. The most potent compound in this series showed a similar fold shift (4.3) of PGE2 EC50 in EP2expressing cells to that of the thiophene-carboxylate lead that we previously identified. These newly developed compounds did not show any potentiation of other tested Gαs-coupled receptors, demonstrating their selectivity to EP2 receptor. Also importantly, these EP2 allosteric potentiators had negligible cellular toxicity and showed higher neuroprotection against NMDA-induced neuronal excitotoxicity in rat primary hippocampal neurons than the thiophene-carboxylate EP2 potentiators. Our results reinforce the notion that EP2 receptor activation by endogenous PGE2 released in an excitotoxic setting can be neuroprotective. These two new classes of allosteric modulators possess CNS drug-like ADME properties and are expected to act on EP2 receptor within the brain. Future study is necessary to better understand the SAR of these two chemical scaffolds, which might yield novel analogs with improved potency. Future efforts should also be directed at determining whether these new EP2 allosteric potentiators with sufficient in vivo half-life and brain-penetration can protect cerebral cortical neurons from excitotoxic and anoxic injury in animal models. Nonetheless, the newly identified compounds in this work are already proving valuable as chemical probes for more detailed investigation of the dichotomous role of the EP2 receptor in the CNS.
METHODS
Cell Culture.
Cell lines stably expressing human EP2 or EP4 receptor were generated by transfecting rat C6 glioma (C6G) cells with the pcDNA3.1 vector containing receptor cDNA controlled by the CMV promoter. Cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) (Invitrogen, Carlsbad, CA), supplemented with 10% (v/v) fetal bovine serum (FBS) (Invitrogen), 100 U/mL penicillin and 100 μg/mL streptomycin (Invitrogen), and 0.5 mg/ mL Geneticin (Invitrogen).
Chemical and Drugs.
PGE2 was purchased from Cayman Chemical (Ann Arbor, MI). Rolipram, doxorubicin, N-methyl-Daspartate (NMDA), and glycine were from Sigma-Aldrich (St. Louis, MO). Positive hit compounds from HTS and derivatives were either obtained through commercial sources [ChemBridge (San Diego, CA) and ChemDiv (San Diego, CA)] or synthesized.
Chemical Design and Synthesis.
The synthesis of the 2piperidine phenyl benzamide hit compounds CID89051 and CID2340325, and their analogues was achieved following Scheme 1. Typically, starting material 2-(4-methylpiperidin-1-yl)aniline (1) was acetylated with commercially available para-fluorophenacyl chloride (3) in the presence of a catalytic amount of N,N-dimethylamino pyridine to obtain the final products in a single step. Similarly, other derivatives were synthesized using corresponding starting materials 2 and 4−6. The 1H NMR data of all compounds was consistent with their structures. For example, TG6–133-1 [4-fluoro-N-(2-(4-methylpiperidin-1-yl)phenyl)benzamide] displayed 1H NMR (400 MHz, CDCl3): δ 9.5 (bs, 1H), 8.5 (d, J = 7.6 Hz, 1H), 7.92 (m, 2H), 7.18 (m, 4H), 7.07 (t, J = 7.2 Hz, 1H), 2.96 (d, J = 12 Hz, 2H), 2.72 (t, J = 12 Hz, 2H), 1.80 (d, J = 12 Hz, 2H), 1.58 (m, 1H), 1.38 (m, 2H), 1.0 (d, J = 6.4 Hz, 3H). LCMS (ESI): >95% purity at λ 254, MS; m/z, 313 [M + H]. Other compounds in this group displayed NMR and mass data consistent with the corresponding structures. The trisubstituted pyrimidine hit compound CID2992168 and its analogues were procured from commercially available sources. CID2992168 [4-(4(3,4-dimethoxyphenyl)-6-(trifluoromethyl)pyrimidin-2-yl)morpholine] displayed 1H NMR (400 MHz, CDCl3): δ 7.65 (d, J = 2.4 Hz, 1H), 7.63 (s, 1H), 7.16 (s, 1H), 6.94 (d, J = 8 Hz, 1H), 3.97 (s, 3H), 3.94 (s, 3H), 3.92 (t, J = 5.2 Hz, 4H), 3.79 (d, J = 5.2 Hz, 4H). LCMS (ESI): >95% purity at λ 254, MS; m/z, 370 [M + H].
Cell-Based cAMP Assay.
Cellular cAMP was measured using a cell-based homogeneous time-resolved fluorescence resonance energy transfer (TR-FRET) method (Cisbio Bioassays, Codolet, France).34,39,40 The assay is based on the generation of a strong FRET signal upon the interaction of two fluorescent molecules: FRET donor (cryptate) coupled to anti-cAMP antibody and FRET acceptor (d2) coupled to cAMP. Endogenous cAMP produced by cells competes with labeled cAMP for binding to the cAMP antibody and thus reduces the FRET signal. Cells were seeded into 384-well plates with 30 μL of complete medium (4000 cells/well) and grown overnight. The medium was completely withdrawn, and 10 μL of Hank’s balanced salt solution (HBSS) (HyClone, Logan, UT) supplemented with 20 μM rolipram was added into the wells to block phosphodiesterases that would metabolize cAMP. The cells were incubated at room temperature for 30 min, and then treated with vehicle or test compound for 5 min before incubation with PGE2 for 40 min. The cells were lysed in 10 μL of lysis buffer containing the FRET acceptor cAMP-d2, and 1 min later another 10 μL of lysis buffer with anti-cAMP-cryptate was added. After incubation for at least 1 h at room temperature, the FRET signal was measured by using a 2103 Envision Multilabel Plate Reader (PerkinElmer Life Sciences, Waltham, MA) with an excitation at 340/25 nm and dual emissions at 665 and 590 nm for d2 and cryptate (100 μs delay), respectively. The FRET signal was expressed as F665/F590 × 104.
Cytotoxicity Assay.
Cytotoxicity of tested compounds was examined with the CellTiter-Glo Luminescent Cell Viability Assay (Promega, Madison, WI) by measuring cellular ATP level, which is believed to correlate with cell viability. Briefly, C6G cells were plated in 384-well plates at 2000 cells/well in 25 μL of DMEM plus test compound and incubated for 2 days. CellTiter-Glo reagent (25 μL) was then added to each well. The contents were mixed for 2 min on an orbital shaker to induce cell lysis and incubated at room temperature for 10 min. Relative viability is proportional to luminescence intensity as measured by a microplate reader (Molecular Devices, Sunnyvale, CA) with an integration time of 1 s.
In vitro Analysis of Neuroprotection.
Hippocampal neurons were isolated from embryonic-day 16−18 (E16−E18) embryos of timed-pregnant Sprague−Dawley rats (Charles River Laboratories, Wilmington, MA), as previously described.2,34 Cells were plated onto poly-D-lysine coated 24-well plates at a density of 150 000 cells/well in Neurobasal medium plus B27 and 5% fetal bovine serum (Invitrogen). Cultures were incubated at 37 ˚C in 95% air/5% CO2 and half of the culture medium was replaced every 4−5 days with serum-free medium. After 14 days in culture, cells were pretreated with tested compounds for 30 min. Excitotoxicity was induced by treating neurons with NMDA (30 μM) plus glycine (10 μM) overnight in the presence of tested drugs. Excitotoxic damage was assessed by measuring the fraction of lactate dehydrogenase (LDH) released into the culture medium (Roche Applied Science, Penzberg, Germany). Released LDH % was calculated as 100 × LDH released/(LDH released + LDH in the cells), where the LDH in the cells was determined in the cell lysate. Relative LDH release % was defined as 100 × (LDH drug − LDH control)/(LDH NMDA − LDH control), where the LDH drug was the released LDH % from wells treated with tested drugs plus NMDA; LDH control was the released LDH % without any treatment; and LDH NMDA was the released LDH % treated by NMDA only. Cultures in which the LDH control was greater than 5% or the (LDH NMDA − LDH control) was less than 3% were discarded.
Statistical Analysis.
The concentration−response curves were generated and EC50/CC50 values were calculated using OriginPro software (OriginLab, Northampton, MA). Statistical analyses were performed using Prism (GraphPad Software, La Jolla, CA) by one-way ANOVA with posthoc Bonferroni test for multiple comparisons. P < 0.05 was considered statistically significant. All data are presented as mean + or ± SEM.
Acknowledgments
Funding
This work was supported by the National Institutes of Health (NIH)/National Institute of Neurological Disorders and Stroke (NINDS) Grants R00NS082379 (J.J.), R01NS100947 (J.J.), R01NS097776 (R.D.), and U01NS074509 (R.D.), and NARSAD Young Investigator Grant 20940 from the Brain & Behavior Research Foundation (BBRF) (J.J.).
ABBREVIATIONS
- ADME
adsorption, distribution, metabolism, and excretion
- C6G
C6 glioma
- cAMP
cyclic adenosine monophosphate
- CNS
central nervous system
- COX
cyclooxygenase
- DMEM
Dulbecco’s modified Eagle’s medium
- EP
prostaglandin E2 receptor
- FBS
fetal bovine serum
- GPCR
G protein-coupled receptor
- HBSS
Hanks’ balanced salt solution
- HTS
highthroughput screening
- LDH
lactate dehydrogenase
- NMDA
N-methyl-D-aspartate
- PGE2
prostaglandin E2
- PKC
protein kinase C
- SAR
structure−activity relationship
- TR-FRET
timeresolved fluorescence energy transfer
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Kaufmann WE, Worley PF, Pegg J, Bremer M, and Isakson P (1996) COX-2, a synaptically induced enzyme, is expressed by excitatory neurons at postsynaptic sites in rat cerebral cortex. Proc. Natl. Acad. Sci. U. S. A 93, 2317–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).McCullough L, Wu L, Haughey N, Liang X, Hand T, Wang Q, Breyer RM, and Andreasson K (2004) Neuroprotective function of the PGE2 EP2 receptor in cerebral ischemia. J. Neurosci 24, 257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Liu D, Wu L, Breyer R, Mattson MP, and Andreasson K (2005) Neuroprotection by the PGE2 EP2 receptor in permanent focal cerebral ischemia. Ann. Neurol 57, 758–761. [DOI] [PubMed] [Google Scholar]
- (4).Serrano GE, Lelutiu N, Rojas A, Cochi S, Shaw R, Makinson CD, Wang D, FitzGerald GA, and Dingledine R (2011) Ablation of cyclooxygenase-2 in forebrain neurons is neuroprotective and dampens brain inflammation after status epilepticus. J. Neurosci 31, 14850–14860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Quan Y, Jiang J, and Dingledine R (2013) EP2 receptor signaling pathways regulate classical activation of microglia. J. Biol. Chem 288, 9293–9302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Jiang J, Yang MS, Quan Y, Gueorguieva P, Ganesh T, and Dingledine R (2015) Therapeutic window for cyclooxygenase-2 related anti-inflammatory therapy after status epilepticus. Neurobiol. Dis 76, 126–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Kirkby NS, Chan MV, Zaiss AK, Garcia-Vaz E, Jiao J, Berglund LM, Verdu EF, Ahmetaj-Shala B, Wallace JL, Herschman HR, Gomez MF, and Mitchell JA (2016) Systematic study of constitutive cyclooxygenase-2 expression: Role of NF-kappaB and NFAT transcriptional pathways. Proc. Natl. Acad. Sci. U. S. A 113, 434–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Lekic T, Krafft PR, Klebe D, Rolland WB, Flores J, Tang J, and Zhang JH (2016) Cyclooxygenase-2 Inhibition Provides Lasting Protection Following Germinal Matrix Hemorrhage in Premature Infant Rats In Brain Edema XVI: Translate Basic Science into Clinical Practice (Applegate RL, Chen G, Feng H, and Zhang JH, Eds.), pp 203–207, Springer International Publishing, Cham. [DOI] [PubMed] [Google Scholar]
- (9).Du Y, Kemper T, Qiu J, and Jiang J (2016) Defining the therapeutic time window for suppressing the inflammatory prostaglandin E2 signaling after status epilepticus. Expert Rev. Neurother 16, 123–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Qiu J, Shi Z, and Jiang J (2017) Cyclooxygenase-2 in glioblastoma multiforme. Drug Discovery Today 22, 148–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Hartings JA, York J, Carroll CP, Hinzman JM, Mahoney E, Krueger B, Winkler MKL, Major S, Horst V, Jahnke P, Woitzik J, Kola V, Du Y, Hagen M, Jiang J, and Dreier JP (2017) Subarachnoid blood acutely induces spreading depolarizations and early cortical infarction. Brain 140, 2673–2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Yousif NM, de Oliveira ACP, Brioschi S, Huell M, Biber K, and Fiebich BL (2017) Activation of EP2 receptor suppresses poly(I: C) and LPS-mediated inflammation in primary microglia and organotypic hippocampal slice cultures: Contributing role for MAPKs. Glia, DOI: 10.1002/glia.23276. [DOI] [PubMed] [Google Scholar]
- (13).Andreasson K (2010) Emerging roles of PGE2 receptors in models of neurological disease. Prostaglandins Other Lipid Mediators 91, 104–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Jiang J, and Dingledine R (2013) Prostaglandin receptor EP2 in the crosshairs of anti-inflammation, anti-cancer, and neuroprotection. Trends Pharmacol. Sci. 34, 413–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Ahmad AS, Zhuang H, Echeverria V, and Dore S (2006) Stimulation of prostaglandin EP2 receptors prevents NMDA-induced excitotoxicity. Journal of neurotrauma 23, 1895–1903. [DOI] [PubMed] [Google Scholar]
- (16).Ahmad M, Saleem S, Shah Z, Maruyama T, Narumiya S, and Dore S (2010) The PGE2 EP2 receptor and its selective activation are beneficial against ischemic stroke,. Exp. Transl. Stroke Med 2, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Mohan S, Narumiya S, and Dore S (2015) Neuroprotective role of prostaglandin PGE2 EP2 receptor in hemin-mediated toxicity. NeuroToxicology 46, 53–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Wu H, Wu T, Han X, Wan J, Jiang C, Chen W, Lu H, Yang Q, and Wang J (2017) Cerebroprotection by the neuronal PGE2 receptor EP2 after intracerebral hemorrhage in middle-aged mice. J. Cereb. Blood Flow Metab 37, 39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Leclerc JL, Lampert AS, Diller MA, Immergluck JB, and Dore S (2015) Prostaglandin E2 EP2 receptor deletion attenuates intracerebral hemorrhage-induced brain injury and improves functional recovery. ASN Neuro, DOI: 10.1177/1759091415578713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Liang X, Wang Q, Hand T, Wu L, Breyer RM, Montine TJ, and Andreasson K (2005) Deletion of the prostaglandin E2 EP2 receptor reduces oxidative damage and amyloid burden in a model of Alzheimer’s disease. J. Neurosci 25, 10180–10187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Jin J, Shie FS, Liu J, Wang Y, Davis J, Schantz AM, Montine KS, Montine TJ, and Zhang J (2007) Prostaglandin E2 receptor subtype 2 (EP2) regulates microglial activation and associated neurotoxicity induced by aggregated alpha-synuclein. J. Neuroinflammation 4, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Liang X, Wang Q, Shi J, Lokteva L, Breyer RM, Montine TJ, and Andreasson K (2008) The prostaglandin E2 EP2 receptor accelerates disease progression and inflammation in a model of amyotrophic lateral sclerosis. Ann. Neurol 64, 304–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Johansson JU, Pradhan S, Lokteva LA, Woodling NS, Ko N, Brown HD, Wang Q, Loh C, Cekanaviciute E, Buckwalter M, Manning-Bog AB, and Andreasson KI (2013) Suppression of inflammation with conditional deletion of the prostaglandin E2 EP2 receptor in macrophages and brain microglia. J. Neurosci 33, 16016–16032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Johansson JU, Woodling NS, Wang Q, Panchal M, Liang X, Trueba-Saiz A, Brown HD, Mhatre SD, Loui T, and Andreasson KI (2015) Prostaglandin signaling suppresses beneficial microglial function in Alzheimer’s disease models. J. Clin. Invest 125, 350–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Jiang J, Ganesh T, Du Y, Quan Y, Serrano G, Qui M, Speigel I, Rojas A, Lelutiu N, and Dingledine R (2012) Small molecule antagonist reveals seizure-induced mediation of neuronal injury by prostaglandin E2 receptor subtype EP2. Proc. Natl. Acad. Sci. U. S. A 109, 3149–3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Jiang J, Quan Y, Ganesh T, Pouliot WA, Dudek FE, and Dingledine R (2013) Inhibition of the prostaglandin receptor EP2 following status epilepticus reduces delayed mortality and brain inflammation. Proc. Natl. Acad. Sci. U. S. A 110, 3591–3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Dey A, Kang X, Qiu J, Du Y, and Jiang J (2016) Anti-Inflammatory Small Molecules To Treat Seizures and Epilepsy: From Bench to Bedside. Trends Pharmacol. Sci 37, 463–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Fu Y, Yang MS, Jiang J, Ganesh T, Joe E, and Dingledine R (2015) EP2 Receptor Signaling Regulates Microglia Death. Mol. Pharmacol 88, 161–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Conn PJ, Lindsley CW, Meiler J, and Niswender CM (2014) Opportunities and challenges in the discovery of allosteric modulators of GPCRs for treating CNS disorders. Nat. Rev. Drug Discovery 13, 692–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Conn PJ, Jones CK, and Lindsley CW (2009) Subtypeselective allosteric modulators of muscarinic receptors for the treatment of CNS disorders. Trends Pharmacol. Sci 30, 148–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Lane JR, Abdul-Ridha A, and Canals M (2013) Regulation of G protein-coupled receptors by allosteric ligands. ACS Chem. Neurosci 4, 527–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Nickols HH, and Conn PJ (2014) Development of allosteric modulators of GPCRs for treatment of CNS disorders. Neurobiol. Dis 61, 55–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Foster DJ, and Conn PJ (2017) Allosteric Modulation of GPCRs: New Insights and Potential Utility for Treatment of Schizophrenia and Other CNS Disorders. Neuron 94, 431–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Jiang J, Ganesh T, Du Y, Thepchatri P, Rojas A, Lewis I, Kurtkaya S, Li L, Qui M, Serrano G, Shaw R, Sun A, and Dingledine R (2010) Neuroprotection by selective allosteric potentiators of the EP2 prostaglandin receptor. Proc. Natl. Acad. Sci. U. S. A 107, 2307–2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Jiang J, Qiu J, Li Q, and Shi Z (2017) Prostaglandin E2 Signaling: Alternative Target for Glioblastoma? Trends in cancer 3, 75–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Shi J, Johansson J, Woodling NS, Wang Q, Montine TJ, and Andreasson K (2010) The prostaglandin E2 E-prostanoid 4 receptor exerts anti-inflammatory effects in brain innate immunity,. J. Immunol 184, 7207–7218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Liang X, Lin L, Woodling NS, Wang Q, Anacker C, Pan T, Merchant M, and Andreasson K (2011) Signaling via the prostaglandin E(2) receptor EP4 exerts neuronal and vascular protection in a mouse model of cerebral ischemia. J. Clin. Invest 121, 4362–4371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Taniguchi H, Anacker C, Wang Q, and Andreasson K (2014) Protection by vascular prostaglandin E2 signaling in hypoxicischemic encephalopathy. Exp. Neurol 255, 30–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Jiang J, and Dingledine R (2013) Role of prostaglandin receptor EP2 in the regulations of cancer cell proliferation, invasion, and inflammation. J. Pharmacol. Exp. Ther 344, 360–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Kang X, Qiu J, Li Q, Bell KA, Du Y, Jung DW, Lee JY, Hao J, and Jiang J (2017) Cyclooxygenase-2 contributes to oxidopamine-mediated neuronal inflammation and injury via the prostaglandin E2 receptor EP2 subtype. Sci. Rep 7, 9459. [DOI] [PMC free article] [PubMed] [Google Scholar]