

Abstract
OBJECTIVE:
To determine whether latency can be established and reversed in both proliferating and non-proliferating CD4+ T cells in the same model in vitro.
METHODS:
Activated CD4+ T cells were infected with either a non-replication competent, luciferase reporter virus or wild-type full-length enhanced green fluorescent protein (EGFP) reporter virus and cultured for 12 days. The cells were then sorted by flow cytometry to obtain two distinct T cell populations that did not express the T cell activation markers, CD69, CD25 and HLA-DR: CD69−CD25−HLA-DR− small cells (non-blasts) that had not proliferated in vitro following mitogen stimulation and CD69−CD25−HLA-DR− large cells (which we here call transitional blasts) that had proliferated. The cells were then reactivated with latency reversing agents (LRAs) and either luciferase or EGFP quantified.
RESULTS:
Inducible luciferase expression, consistent with latent infection, was observed in non-blasts and transitional blasts following stimulation with either phorbol-myristate-acetate/phytohemagglutinin (3.8 ± 1 and 2.9 ± 0.5 fold above DMSO, respectively) or romidepsin (2.1 ± 0.6 and 1.8 ± 0.2 fold above DMSO, respectively). Constitutive expression of luciferase was higher in transitional blasts compared to non-blasts. Using wild-type full-length EGFP reporter virus, inducible virus was observed in non-blasts but not in transitional blasts. No significant difference was observed in the response to LRAs in either non-blasts or transitional blasts.
CONCLUSIONS:
HIV latency can be established in vitro in resting T cells that have not proliferated (non-blasts) and blasts that have proliferated (transitional blasts). This model could potentially be used to assess new strategies to eliminate latency.
Keywords: HIV, latency, latency reversing agents, transitional blasts, proliferating, non-proliferating
BACKGROUND
Despite the successes of antiretroviral therapy (ART), HIV cannot be cured because of the persistence of latently infected resting and proliferating memory T cells [1–5]. One strategy being developed to eliminate latently infected T cells is to stimulate HIV transcription using latency-reversing agents (LRAs) [6]. The response to LRA stimulation has recently been shown to depend on the viral integration site [7], however other factors such as the proliferative history and cellular activation state may also play a role.
HIV latency in primary CD4+ T cells can be established either through direct infection of resting cells (pre-activation latency) [8–11], or through the infection of activated CD4+ T cells that then revert to a ‘resting’ state (post-activation latency) [12–15]. In post-activation models, a ‘resting’ phenotype is often defined by the lack of expression of activation markers CD69, CD25 and HLA-DR [14–17]. In addition, most in vitro models use laboratory-derived virus strains and this may not accurately represent HIV sequences in vivo, which are diverse, especially in individuals who initiate ART during chronic infection [18]. We recently demonstrated that the activity of LRAs was different dependent upon how latency was established [19]. Here we hypothesized that the proliferation history of a latently infected cell will also affect the response to an LRA.
To test this hypothesis, we developed a model of post-activation latency using a luciferase reporter construct that was able to integrate and persist in activated and resting T cells. We confirmed latent infection by the demonstration of inducible luciferase expression in two distinct T cell subsets that did not express activation markers: non-proliferating resting cells (non-blasts) and proliferating cells (CD69−CD25−HLA-DR− transitional blasts). Finally we used this model to assess the response to LRAs following infection with either wild type virus or virus that contained patient-derived long terminal repeat (LTR) sequences.
METHODS
Infection of CD4+ T cells and cell sorting
Resting CD4+ T cells were isolated from healthy donor peripheral blood mononuclear cells (PBMC) (Australian Red Cross Blood Service, Southbank, Australia) as previously described [20]. Resting CD4+ T cells were pre-stained with the proliferation dye, eFluor450 (eBioscience, San Diego, CA) and either left in culture, stimulated with CCL19 (100 nM; R&D Systems, Minneapolis, MN), or activated with phytohemagglutinin (PHA; 10 μg/mL, Sigma-Aldrich, St. Louis, MO) and interleukin-2 (IL-2; 10 IU/mL, Roche Applied Sciences, Penzberg, Germany) at a density of 2 × 106 cells/mL in RF10 media [RPMI1640 (Life Technologies, Carlsbad, CA), 10% fetal bovine serum (Bovogen Biologicals, Melbourne, Australia) and 1× penicillin-streptomycin-glutamine (Life Technologies)]. After 48 hours, cells were infected with NL4.3-EGFP at 50% tissue culture infectious dose (TCID50) of 0.5, or 20× concentrated vesicular stomatitis virus-G (VSV-G) pseudotyped viral supernatant at a ratio of ~10 ng p24 to one million cells. Uninfected ‘mock’ and raltegravir controls were conducted concurrently; raltegravir (1 μM, Selleck Chemicals, Houston, TX) was added five minutes prior to infection. Cells treated with the chemokine CCL19 were cultured in 100 nM CCL19 for 24 hours prior to infection, as previously described [20].
After twelve days, infected cells were stained with anti-CD69-FITC, anti-CD25-APC and anti-HLA-DR-PE (all BD Biosciences, Franklin Lakes, NJ). Cells were then sorted by flow cytometry (MoFlo Astrios, Beckman Coulter, Brea, CA) based on forward- and side-scatter, eFluor, and expression of CD69, CD25 and HLA-DR ± EGFP into three populations: eFluorhigh CD69−CD25−HLA-DR−/EGFP− non-blasts, eFluorlow CD69−CD25−HLA-DR−/EGFP− blasts (transitional blasts), and CD69+CD25+HLA-DR+ blasts (for VSV-G pseudotyped virus only). Cells were harvested for baseline luciferase expression or reactivated with various LRAs.
Cloning of patient LTRs into a vector able to integrate and express luciferase
We amplified and cloned the HIV LTR from integrated virus in memory CD4+ T cells from four HIV-infected individuals prior to and following ART, as previously described [21]. In order to clone patient-derived LTRs into a retroviral vector, we first added the polypurine tract (PPT) to each LTR isolate using an overlap extension PCR (Table S1). A lentiviral vector with a luciferase reporter gene (pGBFM) was then used to evaluate LTR function as previously described [22]. Purified PPT-LTR fragments and the pGBFM vector were digested using KpnI (Promega, Madison, WI) and XbaI restriction enzymes (New England Biolabs), and ligated using T4 DNA ligase (New England Biolabs). The ligation product was transformed into Stbl3 Escherichia Coli competent cells (Life Technologies). Successful clones were identified using colony PCR and confirmed by sequencing (AGRF, Melbourne, Australia).
Production and quantification of VSV-G pseudotyped and NL4.3-EGFP viruses
For VSV-G pseudotyped virus production, 293T cells (5 × 105) were transfected as previously described [22]. Viral production was quantified by measuring p24 levels by ELISA with anti-p24 sheep antibody D7320 (Aalto Bio Reagents, Dublin, Ireland). NL4.3-EGFP was generated and the TCID50 was determined as described in [23].
Reactivation of cells with LRAs
Infected CD4+ T cells were harvested for baseline luciferase expression at day 4 (unsorted population) and day 12 post-infection (unsorted and sorted populations). The unsorted and sorted cell populations (non-blast and blasts) harvested at day 12 were reactivated with the positive control PHA (10 μg/mL) and phorbol myristate acetate (PMA, 10 nM; both Sigma-Aldrich), the vehicle control dimethyl sulfoxide (DMSO; Sigma Aldrich), or a panel of LRAs: romidepsin (40 nM), panobinostat (50 nM), vorinostat (0.5 μM; all Selleck Chemicals), JQ-1 (1 μM) and chaetocin (10 nM; both Sigma-Aldrich). Five hundred thousand cells were plated and treated with raltegravir (1 μM, Selleck Chemicals) for five minutes prior to the addition of LRAs. All cells were reactivated in the presence of 10 IU/mL IL-2 at a cell density of 1 × 106 cells/mL. After 24 hours, 200,000 viable cells were re-plated and luciferase quantified using the Luciferase Assay System (Promega). For infection with the EGFP expressing wild type virus, raltegravir was added to the cultures prior to addition of either romedepsin (40 nM) or PMA/PHA (10 nM/10 μg/mL) and after 24 hours, EGFP was quantified by flow cytometry. Data are shown as fold-change over DMSO control or percent of PMA/PHA stimulation.
Cytotoxicity assay
Resting CD4+ T cells were treated with varying concentrations (range dose 10–100,000 nM for all drugs) of the histone deacetylase inhibitors (HDACi) romidepsin, panobinostat and vorinostat; the bromodomain inhibitor JQ-1, and the histone methyltransferase inhibitor chaetocin. All drugs were diluted in DMSO to establish a final DMSO concentration of <1%. Two hundred thousand resting CD4+ T cells were plated and incubated for 48 hours with the appropriate concentration of LRAs. A negative control with DMSO was included where no drug was added. A positive control was used where NP-40 (Thermo Fisher Scientific) was added to induce cell death. After 48 hours, the MTS proliferation dye (CellTiter 96® Aqueous One Solution Cell Proliferation Assay, Promega) was added to each well and incubated for 6–8 hours. Colorimetric analysis was performed using a plate reader (Thermo Fisher Scientific) measured at OD 492 nm. Data were analyzed using GraphPad Prism software (Version 6, Graphpad, La Jolla, CA) using non-linear regression analysis.
Phenotyping CD69−CD25−HLA-DR− blasts and non-blasts
The negatively sorted CD69−CD25−HLA-DR− blast and non-blasts were stained with anti-CD45RO-FITC, anti-CD45RA-PE-Cy7 (BD Biosciences), anti-CCR7-APC, eFluor-780, anti-CD27 PE-Cy5 (all eBioscience) and anti-CD4-PE-Texas Red (Invitrogen), and analyzed using a flow cytometer (LSRFortessa™, BD Biosciences).
Statistical analysis
A one-way analysis of variance (ANOVA) with Tukey post-test was used to measure the difference in basal luciferase expression between unstimulated cells, CCL19 and PHA/IL-2 stimulated cells; or between different sorted and unsorted cell populations; or between different LRAs. Student t-tests were used to measure differences between DMSO and PMA/PHA-treated populations; or in the PMA/PHA and romidepsin treated non-blast populations.
RESULTS
Establishing latency in vitro using a VSV-G pseudotyped LTR reporter virus
We first developed a VSV-G pseudotyped luciferase reporter virus containing the wild-type NL4–3 LTR that was able to integrate into primary T cells. Using this virus, we were able to infect PHA/IL-2 activated CD4+ T cells but not resting or CCL19 treated CD4+ T cells (Fig. 1A).
Fig. 1. Infection of PHA/IL-2 activated CD4+ T cells with NL4–3 LTR VSV-G pseudotyped pGBFM virus.
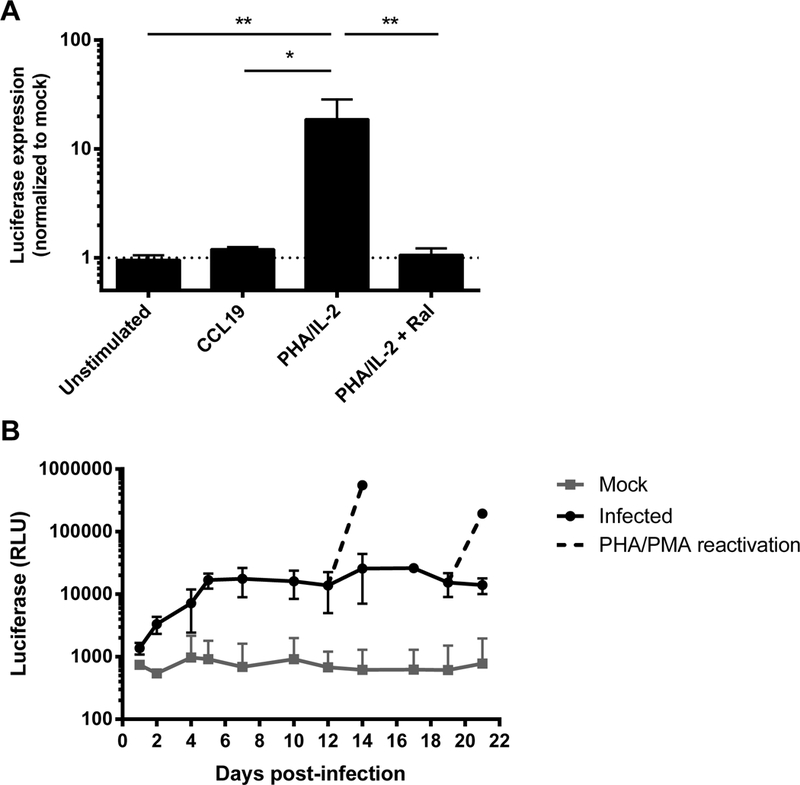
A. Primary resting CD4+ T cells isolated from healthy blood donors were left unstimulated, treated with CCL19, or were activated with PHA in the presence of IL-2 (with or without the integrase inhibitor raltegravir) for 48 hours prior to infection with NL4–3-LTR VSV-G pseudotyped pGBFM virus. Infection was quantified by measuring the level of luciferase produced on day 4. Mean ± SD values are shown from four independent experiments. * p<0.05 ** p<0.01. B. PHA-activated CD4+ T cells were infected as in (A) and the level of luciferase was measured every 1–3 days post-infection. On days 12 and 19 post-infection, cells in culture were further stimulated with PMA/PHA and luciferase measured two days after reactivation. The mean ± SD luciferase expression is shown from three independent donors.
Given this pseudotyped virus was able to infect activated, but not quiescent T cells, we investigated whether latency could be established in primary T cells by allowing activated cells to return to a resting state through long-term culture with 10 IU/ml IL-2. Cells were harvested every 2–3 days to monitor luciferase activity. Luciferase expression increased rapidly during the first four days after infection and plateaued up to day 21 (Fig. 1B), consistent with constitutive expression of luciferase. Because this was a single round non-infectious virus, constitutive expression of luciferase did not result in new rounds of viral replication. The addition of PMA/PHA at days 12 and 19 post-infection, in the presence of raltegravir, led to large increases in luciferase production 48 hours later (mean ± SEM increase above mock of 8.6 ± 0.4 and 11.6 ± 0.9 fold respectively; Fig. 1B). These findings demonstrate that in this culture system, even in the setting of a constitutive luciferase expression, there were cells infected with inducible virus, consistent with latency.
Changes in cell size over time in a model of post-activation latency
To better understand the kinetics of T cell activation and proliferation in this model, we assessed expression of the activation markers CD69, CD25 and HLA-DR as well as history of proliferation using the cytoplasmic dye eFluor. The early activation marker CD69 peaked two days after T cell activation, followed by CD25 at day 4 and then both declined. The late activation marker HLA-DR only began to increase at 3–4 days after and remained elevated to day fourteen (Fig. 2A). Cell viability declined shortly after PHA/IL-2 stimulation and continued to decline over the next 30 days (Fig. 2B). By staining the T cells with eFluor prior to stimulation, we demonstrated the existence of two distinct populations: eFluorlow large cells that had proliferated (blasts); and eFluorhigh small cells (non-blasts) that had never proliferated (Fig. 2C). The proportion of blasts progressively increased over time and plateaued at about 80% (Fig. 2D), while the percentage of non-blasts decreased proportionally.
Fig. 2. The phenotype and viability of CD4+ T cells during long-term culture.
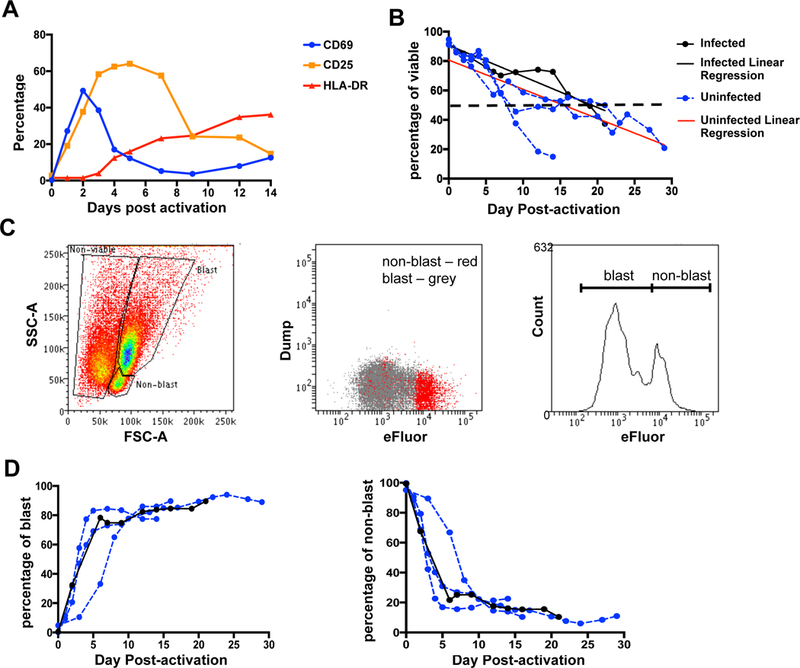
A. Kinetics of expression of T cell activation markers following stimulation with PHA/IL-2. Primary resting CD4+ T cells were activated with PHA in the presence of IL-2 and the percentage of CD69 (blue), CD25 (orange) and HLA-DR (red) in the viable cell population was measured by flow cytometry. Data represent the mean from two individual donors. B. Resting CD4+ T cells were activated with PHA/IL-2 with or without infection with WT-LTR VSV-G pseudotyped pGBFM virus and cell viability was measured by flow cytometry. Linear regression of the uninfected (red line) and infected (black line) donors is also shown. C. Gating strategy to determine the proportion of non-blast, blast and non-viable cells by flow cytometry based on forward- and side-scatter (FSC and SSC, respectively). A representative flow plot of cells collected 12 days post-activation, prior to sorting is shown. Cells stained with the proliferation dye, eFluor, were also used to confirm the gating strategy. D. Percentage of blast and non-blasts over time is shown. Percentage was calculated based on the total number of viable cells. Each line represents an individual donor.
Inducible virus in CD69−CD25−HLA-DR− blasts and non-blasts
To determine which populations of cells were infected with the VSV-G pseudotyped LTR reporter virus, we sorted the cells on day 14 post-activation based on their morphology and expression of CD69, CD25 and HLA-DR (Fig. 3A). Luciferase was measured prior to and following reactivation with PMA/PHA. The non-blasts had minimal constitutive luciferase expression (mean 1.9 ± 0.4 fold increase above mock, Fig. 3B). As expected, constitutive expression of luciferase was highest in the CD69+CD25+HLA-DR+ blasts (60.5 ± 14.3 fold above mock), followed by the unsorted cells (31.9 ± 7.5 fold) and the CD69−CD25−HLA-DR− blasts, that we here call transitional blasts (24.0 ± 4.4 fold) (Fig. 3B).
Fig. 3. Inducible virus is detected in both CD69-CD25-HLA-DR- transitional blasts and non-blasts.
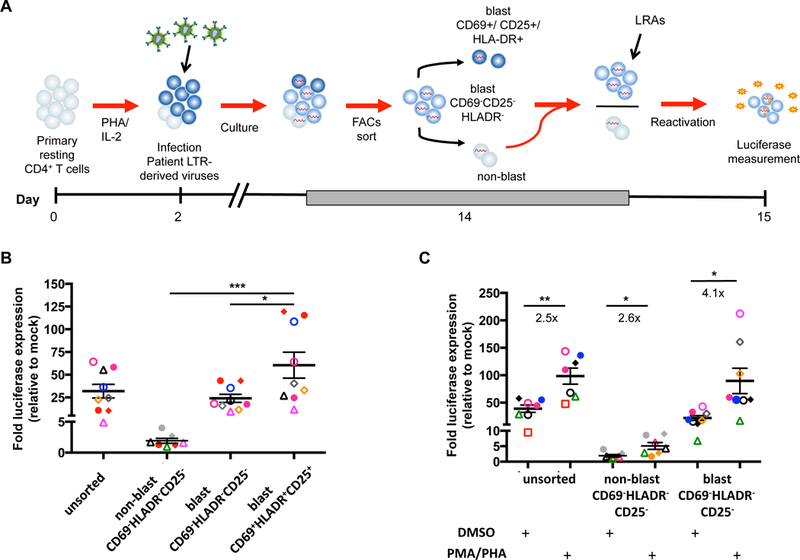
A. Schematic of the method used to establish HIV latency in vitro using a VSV-G pseudotyped reporter virus. PHA/IL-2 activated CD4+ T cells were infected and maintained in culture for 12 days. CD69−CD25−HLA-DR− transitional blast (eFluorlow) and non-blast (eFluorhigh) and CD69+/CD25+/HLA-DR+ cells were sorted and luciferase activity was quantified. Only the activation marker negative cells were then incubated (shown as red arrows) with various LRAs and the change in luciferase activity was quantified again. B. Activated CD4+ T cells were infected with a VSV-G pseudotyped reporter virus (using both patient-derived and NL4–3 LTRs) and luciferase production was measured in the unsorted, CD69−CD25−HLA-DR− transitional blast and non-blast, or the CD69+/CD25+/HLA-DR+ blast populations immediately after sorting. Only statistically significant differences are shown. Each symbol represents a different donor and the black lines represent the mean ± SEM. C. The sorted cells from (B) were either reactivated with PMA and PHA or treated with the negative vehicle control DMSO for 24 hours and luciferase production measured. The fold increase in luciferase following PMA/PHA treatment, relative to DMSO, is shown as the ratio of the arithmetic mean ± SEM. * p<0.05; ** p<0.01; *** p<0.001. Each symbol represents a different virus derived from LTRs isolated from CD4+ T cells from HIV-infected individuals, pre-ART (open) or post-ART (closed). Each color represents an experiment using CD4+ T cells from the same healthy donor.
To identify which subsets of cells contained inducible virus, PMA/PHA was then used to re-stimulate luciferase expression. Upon reactivation with PMA/PHA, a significantly higher fold-increase in luciferase expression was observed in the transitional blasts (4.1 fold, p=0.012), compared to unsorted cells (2.5 fold, p=0.003) or the non-blast population (2.6 fold, p=0.02) (Fig. 3C). The transitional blasts contained a mixed population of central memory (TCM, CD45RA−CCR7+CD27+, 16.9 ± 2.1%), effector memory (TEM, CD45RA−CCR7−CD27−, 4.7 ± 1%), transitional memory (TTM, CD45RA−CCR7−CD27+, 16.7 ± 2.2%), naïve (CD45RA+CCR7+CD27+, 41.5 ± 9.2%) and negligible number of terminally differentiated cells (TTD, CD45RA+CCR7−CD27−, 0.1 ± 0.0%) (Fig. S1A). Interestingly, the non-blasts were also comprised of similar cell subpopulations but with a higher proportion of naïve T cells (51.9 ± 5.5%) cells (Fig. S1B). These data demonstrate that latent infection can be established in vitro in two subsets of cells that don’t express activation markers - non-blasts and transitional blasts
Reversal of latency in non-blasts infected with the replication competent EGFP reporter virus
Given that the pseudotyped virus was a single round virus that also bypassed chemoreceptor signaling because of the VSV-G protein, we repeated the same experiments with a replication-competent full-length wild-type NL4.3 virus that expressed enhanced green fluorescent protein (EGFP) [24]. Following infection, the cells were cultured for 12 days and EGFP− cells sorted into eFluorhigh and eFluorlow cells (Fig. 4A). The sorted cells were incubated with the integrase inhibitor raltegravir to block further rounds of infection and then stimulated with either romidepsin or PMA/PHA. We observed a significant increase in EGFP expression in the non-blasts (Fig. 4B, Fig. S2) but no overall increase in EGFP expression in the transitional blasts.
Fig. 4. Post-activation latency following infection with NL4.3EGFP.
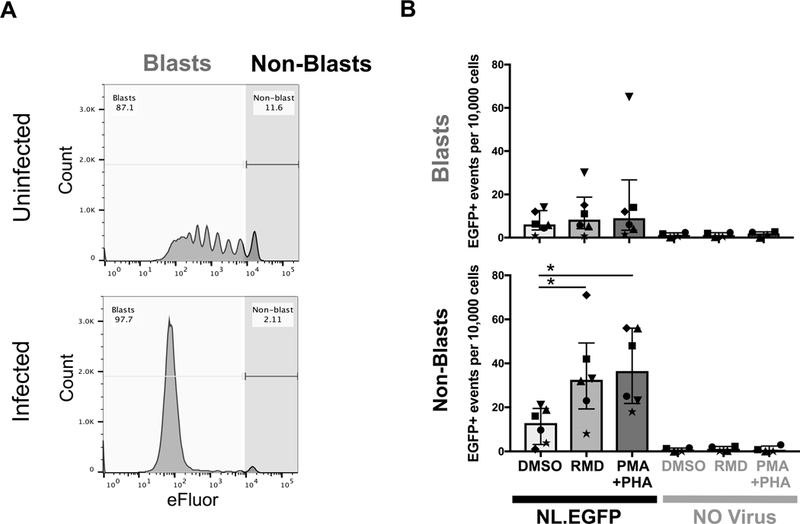
A. Activated CD4+ T cells were infected with NL4.3-EGFP virus and cultured for 12 days to allow cells to return to rest. Cells were sorted at day twelve post-infection for EGFP-negative CD69−CD25−HLA-DR− transitional blasts (eFluorlow) and non-blasts (eFluorhigh) as shown. B. Sorted blasts and non-blasts were reactivated with the HDACi romidepsin (RMD), PMA/PHA or with control DMSO. The number of EGFP+ cells per 10,000 events was quantified by flow cytometry. In each experiment between 10,000 and 100,000 events were collected. Data represent mean ± SEM of six individual donors. Each symbol represents a different donor. * p<0.05
LRAs induced luciferase production from transitional blasts and non-blasts
We next assessed the responsiveness of infected transitional blasts to a panel of LRAs at non-cytotoxic concentrations (Fig. S3) using VSV-G pseudotyped LTR luciferase reporter viruses carrying wild-type NL4–3 or patient-derived LTRs. Using the NL4–3 LTR pseudotyped virus, chaetocin displayed the highest potency (3.1 ± 0.3 fold above DMSO) in transitional blasts (Fig. 5A). Of the HDACi tested, romidepsin induced the highest fold-increase in luciferase activity (1.9 ± 0.7 fold), followed by panobinostat (1.8 ± 0.6 fold), and vorinostat (1.1 ± 0.1 fold). Cells treated with the positive control, PMA/PHA, induced the highest level of luciferase (3.7 ± 1.1 fold) (Fig. 5A). The effect of LRAs was also expressed as a percentage of maximum stimulation with PMA/PHA (Fig. 5B). We then compared the ex vivo response of three patient-derived LTRs (Fig. 5C). Consistent with our previous in vitro model of latency [21], patient-derived LTRs were responsive to LRA stimulation with no significant difference observed across patient-derived LTRs and wild-type NL4–3 (Fig. 5C&D, Fig. S4).
Fig. 5. LRAs induce luciferase expression in both transitional blasts and non-blasts.
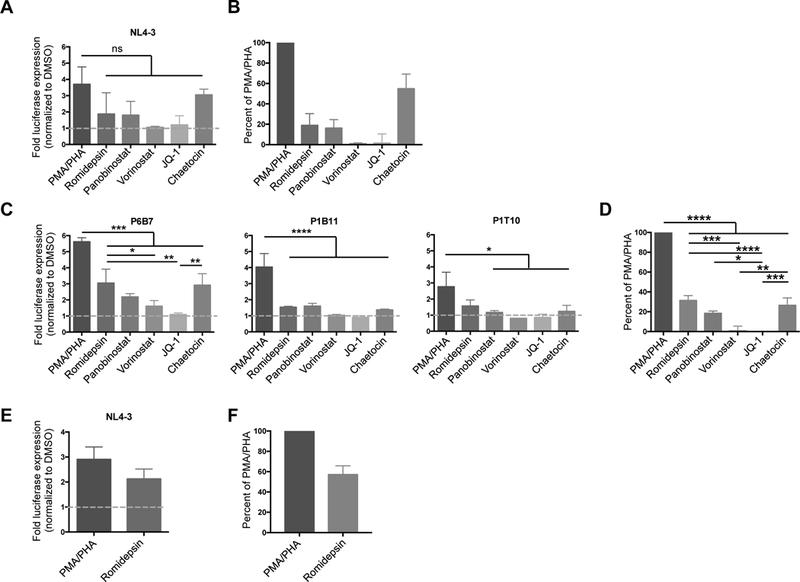
A. Sorted CD69−CD25−HLA-DR− transitional blasts infected with pseudotyped-virus containing NL4–3 LTR were treated with various LRAs including the HDACi romidepsin, panobinostat and vorinostat; the histone-methyltransferase inhibitor chaetocin; and the bromodomain inhibitor JQ-1. The level of luciferase was measured 24 hours after treatment with LRAs. The level of reactivation is expressed as fold change over DMSO (n = 3). B. The level of reactivation from (A) is expressed as a percentage of maximum stimulation using PMA/PHA. C. CD69−CD25−HLA-DR− transitional blasts were infected with pseudotyped-viruses carrying LTRs derived from HIV-infected individuals obtained prior to ART initiation (P1B11 and P6B7) or after full viral suppression on ART (P1T10) and treated with the same panel of LRAs. Data represent mean ± SEM from three independent experiments. D. The level of reactivation from the three patient-derived LTRs is expressed as a percentage of maximum stimulation using PMA/PHA. E. Sorted non-blasts infected with NL4–3 pseudotyped-virus were stimulated with romidepsin and the level of luciferase produced at 24 hour is expressed as fold change over DMSO (n = 3). F. Reactivation is expressed as a percentage of maximum stimulation with PMA/PHA. * p<0.05; ** p<0.01; *** p<0.001; **** p<0.0001
Since HIV latency was detected in the resting non-blast population (Fig. 3C & 4B), we also tested the responsiveness of these cells to the most potent HDACi, romidepsin, using NL4–3 LTR pseudotyped-virus. Following romidepsin stimulation, there was a 2.1 ± 0.4 fold increase in luciferase activity (Fig. 5E), while PMA/PHA induced a 2.9 ± 0.5 fold increase in luciferase over DMSO control. When expressed as percentage of maximal stimulation with PMA/PHA, romidepsin induced 57.1 ± 8.6% increase in luciferase expression (Fig. 5F).
DISCUSSION
We developed a novel model of post-activation latency allowing the assessment of patient-derived LTRs in proliferating and non-proliferating T cell subsets. Using a single round pseudotyped virus, inducible virus was established in CD69−CD25−HLA-DR− T cells that had never proliferated (non-blasts) and in CD69−CD25−HLA-DR− transitional blasts: cells that had proliferated but did not express classical activation markers and retained a blast morphology. Inducible virus was demonstrated in both T cell subsets using single round virus and a panel of LRAs and pseudotyped viruses with patient-derived LTRs. Although an artificial model, these data suggest that virus expression can be induced from both proliferating and non-proliferating cells that don’t express classical activation markers.
Transitional blasts are not found in the peripheral blood but they are readily detected in the lymph node, especially after antigen challenge such as vaccination [25, 26]. In a recent study by Shan et al. [27], a similar transitioning cell was described, where activated CD4+ T cells undergoing effector-to-memory (ETM) transition had downregulation of gene transcription and increased expression of CCR5 with increased permissiveness to latent HIV infection. Similar to the transitional blasts described in our manuscript, these ETM cells had low levels of activation marker expression, had mixed TCM and TEM phenotypes and were transitioning to a smaller cell size. Using a different model, we confirm that latency can be established in cells that have proliferated, have a blast morphology but don’t express activation markers consistent with cells undergoing ETM transition.
Our model is the first to evaluate latency reversal in two distinct cell populations distinguished by activation state, cell size and morphology in primary CD4+ T-cells. Other investigators have assessed cell size in models of post-activation latency [13, 28] but have not identified cells that are negative for all classical activation markers. In a model using anti-CD3/CD28 activation [28], cell size was observed to decrease, although didn’t fully revert to baseline resting size over an 11-day culture period and there was continued expression of the activation marker CD25. In the model developed by Tyagi and colleagues, cells reverted to a resting size after a culture period of up to 49 days [13], however CD25 expression continued to persist in these ‘resting’ cells. In our model, we were unable to culture cells for that duration given the high frequency of cell death. Here we found that a subset of both blasts and non-blasts didn’t express any of the activation markers, CD69, CD25 and HLA-DR.
Using the novel pGBFM luciferase reporter construct, we were able to assess the effect of LRAs on patient-derived HIV LTRs in primary CD4+ T-cells. Use of patient-derived LTRs may provide a more accurate representation of LRA activity, given the heterogeneity of HIV LTR sequences in vivo. This contrasts to other models of latency that use only laboratory-derived strains. The use of primary cells in our model also allows for random integration following infection, as compared to cell line models which use a monoclonal cell population with a single HIV integration site [29, 30]. Our model also provides the potential to evaluate patient-derived HIV LTRs that may be of particular interest e.g. LTRs isolated from patients in clinical trials shown not to respond to LRAs in vivo.
The lack of expression of the activation markers CD69, CD25 and HLA-DR is often used to define a ‘resting’ cell population in models of post-activation latency and when quantifying the frequency of latent infection on ART ex vivo [14–17]. The latently infected transitional blasts identified in our new model expressed persistent low levels of luciferase, suggesting a down-regulated but not fully quiescent transcriptional state. In contrast, the non-blasts had virtually no constitutive luciferase expression. Latently infected cells are often described as being in a state of complete transcriptional dormancy. However, constitutive expression of cell-associated HIV RNA in HIV-infected individuals on ART has been demonstrated by us and other groups [31–33], and expression of viral protein Gag has also been shown to occur in latently infected cells in vitro [34, 35]. This model, using single round LTR luciferase constructs that integrate, may allow for further insights into understanding the spectrum of transcriptional activity in HIV latency.
The establishment of latency in the non-blasts was surprising, given prior evidence has suggested resting T cells to be relatively non-permissive to infection with VSV-G pseudotyped [36] or wild-type virus [20, 37]. Infection of non-blasts could potentially have occurred through several mechanisms. First, it is possible that latent infection was established in a subset of non-blasts that were activated but did not proliferate [38]. This is supported by the finding of a comparable proportion of TCM and TTM phenotypes in the non-blasts compared to transitional blasts. Second, co-culture of these cells with activated proliferating T cells could have facilitated latent infection either through exposure to secreted soluble factors, or through cell-cell transmission of virus [39, 40]. Finally, it is also possible that a proportion of the non-blasts could be T memory stem cells, which possess a CD45RA+CCR7+ phenotype and have been shown to be permissive to infection with VSV-G pseudotyped viruses [41].
We acknowledge that there are several limitations of this model. First we used a single round pseudotyped envelope deleted virus that was unable to induce cell death. We sought to address this by repeating the experiments using a full length EGFP reporter virus. Using this virus we were able to demonstrate inducible EGFP in non-blasts but not in transitional blasts, most likely due to death of the transitional blasts during the prolonged phase of productive infection, when there were no antiretrovirals present. Following stimulation of cells infected with wild type virus, there would be expression of envelope protein, which would also enhance cell death with syncytia formation even in the presence of raltegravir. In addition, we used different methods to quantify the input virus for the wild type and pseudotyped viruses and this could potentially explain the difference in findings for the two viruses. Second, we only measured EGFP expression as a marker of productive infection and didn’t directly measure infectious virus, but based on previous reports of this virus, we assume that EGFP expression correlates with infectious virus [24]. Third, we defined latency here as the demonstration of inducible luciferase. In the setting of high constitutive luciferase expression with a virus that is unable to induce cell death, as seen in the transitional blasts, we cannot rule out productive infection. In addition, inducible luciferase expression does not reflect the frequency of infected cells, but rather the frequency of infected cells with inducible virus. We may have potentially underestimated the frequency of latently infected cells if the virus in some cells did not activate following T-cell stimulation, described in patient-derived cells as “non-induced intact proviruses” [42]. This is clearly a limitation of single reporter viruses that express either luciferase or EGFP. Finally, we acknowledge that in blood, there are no circulating cells with features of transitional blasts (as described here) but these cells could potentially be found in tissue such as lymph node.
CONCLUSION
We show that following T cell activation, latency is established in non-proliferating and proliferating cells that do not express classical activation markers. Both latently infected T cell subsets responded to LRAs with similar efficiency. The contribution of each of these T cell subsets to HIV persistence in vivo in blood and in tissue warrants further evaluation.
Supplementary Material
Primary resting CD4+ T cells were activated with PHA in the presence of IL-2 and cultured for two weeks. CD69-CD25-HLA-DR− transitional blast (eFluorlow, A) and non-blast (eFluorhigh, B) cells were sorted and stained with anti-CD45RA, CD45RO, CCR7, CD27 and CD4 antibodies. Bar graphs represent mean ± SD of two individual donors. Solid symbol: cells infected with VSV-G pseudotyped virus; open symbol: uninfected cells from the same donor. TD: terminally differentiated; CM: central memory; EM: effector memory; TM: transitional memory cells.
CD4+ T cells were activated with PHA and infected with NL4.3-EGFP that expressed enhanced green fluorescent protein (EGFP). After 12 days, cells were sorted into EGFP-negative CD69-CD25-HLA-DR− eFluorlow transitional blasts and EGFP-negative CD69-CD25-HLA-DR− eFluorhigh non-blasts. Cells were stimulated with DMSO, romidepsin (RMD) or PMA/PHA and after 24 hours, EGFP expression quantified.
A. Resting primary CD4+ T cells were cultured with various concentrations of LRAs for 48 hours and the percentage cell viability was determined by MTS assay. The mean ± SEM from four independent experiments performed in duplicates is shown. The dotted line indicates the 50% cellular cytotoxicity concentration value (CC50). B. Cytotoxicity profile of the histone lysine methyltransferase inhibitor chaetocin, which exhibited a ‘double-peak’. Table summarise the CC50 values of LRAs and the concentration used in this study.
Sorted CD69-CD25-HLA-DR− transitional blasts that were infected with pseudotyped virus containing either wild-type NL4-3 LTR or LTRs from CD4+ T cells from HIV-infected individuals were treated with various LRAs for 24 hours and luciferase quantified. The level of reactivation is expressed as a percentage of maximum stimulation using PMA/PHA. Mean ± SEM of 3 independent experiments is shown.
ACKNOWLEDGMENTS
MAM, JLA, LRG, SS, PUC, MJC, SRL, HKL conceived and designed the experiments; MAM, JLA, SA, GK, JJC, AME, WJC, HKL performed experiments; MAM, JLA, GK, JJC, JJZ analyzed the data; JCJ, JJZ, DFJP, PUC, SLR contributed reagents, materials and analysis tools; MAM, SLR, HKL wrote the manuscript and all authors reviewed and approved the manuscript.
We thank the staff of the flow cytometry unit at the Peter Doherty Institute for Infection and Immunity for assistance with sorting and analysis by flow cytometry.
Sources of Funding
SRL is an Australian National Health and Medical Research Council (NHMRC) Practitioner Fellow. This work was supported by a program grant from the Australian NHMRC; the National Institute of Allergy and Infectious Disease (NIAID), US National Institutes of Health (NIH) (Delaney AIDS Research Enterprise, DARE; U19AI096109 and RFA‐AI‐15‐029) and the American Foundation for AIDS Research.
Footnotes
Conflicts of Interest
The authors declare that they have no competing interests.
REFERENCES
- 1.Chun TW, Stuyver L, Mizell SB, Ehler LA, Mican JA, Baseler M, et al. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc Natl Acad Sci U S A 1997,94:13193–13197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Finzi D, Blankson J, Siliciano JD, Margolick JB, Chadwick K, Pierson T, et al. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat Med 1999,5:512–517. [DOI] [PubMed] [Google Scholar]
- 3.Bui JK, Sobolewski MD, Keele BF, Spindler J, Musick A, Wiegand A, et al. Proviruses with identical sequences comprise a large fraction of the replication-competent HIV reservoir. PLoS Pathog 2017,13:e1006283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hosmane NN, Kwon KJ, Bruner KM, Capoferri AA, Beg S, Rosenbloom DI, et al. Proliferation of latently infected CD4+ T cells carrying replication-competent HIV-1: Potential role in latent reservoir dynamics. J Exp Med 2017,214:959–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lorenzi JC, Cohen YZ, Cohn LB, Kreider EF, Barton JP. Paired quantitative and qualitative assessment of the replication-competent HIV-1 reservoir and comparison with integrated proviral DNA. Proc Natl Acad Sci U S A 2016,113:E7908–e7916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pitman MC, Lau JSY, McMahon JH, Lewin SR. Barriers and strategies to achieve a cure for HIV. Lancet HIV 2018,5:e317–e328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen HC, Martinez JP, Zorita E. Position effects influence HIV latency reversal. Nat Struct Mol Biol 2017,24:47–54. [DOI] [PubMed] [Google Scholar]
- 8.Lassen KG, Hebbeler AM, Bhattacharyya D, Lobritz MA, Greene WC. A flexible model of HIV-1 latency permitting evaluation of many primary CD4 T-cell reservoirs. PLoS One 2012,7:e30176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saleh S, Wightman F, Ramanayake S, Alexander M, Kumar N, Khoury G, et al. Expression and reactivation of HIV in a chemokine induced model of HIV latency in primary resting CD4+ T cells. Retrovirology 2011,8:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Swiggard WJ, Baytop C, Yu JJ, Dai J, Li C, Schretzenmair R, et al. Human immunodeficiency virus type 1 can establish latent infection in resting CD4+ T cells in the absence of activating stimuli. J Virol 2005,79:14179–14188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Evans VA, Kumar N, Filali A, Procopio FA, Yegorov O, Goulet JP, et al. Myeloid dendritic cells induce HIV-1 latency in non-proliferating CD4+ T cells. PLoS Pathog 2013,9:e1003799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bosque A, Planelles V. Studies of HIV-1 latency in an ex vivo model that uses primary central memory T cells. Methods 2011,53:54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tyagi M, Pearson RJ, Karn J. Establishment of HIV latency in primary CD4+ cells is due to epigenetic transcriptional silencing and P-TEFb restriction. J Virol 2010,84:6425–6437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marini A, Harper JM, Romerio F. An in vitro system to model the establishment and reactivation of HIV-1 latency. J Immunol 2008,181:7713–7720. [DOI] [PubMed] [Google Scholar]
- 15.Sahu GK, Lee K, Ji J, Braciale V, Baron S, Cloyd MW. A novel in vitro system to generate and study latently HIV-infected long-lived normal CD4+ T-lymphocytes. Virology 2006,355:127–137. [DOI] [PubMed] [Google Scholar]
- 16.Contreras X, Barboric M, Lenasi T, Peterlin BM. HMBA releases P-TEFb from HEXIM1 and 7SK snRNA via PI3K/Akt and activates HIV transcription. PLoS Pathog 2007,3:1459–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang HC, Xing S, Shan L, O’Connell K, Dinoso J, Shen A, et al. Small-molecule screening using a human primary cell model of HIV latency identifies compounds that reverse latency without cellular activation. J Clin Invest 2009,119:3473–3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cuevas JM, Geller R, Garijo R, LÛpez-Aldeguer J, Sanju·n R. Extremely High Mutation Rate of HIV-1 In Vivo. PLoS Biol 2015,13:e1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rezaei SD, Lu HK, Chang JJ, Rhodes A, Lewin SR, Cameron PU. The Pathway To Establishing HIV Latency Is Critical to How Latency Is Maintained and Reversed. J Virol 2018,92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saleh S, Solomon A, Wightman F, Xhilaga M, Cameron PU, Lewin SR. CCR7 ligands CCL19 and CCL21 increase permissiveness of resting memory CD4+ T cells to HIV-1 infection: a novel model of HIV-1 latency. Blood 2007,110:4161–4164. [DOI] [PubMed] [Google Scholar]
- 21.Lu HK, Gray LR, Wightman F, Ellenberg P, Khoury G, Cheng WJ, et al. Ex vivo response to histone deacetylase (HDAC) inhibitors of the HIV long terminal repeat (LTR) derived from HIV-infected patients on antiretroviral therapy. PLoS One 2014,9:e113341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gray LR, On H, Roberts E, Lu HK, Moso MA, Raison JA, et al. Toxicity and in vitro activity of HIV-1 latency-reversing agents in primary CNS cells. J Neurovirol 2016,22:455–463. [DOI] [PubMed] [Google Scholar]
- 23.Anderson JL, Mota TM, Evans VA, Kumar N, Rezaei SD, Cheong K, et al. Understanding Factors That Modulate the Establishment of HIV Latency in Resting CD4+ T-Cells In Vitro. PLoS One 2016,11:e0158778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yamamoto T, Tsunetsugu-Yokota Y, Mitsuki YY, Mizukoshi F, Tsuchiya T, Terahara K, et al. Selective transmission of R5 HIV-1 over X4 HIV-1 at the dendritic cell-T cell infectious synapse is determined by the T cell activation state. PLoS Pathog 2009,5:e1000279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johansen P, Haffner AC, Koch F, Zepter K, Erdmann I, Maloy K, et al. Direct intralymphatic injection of peptide vaccines enhances immunogenicity. Eur J Immunol 2005,35:568–574. [DOI] [PubMed] [Google Scholar]
- 26.Rush C, Mitchell T, Garside P. Efficient priming of CD4+ and CD8+ T cells by DNA vaccination depends on appropriate targeting of sufficient levels of immunologically relevant antigen to appropriate processing pathways. J Immunol 2002,169:4951–4960. [DOI] [PubMed] [Google Scholar]
- 27.Shan L, Deng K, Gao H, Xing S, Capoferri AA, Durand CM, et al. Transcriptional Reprogramming during Effector-to-Memory Transition Renders CD4+ T Cells Permissive for Latent HIV-1 Infection. Immunity 2017,47:766–775.e763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chavez L, Calvanese V, Verdin E. HIV Latency Is Established Directly and Early in Both Resting and Activated Primary CD4 T Cells. PLoS Pathog 2015,11:e1004955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jordan A, Bisgrove D, Verdin E. HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J 2003,22:1868–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jordan A, Defechereux P, Verdin E. The site of HIV-1 integration in the human genome determines basal transcriptional activity and response to Tat transactivation. EMBO J 2001,20:1726–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lewin SR, Vesanen M, Kostrikis L, Hurley A, Duran M, Zhang L, et al. Use of real-time PCR and molecular beacons to detect virus replication in human immunodeficiency virus type 1-infected individuals on prolonged effective antiretroviral therapy. J Virol 1999,73:6099–6103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Elliott JH, Wightman F, Solomon A, Ghneim K, Ahlers J, Cameron MJ, et al. Activation of HIV Transcription with Short-course Vorinostat in HIV- infected Patients on Suppressive Antiretroviral Therapy. PLoS Pathog 2014,in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pasternak AO, DeMaster LK, Kootstra NA, Reiss P, O’Doherty U, Berkhout B. Minor Contribution of Chimeric Host-HIV Readthrough Transcripts to the Level of HIV Cell-Associated gag RNA. J Virol 2016,90:1148–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pace MJ, Graf EH, Agosto LM, Mexas AM, Male F, Brady T, et al. Directly infected resting CD4+T cells can produce HIV Gag without spreading infection in a model of HIV latency. PLoS Pathog 2012,8:e1002818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Passaes CP, Bruel T, Decalf J, David A, Angin M. Ultrasensitive HIV-1 p24 Assay Detects Single Infected Cells and Differences in Reservoir Induction by Latency Reversal Agents. J Virol 2017,91:e02296–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu D, Wang W, Yoder A, Spear M, Wu Y. The HIV envelope but not VSV glycoprotein is capable of mediating HIV latent infection of resting CD4 T cells. PLoS Pathog 2009,5:e1000633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dai J, Agosto LM, Baytop C, Yu JJ, Pace MJ, Liszewski MK, et al. Human immunodeficiency virus integrates directly into naive resting CD4+ T cells but enters naive cells less efficiently than memory cells. J Virol 2009,83:4528–4537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Katzen D, Chu E, Terhost C, Leung DY, Gesner M, Miller RA, et al. Mechanisms of human T cell response to mitogens: IL 2 induces IL 2 receptor expression and proliferation but not IL 2 synthesis in PHA-stimulated T cells. J Immunol 1985,135:1840–1845. [PubMed] [Google Scholar]
- 39.Skinner AM, Chakkaramakkil Verghese S, Kurre P. Cell-cell transmission of VSV-G pseudotyped lentivector particles. PLoS One 2013,8:e74925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pan YW, Scarlett JM, Luoh TT, Kurre P. Prolonged adherence of human immunodeficiency virus-derived vector particles to hematopoietic target cells leads to secondary transduction in vitro and in vivo. J Virol 2007,81:639–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Buzon MJ, Sun H, Li C, Shaw A, Seiss K, Ouyang Z, et al. HIV-1 persistence in CD4+ T cells with stem cell-like properties. Nat Med 2014,20:139–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ho YC, Shan L, Hosmane NN, Wang J, Laskey SB, Rosenbloom DI, et al. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell 2013,155:540–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Primary resting CD4+ T cells were activated with PHA in the presence of IL-2 and cultured for two weeks. CD69-CD25-HLA-DR− transitional blast (eFluorlow, A) and non-blast (eFluorhigh, B) cells were sorted and stained with anti-CD45RA, CD45RO, CCR7, CD27 and CD4 antibodies. Bar graphs represent mean ± SD of two individual donors. Solid symbol: cells infected with VSV-G pseudotyped virus; open symbol: uninfected cells from the same donor. TD: terminally differentiated; CM: central memory; EM: effector memory; TM: transitional memory cells.
CD4+ T cells were activated with PHA and infected with NL4.3-EGFP that expressed enhanced green fluorescent protein (EGFP). After 12 days, cells were sorted into EGFP-negative CD69-CD25-HLA-DR− eFluorlow transitional blasts and EGFP-negative CD69-CD25-HLA-DR− eFluorhigh non-blasts. Cells were stimulated with DMSO, romidepsin (RMD) or PMA/PHA and after 24 hours, EGFP expression quantified.
A. Resting primary CD4+ T cells were cultured with various concentrations of LRAs for 48 hours and the percentage cell viability was determined by MTS assay. The mean ± SEM from four independent experiments performed in duplicates is shown. The dotted line indicates the 50% cellular cytotoxicity concentration value (CC50). B. Cytotoxicity profile of the histone lysine methyltransferase inhibitor chaetocin, which exhibited a ‘double-peak’. Table summarise the CC50 values of LRAs and the concentration used in this study.
Sorted CD69-CD25-HLA-DR− transitional blasts that were infected with pseudotyped virus containing either wild-type NL4-3 LTR or LTRs from CD4+ T cells from HIV-infected individuals were treated with various LRAs for 24 hours and luciferase quantified. The level of reactivation is expressed as a percentage of maximum stimulation using PMA/PHA. Mean ± SEM of 3 independent experiments is shown.