

Abstract
The stimulation of β-adrenergic receptor increases thiazide-sensitive Na-Cl cotransporter (NCC), an effect contributing to salt-sensitive hypertension by sympathetic-stimulation. We now test whether the stimulation of β-adrenergic receptor-induced activation of NCC is achieved through activating basolateral Kir4.1 in the distal convoluted tubule (DCT). Application of norepinephrine (NE) increased the basolateral 40 pS K+ channel (Kir4.1/Kir5.1 heterotetramer) in the DCT. The stimulatory effect of NE on the K+ channel was mimicked by cAMP analogue but abolished by inhibiting protein kinase A (PKA). Also, the effect of NE on the K+ channel in the DCT was recapitulated by isoproterenol (ISO) but not by α−adrenergic agonist and blocked by propranolol, suggesting that NE effect on the K+ channel was mediated by β-adrenergic receptor. The whole-cell recording shows that NE and ISO increased DCT K+ currents and shifted the K+-current (IK) reversal potential to negative range (hyperpolarization). Continuous NE perfusion (7 days) increased DCT K+ currents, hyperpolarized IK reversal potential and increased the expression of total NCC (tNCC)/ phosphorylated NCC (pNCC) but it had no significant effect on the expression of NKCC2 and ENaC-α. Renal clearance study demonstrated that NE perfusion augmented thiazide-induced urinary Na+ excretion only in WT but not in kidney-specific Kir4.1 knockout mice, suggesting that Kir4.1 is required for mediating the effect of NE on NCC. However, NE perfusion did not affect urinary K+ excretion. We conclude that the stimulation of β−adrenergic receptor activates the basolateral Kir4.1 in the DCT and that the activation of Kir4.1 is required for ΝΕ-induced stimulation of NCC.
Keywords: β-adrenoceptor, Kcnj10, sympathetic stimulation, NKCC2, salt-sensitive hypertension, ion transport
Summary
β-adrenergic receptor plays a role in stimulating NCC activity by activating the basolateral Kir4.1 activity in the DCT and that it plays a role in stimulating renal Na+ absorption.
Introduction
Increased renal sympathetic tone in the DOCA-salt rats augmented renal Na+ retention and was associated with the development of hypertension (1). Moreover, Mu et al showed that norepinephrine (NE) or isoproterenol (ISO) infusion significantly raised blood pressure during increased Na+ intake (2). The hypertension induced by sympathetic-stimulation may be partially due to high NCC activity because ISO perfusion increased the thiazide-sensitive NCC activity in the kidney, especially during high salt intake. This notion was also supported by a finding reported by Terker et al demonstrating that the mice acutely treated with NE or ISO increased NCC phosphorylation (an indication of NCC activation). Since the activation of NCC induced by sympathetic-stimulation was also observed in angiotensin II type 1 receptor (AT1R) knockout mice, this indicated that AT1R was not involved in mediating sympathetic stimulation of NCC (3). Furthermore, they have reported that chronic NE infusion caused salt-sensitive hypertension and activated NCC. Although these studies have convincingly established the relationship between sympathetic-stimulation and upregulated NCC activity, the mechanism by which NE infusion activates NCC is not completely understood. It has been reported that the sympathetic-stimulation activated with-no-lysine kinase 4 (WNK4) through epigenetic modulation and that the activation of WNK4 was responsible for the upregulation of NCC (2).
WNK4 is a Cl--sensitive kinase and the basolateral K+ channel activity in the DCT has been shown to affect WNK4 activity by altering the cell membrane potential (4;5). The DCT cell membrane potential is determined by inwardly rectifying K+ channel 4.1 (Kir4.1) encoded by Kcnj10 (6;7). Although Kir4.1 is also expressed in the basolateral membrane of thick ascending limb (TAL), connecting tubule (CNT) and cortical collecting duct (CCD) (8-14), Kir4.1 is the only type of K+ channel in the DCT and it contributes to the basolateral K conductance (6;7). Kir4.1 interacts with Kir5.1 (encoded by Kcnj16) to form a 40 pS heterotetrameric K+ channel under physiological conditions (12;15). While Kir4.1 is responsible for providing the basolateral K+ conductance, the role of Kir5.1 in the regulation of Kir4.1/Kir5.1 heterotetramer is convincingly established as an important regulatory subunit which affects the transport function in the DCT (16-18). Since this 40 pS K+ heterotetramer is the only type of K+ channel expressed in the basolateral membrane of the DCT, the Kir4.1/Kir5.1 channel plays a dominant role in determining the negativity of the membrane potential (9;19;20). The notion that Kir4.1/5.1 heterotetramer plays a key role in regulating NCC activity in the DCT was strongly suggested by the finding that the deletion of kcnj10 strikingly reduced the expression of total NCC (tNCC) and phosphorylated NCC (pNCC) (7;9). Because β-adrenergic receptor is expressed in the DCT (21), it raises the possibility that the activation of the basolateral 40 pS K+ channel in the DCT may be required for NCC activation induced by sympathetic-stimulation. Thus, the aim of the present study is to test the role of Kir4.1 in the DCT in mediating the effect of NE on NCC.
Methods
The authors declare that all supporting data and detailed methods including animal preparation, electrophysiology, western blot and renal clearance method are available within the article (and its online supplementary file).
Animals
C57BL/6 mice (either sex, 12 weeks old) and kidney-specific conditional Kcnj10−/− or Kir4.1 knockout (Ks-Kir4.1 KO) mice were used in the present study. C57/BL/6 mice were purchased from the Second Hospital of Harbin Medical University or Jackson Laboratory (Bar Harbor, ME). KS-Kir4.1 KO mice were bred at New York Medical College for the experiments and the procedure for generating Ks-Kir4.1 KO mice are described in the on-line supplemental material. The mice were fed a normal K+ diet (1% KCl) and had free access to water. In some experiments, the mice received NE perfusion for 7 days (2.5 mg/kg/day) through a subcutaneously installed osmotic mini-pump. The preparation of DCT for the patch-clamp experiments has been described in detail in the supplemental material.
To examine the effect of NE on the basolateral Kir4.1/5.1 in the DCT, we employed both single-channel and whole-cell recording techniques. For single-channel recording, the experiments were performed in both early part of the DCT (DCT1) and the late part of the DCT (DCT2). For the whole-cell recording, the experiments were performed in the DCT1 to avoid the contamination of ROMK (Kir1.1) contamination. As shown in Figs1A, ROMK channel activity is expressed only in the DCT2 but not in the DCT1. This is confirmed by the patch-clamp experiments in which TPNQ (a ROMK inhibitor)-sensitive K currents were measured at −40 mV in the DCT1 and DCT2 with the whole-cell recording. From the inspection of Fig.s1B and Fig.s1C, it is apparent that TPNQ (400 nM)-sensitive ROMK currents were only detected in the DCT2 (1030±70 pA) but not in the DCT1 (n=6). Thus, the whole-cell K+ currents measured in the DCT1 is completely composed of Kir4.1 channel activity. Morphological appearance of DCT1 is slightly different from that of DCT2. Fig.s1A shows the image of the isolated DCT demonstrating that the tubule surface of mouse DCT2 appears to be less smooth than that of DCT1. Thus, we are able to clearly identify the DCT1 and DCT2 under microscope and to patch on corresponding DCT segment.
Electrophysiology
A Narishige electrode puller (Narishige, Japan) was used to manufacture the patch-clamp pipettes from Borosilicate glass (1.7-mm OD). The resistance of the pipette was 5 MΩ (for single channel recording) or 2 MΩ (for whole cell-recording) when it was filled with solution containing (in mmol/l) 140 KCl, 1.8 MgCl2 and 10 HEPES (titrated with KOH to pH 7.4). The details for the single channel and whole-cell recordings are described in the on-line supplemental material.
Immunoblotting
Whole kidney protein extract was obtained from frozen kidney homogenized in a buffer containing (in mmol/l) 250 sucrose, 50 Tris-HCl (pH 7.5), 1 EDTA, 1 EGTA, 1 DTT supplemented with phosphatase and protease inhibitor cocktails (pH 7.6). Protein (60-80 μg) was separated on 8-12% (wt/vol) Tris-Glycine geland transferred to nitrocellulose membrane. The membranes were incubated 2 hour with 5% nonfat dry milk in TBS-T and then incubated overnight at 4ºC with anti-NCC (1:1000, No:AB3553, EMD Milipore), anti-pNCC at threonine-53 (1:1000, No: p1311-53. Phosphosolutions), anti-NKCC2 (1:1000, No:AB171747, Abcam) and anti-ENaC-α(1:1000, No:SAB5200105, sigma)antibodies. Protein Simple infrared imaging system (FlourChem R) was used to capture the images.
Material and statistical analysis
All chemicals including norepinephrine, phenylephrine, isoprotereno, epinephrine, propranolol, db-cAMP, H-89 and polyclone antibodies for ENaC-α were purchased from Sigma Chemicals (St Louis, MO). NCC, pNCC and NKCC2 were purchased from EMD Millipore, Phosphosolutions and Abcam, respectively. Data were analyzed using student t test for comparisons between two groups or one-way ANOVA for comparisons among more than 2 groups. P-values <0.05 were considered statistically significant. Data are presented as the mean ± SEM.
Results
We first used the single channel recording to examine the effect of NE on the basolateral 40 pS K+ channel, which is known as a Kir4.1 and Kir5.1 heterotetramer (9). Although a 20 pS K+ channel (Kir4.1 homotetramer) could also be detected in less than 1% patches, the 40 pS K+ channels are overwhelmingly the main type of K+ channel in the basolateral membrane of the DCT. Thus, no efforts were made to examine the effect of NE on the 20 pS K+ Channel in the basolateral membrane of the DCT in the present study. Fig. 1A is a typical single-channel recording showing the effect of NE on the K+ channel in a cell-attached patch. The application of NE at 1 nmol/l slightly increased the K+ channel activity (defined by NPo) in the DCT (control, 1.62±0.1, n=11 and 1 nmol/l NE,1.9±0.2, n=5), although it was not significant. However, 10 nmol/l NE significantly stimulated the basolateral 40 pS K+ channels and increased NPo to 2.2±0.1 (n=5) (Fig.1B). Results summarized in Fig.1B show that the effect of NE on the K+ channel was dose-dependent and that raising NE concentrations to 0.1, 1 and 10 μmol/l increased the basolateral 40 pS K+ channel activity to 2.92±0.2 (n=10), 3.23±0.2 (n=5) and 3.35±0.2 (n=4), respectively. A previous study had demonstrated that the stimulation of protein kinase A (PKA) activated the basolateral 40 pS K+ channel in the TAL (22). Since this 40 pS K+ channel in the TAL was also a Kir4.1/Kir5.1 heterotetramer (8), we suspected that PKA might also stimulate the basolateral K+ channel in the DCT. From the inspection of Fig.s2, it is apparent that db-cAMP (500 μmol/l), a membrane-permeable cAMP analogue, significantly stimulated the K+ channel in the DCT and increased NPo from 1.70±0.16 to 2.97±0.2 (n=8).
Fig 1. NE stimulates the basolateral Kir4.1 in the DCT.
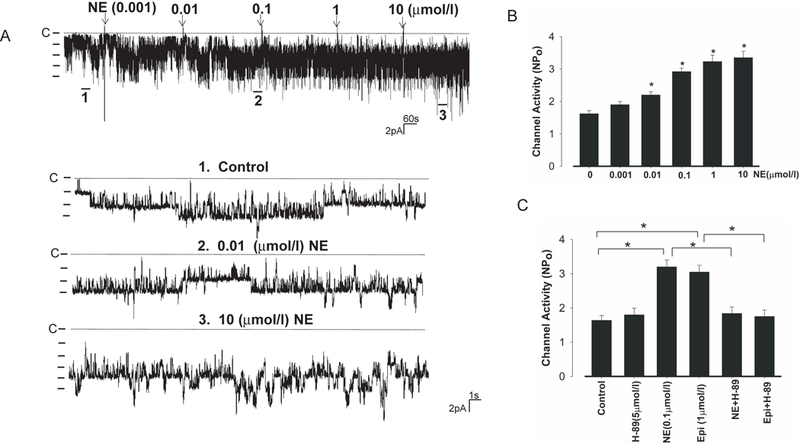
(A) A single channel recording shows the effect of NE from 1 nmol/l to 10 μmol/l on the basolateral 40 pS K+ channel in the DCT. The top trace illustrates the time course of the experiments. Three parts of the recording indicated by numbers are extended to demonstrate the fast time resolution. The experiments were performed in cell-attached patches with 140 mmol/l NaCl and 5 mmol/l KCl in the bath. The pipette holding potential was 0 mV and the channel closed level is indicated by a dotted line. (B) A bar graph summarizing the results of experiments in which the patch-clamp technique was used to test the effect of NE on the basolateral K channels at different doses. “*” indicates the significant difference in comparison to the control value. (C) A bar graph summarizing the results of experiments in which the patch-clamp technique was used to test the effect of NE (0.1μmol/l) and 1μmol epinephrine (Epi) on the basolateral K+ channel in the presence of or absence of H89 (5 μmol). “*” indicates the significant difference (p<0.05).
To examine whether the stimulatory effect of NE on the K+ channel in the DCT was also mediated by PKA, we examined the effect of 0.1 μmol/l NE on the basolateral 40 pS K+ channel in the presence of (5 μmol/l) H89, an inhibitor of PKA (Fig. s3). While the inhibition of PKA with H89 (5 μmol/l) had no significant effect on the basolateral 40 pS K+ channel activity, it abolished the effect of NE (0.1 μmol/l) on the K+ channel. Results summarized in Fig.1C show that mean NPo of the K+ channel was 1.84±0.19 (n=7) in the group treated with H89+NE and it was not significantly different from the control (1.64±0.14) or H89 alone (1.8±0.19) (Fig.1C). The notion that PKA mediates the effect of sympathetic-stimulation of the basolateral 40 pS K+ channel was also supported by the finding that inhibition of PKA abolished the effect of epinephrine (Epi) on the basolateral 40 pS K+ channel in the DCT. Fig.1C shows that application of Epi (1μmol/l) increased K+ channel activity to 3.05±0.2 (n=7) but it failed to stimulate K+ channel in the presence of H89 (NP0=1.75±0.19).
The notion that β-adrenergic receptor mediated the stimulatory effect of NE on the basolateral K+ channel in the DCT was further suggested by experiments in which the effect of NE on the basolateral K+ channel activity was examined in the DCT treated with 10 μmol/l propranolol (Fig.s4A). Although propranolol alone did not significantly affect the channel activity (control NPo,1.65±0.22; propranolol, 1.64±0.21), the application of β-adrenergic antagonist abolished the effect of NE. Results from 6 experiments are summarized in Fig.s4B showing that NE failed to stimulate the 40 pS K+ channel in the DCT treated with propranolol (NPo, 1.74±0.15). Also, 100 nmol/l isoproterenol (ISO), a specific β-adrenergic agonist, mimicked the effect of NE and stimulated the basolateral K+ channels in the DCT (Fig.2A) and increased NPo to 2.9±0.2 (n=8) (Fig.2B). In contrast, phenylephrine (5 μmol/l), a specific α-adrenergic agonist, failed to increase the K+ channel activity (Fig.2C) and NPo was 1.88±0.2 (n=5) which was not significantly different from the control (Fig.2B). Also, the stimulatory effect of ISO was blocked by propranolol (Fig. 2D) since ISO failed to increase the channel activity in the DCT treated with propranolol (NPo, 1.75±0.21) (n=6), a value not significantly different from the control (1.65±0.22) and propranolol (1.64±0.21). Thus, the effect of NE on the basolateral 40 pS K+ channel in the DCT was mediated by β-adrenergic receptor and cAMP-dependent pathway.
Fig.2. β−Adrenergic receptor agonist stimulates the basolateral Kir4.1 in the DCT.
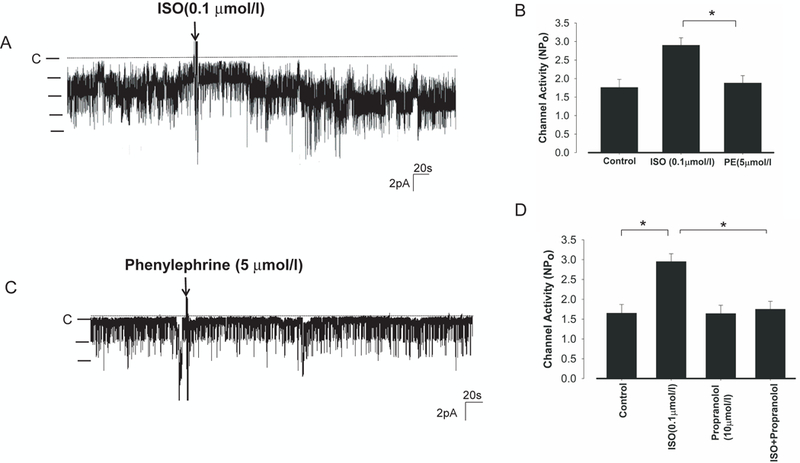
(A) A recording showing the effect of 0.1 μmol/l isoproterenol (ISO) on the basolateral 40 pS K+ channel in the DCT. The experiments were performed in cell-attached patches with 140 mmol/l NaCl and 5 mmol/l KCl. The pipette holding potential was 0 mV and the channel closed level is indicated by a dotted line. (B) A bar graph summarizes the results of experiments in which the effect of ISO or PE on the K+ channel activity was examined. “*” indicates the significant difference (p<0.05). (C) A recording showing the effect of 5 μmol/l phenylephrine (PE) on the basolateral 40 pS K+ channel in the DCT. (D) A bar graph summarizes the results of experiments in which effect of ISO on the K+ channel activity was examined in the presence or in the absence of propranolol (10 μmol/l). “*” indicates the significant difference (p<0.05).
NE-induced activation of the 40 pS K+ channel is expected to increase the basolateral K+ conductance in the DCT. Thus, we used whole-cell recording technique to measure the K+ currents in the early portion of the DCT (DCT1). As mentioned above, the reason to measure K+ conductance in the DCT1 is due to the absence of ROMK channel activity in the apical membrane of the DCT1 (Fig.s1). Moreover, the Kir4.1 is the only type of K+ channel providing the basolateral K+ conductance along the whole DCT (6;7). Thus, the whole-cell K+ conductance would be identical to Kir4.1 activity. Since the deletion of Kir4.1 inhibited the expression of NCC along whole DCT (7), this indicates that Kir4.1 activity regulates NCC activity in the whole DCT segment. Thus, the observation made in the DCT1 about Kir4.1 channel should also apply to the DCT2. Fig. 3A is a set of recordings showing the effect of 0.1 μmol/l NE or ISO on the whole-cell K+ currents in the DCT using step protocol (clamped from −60 to 60 mV at 20 mV step). It is apparent that NE (n=8) and ISO (n=8) significantly increased the whole-cell K+ conductance (at −60mV) from 1010±32 pA to 1440±40 pA (NE) or 1645±113 pA (ISO), respectively (Fig.3C). Not only NE acutely increased the basolateral K+ conductance in the DCT, but also it chronically increased the whole-cell K+ conductance when the mice were infused with NE (2.5 mg/kg/day) by an osmotic pump (7 days). Fig.3B is a recording showing the Ba2+-sensitive K+ currents measured with whole-cell-recording in the DCT1 of mice treated with NE for 7 days and NE infusion increased K+ currents to 1890±95pA at −60 mV (n=6) (Fig.3B).
Fig.3. Sympathetic agonists increase the DCT K+ currents.
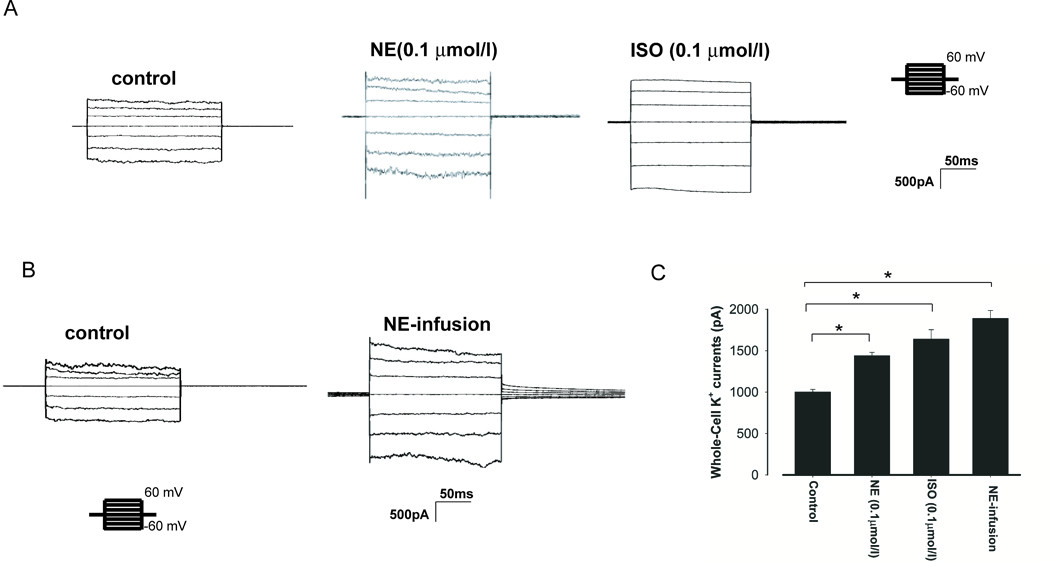
(A) A whole-cell recording showing Ba2+ -sensitive K+ currents in the DCT treated with NE or ISO, respectively. The K+ currents were measured with a step protocol from −60 to 60 mV using symmetrical 140 mmol/l KCl solution in the bath and pipette. (B) A whole-cell recording showing Ba2+ -sensitive K+ currents in the DCT of the mice treated with vehicle (control) or NE infusion by an osmotic pump for seven days. (C) A bar graph summarizes the results of experiments in which Ba2+-sensitive K+ currents of the DCT were measured at −60 mV. “*” indicates the significant difference (p<0.05).
Since the basolateral K+ channel plays a key role in determining the negativity of the DCT membrane, K+ channel activation induced by sympathetic-stimulation should increase the negativity of the membrane (hyperpolarization). Thus, we used perforated whole-cell recording to measure K+ current (IK) reversal potential in the DCT (an index of membrane potential). Fig. 4A is a trace showing the effect of 0.1 μmol/l NE or ISO on the IK reversal potential and Fig. 4B is a trace showing the effect of NE-infusion for 7 days on the IK reversal potential. It is apparent that sympathetic-stimulation increased the negativity of the IK reversal potential. Results from 6 experiments summarized in Fig. 4C show that NE (0.1 μmol/l), ISO (0.1 μmol/l), and NE-infusion hyperpolarized the DCT membrane from the control (63±1) to 70±1 (NE), 72±1 (ISO) and 73±2 mV (NE infusion), respectively. Previous studies have demonstrated that a hyperpolarization in the DCT membrane was associated with the stimulation of NCC activity by activating Cl--sensitive WNK (5). Thus, we examined the effect of NE-infusion on the expression of tNCC and pNCC. From the inspection of Fig. 4D, it is apparent that NE infusion significantly increased the expression of pNCC (170± 9%) and tNCC (150± 7%) (n=6). The effect of NE-infusion on NCC was specific because it had no significant effect on the expression of NKCC2 (n=6) in the mice treated with NE for 30 min (peritoneal injection) (Fig. s5A) or for 7-days (Fig.s5B). Also, neither acute NE treatment (30 min) (Fig.s6A) nor 7-days NE infusion (Fig.s6B) has significantly affect the expression of full-length ENaC-α or cleaved ENaC-α (n=6), although it had a trend to slightly increase their expression.
Fig.4. Sympathetic agonists hyperpolarize DCT membrane.
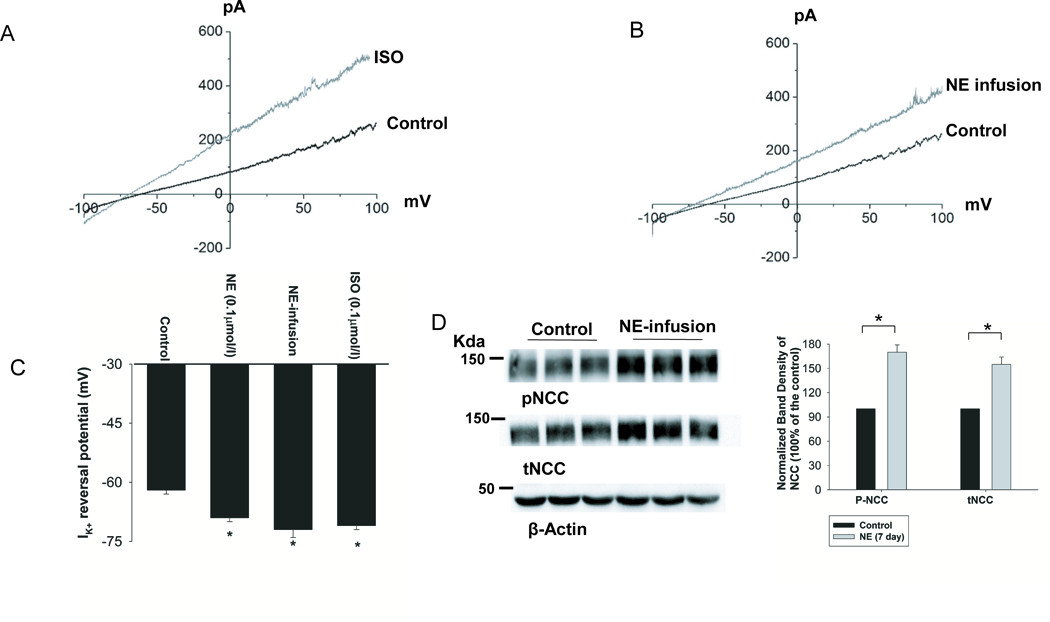
(A) K+-current (IK) reversal potential of a DCT cell measured with perforated whole-cell recording in the isolated tubule treated with 0.1 μmol/l ISO. (B) IK reversal potential of DCT measured with perforated whole-cell recording in the mice treated with NE through an osmatic pump for three days. The bath solution contains (in mmol/L) 140 NaCl and 5 KCl while the pipette solution has 140 KCl. (C) A bar graph summarizes the results of above experiments. “*” means that the difference is significant in comparison to the control value (P<0.05). (D) Western blots showing the effect of NE infusion on the expression of phospho-NCC (pNCC) and total NCC (tNCC). The normalized band density is shown in the right panel. “*” indicates the significant difference (p<0.05).
The notion that NE-infusion stimulates NCC activity was also supported by renal clearance studies in which the effect of NE-infusion on HCTZ-induced natriuresis was examined. Fig.5A is a line graph showing the results of each experiment in which urinary Na+ excretion (ENa) was measured before and after a single dose of HCTZ (25 mg/kg BW) in WT mice with or without NE infusion for 7 days. A bar graph summarizing the results of 5 experiments shows that NE-infusion significantly increased HCTZ-induced ENa (from 0.30±0.05 to 2.7±0.13 μEqmol/l per min per 100g BW). In contrast, HCTZ increased ENa in the untreated mice from 0.67±0.06 to 2.31±0.10 μEqmol/l per min per 100g BW (Fig.5B). Thus, HCTZ-induced ENa was significantly larger in the NE-infused mice than those in untreated mice. However, NE-induced stimulation of ENa was completely absent in Ks-Kcnj10−/− mice which had renal phenotypes of human EAST/Sesame syndrome including metabolic alkalosis, hypokalemia and hypotension (7). In addition, the Ks-Kir4.1 KO mice had a high basal level of renal Na+ excretion due to the inhibition of NCC (7). In untreated Ks-Kcnj10−/− mice, ENa was 1.22±0.09 (basal) and 1.43±0.11 (HCTZ) μEqmol/l per min per 100g BW (n=5), respectively (Fig.5B). In NE-infused Ks-Kcnj10−/− mice, ENa was 1.14±0.2 (basal) and 1.32±0.2 (HCTZ) μEqmol/l per min per 100g BW (n=5), respectively. Also, plasma Na+ concentrations were unchanged in NE-treated or untreated WT and Ks- Kcnj10−/− mice (Fig.s7). Thus, the deletion of Kir4.1 abolished NE-induced stimulation of NCC activity.
Fig.5. NE-induced stimulation of NCC activity depends on the presence of Kir4.1.
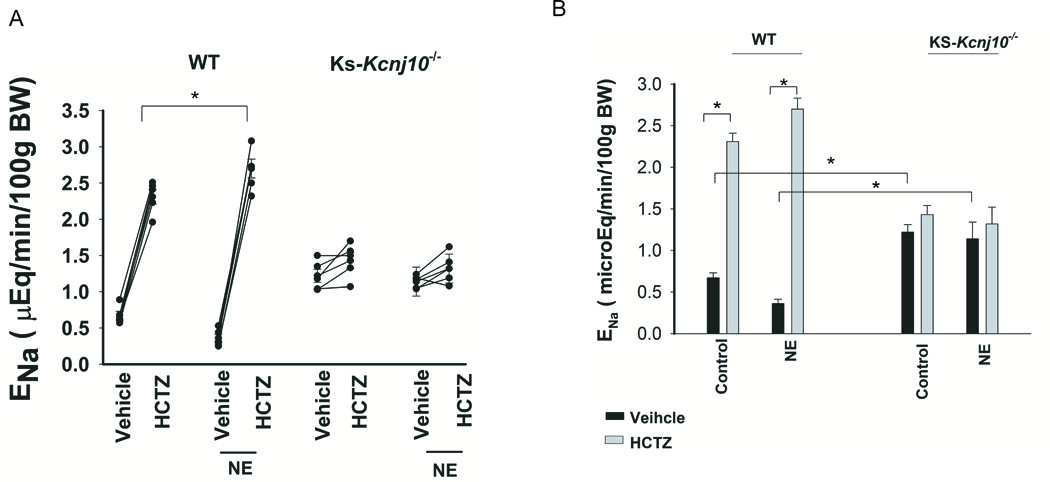
(A) A line graph shows the results of each experiment in which urinary sodium excretion (ENa) was measured before and after a single dose of HCTZ (25 mg/kg BW) in WT mice and KS-Kir4.1 KO mice with or without NE infusion for 7 days. “*” indicates the significant difference between two groups (p<0.05). (B) A bar graph summarizes the results of experiments in which HCTZ-induced natriuretic effect was examined between control and NE treated WT or KS-Kir4.1 KO mice. NE was continuously infused for 7 days (2.5 mg/kg/day) through a subcutaneously installed osmotic mini-pump. “*” indicates the significant difference (p<0.05).
We have also examined the effect of NE-infusion on urinary K+ excretion (EK) in WT and in Ks-Kcnj10−/− mice using renal clearance method. Fig. 6A is a line graph showing the results of each experiment in which EK was measured before and after a single dose of HCTZ (25 mg/kg BW) in WT mice and Ks-Kcnj10−/− mice with or without NE treatment for 7 days. Fig.6B is a bar graph summarizing the results of 5 similar experiments. Although NE-infusion stimulated NCC activity, NE infusion had no significant effect on EK in WT or in Ks-Kcnj10−/− mice. In untreated WT mice, EK was 0.61±0.04 (basal) and 1.16±0.04 (HCTZ) μEqmol/l per min per 100g BW. These values were not significantly different from the NE-treated mice and EK was 0.54±0.03 (basal) and 1.14±0.04 (HCTZ) μEqmol/l per min per 100g BW. Also, plasma K+ levels were similar between untreated and NE-treated mice (Fig. s7). As reported previously, Ks-Kcnj10−/− mice were K+ wasting (7), basal EK was 1.02±0.05 (untreated) and 0.96±0.03 μEqmol/l per min per 100g BW (NE-infused mice). Also, HCTZ did not significantly alter EK in untreated (1.07±0.05) or NE-treated Ks-Kcnj10−/−mice (0.98±0.04 μEqmol/l per min per 100g BW). In addition, untreated and NE-treated Ks-Kcnj10−/− mice were similarly hypokalemic (Fig.s7). Thus, we confirm that NE stimulates NCC through β-adrenergic receptor and that Kir4.1 activity is required for NE-induced stimulation of NCC.
Figure 6. NE infusion does not affect urinary K excretion.
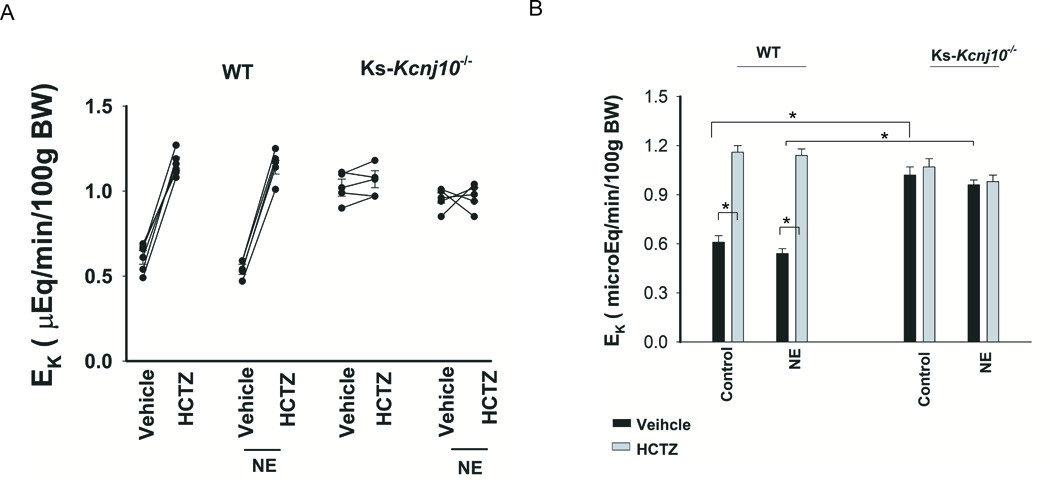
(A) A line graph shows the results of each experiment in which urinary potassium excretion (EK) was measured before and after a single dose of HCTZ (25 mg/kg BW) in WT mice and KS-Kir4.1 KO mice with or without NE treatment for 7 days. (B) A bar graph summarizes the results of experiments in which HCTZ-induced natriuretic effect was examined between control and NE treated WT or KS-Kir4.1 KO mice. NE was continuously infused for 7 days (2.5 mg/kg/day) through a subcutaneously installed osmotic mini-pump. “*” indicates the significant difference (p<0.05).
Discussion
The main finding of the present study is that NE stimulates the basolateral 40 pS K+ channels and NCC in the DCT. Three lines of evidence indicate that the effect of NE on the basolateral 40 pS K+ channel was mediated by stimulating β-adrenoceptor: 1) Specific β-adrenergic receptor agonist but not α-adrenergic receptor agonist mimicked the effect of NE on the K+ channel in the DCT; 2) The stimulatory effect of NE on the K+ channel was blocked by propranolol; and 3) NE-induced stimulation of the basolateral 40 pS K+ channel was mimicked by membrane-permeable cAMP analogue and inhibited by H89. Thus, our present finding strongly suggests the role of β-adrenoceptor in the stimulation of the basolateral 40 pS K+ channel in the DCT.
Since this 40 pS K+ channel is the only type of K+ channel expressed in the basolateral membrane of the DCT (7), it is conceivable that NE-mediated stimulation of the 40 pS K+ channel activity should have a significant effect on the basolateral K+ conductance and the membrane potential. Indeed, we observed that NE or ISO treatment not only increased the basolateral K+ conductance but also hyperpolarized the DCT membrane. This suggests that the β-adrenoceptor pathway is involved in tonic stimulation of the membrane potential in the DCT. It is well documented that the basolateral 40 pS K+ channel in the DCT is composed of Kir4.1 and Kir5.1 (encoded by Kcnj16) and that Kir4.1 is a pore-forming component for the Kir4.1/Kir5.1 heterotetramer (7;12;15). Thus, the present study indicates that β-adrenoceptor in the DCT plays a role in stimulating Kir4.1/Kir5.1 activity.
Not only stimulating Kir4.1 in the DCT, NE has been shown to increase NCC activity (2;3). This finding was also confirmed by our present experiments demonstrating that NE-treatment significantly increased the expression of tNCC and pNCC. Furthermore, Mu et al have shown that the effect of NE on NCC was absent in the mice treated with propranolol, suggesting that the effect of NE on NCC was mediated by stimulating the β-adrenoceptor pathway (2). In addition, Terker et al have shown that the stimulatory effect of NE on NCC was diminished in kidney-specific oxidative stress-response kinase 1 (OxSR1) knockout mice, suggesting the role of OxSR1 in mediating the effect of NE on NCC (3). Finally, Mu et al have shown that sympathetic activation during increased dietary Na+ intake increased WNK4 activity through histone modification (2). Thus, these studies have convincingly suggested that sympathetic-stimulation induced activation of NCC is mediated by WNK4-OxSR pathways.
Two lines of evidence suggest the possibility that Kir4.1/Kir5.1 activity is also essential for the effect of NE on NCC, although we could not completely rule out the direct effect of NE on NCC. First, NE-induced stimulation of NCC activity was absent in Ks-Kcnj10−/− mice. Second, NE-induced stimulation of NCC was closely correlated with NE-induced hyperpolarization of DCT membrane. Thus, we speculate that NE-induced stimulation of NCC activity may be, at least in part, the result of NE-induced activation of basolateral Kir4.1/Kir5.1 and hyperpolarization. A large body of evidence has suggested the membrane potential in the DCT plays an important role in the regulation of NCC activity such that an increase in the membrane negativity (hyperpolarization) stimulates, whereas a decrease in membrane negativity (depolarization) inhibits NCC activity(5;7;9). The mechanism by which Kir4.1/Kir5.1 activity regulates NCC activity in the DCT is possibly mediated by a Cl- -sensitive WNK-SPAK (ste20 proline-alanine rich kinase)/OxRS pathway because the membrane potential of the DCT could affect the intracellular Cl- levels. (4;5;23). We have previously demonstrated that the deletion of Kir4.1 in the DCT depolarized the basolateral membrane (7;9). Because the DCT membrane potential provides the driving force for Cl- exit across the basolateral membrane through ClC-Kb (24;25), alteration of Kir4.1/Kir5.1 activity should affect Cl- exit thereby changing intracellular Cl- (Cli-) concentrations and altering WNK and SPAK or OxSR activity (4). Indeed, our previous study demonstrated that the deletion of Kir4.1 in the kidney inhibited the SPAK activity (7). Since WNK and SPAK/OxSR are required for controlling NCC activity (26-32), the stimulation of WNK should increase while the inhibition of WNK should decrease NCC activity. Thus, NE-induced stimulation of the basolateral Kir4.1/Kir5.1 should decrease the intracellular Cl- levels thereby activating WNK-SPAK/OxSR pathway. Although it is possible that the sympathetic stimulation may have a direct effect on WNK4 activity through histone modification (2), the NE-induced stimulation of Kir4.1/Kir5.1 should synergize the effect of NE on WNK4.
NE-induced stimulation of NCC is expected to decrease Na+ and volume delivery to the distal nephron segment thereby affecting K+ excretion. However, neither renal K+ excretion rate nor plasma K+ level was changed in the mice treated with NE-infusion for 7 days. We speculate that ENaC activity in the aldosterone-sensitive distal nephron may also be upregulated in NE-treated mice thereby enhancing K+ secretion. Although the expression of ENaC-α was not statistically different in comparison to untreated mice, it is possible that NE-treatment may functionally activate ENaC by PKA. It has been shown that NCC activity is functionally coupled with ENaC by PKA (33), PKA may stimulate ENaC thereby enhancing renal K+ excretion. Further experiments were needed to check this notion. Also, NE-infusion was expected to stimulate β-adrenoceptor in the skeletal muscle thereby stimulating K+ uptake into the cells and the K+ movement should play a role in maintaining plasma K+ level in the normal range in NE-treated mice (34).
Perspective
The main finding of the present study is to demonstrate that the stimulation of β-adrenergic receptor activates the basolateral Kir4.1/Kir5.1 in the DCT and that the activation of Kir4.1 is essential for sympathetic stimulation of NCC activity. Thus, the basolateral Kir4.1 in the DCT serves as a gateway for sympathetic-stimulation-induced activation of NCC. It is well documented that sympathetic-stimulation is one important factor causing salt-sensitive hypertension and that NE-induced stimulation of NCC contributes the salt-sensitive hypertension by sympathetic-stimulation (2;3;35). Thus, our study provided an integrated mechanism by which sympathetic stimulation increases kidney Na+ absorption. In addition, our present finding has further suggested the role in Kir4.1/Kir5.1 in mediating the effects of a variety of factors/hormones on NCC and transepithelial Na+ transport in the DCT. For instance, our previous studies have demonstrated stimulation of angiotensin type II receptor inhibits NCC by suppressing Kir4.1/Kir5.1 in the DCT (36). These data suggest that Kir4.1/Kir5.1 in the DCT is a key target for controlling transepithelial Na+ transport. In conclusion, the stimulation of β-adrenoceptor in the DCT stimulates Kir4.1/Kir5.1 and sympathetic-stimulation of basolateral Kir4.1 is essential for the activation of NCC by NE.
Supplementary Material
Novelty and Significance:
1). What is New?
Norepinephrine (NE) stimulates the basolateral Kir4.1 in the DCT by activating β-adrenergic receptor and hyperpolarizes DCT membrane.
NE application increases renal Na+ absorption and stimulates NCC activity. This anti-natriuresis effect of NE depends on the presence of Kir4.1 in the DCT.
2). What is relevant?
The finding that NE stimulates Kir4.1 through β-adrenoceptor is relevant for understanding an integrated mechanism of sympathetic-stimulation of NCC.
The finding that NE stimulates NCC and Kir4.1 in the DCT is highly relevant for understanding the mechanism of sympathetic stimulation-induced salt-sensitive hypertension.
Acknowledgment:
Authors thank Ms.Gail Anderson for her assistance in preparing the manuscript.
Source of Funding:
The work is supported by Chinese National Natural Science Foundation #31671196 (RMG), postgraduate innovation fund #YJSCX2015-11HYD(XPD), NIH grant DK 54983 (WHW) and DK 115366 (LDH).
Footnotes
Conflict(s) of Interest/Disclosure:
None.
References
- (1).Katholi RE, Natulan AJ, Oparil S. Importance of renal sympathetic tone in the development of DOCA-salt hypertension in the rat. Hypertension 1980; 2:266–273. [DOI] [PubMed] [Google Scholar]
- (2).Mu S, Shimosawa T, Ogura S, Wang H, Uetake Y, Kawakami-Mori F, Marumo T, Yatomi Y, Geller DS, Tanaka H, Fujita T. Epigenetic modulation of the renal β−adrenergic−WNK4 pathway in salt-sensitive hypertension. Nature Medicine 2011; 17:573–580. [DOI] [PubMed] [Google Scholar]
- (3).Terker AS, Yang CL, McCormick JA, Meermeier NP, Rogers SL, Grossmann S, Trompf K, Delpire E, Loffing J, Ellison DH. Sympathetic Stimulation of Thiazide-Sensitive Sodium Chloride Cotransport in the Generation of Salt-Sensitive Hypertension. Hypertension 2014; 64(1):178–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).piala AT, Moon TM, Akella R, He HX, Cobb MH, Goldsmith EJ. Chloride sensing by WNK1 involves inhibition of autophosphosphorylation. Sciencesignaling 2014; 7(324):ra41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Terker A-S, Zhang C, McCormick J-A, Lazelle R-A, Zhang C, Meermeier N-P, Siler D-A, Park H-J, Fu Y, Cohen D-M, Weinstein A-M, Wang WH, Yang CL, Ellison D-H. Potassium Modulates Electrolyte Balance and Blood Pressure through Effects on Distal Cell Voltage and Chloride. Cell Metabolism 2015; 21(1):39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Wang MX, Cuevas-Gallardo C, Su XT, Wu P, Gao Z-X, Lin DH, McCormick JA, Yang CL, Wang WH, Ellison DH. Potassium (K+) intake modulates NCC activity via the K+ channel. Kir4.1. Kid Int 2018; 93(4):893–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Cuevas CA, Su XT, Wang MX, Terker AS, Lin DH, McCormick JA, Yang C-L, Ellison DH, Wang WH. Potassium sensing by renal distal tubules requires Kir4.1. J Am Soc Nephrol 2017; 28:1814–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Zhang C, Wang L, Su XT, Lin DH, Wang WH. KCNJ10 (Kir4.1) is expressed in the basolateral membrane of the cortical thick ascending limb. American Journal of Physiology - Renal Physiology 2015; 308:F1288–F1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Zhang C, Wang L, Zhang J, Su X-T, Lin DH, Scholl UI, Giebisch G, Lifton RP, Wang WH. KCNJ10 determines the expression of the apical Na-Cl cotransporter (NCC) in the early distal convoluted tubule (DCT1). Proc Natl Acad Sci USA 2014; 111:11864–11869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Su XT, Zhang C, Wang L, Gu R, Lin DH, Wang WH. The disruption of KCNJ10 (Kir4.1) stimulates the expression of ENaC in the collecting duct. American Journal of Physiology - Renal Physiology 2016; 310:F985–F993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Lachheb S, Cluzeaud F, Bens M, Genete M, Hibino H, Lourdel S, Kurachi Y, Vandewalle A, Teulon J, Paulais M. Kir4.1/Kir5.1 channel forms the major K+ channel in the basolateral membrane of mouse renal collecting duct principal cells. AJP - Renal Physiology 2008; 294(6):F1398–F1407. [DOI] [PubMed] [Google Scholar]
- (12).Lourdel S, Paulais M, Cluzeaud F, Bens M, Tanemoto M, Kurachi Y, Vandewalle A, Teulon J. An inward rectifier K+ channel at the basolateral membrane of the mouse distal convoluted tubule: similarities with Kir4-Kir5.1 heteromeric channels. J Physiol 2002; 538(Pt 2):391–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Zaika OL, Mamenko M, Palygin O, Boukelmoune N, Staruschenko A, Pochynyuk O. Direct inhibition of basolateral Kir4.1/5.1 and Kir4.1 channels in the cortical collecting duct by dopamine. American Journal of Physiology - Renal Physiology 2013; 305(9):F1277–F1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Huang C, Sindic A, Hill CE, Hujer KM, Chan KW, Sassen M, Wu Z, Kurachi Y, Nielsen S, Romero MF, Miller RT. Interaction of the Ca2+-sensing receptor with the inwardly rectifying potassium channels Kir4.1 and Kir4.2 results in inhibition of channel function. AJP - Renal Physiology 2007; 292(3):F1073–F1081. [DOI] [PubMed] [Google Scholar]
- (15).Pessia M, Tucker SJ, Lee K, Bond CT, Adelman JP. Subunit positional effects revealed by novel heteromeric inwardly rectifying K+ channels. EMBO J 1996; 15(12):2980–2987. [PMC free article] [PubMed] [Google Scholar]
- (16).Palygin O, Levchenko V, Ilatovskaya DV, Pavlov TS, Pochynyuk OM, Jacob HJ, Geurts AM, Hodges MR, Staruschenko A. Essential role of Kir5.1 channels in renal salt handling and blood pressure control. JCI Insight 2017; 2(18) e9233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Wang MX, Su XT, Wu P, Gao ZX, Wang WH, Staub O, Lin DH. Kir5.1 regulates Nedd4–2-mediated ubiquitination of Kir4.1 in distal nephron. American Journal of Physiology-Renal Physiology 2018. Published on line. DOI: 10.1152/ajprenal.00059.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Paulais M, Bloch-Faure M, Picard N, Jacques T, Ramakrishnan SK, Keck M, Sohet F, Eladari D, Houillier P, Lourdel Sp, Teulon J, Tucker SJ. Renal phenotype in mice lacking the Kir5.1 (Kcnj16) K+ channel subunit contrasts with that observed in SeSAME/EAST syndrome. Proceedings of the National Academy of Sciences 2011; 108(25):10361–10366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Zhang C, Wang L, Thomas S, Wang K, Lin DH, Rinehart J, Wang WH. Src-family protein tyrosine kinase regulates the basolateral K channel in the distal convoluted tubule (DCT) by phosphorylation of KCNJ10. Journal of Biological Chemistry 2013; 288:26135–26146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Wang L, Zhang C, Su X, Lin DH, Wang W. Caveolin-1 Deficiency Inhibits the Basolateral K+ Channels in the Distal Convoluted Tubule and Impairs Renal K+ and Mg2+ Transport. Journal of the American Society of nephrology 2015; 26:2678–2690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Summers R, Stephenson JA, Kuhar MJ. LOcalization of beta adreceptor subtypes in rat kidney by light microscopic autoradiography. Journal of Pharmacology And Experimental Therapeutics 1985; 232:561–569. [PubMed] [Google Scholar]
- (22).Fan L, Wang X, Zhang D, Duan X, Zhao C, Zu M, Meng X, Zhang C, Su XT, Wang MX, Wang WH, Gu R. Vasopressin-induced stimulation of the Na+-activated K+ channels is responsible for maintaining the basolateral K+ conductance of the thick ascending limb (TAL) in EAST/SeSAME syndrome. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 2015; 1852(11):2554–2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Bazua-Valenti S, Chavez-Canales M, Rojas-Vega L, Gonzalez-Rodriguez X, Vazquez N, Rodriguez-Gama A, Argaiz ER, Melo Z, Plata C, Ellison DH, Garcia-Valdes J, Hadchouel J, Gamba G. The Effect of WNK4 on the Na+Cl Cotransporter Is Modulated by Intracellular Chloride. J Am Soc Nephrol 2014; 26:1781–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Nissant A, Lourdel S, Baillet S, Paulais M, Marvao P, Teulon J, Imbert-Teboul M. Heterogeneous distribution of chloride channels along the distal convoluted tubule probed by single-cell RT-PCR and patch clamp. AJP - Renal Physiology 2004; 287(6):F1233–F1243. [DOI] [PubMed] [Google Scholar]
- (25).Hennings JC, Andrini O, Picard N, Paulais M, Huebner AK, Cayuqueo IKL, Bignon Y, Keck M, Cornire N, Boehm D, Jentsch TJ, Chambrey R, Teulon J, Hubner CA, Eladari D. The ClC-K2 Chloride Channel Is Critical for Salt Handling in the Distal Nephron. J Am Soc Nephrol 2017; 28(1):209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Subramanya AR, Yang CL, McCormick JA, Ellison DH. WNK kinases regulate sodium chloride and potassium transport by the aldosterone-sensitive distal nephron. Kidney Int 2006; 70(4):630–634. [DOI] [PubMed] [Google Scholar]
- (27).McCormick JA, Mutig K, Nelson JH, Saritas T, Hoorn EJ, Yang C-L, Rogers S, Curry J, Delpire E, Bachmann S, Ellison DH. A SPAK isoform switch modulates renal salt transport and blood pressure. Cell Metabolism 2011; 14:352–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Chiga M, Rai T, Yang SS, Ohta A, Takizawa T, Sasaki S, Uchida S. Dietary salt regulates the phosphorylation of OSR1//SPAK kinases and the sodium chloride cotransporter through aldosterone. Kidney Int 2008; 74(11):1403–1409. [DOI] [PubMed] [Google Scholar]
- (29).Bazua-Valenti S, Gamba G. Revisiting the NaCl cotransporter regulation by with-no-lysine kinases. American Journal of Physiology - Cell Physiology 2015; 308(10):C779–C791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Liu Z, Xie J, Wu T, Truong T, Auchus RJ, Huang CL. Downregulation of NCC and NKCC2 cotransporters by kidney-specific WNK1 revealed by gene disruption and transgenic mouse models. Human Molecular Genetics 2011; 20(5):855–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Grimm PR, Taneja TK, Liu J, Coleman R, Chen YY, Delpire E, Wade JB, Welling PA. SPAK Isoforms and OSR1 Regulate Sodium-Chloride Co-transporters in a Nephron-specific Manner. J Biol Chem 2012; 287(45):37673–37690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Ponce-Coria J, Markadieu N, Austin TM, Flammang L, Rios K, Welling PA, Delpire E. A Novel Ste20-related Proline/Alanine-rich Kinase (SPAK)-independent Pathway Involving Calcium-binding Protein 39 (Cab39) and Serine Threonine Kinase with No Lysine Member 4 (WNK4) in the Activation of Na-K-Cl Cotransporters. J Biol Chem 2014; 289(25):17680–17688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Mistry AC, Wynne BM, Yu L, Tomilin V, Yue Q, Zhou Y, Al-Khalili O, Mallick R, Cai H, Alli AA, Ko B, Mattheyses A, Bao HF, Pochynyuk O, Theilig F, Eaton DC, Hoover RS. The sodium chloride cotransporter (NCC) and epithelial sodium channel (ENaC) associate. Biochem J 2016; 473(19):3237–3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Palmer BF. Regulation of Potassium Homeostasis. Clinical Journal of the American Society of Nephrology 2015. 10:1050–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Foss JD, Fink GD, Osborn JW. Reversal of Genetic Salt-Sensitive Hypertension by Targeted Sympathetic Ablation. Hypertension 2013; 61(4):806–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Wu P, Gao Z-X, Duan X, Su X- T, Wang MX, Lin D- H, Gu RM, Wang WH. AT2R-mediated regulation of Na-Cl cotransporter (NCC) and renal K excretion depends on the K channel, Kir4.1. Hypertension 2018; 71:622–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.