

Abstract
Elevated low-density lipoprotein cholesterol (LDL-C) is one of the major contributors to cardiovascular heart disease (CHD), the leading cause of death worldwide. Due to severe side effects of statins, alternative treatment strategies are required for statin-intolerant patients. Monoclonal antibodies (mAbs) targeting proprotein convertase subtilisin/kexin type 9 (PCSK9) have shown great efficacy in LDL-C reduction. Limitations for this approach include the need for multiple injections as well as increased costs associated with patient management. Here, we engineered a DNA-encoded mAb (DMAb) targeting PCSK9 (daPCSK9), as an alternative approach to protein-based lipid-lowering therapeutics, and we characterized its expression and activity. A single intramuscular administration of mouse daPCSK9 generated expression in vivo for over 42 days that corresponded with a substantial decrease of 28.6% in non-high-density lipoprotein cholesterol (non-HDL-C) and 10.3% in total cholesterol by day 7 in wild-type mice. Repeated administrations of the DMAb plasmid led to increasing expression, with DMAb levels of 7.5 μg/mL at day 62. daPCSK9 therapeutics may provide a novel, simple, less frequent, cost-effective approach to reducing LDL-C, either as a stand-alone therapy or in combination with other LDL-lowering therapeutics for synergistic effect.
Keywords: gene therapy, cardiovascular disease, PCSK9 inhibitor
Graphical Abstract
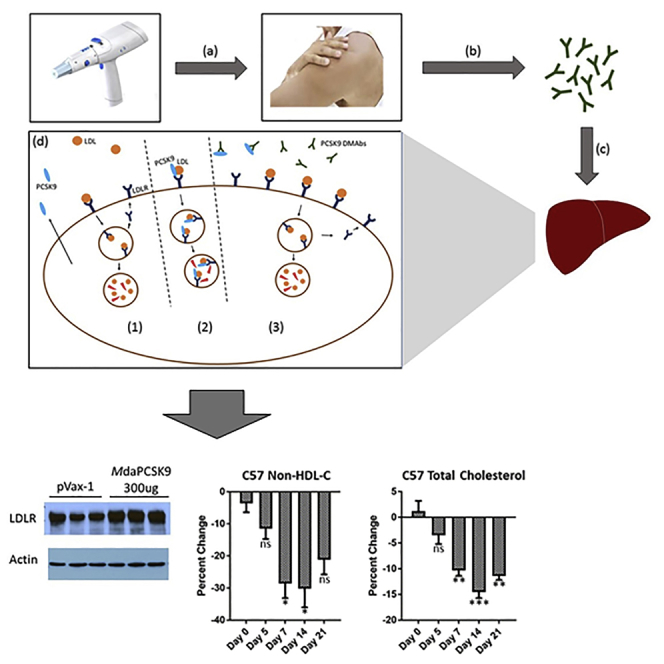
PCSK9 inhibitors have emerged as an important therapeutic approach for reducing LDL cholesterol. Recombinant anti-PCSK9 monoclonal antibodies have shown promise, however, recombinant antibody production can be challenging and costly. Khoshnejad et al. developed a DNA-encoded anti-PCSK9 mAb (DMAb) platform for intramuscular production of PCSK9 mAbs, demonstrating important and direct reduction in LDL cholesterol from a single administration.
Introduction
Cardiovascular disease is the most common cause of death, accounting for more than 800,000 deaths in the United States and 17.3 million deaths globally.1 Elevated low-density lipoprotein cholesterol (LDL-C) has been found to be one of the major contributors to atherosclerosis and cardiovascular heart disease (CHD).2, 3, 4 Familial hypercholesterolemia (FH) is an autosomal dominant disorder characterized by elevated total cholesterol that is primarily LDL-C. It has been associated with premature cardiovascular disease, often resulting in death at a young age. Lipid-lowering medications and LDL apheresis, especially for statin-intolerant patients, have been used for the management of familial hypercholesterolemia.5, 6 Better pharmacological therapeutics are needed to achieve and maintain the desired target LDL-C levels.
Proprotein convertases are secretory mammalian serine proteases that induce site-specific proteolytic cleavage in proteins. This is a form of irreversible post-translational modification, which can lead to the activation or inactivation of proteins, such as growth factors, receptors, hormones, transcription factors, bacterial toxins, and viral glycoproteins. They have major implications in health and for disease due to their broad biological roles.7 Several modulators and inhibitors of proprotein convertases are being developed for hypercholesterolemia (proprotein convertase subtilisin/kexin type 9 [PCSK9] inhibitors8, 9, 10, 11, 12, 13, 14, 15), viral infections (Furin, PACE4, and SKI-1 inhibitors16, 17, 18, 19, 20, 21, 22, 23, 24), and cancer immunotherapy (Furin and PACE4 inhibitors25, 26, 27, 28, 29, 30), as well as for use as a non-steroidal male contraceptive (PC4 inhibitors31, 32).
PCSK9 is a serine protease that belongs to the family of mammalian proprotein convertases. PCSK9 cleaves itself to form the catalytically inactive PCSK9 protein and a prosegment, which remains associated as a complex. The PCSK9-prosegment complex interacts with low-density lipoprotein receptor (LDLR), leading to its internalization and degradation. This decrease of LDLR on the hepatocyte surface results in decreased clearance of LDL particles from the circulation.33, 34, 35, 36, 37 Gain-of-function (GOF) mutations in PCSK9 leading to higher LDL-C levels have been reported to be associated with hypercholesterolemia and with an increased risk of CHD.34, 38, 39 Loss-of-function (LOF) mutations, on the other hand, have been correlated with decreased LDL-C and reduced CHD risk. In rare cases, severe LOF mutations that produce no detectable PCSK9 protein have been reported, exhibiting LDL-C levels around 15 mg/dL, and, interestingly, such individuals are identified without negative effects.7, 40, 41 Such individuals validate PCSK9 as a therapeutic target for pharmacological intervention.
PCSK9 inhibitors have emerged as a novel approach for the treatment of dyslipidemia. They are primarily being used for high-risk cardiovascular patients, including familial hypercholesterolemia cases, those who have managed LDL-C but are at high risk of plaque progression, and people who are statin intolerant. Around 5%–10% of patients treated with statins exhibit adverse side effects, such as muscle pain, weakness, and rhabdomyolysis.42, 43, 44, 45 Some approaches for PCSK9 inhibition involve monoclonal antibodies,46, 47, 48, 49, 50, 51 small interfering RNAs (siRNAs),13, 52 anti-sense oligonucleotides,53, 54 adnectins,55 small molecules,56, 57, 58, 59 peptide mimetics,60, 61, 62 and vaccines.63, 64, 65
Currently there are two US Food and Drug Administration (FDA)-approved PCSK9 inhibitors, alirocumab (Regeneron and Sanofi) and evolocumab (Amgen), both of which are fully human monoclonal antibodies (mAbs) targeting PCSK9. Clinical trials on high-risk cardiovascular patients have demonstrated their efficacy in reducing LDL-C. In the RUTHERFORD-2 clinical trial on heterozygous familial hypercholesterolemia patients, treatment with evolocumab (140 mg/2 weeks or 420 mg/month) resulted in a 60%–65% decrease in LDL-C levels. More than 60% of the patients were able to reach their LDL-C goal of 70 mg/dL.66 In the ODYSSEY HIGH clinical trial on familial hypercholesterolemia patients treated with alirocumab (75/150 mg every 2 weeks), 57% of those with difficult-to-treat severe familial hypercholesterolemia were able to reach their LDL-C goal of 100 mg/dL.67 Moreover, when alirocumab was administered to statin-treated patients, they achieved significantly greater reductions in LDL-C than those who doubled their dose of statin or received the addition of ezetimibe.67, 68, 69, 70
Cost is a major limitation in the clinical management of high-risk cardiovascular patients. Biological products such as recombinant mAbs are complex and expensive to manufacture. Evolocumab and alirocumab are projected to cost around $14,350 per year per patient.71 A recent analysis on the cost-effectiveness of PCSK9 inhibitors for use in atherosclerotic cardiovascular disease (ASCVD) patients found that these inhibitors are poorly cost-effective and that a 71% reduction in price is needed to reach the patient willingness-to-pay threshold of $100,000/quality-adjusted life-year (QALY).72 In addition to the high cost, current anti-PCSK9 recombinant antibodies require frequent administration, such as injections every 2 weeks. Development of more cost-effective, longer-lived, and potent pharmacologic interventions would provide an avenue for making such therapeutics more widely available for high-risk cardiovascular patients.
Compared to other therapeutic approaches, with in vivo electroporation technology, the plasmid DNA is delivered transiently and is not integrated, therefore allowing for repeat administration without any adverse consequences. Furthermore, with the recombinant protein therapy approach, numerous repeat administrations are required, which generally come at a high financial cost to the patient. With the DNA-encoded mAb (DMAb) platform it is possible to administer therapeutics repeatedly without facing challenges such as pre-existing immunity, difficult manufacturing processes, or high cost. DMAb technology represents a new approach for a direct in vivo production of biologically active immunoglobulins in a patient in a non-permanent fashion. Low-voltage electropermeabilization allows for robust plasmid DNA delivery into tissue where the delivered plasmid containing an encoded antibody sequence can now drive transcription and in vivo translation of the desired protein. While there have been a few recent reports describing adaptations of this technology as an approach to specific infectious diseases or cancer targets,73, 74, 75, 76, 77 there have not been reports of utilizing this approach for in vivo chronic disease therapy.
Here we developed a DMAb approach (Figure 1A) targeting PCSK9 based on engineering a plasmid DNA to encode a novel anti-PCSK9 mAb as a single insert expression cassette, named daPCSK9 (labeled HdaPCSK9 [human] or MdaPCSK9 [mouse], respectively bearing human or mouse Fc regions). In vivo daPCSK9 plasmid administration using electroporation-enhanced intramuscular injection was performed to evaluate anti-PCSK9 DMAb production kinetics and the efficacy of PCSK9 inhibition on lipid reduction in vivo. Robust production of anti-PCSK9 DMAb began within days of administration, with peak levels of antibody achieved by 1 week post-injection. PCSK9 DMAb could be detected in sera for up to 2 months, and levels could be boosted with additional administrations. In vivo-produced anti-PCSK9 DMAb retained its ability to bind to its target. An increase in LDLR was detected upon in vivo administration and expression of daPCSK9. This was accompanied by significant reductions in non-high-density lipoprotein cholesterol (non-HDL-C) and total cholesterol levels in both C57BL/6 and nude mice studies. These observations support an exciting proof of principle for further study utilizing daPCSK9 DMAb approaches for the treatment of dyslipidemic disorders.
Figure 1.

Illustration of DMAb Approach
(A) Introduction of anti-PCSK9 DMAbs and mechanism of action. (a) In vivo electroporation device is used for intracellular delivery of anti-PCSK9 DMAb plasmids. (b) Intramuscular expression of DNA-encoded anti-PCSK9 mAbs and their subsequent release into the circulation. (c) Anti-PCSK9 DMAbs migrate to the liver where they bind and inhibit PCSK9. (d) Anti-PCSK9 DMAb mechanism of action. (1) LDL binding to LDLR results in uptake and degradation of LDL and recycling of LDLR. (2) PCSK9 binds LDLR, resulting in LDLR degradation. (3) Anti-PCSK9 DMAb inhibits PCSK9, allowing for increased LDLR recycling and display on the cell surface. (B) Schematic of the anti-PCSK9 DMAb bicistronic plasmid consisting of antibody heavy- and light-chain sequences separated by furin and P2A cleavage sites.
Results
Construction of Mouse Anti-PCSK9 DMAb Plasmids and In Vitro Characterization
The mouse anti-PCSK9 (IgG2a) was designed in a single linked heavy- and light-chain cassette to be expressed from a single open reading frame, with the heavy- and light-chain genes separated by a previously developed furin/P2A cleavage site (Figure 1B) to provide efficient protein processing necessary for immunoglobulin G (IgG) production in vivo. A cytomegalovirus (CMV) promoter in the pVax-1 vector was used to drive the expression of the antibodies, and the final construct was designated MdaPCSK9. At 72 hr after plasmid transfection into HEK293T cells, secreted antibody expression in supernatants was determined to be 831 ng/mL for MdaPCSK9 (Figure 2A). Western blot-binding studies, using supernatants from MdaPCSK9 DMAb-transfected cells, showed specific binding to both human and mouse recombinant PCSK9 antigens, with greater binding detected to human PCSK9 (Figure 2B). This showed proper binding activity of MdaPCSK9 against the PCSK9 antigen. Fluorescence microscopy was also used to demonstrate MdaPCSK9 expression in Hepa1-6 cells (Figure 2C), and it showed a strong expression of MdaPCSK9 in DMAb plasmid-transfected, but not control, pVax1-transfected cells.
Figure 2.

In Vitro Evaluation of MdaPCSK9
(A) In vitro expression of MdaPCSK9 compared to empty backbone pVax-1 plasmid in HEK293 cells. Supernatants were harvested at 24, 48, and 72 hr post-transfection and analyzed by quantitative ELISA. (B) Binding western blot analysis of MdaPCSK9 from cellular supernatants. Binding of MdaPCSK9 obtained from HEK293T transfected cells was evaluated against recombinant mouse and human PCSK9 proteins. Membranes were stained with supernatant from MdaPCSK9- or pVax-1-transfected cells at 72 hr post-transfection. (C) Immunofluorescence staining of mouse Hepa1-6 cells transfected with MdaPCSK9 plasmids. Cells were fixed 48 hr after transfection. Cells were stained with anti-mouse IgG-FITC and DAPI nuclear stain. pVax-1-transfected cells were used as a negative control.
In Vivo Expression and Kinetics of MdaPCSK9
The expression level and duration of the daPCSK9 DMAbs were evaluated in C57BL/6J wild-type and nude B6.Cg-foxn1nu/J mice (Jackson Laboratory) (Figure 3). Plasmid DNA (100 or 300 μg) was injected intramuscularly into the tibialis anterior muscle, followed by electroporation (IM-EP). daPCSK9 with a mouse backbone demonstrated high expression with a long duration. MdaPCSK9 levels reached a maximum mean of 2,902 ng/mL at day 14, and they persisted for over 6 weeks, with blood levels at 1,649 ng/mL on day 42. In nude mice, the MdaPCSK9 expression was higher, reaching a maximum mean of 4,923 ng/mL at day 21, and expression persisted through the last bleed at day 42. Sequential administration of mouse DMAbs at days 0, 21, and 42 resulted in continuous increases in DMAb expression, with MdaPCSK9 levels reaching 7,521 ng/mL (3 × 100 μg doses) on day 62 (Figure 3C). This highlights the sustainability of DMAb expression over long periods. Improved MdaPCSK9 expression was observed when doses were spread out over several-week periods, simplifying administration.
Figure 3.

In Vivo Expression and Kinetics of MdaPCSK9
(A) Quantitative ELISA analysis for MdaPCSK9 following a single intramuscular injection of DMAb plasmids in B6.Cg-foxn1nu/J nude mice. (B) Quantitative ELISA analysis for MdaPCSK9 following a single intramuscular injection of DMAb plasmids in C57BL/6J wild-type mice. (C) Sequential administrations of MdaPCSK9 by intramuscular injection in C57BL/6J wild-type mice at days 0, 21, and 42. Quantitative ELISA analysis was performed for MdaPCSK9 at the indicated time points. Plates were coated with recombinant mouse PCSK9 protein, followed by incubation with anti-mouse IgG-HRP for the detection of MdaPCSK9. Values represent mean expression in each group (n = 5) ± SEM.
PCSK9 Protein Binding and Inhibition by DMAbs
PCSK9 inhibition was determined using western blot analysis of LDLR expression levels in mouse liver sections (Figure 4A). Mouse livers were harvested at day 5, and the LDLR expression was evaluated by comparing the human and MdaPCSK9-treated mice to pVax-1 control mice. Western blot analysis showed that LDLR expression levels were significantly higher for PCSK9 DMAb-treated mice than control mice. ELISA protein quantification was used to detect the presence of DMAbs in the livers of treated mice. DMAb levels were 185 ng/mg tissue for MdaPCSK9 (Figure 4B). Binding western blot analysis was used to evaluate the binding of DMAbs from serum samples of DMAb-treated and untreated mice to human and mouse PCSK9 protein (Figure 4C). After incubation of the serum samples with blots containing the recombinant mouse or human PCSK9 proteins, the sera of DMAb-treated mice reacted with the PCSK9 protein, but sera from the pVax-1-treated mice did not. Furthermore, MdaPCSK9 were evaluated on a human liver cell line, Huh7; 72 hr after transfection, cells were harvested and analyzed by flow cytometry (Figure 4D). There were significant increases in LDLR levels in the MdaPCSK9-treated group, with mean fluorescence intensity (MFI) increasing from 1,573 in the control group to 11,166 in the DMAb-treated group. The results demonstrate the functional efficacy of daPCSK9 at inhibiting PCSK9 protein from inducing LDLR degradation in hepatocytes.
Figure 4.

Evaluation of Binding and Functional Activity of MdaPCSK9
(A) Western analysis of liver LDLR expression. Livers of control- and DMAb-treated C57B/6J wild-type mice were harvested at day 5 post-treatment. Levels of LDLR expression in tissue lysates were evaluated by staining for anti-mouse LDLR antibody. (B) Quantitative ELISA for MdaPCSK9 was performed on liver lysates at day 5 post-treatment. (C) Binding western blot analysis of anti-PCSK9 DMAbs from mouse sera. Anti-PCSK9 DMAbs obtained from mouse sera at 5 days post-treatment were evaluated for binding against recombinant mouse and human PCSK9 proteins. (D–F) Flow cytometry analysis was performed for LDLR expression in Huh7 cells. Human hepatoma Huh7 cells were treated with pVax-1 control DNA or MdaPCSK9 and harvested at 72 hr post-treatment. The level of LDLR expression was evaluated by flow cytometry by staining with anti-LDL receptor antibody (ab52818), followed by anti-rabbit IgG-FITC (sc-2012). (D) Histogram, (E) dot plot, and (F) mean fluorescence intensity (MFI) of Huh7 cells treated with pVax-1 or MdaPCSK9 are shown.
Lipid-Lowering Capability of Mouse Anti-PCSK9 DMAb
The lipid-lowering capability of daPCSK9 was evaluated in C57BL/6J wild-type and B6.Cg-foxn1nu/J nude mice (Figure 5). For accurate calculations of lipid reductions in the treated group, percentage decreases were analyzed against the control group for each day. In C57BL/6J mice, a significant 28.6% decrease in non-HDL-C was detected on day 7 for the MdaPCSK9 group. By day 14, there was a 30% decrease in non-HDL-C for the MdaPCSK9 group. Total cholesterol reductions were also observed for the MdaPCSK9 group, with a 14% decrease on day 14. In nude mice, there was a significant 32.5% reduction in non-HDL-C on day 14 for those treated with MdaPCSK9. This reduction persisted until day 21. There were similar reductions in total cholesterol that persisted to day 21, with a 14.3% decrease. The persistent reduction of non-HDL-C and total cholesterol in the mouse models correlates with the DMAb expression in the circulation. Repeated administrations of MdaPCSK9 led to increasing DMAb expression kinetics over a period of 62 days, with reductions in non-HDL-C of 20.9% from pooled sera on days 56 and 62 (Figures 5E and 5F).
Figure 5.

In Vivo Lipid Panel Analysis of MdaPCSK9
(A–F) B6.Cg-foxn1nu/J nude mice (A and B) and C57B/6J wild-type mice (C and D) were bled at the indicated time points following a single intramuscular administration of 300 μg MdaPCSK9 plasmid. (E and F) C57B/6J wild-type mice were sequentially administered 100 μg MdaPCSK9 plasmid by intramuscular injection. Lipid panel (A, C, and E, non-HDL-C; and B, D, F, total cholesterol) analyses were carried out on mice sera using the VITROS 350 Clinical Chemistry Analyzer. Percent changes were calculated for each day relative to control pVax-1-treated mice. Statistical differences are indicated relative to day 0 of treatment. Data are expressed as ± SEM (n = 5). Statistical differences were measured using two-way ANOVA tests (*p < 0.05, **p < 0.01, and ***p < 0.001; n.s., not significant).
Construction and Evaluation of Human Anti-PCSK9 DMAb
daPCSK9 plasmid engineered to express either human or mouse Fc regions (labeled HdaPCSK9 and MdaPCSK9, respectively) were designed to evaluate the effect on expression levels and kinetics in vivo. Evaluation of HdaPCSK9 in HEK293T cells showed the DMAb expression levels at 1,349 ng/mL (Figure S1). Strong expression for HdaPCSK9 was also confirmed in Hepa1-6 cells by using fluorescence microscopy after intracellular staining with anti-human IgG-fluorescein isothiocyanate (FITC). Western blot-binding studies with the HdaPCSK9 demonstrated binding to both recombinant human and mouse PCSK9 proteins.
HdaPCSK9 expression levels and kinetics were analyzed in vivo in wild-type C57BL/6J and nude B6.Cg-foxn1nu/J mice (Figures 6A and 6B). After a single intramuscular administration of 300 μg plasmid DNA, the HdaPCSK9 reached a maximum mean of 2,224 ng/mL protein mAbs at day 14 in nude mice. In wild-type mice, there was an acute immune reaction to the DMAbs containing human Fc, resulting in drops in DMAb expression by day 7. Maximum expression was observed at day 5, with a mean of around 2,485 ng/mL for the 300 μg dose of HdaPCSK9. The results of human DMAb studies in wild-type mice demonstrate the importance of considering intraspecies differences in antibody evaluation and how this may impact translation to other models. The reason for the resulting drop in expression of human PCSK9 DMAb is interesting. It is possible that immunogenicity or other factors could be involved, which will be investigated in future studies. The functional activity of sera DMAbs was confirmed by binding western blot analysis, showing binding to both recombinant mouse and human PCSK9 proteins (Figure 6C).
Figure 6.

In Vivo Expression and Kinetics of HdaPCSK9
(A and B) Quantitative ELISA analysis for MdaPCSK9 following single intramuscular injection of DMAb plasmids in (A) B6.Cg-foxn1nu/J nude mice and (B) C57BL/6J wild-type mice. Quantitative ELISAs were performed for anti-hPCSK9 DMAb at the indicated time points. Plates were coated with anti-human Fc antibody, followed by incubation with anti-human IgG-HRP antibody for the detection of HdaPCSK9. Values represent mean expression in each group (n = 5) ± SEM. (C) Binding western blot analysis of HdaPCSK9 from mouse sera. Anti-PCSK9 DMAbs obtained from mouse sera at 5 days post-treatment were evaluated for binding against recombinant mouse and human PCSK9 proteins. (D) Histology and immunohistochemistry of HdaPCSK9 in mouse muscle (magnification, 20×; scale bars, 100 μm). Immunohistochemistry of PCSK9 using anti-human IgG-HRP for mouse muscle sections at day 5 in C57BL/6J wild-type is shown. Insets show 30% magnification of the images. Prominent browning is detected in HdaPCSK9-treated muscle. H&E-stained sections of treated muscle show no observable tissue damage. The images were taken from the muscle mid-section, away from the needle tract area.
Anti-PCSK9 DMAb expression was also confirmed in vivo by immunohistochemistry analysis of mouse muscle tissue at day 5. As shown in Figure 6D, immunohistochemistry using HdaPCSK9 immunostained with anti-human IgG-horseradish peroxidase (HRP) indicates positive browning in the mouse tibialis anterior muscle section, compared to no browning in the control pVax-1 muscle sections. H&E staining of the DMAb-treated and control groups showed no signs of tissue damage. This confirms the expression of anti-PCSK9 DMAbs and the lack of tissue damage caused by their administration.
Lipid-Lowering Capability of Human Anti-PCSK9 DMAbs
The HdaPCSK9 performed similarly to MdaPCSK9 in its capacity at lipid reduction in C57BL/6J wild-type and B6.Cg-foxn1nu/J nude mice (Figure S2). In wild-type mice, there was a significant 23.8% decrease in non-HDL-C detected on day 7 for the HdaPCSK9 group and a 30% reduction by day 14. Total cholesterol reduction was also observed, with a 14% decrease on day 14. Compared to HdaPCSK9, MdaPCSK9 demonstrated better reductions of both non-HDL-C and total cholesterol, which correlates with its superior expression levels and duration in the circulation in the matched species. In nude mice, there were significant reductions in non-HDL-C on day 14 for both species’ antibodies, with 28.6% for human and 32.5% for mouse anti-PCSK9 DMAb groups. This reduction persisted until the final endpoint at day 21. There were similar reductions in total cholesterol that persisted until day 21, with a 15.2% decrease for the HdaPCSK9. Although they were not expressed as well in mouse as MdaPCSK9, the human DMAb performed well at lipid reduction in this short-term study supporting potential application of the human Fc DMAbs in a matched species setting.
Discussion
Individuals with elevated LDL-C or those afflicted with hereditary familial hypercholesterolemia are at increased risk for CHD. LDL-C reduction therapy with 3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG-CoA) reductase inhibitors, such as statins, has been a standard treatment for reducing the risk of CHD, but intolerable musculoskeletal symptoms can prevent their use.78 Alternative therapies are needed when the gold standard for reducing high LDL-C causes such adverse effects. PCSK9 inhibitors are emerging as a potent important new approach for reducing LDL-C by increasing its hepatic clearance via the LDLR.
Several strategies have been taken to inhibit PCSK9, with mAbs achieving recent success. Evolocumab and alirocumab are two FDA-approved anti-PCSK9 recombinant mAbs that have shown significant LDL-C reduction. Various recombinant anti-PCSK9 antibodies have been investigated in mice for their lipid-lowering capabilities. Zhang et al.9 developed a transgenic cholesteryl ester transfer protein (CETP)/LDLR-hemi mouse model that expressed human CETP transgene and one copy of the LDLR, to mimic healthy human lipid levels in mice. After 48 hr of a single intravenous administration of the anti-PCSK9 antibody at 1.1 mg/kg, there was a 29% reduction from the baseline in LDL-C, with serum antibody levels of 3.5 μg/mL. This correlates with expression levels and the lipid reduction capability observed with the DMAbs. There was even a significant 50%–70% reduction in LDL-C at higher administered recombinant antibody doses of 3 mg/kg and 10 mg/kg.9 Chan et al. evaluated an anti-PCSK9 antibody in wild-type mice and LDLR-knockout mice, and they observed a 36% reduction in total cholesterol 3 days after administration to the wild-type mice, while detecting no change in the LDLR-knockout mice.8 Another group examined the effect of an anti-PCSK9 antibody on humanized hyperlipidemic Pcsk9hum/hum Ldlr−/+ mice on a carbohydrate-rich diet. The anti-PCSK9 antibody REGN727 reduced LDL-C to a pre-diet level within 24 hr. It was also found to be effective in cynomolgus monkeys, with up to a 75% reduction in LDL-C for over 20 days after a single intravenous administration.79
Recombinant anti-PCSK9 mAbs have shown success, but a major limiting factor to their use is their high cost. Moreover, they require frequent dosing, such as every 2 weeks used for recombinant anti-PCSK9 antibody therapeutics. Here we developed a novel approach to inhibit PCSK9 using DMAb technology. DMAbs have several advantages that make them an important new technology for the development of protein-based therapeutics, such as cost-effectiveness, ease of manufacturing, stability, and no requirement for a cold chain and rapid development and engineering. DMAbs can be delivered via intramuscular or intradermal routes via in vivo electroporation of plasmid DNA. Low-voltage electroporation leads to consistent intracellular delivery of the injected plasmid DNA. Our DMAb platform has shown protective efficacy against several viral pathogens, including HIV, Middle East Respiratory Syndrome (MERS), and Zika among others, and it is currently under evaluation for immunotherapeutics against cancers.73, 74, 75, 76, 77
In this study, we used a CELLECTRA in vivo electroporation device, which utilizes an adaptive constant current electroporation system that can measure tissue resistance and adjust in real time to prevent any tissue damage and enhance DNA uptake. For DNA delivery, electroporation has been able to allow 1,000-fold enhancement in gene expression in vivo. There have been several impactful clinical studies performed using this in vivo electroporation technology. One example is the human papilloma virus (HPV) VGX-3100 DNA vaccine, which showed efficacy in phase IIb clinical trials, demonstrating robust antigen-specific T cell response, as well as inducing clearance of the HPV virus and lesions.80 Another example is the development of the Zika virus DNA vaccine reported in the New England Journal of Medicine in 2017.81 In a phase I clinical trial evaluating the safety and immunogenicity of DNA vaccine GLS-5700, healthy adults demonstrated development of both binding and neutralizing antibodies, associated with protection in animal models.81
In our study, the mouse PCSK9 DMAb was capable of expression for over 42 days in wild-type mice. Expression for several months has been demonstrated with several DMAbs, such as Anti-Dengue,73 Anit-Ebola (A.P., unpublished data), and the immunoadhesin eCD4-Ig.82 These studies support the potential of DMAbs, some lasting several months, to be administered as a possible treatment for atherosclerotic cardiovascular disease. Here we evaluated the efficacy of the anti-PCSK9 DMAb platform for its ability to express anti-PCSK9 antibodies directly in vivo and to exhibit lipid-lowering properties in mice. An anti-PCSK9 DMAb (MdaPCSK9) that used a mouse-matched backbone was found to express for at least 6 weeks in vivo at reasonable levels. Importantly, inhibition of PCSK9 was observed in vivo, showing elevated liver LDLR expression 5 days after treatment. With a single intramuscular administration of anti-PCSK9 DMAbs (300 μg plasmid DNA), we observed reductions in non-HDL-C and total cholesterol with both mouse and human anti-PCSK9 antibodies. Significant reductions in non-HDL-C were observed with MdaPCSK9 as early as day 7 post-administration in wild-type and nude mice. Compared to wild-type pVax-1 negative controls, there was a 28.6% reduction on day 7 for the MdaPCSK9 group. Both human and mouse anti-PCSK9 DMAbs led to 30% decreases in non-HDL-C and 14% reductions in total cholesterol by day 14. Wild-type mice, which are normocholesterolemic, were used in the current study, and it is difficult to observe significant decreases in LDL levels in these mice as compared to transgenic hypercholesterolemic mice. Future studies with transgenic hypercholesterolemic mice are important for evaluation of the PCSK9 DMAbs.
In conclusion, these results demonstrate the efficacy of highly novel anti-PCSK9 DMAbs at reducing non-HDL-C and total cholesterol, and they suggest the potential of this new technology in the treatment of atherosclerotic cardiovascular disease and familial hypercholesterolemia patients. These novel DNA-produced, antibody-based therapeutics could be valuable as an alternative method of therapy of treatment for hypercholesterolemia and other cardiovascular diseases.
Materials and Methods
Cell Culture
HEK293T, mouse hepatoma Hepa1-6, and human Huh-7 cells were maintained in DMEM (Gibco-Life Technologies) supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 μg/mL streptomycin (Life Technologies).
Anti-PCSK9 DMAb Plasmid Design and Construction
DMAb plasmids were designed to encode human or mouse anti-PCSK9 mAbs. DMAb transgenes were designed in a single open reading frame, with the variable and constant heavy chains separated from variable and constant light chains by a furin/P2A cleavage site. Two constructs were developed: a human anti-PCSK9 DMAb (HdaPCSK9) on an IgG1 backbone, and a mouse anti-PCSK9 DMAb (MdaPCSK9) on an IgG2a backbone. DMAb transgenes were flanked with BamHI and XhoI restriction enzyme sites and synthesized by GenScript for subcloning into the corresponding restriction enzyme sites in a pVax-1 mammalian expression vector. Human CMV immediate-early promoter in the pVax-1 vector was used to drive the expression of the antibodies.
In Vitro Transfection of DMAb Plasmids
HEK293T, Hepa1-6, or Huh7 cells were transfected with anti-PCSK9 DMAb plasmids using lipofectamine LTX reagent and following the manufacturer’s protocol. Briefly, 5 × 105 cells were seeded in a 12-well plate and incubated in a 37°C incubator with 5% CO2. Cells were transfected with 1 μg DMAb plasmid at 70% confluence and incubated for a maximum of 72 hr. DMAb expression following transfection was analyzed by western blot, ELISA, and immunofluorescence microscopy.
Western Blot and ELISA Analyses
A binding western blot analysis was performed to evaluate anti-PCSK9 DMAb-binding capability. Recombinant human or mouse PCSK9 protein (0.5–5 μg) was run in precast Bis-Tris gels (Invitrogen) under nonreducing conditions. Gels were transferred to Immobilon-FL polyvinylidene fluoride (PVDF) transfer membranes (Millipore). The membranes were blocked in Odyssey Blocking Buffer (LI-COR Biosciences) for 1 hr, then stained overnight at 4°C with either supernatant from DMAb-transfected cells or serum from DMAb-treated mice, and compared to those of pVax-1-negative control samples. Membranes were washed with PBST (PBS with 0.05% Tween 20). Subsequently, membranes were stained with either anti-human or anti-mouse IgG 680RD antibody (LI-COR Biosciences) for 1 hr at room temperature (RT) and washed in PBST. Membranes were scanned on a LI-COR Odyssey CLx imager. 50 μg total sample protein was loaded in gels and run as described above for the analysis of liver lysates for LDLR expression. Membranes were stained with an anti-mLDLR primary antibody (AF2255; R&D Systems). Actin was stained using an anti-actin primary antibody (ab198991; Abcam) and an anti-rabbit IgG-HRP secondary antibody.
DMAbs with human backbones were quantified by an ELISA for total human IgG. ELISA MaxiSorp plates were coated with 1 μg/well anti-human IgG-Fc fragment antibody (A80-104A; Bethyl Laboratories) overnight at 4°C. Plates were blocked in 10% FBS in PBS for 1 hr at RT. Samples and recombinant human IgG standards were added to the plate for 1 hr at RT. After washing the plates, anti-human IgG-Fc fragment antibody-HRP (A80-104P; Bethyl Laboratories) was added for 1 hr at RT. SigmaFast OPD (Sigma-Aldrich) substrate solution was used for sample detection. Optical density was measured at 450 nm using a GloMax 96 Microplate Luminometer (Promega). For quantification of DMAbs with mouse backbones from cell supernatants, plates were coated with anti-mouse IgG-Fc fragment antibody (A90-131A; Bethyl Laboratories). Anti-mouse IgG-HRP (62-6520; Invitrogen) was used for detection. Serum samples of DMAbs with a mouse backbone were differentiated from background mouse IgGs by coating ELISA plates with recombinant PCSK9 protein. Purified recombinant anti-PCSK9 mAb was used as the standard. Quantitative ELISA data are expressed as ± SEM (n = 3).
Immunofluorescence Analysis
Hepa1-6 cells were transfected with anti-PCSK9 DMAbs using lipofectamine LTX reagent as described above. 48 hr after transfection, cells were washed in PBS and fixed in 4% paraformaldehyde for 10 min at RT. Cells were then washed and incubated with anti-human IgG-FITC (A80-319F; Bethyl Laboratories) for 1 hr. After washing, cells were stained with DAPI nuclear stain and imaged on an EVOS FL Imaging System.
Flow Cytometry Analysis
Flow cytometry was used for the detection of LDLR on Huh7 cells transfected with control pVax-1 or anti-PCSK9 DMAb plasmids. 72 hr post-transfection, cells were washed with fluorescence-activated cell sorting (FACS) buffer (3% FBS in PBS) and stained with anti-LDLR primary antibody (ab52818; Abcam) for 1 hr at 4°C, followed by anti-rabbit IgG-FITC secondary antibody (ab6717; Abcam) for 1 hr at 4°C. Cells were washed 3 times in FACS buffer and fixed in 2% paraformaldehyde. Cells were analyzed on an LSR18 flow cytometer (BD Biosciences).
Immunohistochemistry of Mouse Muscle
Immunohistochemistry was performed by The Wistar Institute Histotechnology Facility. Briefly, mouse anterior tibialis muscle sections were resected and embedded in paraffin. Paraffin-embedded samples were treated with antigen retrieval reagent and deparaffinized. Slides were then fixed with acetone and washed with PBS, followed by blocking and staining the sections with anti-human IgG antibody-HRP.
In Vivo DMAb Plasmid Administration
Evaluation of DMAbs was carried out in 6- to 8-week-old C57BL/6J wild-type and nude B6.Cg-foxn1nu/J mice. Animal experiments were carried out in accordance with the guidelines of the NIH and The Wistar Institute Institutional Animal Care and Use Committee (IACUC). Mice were injected with plasmid DNA in the tibialis anterior or quadricep muscles. In vivo electroporation was performed using a CELLECTRA adaptive constant-current EP device (Inovio Pharmaceuticals). Triangular 3-electrode arrays consisting of 26G solid stainless steel electrodes were used to deliver square-wave pulses. Two constant-current pulses were delivered at 0.1 A for 52 ms/pulse, separated with a 1-s delay between pulses. Mice were given either 100 or 300 μg plasmid DNA resuspended in water. The plasmids were co-formulated with recombinant hyaluronidase to enhance their intramuscular distribution. A maximum of 30 μL volume was used at each injection site. pVax-1 empty vector was used as a negative control for DMAbs. Blood was collected by submandibular bleeding for DMAb quantification and lipid panel analysis. Samples were collected until day 42 or 62.
Mouse Lipid Panel Analysis
Lipid panel analysis was carried out using a VITROS 350 Clinical Chemistry Analyzer. Mice were fasted for 4 hr and bled by submandibular bleeding. Serum samples collected from whole blood were used for the lipid panel analysis. Mouse non-HDL-C levels were calculated by subtracting HDL-C from total cholesterol.
Statistics
Experimental data were analyzed using two-way ANOVA tests. Differences were deemed significant at p values < 0.05. All graphs were prepared using GraphPad Prism 6 software.
Author Contributions
M.K. performed the experiments, analyzed data, and wrote the manuscript. M.K., A.P., K.M., and D.B.W. were involved in study conception and design. A.P., K.W., S.B.K., L.H., N.N.L., and D.J.R. were involved in experimental design and data analysis and contributed intellectually to the research. All authors contributed to manuscript editing.
Conflicts of Interest
L.H. is an employee of Inovio Pharmaceuticals and as such receives salary and benefits, including ownership of stock and stock options. D.B.W. discloses grant funding, industry collaborations, speaking honoraria, and fees or stock for consulting. His services include serving on scientific review committees and advisory boards. Remuneration includes direct payments and/or stock or stock options. K.M. reports receiving grants from Inovio and receiving consulting fees from Inovio related to DNA vaccine development. The other authors declare no competing financial interests.
Acknowledgments
This work was supported by funding from Inovio Pharmaceuticals to M.K., A.P., K.M., and D.B.W.
Footnotes
Supplemental Information includes two figures and can be found with this article online at https://doi.org/10.1016/j.ymthe.2018.10.016.
Supplemental Information
References
- 1.Benjamin E.J., Blaha M.J., Chiuve S.E., Cushman M., Das S.R., Deo R., de Ferranti S.D., Floyd J., Fornage M., Gillespie C., American Heart Association Statistics Committee and Stroke Statistics Subcommittee Heart Disease and Stroke Statistics-2017 Update: A Report From the American Heart Association. Circulation. 2017;135:e146–e603. doi: 10.1161/CIR.0000000000000485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nelson R.H. Hyperlipidemia as a risk factor for cardiovascular disease. Prim. Care. 2013;40:195–211. doi: 10.1016/j.pop.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roy S. Atherosclerotic cardiovascular disease risk and evidence-based management of cholesterol. N. Am. J. Med. Sci. 2014;6:191–198. doi: 10.4103/1947-2714.132916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goldstein J.L., Brown M.S. A century of cholesterol and coronaries: from plaques to genes to statins. Cell. 2015;161:161–172. doi: 10.1016/j.cell.2015.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Repas T.B., Tanner J.R. Preventing early cardiovascular death in patients with familial hypercholesterolemia. J. Am. Osteopath. Assoc. 2014;114:99–108. doi: 10.7556/jaoa.2014.023. [DOI] [PubMed] [Google Scholar]
- 6.Perak A.M., Ning H., de Ferranti S.D., Gooding H.C., Wilkins J.T., Lloyd-Jones D.M. Long-term risk of atherosclerotic cardiovascular disease in US adults with the familial hypercholesterolemia phenotype. Circulation. 2016;134:9–19. doi: 10.1161/CIRCULATIONAHA.116.022335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seidah N.G., Prat A. The biology and therapeutic targeting of the proprotein convertases. Nat. Rev. Drug Discov. 2012;11:367–383. doi: 10.1038/nrd3699. [DOI] [PubMed] [Google Scholar]
- 8.Chan J.C.Y., Piper D.E., Cao Q., Liu D., King C., Wang W., Tang J., Liu Q., Higbee J., Xia Z. A proprotein convertase subtilisin/kexin type 9 neutralizing antibody reduces serum cholesterol in mice and nonhuman primates. Proc. Natl. Acad. Sci. USA. 2009;106:9820–9825. doi: 10.1073/pnas.0903849106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang L., McCabe T., Condra J.H., Ni Y.G., Peterson L.B., Wang W., Strack A.M., Wang F., Pandit S., Hammond H. An anti-PCSK9 antibody reduces LDL-cholesterol on top of a statin and suppresses hepatocyte SREBP-regulated genes. Int. J. Biol. Sci. 2012;8:310–327. doi: 10.7150/ijbs.3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ni Y.G., Condra J.H., Orsatti L., Shen X., Di Marco S., Pandit S., Bottomley M.J., Ruggeri L., Cummings R.T., Cubbon R.M. A proprotein convertase subtilisin-like/kexin type 9 (PCSK9) C-terminal domain antibody antigen-binding fragment inhibits PCSK9 internalization and restores low density lipoprotein uptake. J. Biol. Chem. 2010;285:12882–12891. doi: 10.1074/jbc.M110.113035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ni Y.G., Di Marco S., Condra J.H., Peterson L.B., Wang W., Wang F., Pandit S., Hammond H.A., Rosa R., Cummings R.T. A PCSK9-binding antibody that structurally mimics the EGF(A) domain of LDL-receptor reduces LDL cholesterol in vivo. J. Lipid Res. 2011;52:78–86. doi: 10.1194/jlr.M011445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liang H., Chaparro-Riggers J., Strop P., Geng T., Sutton J.E., Tsai D., Bai L., Abdiche Y., Dilley J., Yu J. Proprotein convertase substilisin/kexin type 9 antagonism reduces low-density lipoprotein cholesterol in statin-treated hypercholesterolemic nonhuman primates. J. Pharmacol. Exp. Ther. 2012;340:228–236. doi: 10.1124/jpet.111.187419. [DOI] [PubMed] [Google Scholar]
- 13.Frank-Kamenetsky M., Grefhorst A., Anderson N.N., Racie T.S., Bramlage B., Akinc A., Butler D., Charisse K., Dorkin R., Fan Y. Therapeutic RNAi targeting PCSK9 acutely lowers plasma cholesterol in rodents and LDL cholesterol in nonhuman primates. Proc. Natl. Acad. Sci. USA. 2008;105:11915–11920. doi: 10.1073/pnas.0805434105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gupta N., Fisker N., Asselin M.C., Lindholm M., Rosenbohm C., Ørum H., Elmén J., Seidah N.G., Straarup E.M. A locked nucleic acid antisense oligonucleotide (LNA) silences PCSK9 and enhances LDLR expression in vitro and in vivo. PLoS ONE. 2010;5:e10682. doi: 10.1371/journal.pone.0010682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lindholm M.W., Elmén J., Fisker N., Hansen H.F., Persson R., Møller M.R., Rosenbohm C., Ørum H., Straarup E.M., Koch T. PCSK9 LNA antisense oligonucleotides induce sustained reduction of LDL cholesterol in nonhuman primates. Mol. Ther. 2012;20:376–381. doi: 10.1038/mt.2011.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Becker G.L., Lu Y., Hardes K., Strehlow B., Levesque C., Lindberg I., Sandvig K., Bakowsky U., Day R., Garten W., Steinmetzer T. Highly potent inhibitors of proprotein convertase furin as potential drugs for treatment of infectious diseases. J. Biol. Chem. 2012;287:21992–22003. doi: 10.1074/jbc.M111.332643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hardes K., Becker G.L., Lu Y., Dahms S.O., Köhler S., Beyer W., Sandvig K., Yamamoto H., Lindberg I., Walz L. Novel Furin Inhibitors with Potent Anti-infectious Activity. ChemMedChem. 2015;10:1218–1231. doi: 10.1002/cmdc.201500103. [DOI] [PubMed] [Google Scholar]
- 18.Inocencio N.M., Sucic J.F., Moehring J.M., Spence M.J., Moehring T.J. Endoprotease activities other than furin and PACE4 with a role in processing of HIV-I gp160 glycoproteins in CHO-K1 cells. J. Biol. Chem. 1997;272:1344–1348. doi: 10.1074/jbc.272.2.1344. [DOI] [PubMed] [Google Scholar]
- 19.Watanabe M., Hirano A., Stenglein S., Nelson J., Thomas G., Wong T.C. Engineered serine protease inhibitor prevents furin-catalyzed activation of the fusion glycoprotein and production of infectious measles virus. J. Virol. 1995;69:3206–3210. doi: 10.1128/jvi.69.5.3206-3210.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ozden S., Lucas-Hourani M., Ceccaldi P.E., Basak A., Valentine M., Benjannet S., Hamelin J., Jacob Y., Mamchaoui K., Mouly V. Inhibition of Chikungunya virus infection in cultured human muscle cells by furin inhibitors: impairment of the maturation of the E2 surface glycoprotein. J. Biol. Chem. 2008;283:21899–21908. doi: 10.1074/jbc.M802444200. [DOI] [PubMed] [Google Scholar]
- 21.Becker G.L., Sielaff F., Than M.E., Lindberg I., Routhier S., Day R., Lu Y., Garten W., Steinmetzer T. Potent inhibitors of furin and furin-like proprotein convertases containing decarboxylated P1 arginine mimetics. J. Med. Chem. 2010;53:1067–1075. doi: 10.1021/jm9012455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Olmstead A.D., Knecht W., Lazarov I., Dixit S.B., Jean F. Human subtilase SKI-1/S1P is a master regulator of the HCV Lifecycle and a potential host cell target for developing indirect-acting antiviral agents. PLoS Pathog. 2012;8:e1002468. doi: 10.1371/journal.ppat.1002468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blanchet M., Sureau C., Guévin C., Seidah N.G., Labonté P. SKI-1/S1P inhibitor PF-429242 impairs the onset of HCV infection. Antiviral Res. 2015;115:94–104. doi: 10.1016/j.antiviral.2014.12.017. [DOI] [PubMed] [Google Scholar]
- 24.Hyrina A., Meng F., McArthur S.J., Eivemark S., Nabi I.R., Jean F. Human Subtilisin Kexin Isozyme-1 (SKI-1)/Site-1 Protease (S1P) regulates cytoplasmic lipid droplet abundance: A potential target for indirect-acting anti-dengue virus agents. PLoS ONE. 2017;12:e0174483. doi: 10.1371/journal.pone.0174483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coppola J.M., Bhojani M.S., Ross B.D., Rehemtulla A. A small-molecule furin inhibitor inhibits cancer cell motility and invasiveness. Neoplasia. 2008;10:363–370. doi: 10.1593/neo.08166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Levesque C., Couture F., Kwiatkowska A., Desjardins R., Guérin B., Neugebauer W.A., Day R. PACE4 inhibitors and their peptidomimetic analogs block prostate cancer tumor progression through quiescence induction, increased apoptosis and impaired neovascularisation. Oncotarget. 2015;6:3680–3693. doi: 10.18632/oncotarget.2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Levesque C., Fugère M., Kwiatkowska A., Couture F., Desjardins R., Routhier S., Moussette P., Prahl A., Lammek B., Appel J.R. The Multi-Leu peptide inhibitor discriminates between PACE4 and furin and exhibits antiproliferative effects on prostate cancer cells. J. Med. Chem. 2012;55:10501–10511. doi: 10.1021/jm3011178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bassi D.E., Zhang J., Cenna J., Litwin S., Cukierman E., Klein-Szanto A.J.P. Proprotein convertase inhibition results in decreased skin cell proliferation, tumorigenesis, and metastasis. Neoplasia. 2010;12:516–526. doi: 10.1593/neo.92030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scamuffa N., Siegfried G., Bontemps Y., Ma L., Basak A., Cherel G., Calvo F., Seidah N.G., Khatib A.M. Selective inhibition of proprotein convertases represses the metastatic potential of human colorectal tumor cells. J. Clin. Invest. 2008;118:352–363. doi: 10.1172/JCI32040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ma Y.C., Fan W.J., Rao S.M., Gao L., Bei Z.Y., Xu S.T. Effect of Furin inhibitor on lung adenocarcinoma cell growth and metastasis. Cancer Cell Int. 2014;14:43. doi: 10.1186/1475-2867-14-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Basak A., Shervani N.J., Mbikay M., Kolajova M. Recombinant proprotein convertase 4 (PC4) from Leishmania tarentolae expression system: purification, biochemical study and inhibitor design. Protein Expr. Purif. 2008;60:117–126. doi: 10.1016/j.pep.2008.03.013. [DOI] [PubMed] [Google Scholar]
- 32.Mbikay M., Tadros H., Ishida N., Lerner C.P., De Lamirande E., Chen A., El-Alfy M., Clermont Y., Seidah N.G., Chrétien M. Impaired fertility in mice deficient for the testicular germ-cell protease PC4. Proc. Natl. Acad. Sci. USA. 1997;94:6842–6846. doi: 10.1073/pnas.94.13.6842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maxwell K.N., Breslow J.L. Adenoviral-mediated expression of Pcsk9 in mice results in a low-density lipoprotein receptor knockout phenotype. Proc. Natl. Acad. Sci. USA. 2004;101:7100–7105. doi: 10.1073/pnas.0402133101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Benjannet S., Rhainds D., Essalmani R., Mayne J., Wickham L., Jin W., Asselin M.C., Hamelin J., Varret M., Allard D. NARC-1/PCSK9 and its natural mutants: zymogen cleavage and effects on the low density lipoprotein (LDL) receptor and LDL cholesterol. J. Biol. Chem. 2004;279:48865–48875. doi: 10.1074/jbc.M409699200. [DOI] [PubMed] [Google Scholar]
- 35.Park S.W., Moon Y.A., Horton J.D. Post-transcriptional regulation of low density lipoprotein receptor protein by proprotein convertase subtilisin/kexin type 9a in mouse liver. J. Biol. Chem. 2004;279:50630–50638. doi: 10.1074/jbc.M410077200. [DOI] [PubMed] [Google Scholar]
- 36.Seidah N.G., Benjannet S., Wickham L., Marcinkiewicz J., Jasmin S.B., Stifani S., Basak A., Prat A., Chretien M. The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): liver regeneration and neuronal differentiation. Proc. Natl. Acad. Sci. USA. 2003;100:928–933. doi: 10.1073/pnas.0335507100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cunningham D., Danley D.E., Geoghegan K.F., Griffor M.C., Hawkins J.L., Subashi T.A., Varghese A.H., Ammirati M.J., Culp J.S., Hoth L.R. Structural and biophysical studies of PCSK9 and its mutants linked to familial hypercholesterolemia. Nat. Struct. Mol. Biol. 2007;14:413–419. doi: 10.1038/nsmb1235. [DOI] [PubMed] [Google Scholar]
- 38.Abifadel M., Varret M., Rabès J.P., Allard D., Ouguerram K., Devillers M., Cruaud C., Benjannet S., Wickham L., Erlich D. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat. Genet. 2003;34:154–156. doi: 10.1038/ng1161. [DOI] [PubMed] [Google Scholar]
- 39.Timms K.M., Wagner S., Samuels M.E., Forbey K., Goldfine H., Jammulapati S., Skolnick M.H., Hopkins P.N., Hunt S.C., Shattuck D.M. A mutation in PCSK9 causing autosomal-dominant hypercholesterolemia in a Utah pedigree. Hum. Genet. 2004;114:349–353. doi: 10.1007/s00439-003-1071-9. [DOI] [PubMed] [Google Scholar]
- 40.Cohen J., Pertsemlidis A., Kotowski I.K., Graham R., Garcia C.K., Hobbs H.H. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat. Genet. 2005;37:161–165. doi: 10.1038/ng1509. [DOI] [PubMed] [Google Scholar]
- 41.Kotowski I.K., Pertsemlidis A., Luke A., Cooper R.S., Vega G.L., Cohen J.C., Hobbs H.H. A spectrum of PCSK9 alleles contributes to plasma levels of low-density lipoprotein cholesterol. Am. J. Hum. Genet. 2006;78:410–422. doi: 10.1086/500615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nissen S.E., Dent-Acosta R.E., Rosenson R.S., Stroes E., Sattar N., Preiss D., Mancini G.B., Ballantyne C.M., Catapano A., Gouni-Berthold I., GAUSS-3 Investigators Comparison of PCSK9 Inhibitor Evolocumab vs Ezetimibe in Statin-Intolerant Patients: Design of the Goal Achievement After Utilizing an Anti-PCSK9 Antibody in Statin-Intolerant Subjects 3 (GAUSS-3) Trial. Clin. Cardiol. 2016;39:137–144. doi: 10.1002/clc.22518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang H., Plutzky J., Skentzos S., Morrison F., Mar P., Shubina M., Turchin A. Discontinuation of statins in routine care settings: a cohort study. Ann. Intern. Med. 2013;158:526–534. doi: 10.7326/0003-4819-158-7-201304020-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thompson P.D., Clarkson P., Karas R.H. Statin-associated myopathy. JAMA. 2003;289:1681–1690. doi: 10.1001/jama.289.13.1681. [DOI] [PubMed] [Google Scholar]
- 45.Bruckert E., Hayem G., Dejager S., Yau C., Bégaud B. Mild to moderate muscular symptoms with high-dosage statin therapy in hyperlipidemic patients--the PRIMO study. Cardiovasc. Drugs Ther. 2005;19:403–414. doi: 10.1007/s10557-005-5686-z. [DOI] [PubMed] [Google Scholar]
- 46.Stein E.A., Mellis S., Yancopoulos G.D., Stahl N., Logan D., Smith W.B., Lisbon E., Gutierrez M., Webb C., Wu R. Effect of a monoclonal antibody to PCSK9 on LDL cholesterol. N. Engl. J. Med. 2012;366:1108–1118. doi: 10.1056/NEJMoa1105803. [DOI] [PubMed] [Google Scholar]
- 47.Raal F.J., Giugliano R.P., Sabatine M.S., Koren M.J., Langslet G., Bays H., Blom D., Eriksson M., Dent R., Wasserman S.M. Reduction in lipoprotein(a) with PCSK9 monoclonal antibody evolocumab (AMG 145): a pooled analysis of more than 1,300 patients in 4 phase II trials. J. Am. Coll. Cardiol. 2014;63:1278–1288. doi: 10.1016/j.jacc.2014.01.006. [DOI] [PubMed] [Google Scholar]
- 48.Colhoun H.M., Robinson J.G., Farnier M., Cariou B., Blom D., Kereiakes D.J., Lorenzato C., Pordy R., Chaudhari U. Efficacy and safety of alirocumab, a fully human PCSK9 monoclonal antibody, in high cardiovascular risk patients with poorly controlled hypercholesterolemia on maximally tolerated doses of statins: rationale and design of the ODYSSEY COMBO I and II trials. BMC Cardiovasc. Disord. 2014;14:121. doi: 10.1186/1471-2261-14-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stein E.A., Honarpour N., Wasserman S.M., Xu F., Scott R., Raal F.J. Effect of the proprotein convertase subtilisin/kexin 9 monoclonal antibody, AMG 145, in homozygous familial hypercholesterolemia. Circulation. 2013;128:2113–2120. doi: 10.1161/CIRCULATIONAHA.113.004678. [DOI] [PubMed] [Google Scholar]
- 50.Ridker P.M., Amarenco P., Brunell R., Glynn R.J., Jukema J.W., Kastelein J.J.P., Koenig W., Nissen S., Revkin J., Santos R.D., Studies of PCSK9 Inhibition and the Reduction of vascular Events (SPIRE) Investigators Evaluating bococizumab, a monoclonal antibody to PCSK9, on lipid levels and clinical events in broad patient groups with and without prior cardiovascular events: Rationale and design of the Studies of PCSK9 Inhibition and the Reduction of vascular Events (SPIRE) Lipid Lowering and SPIRE Cardiovascular Outcomes Trials. Am. Heart J. 2016;178:135–144. doi: 10.1016/j.ahj.2016.05.010. [DOI] [PubMed] [Google Scholar]
- 51.Qian L.J., Gao Y., Zhang Y.M., Chu M., Yao J., Xu D. Therapeutic efficacy and safety of PCSK9-monoclonal antibodies on familial hypercholesterolemia and statin-intolerant patients: A meta-analysis of 15 randomized controlled trials. Sci. Rep. 2017;7:238. doi: 10.1038/s41598-017-00316-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fitzgerald K., White S., Borodovsky A., Bettencourt B.R., Strahs A., Clausen V., Wijngaard P., Horton J.D., Taubel J., Brooks A. A Highly Durable RNAi Therapeutic Inhibitor of PCSK9. N. Engl. J. Med. 2017;376:41–51. doi: 10.1056/NEJMoa1609243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.van Poelgeest E.P., Hodges M.R., Moerland M., Tessier Y., Levin A.A., Persson R., Lindholm M.W., Dumong Erichsen K., Ørum H., Cohen A.F., Burggraaf J. Antisense-mediated reduction of proprotein convertase subtilisin/kexin type 9 (PCSK9): a first-in-human randomized, placebo-controlled trial. Br. J. Clin. Pharmacol. 2015;80:1350–1361. doi: 10.1111/bcp.12738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yamamoto T., Harada-Shiba M., Nakatani M., Wada S., Yasuhara H., Narukawa K., Sasaki K., Shibata M.A., Torigoe H., Yamaoka T. Cholesterol-lowering Action of BNA-based Antisense Oligonucleotides Targeting PCSK9 in Atherogenic Diet-induced Hypercholesterolemic Mice. Mol. Ther. Nucleic Acids. 2012;1:e22. doi: 10.1038/mtna.2012.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mitchell T., Chao G., Sitkoff D., Lo F., Monshizadegan H., Meyers D., Low S., Russo K., DiBella R., Denhez F. Pharmacologic profile of the Adnectin BMS-962476, a small protein biologic alternative to PCSK9 antibodies for low-density lipoprotein lowering. J. Pharmacol. Exp. Ther. 2014;350:412–424. doi: 10.1124/jpet.114.214221. [DOI] [PubMed] [Google Scholar]
- 56.Cameron J., Ranheim T., Kulseth M.A., Leren T.P., Berge K.E. Berberine decreases PCSK9 expression in HepG2 cells. Atherosclerosis. 2008;201:266–273. doi: 10.1016/j.atherosclerosis.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 57.Dong B., Li H., Singh A.B., Cao A., Liu J. Inhibition of PCSK9 transcription by berberine involves down-regulation of hepatic HNF1α protein expression through the ubiquitin-proteasome degradation pathway. J. Biol. Chem. 2015;290:4047–4058. doi: 10.1074/jbc.M114.597229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li H., Dong B., Park S.W., Lee H.S., Chen W., Liu J. Hepatocyte nuclear factor 1alpha plays a critical role in PCSK9 gene transcription and regulation by the natural hypocholesterolemic compound berberine. J. Biol. Chem. 2009;284:28885–28895. doi: 10.1074/jbc.M109.052407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Petersen D.N., Hawkins J., Ruangsiriluk W., Stevens K.A., Maguire B.A., O’Connell T.N., Rocke B.N., Boehm M., Ruggeri R.B., Rolph T. A Small-Molecule Anti-secretagogue of PCSK9 Targets the 80S Ribosome to Inhibit PCSK9 Protein Translation. Cell Chem. Biol. 2016;23:1362–1371. doi: 10.1016/j.chembiol.2016.08.016. [DOI] [PubMed] [Google Scholar]
- 60.Shan L., Pang L., Zhang R., Murgolo N.J., Lan H., Hedrick J.A. PCSK9 binds to multiple receptors and can be functionally inhibited by an EGF-A peptide. Biochem. Biophys. Res. Commun. 2008;375:69–73. doi: 10.1016/j.bbrc.2008.07.106. [DOI] [PubMed] [Google Scholar]
- 61.Alghamdi R.H., O’Reilly P., Lu C., Gomes J., Lagace T.A., Basak A. LDL-R promoting activity of peptides derived from human PCSK9 catalytic domain (153-421): design, synthesis and biochemical evaluation. Eur. J. Med. Chem. 2015;92:890–907. doi: 10.1016/j.ejmech.2015.01.022. [DOI] [PubMed] [Google Scholar]
- 62.Zhang Y., Eigenbrot C., Zhou L., Shia S., Li W., Quan C., Tom J., Moran P., Di Lello P., Skelton N.J. Identification of a small peptide that inhibits PCSK9 protein binding to the low density lipoprotein receptor. J. Biol. Chem. 2014;289:942–955. doi: 10.1074/jbc.M113.514067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Landlinger C., Pouwer M.G., Juno C., van der Hoorn J.W.A., Pieterman E.J., Jukema J.W., Staffler G., Princen H.M.G., Galabova G. The AT04A vaccine against proprotein convertase subtilisin/kexin type 9 reduces total cholesterol, vascular inflammation, and atherosclerosis in APOE*3Leiden.CETP mice. Eur. Heart J. 2017;38:2499–2507. doi: 10.1093/eurheartj/ehx260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Crossey E., Amar M.J.A., Sampson M., Peabody J., Schiller J.T., Chackerian B., Remaley A.T. A cholesterol-lowering VLP vaccine that targets PCSK9. Vaccine. 2015;33:5747–5755. doi: 10.1016/j.vaccine.2015.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Galabova G., Brunner S., Winsauer G., Juno C., Wanko B., Mairhofer A., Lührs P., Schneeberger A., von Bonin A., Mattner F. Peptide-based anti-PCSK9 vaccines - an approach for long-term LDLc management. PLoS ONE. 2014;9:e114469. doi: 10.1371/journal.pone.0114469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Raal F.J., Stein E.A., Dufour R., Turner T., Civeira F., Burgess L., Langslet G., Scott R., Olsson A.G., Sullivan D., RUTHERFORD-2 Investigators PCSK9 inhibition with evolocumab (AMG 145) in heterozygous familial hypercholesterolaemia (RUTHERFORD-2): a randomised, double-blind, placebo-controlled trial. Lancet. 2015;385:331–340. doi: 10.1016/S0140-6736(14)61399-4. [DOI] [PubMed] [Google Scholar]
- 67.Ginsberg H.N., Rader D.J., Raal F.J., Guyton J.R., Baccara-Dinet M.T., Lorenzato C., Pordy R., Stroes E. Efficacy and Safety of Alirocumab in Patients with Heterozygous Familial Hypercholesterolemia and LDL-C of 160 mg/dl or Higher. Cardiovasc. Drugs Ther. 2016;30:473–483. doi: 10.1007/s10557-016-6685-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Farnier M., Jones P., Severance R., Averna M., Steinhagen-Thiessen E., Colhoun H.M., Du Y., Hanotin C., Donahue S. Efficacy and safety of adding alirocumab to rosuvastatin versus adding ezetimibe or doubling the rosuvastatin dose in high cardiovascular-risk patients: The ODYSSEY OPTIONS II randomized trial. Atherosclerosis. 2016;244:138–146. doi: 10.1016/j.atherosclerosis.2015.11.010. [DOI] [PubMed] [Google Scholar]
- 69.Cannon C.P., Cariou B., Blom D., McKenney J.M., Lorenzato C., Pordy R., Chaudhari U., Colhoun H.M., ODYSSEY COMBO II Investigators Efficacy and safety of alirocumab in high cardiovascular risk patients with inadequately controlled hypercholesterolaemia on maximally tolerated doses of statins: the ODYSSEY COMBO II randomized controlled trial. Eur. Heart J. 2015;36:1186–1194. doi: 10.1093/eurheartj/ehv028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bays H., Gaudet D., Weiss R., Ruiz J.L., Watts G.F., Gouni-Berthold I., Robinson J., Zhao J., Hanotin C., Donahue S. Alirocumab as add-on to atorvastatin versus other lipid treatment strategies: ODYSSEY OPTIONS I randomized trial. J. Clin. Endocrinol. Metab. 2015;100:3140–3148. doi: 10.1210/jc.2015-1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Institute for Clinical and Economic Review PCSK9 Inhibitor Therapies for High Cholesterol: Effectiveness, Value, and Value-Based Price Benchmarks. Draft Report. 2015 (ICER) [Google Scholar]
- 72.Arrieta A., Hong J.C., Khera R., Virani S.S., Krumholz H.M., Nasir K. Updated cost-effectiveness assessments of PCSK9 inhibitors from the perspectives of the health system and private payers: Insights derived from the FOURIER Trial. JAMA Cardiol. 2017;2:1369–1374. doi: 10.1001/jamacardio.2017.3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Flingai S., Plummer E.M., Patel A., Shresta S., Mendoza J.M., Broderick K.E., Sardesai N.Y., Muthumani K., Weiner D.B. Protection against dengue disease by synthetic nucleic acid antibody prophylaxis/immunotherapy. Sci. Rep. 2015;5:12616. doi: 10.1038/srep12616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Elliott S.T.C., Kallewaard N.L., Benjamin E., Wachter-Rosati L., McAuliffe J.M., Patel A., Smith T.R.F., Schultheis K., Park D.H., Flingai S. DMAb inoculation of synthetic cross reactive antibodies protects against lethal influenza A and B infections. npj Vaccines. 2017;2:18. doi: 10.1038/s41541-017-0020-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Muthumani K., Block P., Flingai S., Muruganantham N., Chaaithanya I.K., Tingey C., Wise M., Reuschel E.L., Chung C., Muthumani A. Rapid and Long-Term Immunity Elicited by DNA-Encoded Antibody Prophylaxis and DNA Vaccination Against Chikungunya Virus. J. Infect. Dis. 2016;214:369–378. doi: 10.1093/infdis/jiw111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Patel A., DiGiandomenico A., Keller A.E., Smith T.R.F., Park D.H., Ramos S., Schultheis K., Elliott S.T.C., Mendoza J., Broderick K.E. An engineered bispecific DNA-encoded IgG antibody protects against Pseudomonas aeruginosa in a pneumonia challenge model. Nat. Commun. 2017;8:637. doi: 10.1038/s41467-017-00576-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Muthumani K., Marnin L., Kudchodkar S.B., Perales-Puchalt A., Choi H., Agarwal S., Scott V.L., Reuschel E.L., Zaidi F.I., Duperret E.K. Novel prostate cancer immunotherapy with a DNA-encoded anti-prostate-specific membrane antigen monoclonal antibody. Cancer Immunol. Immunother. 2017;66:1577–1588. doi: 10.1007/s00262-017-2042-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Backes J.M., Ruisinger J.F., Gibson C.A., Moriarty P.M. Statin-associated muscle symptoms-Managing the highly intolerant. J. Clin. Lipidol. 2017;11:24–33. doi: 10.1016/j.jacl.2017.01.006. [DOI] [PubMed] [Google Scholar]
- 79.Gusarova V., Howard V.G., Okamoto H., Koehler-Stec E.-M., Papadopoulos N., Murphy A.J., Yancopoulos G.D., Stahl N., Sleeman M.W. Reduction of LDL cholesterol by a monoclonal antibody to PCSK9 in rodents and nonhuman primates. Clin. Lipidol. 2012;7:737–743. [Google Scholar]
- 80.Trimble C.L., Morrow M.P., Kraynyak K.A., Shen X., Dallas M., Yan J., Edwards L., Parker R.L., Denny L., Giffear M. Safety, efficacy, and immunogenicity of VGX-3100, a therapeutic synthetic DNA vaccine targeting human papillomavirus 16 and 18 E6 and E7 proteins for cervical intraepithelial neoplasia 2/3: a randomised, double-blind, placebo-controlled phase 2b trial. Lancet. 2015;386:2078–2088. doi: 10.1016/S0140-6736(15)00239-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tebas P., Roberts C.C., Muthumani K., Reuschel E.L., Kudchodkar S.B., Zaidi F.I., White S., Khan A.S., Racine T., Choi H. Safety and immunogenicity of an anti–Zika virus DNA vaccine — preliminary report. N. Engl. J. Med. 2017 doi: 10.1056/NEJMoa1708120. Published online October 4, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Xu Z., Wise M.C., Choi H., Perales-Puchalt A., Patel A., Tello-Ruiz E., Chu J.D., Muthumani K., Weiner D.B. Synthetic DNA delivery by electroporation promotes robust in vivo sulfation of broadly neutralizing anti-HIV immunoadhesin eCD4-Ig. EBioMedicine. 2018;35:97–105. doi: 10.1016/j.ebiom.2018.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.