

Cases of a distinct congenital ichthyosiform erythroderma with evolving white spots were first reported in 1984 and have been assigned a variety of names including ichthyosis en confetti, ichthyosis variegata, congenital reticular ichthyosiform erythroderma (CRIE), and MAUIE syndrome (micropinnae, alopecia universalis, congenital ichthyosis, and ectropion). The first report of this form of heritable ichthyosis was made in 1984 by French dermatologists Camenzind et. al. as “ichthyose en confettis” and two patients were described: an unrelated 14 year-old boy and a 12 year-old girl who had been born erythrodermic and had widespread ichthyosiform erythroderma as a children, but at the age of 10 years began developing patches of normal appearing skin on the trunk which enlarged slowly.1 Biopsy of affected skin showed parakeratosis, acanthosis, and vacuolization of keratinocytes in the upper layers of the epidermis. The female patient was reported in two later publications which noted hypertrichosis and continued enlargement of areas of apparently normal skin with slow development of areas of hyperpigmentation limited to erythrodermic skin. On histopathologic examination, these hyperpigmented macules showed an increased number of dendritic melanocytes.2,3 In another German report of a 57 year old woman with icthyosiform erythroderma and evolving white spots, a diagnosis of CRIE was made on the basis of histology of affected skin and clinical presentation, and biopsy of one of her white spots provided the first evidence that white spots were, in fact, normal skin and normal keratinization was identified.4 Elbaum et. al. and Hendrix et. al. subsequently described two cases of what they coined MAUIE syndrome which is characterized by congenital ichthyosiform erythroderma with evolving white spots, micropinnae, squamous cell skin cancers, and ectropion.5,6 Notably, the Hendrix report again showed that biopsy of a white spot revealed normal skin histology. In 2003, Krunic et. al. described a single case of CRIE with a slightly different phenotype showing small, reticulate areas of apparently normal skin on a background of erythroderma with no other associated clinical findings described.7 These initial case reports were linked by the common features of erythroderma and evolving white spots but differed with respect to white spot size and associated clinical findings. Subsequent reports, including the case series reported by Spoerri, et al. in this issue, have further expanded the phenotypic spectrum of ichthyosis with confetti (IWC).
Unlike other disorders of keratinization in which large kindreds with multiple affected individuals had permitted linkage analysis and positional cloning of the disease-causing genes, IWC appeared to be a very rare sporadic disorder, with one affected index case in each kindred. As such, traditional genetic tools such as linkage analysis were of limited utility in gene identification. Clinical insight, however, from our 15 years of following such patients and from prior clinical reports, provided the basis for a novel approach to genetic investigation.
While the ichthyosiform erythroderma of IWC is congenital, its white spots are not, and we found that in each of our subjects new white spots appeared and expanded over time. These spots showed normal histology and a phylloid morphology previously described in patches of normal skin in other revertant mosaic disorders.8 We recognized that IWC is a disorder with widespread somatic mosaicism and that while multiple mechanisms of reversion are possible, including gene conversion, second site mutation, and back-mutation, the most common type of somatic mutation is loss of heterozygosity (LOH), We predicted that revertant spots resulted from loss of the mutant allele, and then mapped the IWC locus by assessing multiple white spots for allelic loss. We found that all white spots from six independent patients showed large, overlapping regions of copy neutral LOH occurring on chromosome 17q. In each case, LOH extended from a proximal breakpoint to the telomere (Figure 1), supporting mitotic recombination as the mechanism by which the entire chromosome arm carrying the mutant allele is replaced with a copy of the arm carrying the wild-type allele. Subsequent long range amplicon-based sequencing of the 3.2 megabase proximal portion of this locus in blood of two affected individuals and their unaffected parents showed that IWC resulted from de novo frameshift mutations in KRT10 which replace its glycine/serine rich tail with an arginine-rich motif, leading to protein mislocalization to the nucleus and intermediate filament network collapse. Ultimately, all seven of our kindreds would show KRT10 mutations, with all encoding an arginine-rich carboxy-terminal tail of variable length.9
Overlapping Independent Loss of Heterozygosity Events on Chromosome 17 in IWC White Spots.
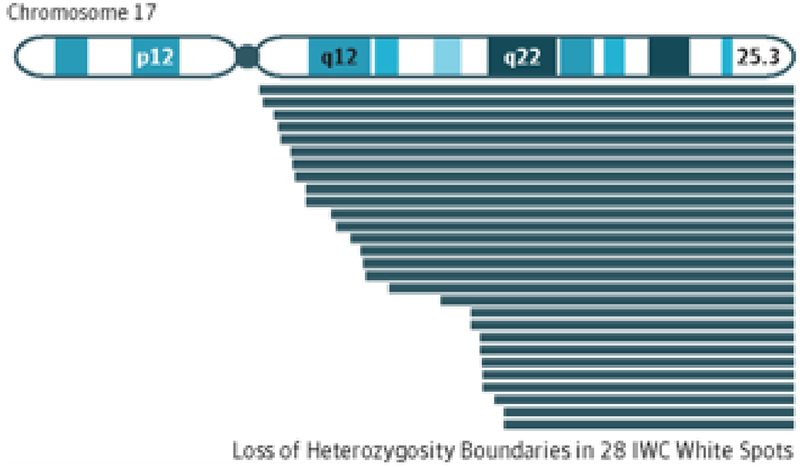
Using high-density single nucleotide polymorphism genotyping platforms, DNA from 28 white spots was run in parallel with peripheral blood genomic DNA. In paired analysis, all samples showed overlapping loss of heterozygosity of a single region of chromosome 17q (blue lines).
Overlapping breakpoints identified the proximal boundary for the ichthyosis with confetti (IWC) losus.
The relationship between KRT10 mutations which cause IWC and the frequent appearance of revertant clones in this disorder remains a mystery. KRT10 mutations that do not result in an arginine-rich tail have never been associated with revertant mosaicism. Arginine-rich motifs encode nuclear localization sequences which lead both to RNA binding and to nuclear entry, and mutant keratin10 was shown to both disrupt the normal intermediate filament network and to accumulate within the nucleus, specifically within nucleoli. Notably, carboxy-terminal frameshift mutations in loricrin causing Vohwinkel syndrome encode a frameshifted protein with a similar arginine-rich motif, but this disorder shows no evidence of reversion.10 While we presume that mutant keratin10 protein contributes to selective growth advantage for revertant clones, whether it acts directly or indirectly in inducing mitotic recombination is yet unknown.
The ability to genetically define this group of patients has enabled identification of a larger cohort of subhect, permitting careful clinical characterization of the disease phenotype. In this issue, Spoerri, et al. identify seven IWC subjects, all with KRT10 frameshift mutations, and have performed systematic phenotyping of 6 members of this cohort. Consistent findings include erythroderma from birth, ear malformation, hypoplasia of the mammillae, short stature/low weight, and dorsal acral hypertrichosis. Findings present in a smaller fraction of their cohort include ectropion, hypercurvature of the fingernails, joint contractures, strabismus, lash hypoplasia and other clinical features. There is no apparent correlation between identified KRT10 mutations and phenotype.
In our own IWC cohort of 20 individuals in 18 kindreds, some of which are multi-generational, we find a similar spectrum of clinical findings. All affected individuals have KRT10 mutations resulting in frameshift into an arginine-rich reading frame, with 66% having splice site mutations. 8 of our index cases have contractures with gait abnormality; in three cases these have progressed to cause marked disability. In four subjects with moderately severe contractures, neurologic evaluation has revealed peripheral hyper-reflexia which would not alone be considered sufficient to cause contracture, and other age-matched subjects with an identical mutation have no contractures at all. Given the high incidence of contractures in our cohort and that reported by Spoerri et. al., evidence of progression to disability, and lack of a clear genotype-phenotype correlation, we advocate that all subjects with IWC should be evaluated by a neurologist for gait abnormality, contractures, and hyper-reflexia and that physical and occupational therapy be instituted for subjects with these findings.
Of the subjects in our cohort who are greater than 30 years of age, 3/6 have had at least one non-melanoma skin cancer (NMSC). Frequent, ultimately lethal, squamous cell carcinomas also appear to be a feature of MAUIE syndrome which features phylloid white spots and other features of IWC, though it has yet to be demonstrated whether this syndrome has the same genetic basis.5,6.Since revertant spots in IWC occur via mitotic recombination, it is possible that LOH events leading to reversion will also reduce a heterozygous tumor suppressor mutation to homozygosity. TP53 and other tumor suppressors lie on 17q distal to KRT10, and protein-damaging mutations in these genes on the non-mutant KRT10 allele could increase NMSC risk, with more severe mutations possibly causing the MAUIE syndrome phenotype. As a part of routine care, IWC patients should therefore have regular examination for skin cancer.
Altogether, reported cases of IWC define a broad phenotypic spectrum without clear genotype-phenotype correlation. The determinants of the marked clinical variation in these subjects are yet to be identified, though there are likely both genetic and enironmental modifiers. Understanding of IWC phenotypic variation will enable accurate diagnosis, and, possibly, early intervention for some its potentially more severe complications including contractures and malignancy.
References
- 1.Camenzind M, Harms M, Chavaz P, Saurat JH. [Confetti ichthyosis]. Annales de dermatologie et de venereologie. 1984;111(8):675–676. [PubMed] [Google Scholar]
- 2.Brusasco A, Tadini G, Cambiaghi S, Ermacora E, Grimalt R, Caputo R. A case of congenital reticular ichthyosiform erythroderma--ichthyosis ‘en confettis’. Dermatology. 1994;188(1):40–45. [DOI] [PubMed] [Google Scholar]
- 3.Brusasco A, Cambiaghi S, Tadini G, Berti E, Caputo R. Unusual hyperpigmentation developing in congenital reticular ichthyosiform erythroderma (ichthyosis variegata). The British journal of dermatology. November 1998;139(5):893–896. [DOI] [PubMed] [Google Scholar]
- 4.Marghescu S, Anton-Lamprecht I, Rudolph PO, Kaste R. [Congenital reticular ichthyosiform erythroderma]. Der Hautarzt; Zeitschrift fur Dermatologie, Venerologie, und verwandte Gebiete. October 1984;35(10):522–529. [PubMed] [Google Scholar]
- 5.Hendrix JD Jr., Patterson JW, Greer KE. Skin cancer associated with ichthyosis: the MAUIE syndrome. Journal of the American Academy of Dermatology. December 1997;37(6):1000–1002. [DOI] [PubMed] [Google Scholar]
- 6.Elbaum DJ, Kurz G, MacDuff M. Increased incidence of cutaneous carcinomas in patients with congenital ichthyosis. Journal of the American Academy of Dermatology. November 1995;33(5 Pt 2):884–886. [DOI] [PubMed] [Google Scholar]
- 7.Krunic AL, Palcesky D, Busbey S, Medenica M. Congenital reticular ichthyosiform erythroderma--ichthyosis variegata: a case report and review of the literature. Acta dermato-venereologica. 2003;83(1):36–39. [DOI] [PubMed] [Google Scholar]
- 8.Jonkman MF, Scheffer H, Stulp R, et al. Revertant mosaicism in epidermolysis bullosa caused by mitotic gene conversion. Cell. February 21 1997;88(4):543–551. [DOI] [PubMed] [Google Scholar]
- 9.Choate KA, Lu Y, Zhou J, et al. Mitotic recombination in patients with ichthyosis causes reversion of dominant mutations in KRT10. Science. October 1 2010;330(6000):94–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maestrini E, Monaco AP, McGrath JA, et al. A molecular defect in loricrin, the major component of the cornified cell envelope, underlies Vohwinkel’s syndrome. Nature genetics. May 1996;13(1):70–77. [DOI] [PubMed] [Google Scholar]