

Insulin resistance has long been established as the key metabolic defect linking obesity with its related cardiometabolic diseases, including type 2 diabetes, hypertension, dyslipidemia, accelerated atherosclerosis and even cancers. In recent years, it has also been increasingly recognized that insulin resistance in obesity is closely related to adipose tissue inflammation, after the demonstration of macrophage infiltration of the adipose tissue in obese rodents and humans. In one of the seminal studies reporting this observation,1 infiltrating macrophages were shown to be the predominant source of the pro‐inflammatory cytokine, tumor necrosis factor‐alpha (TNF‐α), which in mice deficiency in both TNF‐α and its receptors, was shown to play a key role in obesity‐induced insulin resistance. This has led to the concept that macrophage infiltration, leading to adipose tissue inflammation, is a major event in the development of obesity‐related insulin resistance. However, whether adipose tissue macrophage (ATM) infiltration is crucial to the initiation of insulin resistance in the obese state remains a controversial issue.
A recent study by Shimobayashi et al.2 has provided convincing evidence that insulin resistance precedes the accumulation of classically activated or pro‐inflammatory macrophages, also known as M1 macrophages, in the epididymal white adipose tissue (eWAT) in mice with diet‐induced obesity (DIO). In their study, insulin resistance, assessed by insulin‐stimulated glucose uptake and insulin tolerance test, was evident by week 4 of a high‐fat diet (HFD). However, M1 macrophage accumulation in eWAT was only mildly increased by week 10, when a significant increase in TNF‐α expression was not yet evident. The ATMs were likely recruited to the eWAT by the chemokine monocyte chemoattractant protein 1 (MCP1), the expression of which in the eWAT was significantly elevated by 10 weeks, but not at 4 weeks of a HFD. Their findings agreed with those of a previous study that reported the presence of insulin resistance in eWAT of DIO mice after 1 week of a HFD, whereas significant elevation in the messenger ribonucleic acid levels of macrophage‐specific markers, such as F4/80, in eWAT was only evident at 16 weeks. The temporal relationship of insulin resistance to ATM infiltration in these studies clearly suggests that insulin resistance can lead to ATM accumulation and adipose tissue inflammation.
So how does insulin resistance lead to ATM infiltration? Shimobayashi et al.2 showed that the mammalian target of rapamycin complex 2 (mTORC2) knockout mice, with genetically induced adipose‐specific insulin resistance, developed earlier and greater ATM infiltration on HFD, starting at 6 weeks of HFD, compared with the wild‐type mice. This increase in ATM accumulation could be inhibited by treatment with a MCP1‐neutralizing antibody, and was likely mediated by the increased adipocyte expression of MCP1 in the eWAT of the knockout mice, which also had raised MCP1 circulating levels. The ability of insulin to suppress MCP1 expression in normal adipocytes further supported the induction of ATM infiltration by insulin resistance in obesity. It was also noted that the difference in MCP1 expression between the knockout and wild‐type mice was only seen when the mice were on a HFD and, in vitro, could be blocked by treatment of the adipocytes with a c‐Jun NH2‐terminal kinase (JNK) inhibitor, suggesting that JNK stimulation by obesity is required to stimulate MCP1 expression, in conjunction with impaired mTORC2 signaling. Furthermore, in human omental fat, the authors showed that serine phosphorylation of protein kinase B beta, downstream of mTORC2 signaling, was reduced in obese individuals who also had higher MCP1 expression. Thus, in obesity, adipocyte‐specific insulin resistance can initiate ATM recruitment through reduced mTORC2‐mediated repression of MCP1 expression in the adipocytes. The authors further cited other studies in which different steps along the insulin signaling pathway, when enhanced, could protect against ATM infiltration on a HFD. For instance, in the study by Morley et al.,3 mice with adipocyte‐specific deletion of the protein phosphatase and tensin homolog, a phosphatase critically involved in turning off the insulin signal transduction cascade, had enhanced insulin signaling specifically in mature adipocytes, and were protected against ATM infiltration, despite weight gain, on a long‐term HFD. They also cited the study by McCurdy et al 4, which showed that impaired insulin signaling at the level of phosphoinositide 3‐kinase could be another key step for the initiation and propagation of ATM accumulation in mice with DIO.
Thus, there are multiple steps in the insulin signaling cascade at which adipocyte‐specific insulin resistance has been shown to initiate ATM infiltration in mice with DIO. How then does obesity lead to insulin resistance in the first place? Again, multiple mechanisms might be involved. First, lipotoxicity is known to induce insulin resistance. Excess intake of saturated fatty acids, such as palmitate, has been implicated in endoplasmic reticulum stress, as well as activation of JNK and activator protein‐1‐mediated inflammation, which can all lead to insulin resistance. Palmitate has also been shown to contribute to the synthesis of diacylglycerol and ceramide, which activate the stress kinases including nuclear factor kappa B and JNK, and cause insulin resistance, through the inhibition of insulin receptor substrate‐1 and protein kinase B, respectively. Indeed, increased ceramide and sphingomyelin species were found in the adipose tissue at the early stages of insulin resistance in DIO mice5, and increased sphingolipids were present in the adipose tissue of obese humans. Increased ceramide, diacylglycerol and non‐esterified fatty acids were also found in muscle and liver by 3 days of a HFD in mice, accompanied by impaired glucose tolerance, and were not affected by macrophage depletion.6 In contrast, chronic nutrient overload causes pathological expansion of the adipose tissue, resulting in tissue hypoxia, adipocyte cell death, oxidative and endoplasmic reticulum stress, which can all promote insulin resistance. Hypoxia also contributes to the dysregulated secretion of adipokines, with the enhanced secretion of pro‐inflammatory adipokines, such as interleukin‐6, adipocyte fatty acid binding protein, leptin and MCP‐1, and reduced secretion of anti‐inflammatory adipokines, such as adiponectin. Such dysregulation leads, on the one hand, to impaired insulin signaling and, on the other hand, to recruitment of ATM. The production by the ATMs of pro‐inflammatory cytokines, such as TNF‐α, interleukin‐6, adipocyte fatty acid‐binding protein and MCP‐1, further aggravates insulin resistance as well as adipose tissue inflammation, directly and through activating inflammatory pathways, such as JNK and nuclear factor kappa B, setting up a vicious cycle. Thus, obesity‐related insulin resistance likely occurs early in the course of DIO, independent of ATM infiltration, and can initiate ATM recruitment, together with other obesity‐induced stress pathways, which contribute to both insulin resistance and ATM recruitment.2 However, the adipose tissue inflammation that ensues does play a significant role in perpetuating long‐term insulin resistance,6 in the adipose tissue as well as in other tissues involved in glucose metabolism.7 Downregulation of JNK in the adipose tissue, which led to marked reduction in ATM infiltration, was shown to result in reduced circulating levels of pro‐inflammatory cytokines, but raised adiponectin levels, and beneficial effects on hepatic steatosis as well as hepatic, muscle and systemic insulin resistance.7
Over the past few decades, the obesogenic environment has pushed the prevalence of various insulin‐resistant disorders, such as non‐alcoholic fatty liver disease and type 2 diabetes, to global epidemic levels. Although emerging evidence has confirmed the causal role of insulin resistance in the development of inflammation in the adipose tissue, the impact of adipose tissue insulin resistance on whole‐body glucose metabolism might be mediated through adipose tissue inflammation.7 In the adipose tissue, it is likely that obesity‐related cellular perturbations activate the cascade of events leading to insulin resistance, and thereby initiate and potentiate local tissue inflammation. Subsequently, the latter maintains and perpetuates adipose tissue and insulin resistance, and likely impairs whole‐body insulin sensitivity, through inducing chronic subclinical systemic inflammation (Figure 1), the role of which in disorders of insulin resistance has led to clinical trials with anti‐inflammatory drugs. Examples of such trials include the use of salsalate in type 2 diabetes and monoclonal antibody against interleukin‐1 beta in cardiovascular diseases, the definitive conclusions from which are as yet unavailable. In contrast, the importance of obesity per se in both the initiation of insulin resistance and adipose tissue inflammation would suggest that public health policies to promote healthy diet with avoidance of excess calories and saturated fats, and regular physical activity to maintain optimal bodyweight, should be the best way to reduce the cardiometabolic consequences of obesity.
Figure 1.
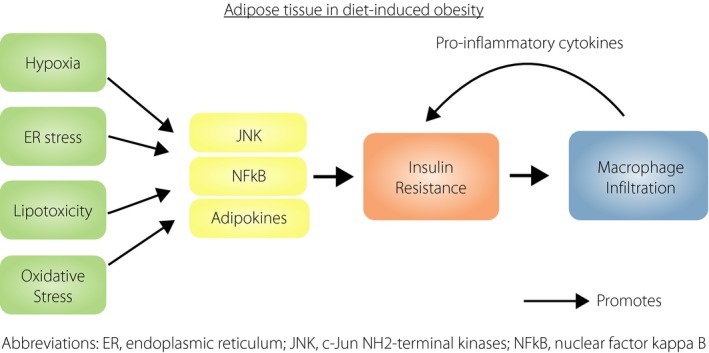
The relationship between adipose tissue insulin resistance and inflammation in diet‐induced obesity. ER, endoplasmic reticulum; JNK, c‐Jun NH2‐terminal kinase; NFκB, nuclear factor kappa B.
Disclosure
The authors declare no conflict of interest.
References
- 1. Weisberg SP, McCann D, Desai M, et al Obesity is associated with macrophage accumulation in adipose tissue. J Clin Investig 2003; 112: 1796–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shimobayashi M, Albert V, Woelnerhanssen B, et al Insulin resistance causes inflammation in adipose tissue. J Clin Investig 2018; 128: 1538–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Morley TS, Xia JY, Scherer PE. Selective enhancement of insulin sensitivity in the mature adipocyte is sufficient for systemic metabolic improvements. Nat Commun 2015; 6: 7906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. McCurdy CE, Schenk S, Holliday MJ, et al Attenuated Pik3r1 expression prevents insulin resistance and adipose tissue macrophage accumulation in diet‐induced obese mice. Diabetes 2012; 61: 2495–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Turner N, Kowalski GM, Leslie SJ, et al Distinct patterns of tissue‐specific lipid accumulation during the induction of insulin resistance in mice by high‐fat feeding. Diabetologia 2013; 56: 1638–1648. [DOI] [PubMed] [Google Scholar]
- 6. Lee YS, Li P, Huh JY, et al Inflammation is necessary for long‐term but not short‐term high‐fat diet‐induced insulin resistance. Diabetes 2011; 60: 2474–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhang X, Xu A, Chung SK, et al Selective inactivation of c‐Jun NH2‐terminal kinase in adipose tissue protects against diet‐induced obesity and improves insulin sensitivity in both liver and skeletal muscle in mice. Diabetes 2011; 60: 486–495. [DOI] [PMC free article] [PubMed] [Google Scholar]