

Supplemental Digital Content is available in the text.
Keywords: cigarette smoking; coronary disease; gene-environment interaction; genetic predisposition to disease; ppolymorphism, genetic; smoking
Abstract
Background:
Coronary heart disease (CHD) is a multifactorial disease with both genetic and environmental components. Smoking is the most important modifiable risk factor for CHD. Our aim was to test whether the increased CHD incidence by smoking is modified by genetic predisposition to CHD.
Methods and Results:
Our study included 24 443 individuals from the MDCS (Malmö Diet and Cancer Study). A weighted polygenic risk score (PRS) was created by summing the number of risk alleles for 50 single-nucleotide polymorphisms associated with CHD. Individuals were classified as current, former, or never smokers. Interactions were primarily tested between smoking status and PRS and secondarily with individual single-nucleotide polymorphisms. Then, the predictive use of PRS for CHD incidence was tested among different smoking categories. During a median follow-up time of 19.4 years, 3217 incident CHD cases were recorded. The association between smoking and CHD was modified by the PRS (Pinteraction=0.005). The magnitude of increased incidence of CHD by smoking was highest among individuals in the lowest tertile of PRS (odds ratio, 1.42; 95% confidence interval, 1.29–1.56 per smoking risk category) compared with the highest tertile (odds ratio, 1.20; 95% confidence interval, 1.11–1.30 per smoking risk category). This interaction was stronger among men (Pinteraction=0.001) compared with women (Pinteraction=0.44). The PRS provided a significantly better net reclassification and discrimination on top of traditional risk factors among never smokers compared with current smokers (P<0.001).
Conclusions:
Genetic predisposition to CHD modifies the associated increased CHD risk by smoking. The PRS has a better predictive use among never smokers compared with smokers.
See Editorial by Elosua
Clinical Perspective
Cigarette smoking is the most important modifiable cause of coronary heart disease (CHD). However, it is not known whether the risk increase by smoking is uniform among all individuals. Therefore, we tested the association between smoking and CHD among individuals with different levels of genetic predisposition for CHD, measured as a polygenic risk score constructed from 50 genetic variants known to be associated with CHD. In around 24 000 middle-aged Swedish participants followed up for around 20 years, our study found that the magnitude of CHD risk increase by smoking is not uniform among all individuals and that the relative risk increase is higher among individuals with lower polygenic risk burden compared with those with higher burden. This observation indicates that smoking cessation may provide higher relative risk reduction in CHD among individuals with low polygenic risk when compared with those with high polygenic risk. In addition, our results highlight a similar 2-fold risk increase in CHD among never smokers with high polygenic risk burden and current smokers with low polygenic risk burden, compared with never smokers with a low polygenic risk burden. Finally, our study indicates that the clinical use of polygenic risk scores in prediction of CHD may be dependent on certain environmental factors as we found that the polygenic risk for CHD can provide better risk prediction, discrimination, and net reclassification on top of traditional risk factors among never smokers compared with current smokers.
Cigarette smoking is the strongest modifiable risk factor for cardiovascular morbidity and mortality,1 and smoking cessation decreases the risk for coronary heart disease (CHD). Former smokers have a lower CHD risk than current smokers, and the difference increases with time since quitting smoking.2 However, the magnitude of deleterious effects of smoking on cardiovascular health may differ between individuals.
During the past decade, genome-wide association studies have led to the discovery of >50 single-nucleotide polymorphisms (SNPs) that associate with CHD. Although some of these variants have been shown to associate with blood lipid traits and blood pressure, the functional consequences of the majority of the associated loci are still unknown,3,4 which highlights the importance of undiscovered pathways in the development of CHD. Polygenic risk scores (PRSs) based on SNPs associated with an increased risk for CHD have been reported to modestly improve CHD risk prediction on the top of conventional risk factors.5–7 A recent study by Tada et al8 showed that adding a PRS to a model based on established risk factors for CHD, including self-reported family history, improved reclassification and discrimination.
We have previously reported that cigarette smoking modifies the effect of the top SNP (rs4977574) at the chromosome 9p21 locus, which is the strongest CHD-associated locus. We observed that smoking attenuated the increased risk for CHD associated with having 1 or 2 risk alleles.9 However, to the best of our knowledge, no studies have tested interaction between smoking and a PRS for CHD, although such studies have been conducted in the context of other phenotypes.10–12 In addition, in a recent study involving 55 685 participants, a PRS for CHD and a favorable lifestyle, defined as adherence to at least 3 of the 4 factors (no current smoking, no obesity, regular physical activity, and a healthy diet), were independently associated with susceptibility to CHD.13 However, this study did not address nor answer the question whether the individual lifestyle factors may interact with a PRS on the risk of CHD.
The aim of our study was to challenge the question whether smoking, the strongest individual risk factor for CHD, interacts with PRS for CHD. To investigate that, we studied associations between smoking and incidence of CHD stratified by PRS. We then investigated whether genetic prediction of CHD differs according to smoking behavior. Finally, we tested interactions between each of the CHD-associated SNPs and smoking on the risk of CHD.
Methods
The data, analytic methods, and study materials will not be made available to other researchers for purposes of reproducing the results or replicating the procedure. The authors confirm that some access restrictions apply to the data underlying the findings as the consent signed by study participants does not allow the public release of their data. However, data for the MDCS (Malmö Diet and Cancer Study) are available from the Board of MDCS at the Lund University for researchers who meet the criteria for access to confidential data.
Study Population
The MDCS is a population-based prospective study that recruited 30 447 subjects from a source population of 74 138 individuals born between 1923 and 1950 and living in the city of Malmö in southern Sweden. Baseline data were collected between 1991 and 1996: all subjects completed a questionnaire about lifestyle and socioeconomic factors, such as smoking habits, physical activity, education, and medication history. In addition, nurses drew nonfasting blood samples for storage and measured anthropometric factors and blood pressure. In addition, a modified diet history methodology consisting of a 7-day food diary, a 168-item questionnaire and a diet history interview was used to collect information on dietary habits.14,15 Details of the design of MDCS have been reported elsewhere.16 After excluding subjects with prevalent CHD (n=774), those with missing data on smoking (n=1965), and individuals with missing or bad quality DNA, defined as those with missing genotypes for >10 of the 50 SNPs making up the PRS, our study population included 24 443 individuals.
The study participants were stratified into 3 smoking categories based on self-reported smoking status: never smokers, former smokers, and current smokers (defined as reporting cigarette use during the last year). Blood pressure measurements were performed using a mercury column sphygmomanometer in the supine position after 10 minutes of rest. Body weight (kg) was measured using a balance beam scale with subjects wearing light clothing and no shoes. Height (cm) was measured using a fixed stadiometer. Body mass index was calculated as the ratio of weight in kilograms to height in meters squared. Total energy and nutrient intakes were calculated from the average daily consumption of food groups using the Malmö Diet and Cancer Food and Nutrient Database which was designed specifically for MDCS and was derived from the Swedish Food Database PC KOST2-93 of the Swedish National Food Administration.14,15 Family history was based on the self-reported response of individuals to a questionnaire on whether their father, mother, or siblings had a history of myocardial infarction. The education variable was created by assessing each participant’s highest educational level and creating groups of ≤8 years, 9 to 10 years, 11 to 13 years, and university level. Physical activity was measured through an extensive lifestyle questionnaire based on the Minnesota Leisure Time Physical Activity Questionnaire. A leisure-time physical activity score was created and divided into tertiles. An alcohol intake variable was created classifying individuals into 4 categories based on sex-specific grams of total alcohol consumed per day: abstainers, low consumption (<15 g/d in women or <20 g/d in men), medium consumption (15–30 g/d in women or 20–40 g/d in men) or high consumption (>30 g/d in women or >40 g/d in men). The levels of Apo (apolipoprotein) AI and ApoB were measured in nonfasted plasma samples of the entire MDCS by Quest Diagnostics (San Juan Capistrano, CA), blinded to case–control status, using an immunonephelometric assay run on the Siemens BN II (Siemens, Newark, DE). The interassay variability was <4.0% for both Apo AI and Apo B. Prevalent diabetes mellitus was defined as self-reported physician diagnosis or use of antidiabetic medication, fasting whole blood glucose of ≥6.1 mmol/L (corresponding to fasting plasma glucose concentration of ≥7.0 mmol/L), or by linkage to the local or national diabetes mellitus registries.17–21 The MDCS protocols were approved by the ethics committee at Lund University. All participants provided written informed consent.
Ascertainment of CHD
CHD was defined as fatal or nonfatal myocardial infarction, coronary artery bypass graft, percutaneous coronary intervention, or death because of ischemic heart disease. Three previously validated19,22 Swedish registers were used to ascertain cases: the Swedish Hospital Discharge Register, the Swedish Cause of Death Register and the Swedish Coronary Angiography and Angioplasty Registry. Myocardial infarction was defined based on either International Classification of Diseases, Ninth Revision, code 410 or International Classification of Diseases, Tenth Revision, code I21. Information about coronary artery bypass graft was identified from the national Swedish classification systems of surgical procedures and defined as procedure codes 3065, 3066, 3068, 3080, 3092, 3105, 3127, or 3158 (the Op6 system) or procedure code FN (the KKÅ97 system). Percutaneous coronary intervention was identified from the Swedish Coronary Angiography and Angioplasty Registry. Death because of ischemic heart disease was defined as International Classification of Diseases, Ninth Revision, codes 412 and 414 or International Classification of Diseases, Tenth Revision, codes I22, I23, and I25.
Genotyping and Construction of PRSs
The participants of MDCS were genotyped using a multiplex method that combines polymerase chain reaction, allele-specific oligonucleotide ligation assays, and hybridization to oligonucleotides coupled to Luminex 100TM xMAPTM microspheres (Luminex, Austin, TX).8 An automated clustering algorithm was used to call genotypes, and the resulting clusters were further visually inspected for outliers that were manually called. The resulting genotypes had >99% concordance with a second method using real-time allele-specific polymerase chain reaction.23 All SNPs passed the Hardy–Weinberg equilibrium test with a P>0.001 (0.05/50), and the SNP call rates ranged between 95.7% and 99.6%. Of all the 27 754 individuals that were genotyped, 1040 (3.7%) were excluded because of missing genotypes for ≥11 SNPs.
We used a PRS described previously by Tada et al8 and comprising 50 SNPs. In brief, we included all SNPs or their proxies (r2≥0.86) that were reported to pass genome-wide significance in genome-wide association studies24–27 published before and including the CARDIoGRAMplusC4D meta-analysis in 2013.4 Table I in the Data Supplement provides detailed information on each included SNP and reference to the risk estimate used to create PRS. To compute the PRS, the risk estimate per risk allele of each SNP was log transformed and multiplied by the amount of risk alleles (0, 1, or 2) and then summing the products. We imputed genotypes for samples missing ≤10 SNPs using a random sampling method that generates genotypes based on frequencies among the group of participants with CHD events and among the participants without CHD events. For a sensitivity analysis, we additionally created a 49 SNP PRS after excluding the strongest CHD-associated SNP (rs4977574) at the 9p21 locus.
Statistical Analysis
Baseline characteristics were created for all individuals and for strata defined by tertiles of PRS or by smoking status. As the assumption of proportionality was violated in Cox regression analyses, the main effects and interactions were assessed by constructing logistic regression models. Smoking status was modeled as a categorical or continuous variable with never smokers coded as 0, former smokers as 1 and current smokers as 2. The PRS was stratified into tertiles and modeled as a categorical or a continuous variable. Interactions were tested by including the continuous smoking variable and the z score transformed continuous PRS variable together with a multiplicative term of these variables. Two models were tested: the first including baseline age, sex, education, total energy intake, physical activity, and alcohol consumption (model 1), and the second including model 1 with further adjustments for CHD traditional risk factors including ApoB as a proxy for LDL (low-density lipoprotein) cholesterol, Apo AI as a proxy for HDL (high-density lipoprotein) cholesterol, systolic blood pressure, antihypertensive medication, diabetes mellitus at baseline, and family history of myocardial infarction (model 2). As both the risk of CHD and smoking behavior markedly differ between men and women, we additionally performed all analyses stratified by sex. Interactions were also tested between smoking and family history of myocardial infarction, to investigate whether family history reflects the results with the PRS. Sensitivity analyses were performed excluding rs4977574 at the 9p21 locus, which is the strongest locus associated with CHD and has previously been reported to interact with smoking.9 Additional sensitivity analysis was done after censoring at 10 years of follow-up to account for changes in smoking behavior during longer periods of follow-up. Although our primary hypothesis concerned interaction between the CHD PRS and smoking, we additionally performed interaction analyses for each of the 50 SNPs included in the PRS among all individuals, and among men and women separately, to provide information about the contribution of each individual SNP. To investigate whether the interaction could be driven by SNPs associated with specific known risk factors, we created 3 trait-specific PRSs using CHD SNPs that were previously associated with lipid traits, blood pressure, or type 2 diabetes mellitus. We investigated the associations of each of the 50 SNPs with lipids, blood pressure, and type 2 diabetes mellitus in previous genome-wide association studies using phenoscanner (http://www.phenoscanner.medschl.cam.ac.uk/). For the lipids PRS, we included 17 CHD SNPs that showed associations with total, LDL or HDL cholesterol, triglycerides or dyslipidemia at P<0.001 (0.05/50). For the blood pressure PRS, we included 8 CHD SNPs that previously showed associations with systolic blood pressure, diastolic blood pressure, or hypertension at P<0.001. Finally, for the type 2 diabetes mellitus PRS, we included 4 CHD that showed association with type 2 diabetes mellitus at P<0.001 (Table I in the Data Supplement).
We then performed C statistics and net reclassification improvement (NRI) analyses to evaluate whether the PRS provides different risk predictions among individuals with different smoking status. We tested the NRI and integrated discrimination improvement (IDI) when adding the PRS to a risk prediction model based on traditional risk factors. The model comprised age, sex, ApoB, Apo AI, systolic blood pressure, antihypertensive medication, diabetes mellitus at baseline, and family history of myocardial infarction. These analyses were also performed among men and women separately. Finally, we performed NRI analysis among current and never smokers by adding the interaction term between smoking and PRS to the prediction model including age, sex, ApoB, Apo AI, systolic blood pressure, antihypertensive medication, diabetes mellitus at baseline, family history of myocardial infarction, smoking, and PRS. All analyses were conducted using STATA SE 13.1 (StataCorp LP).
Results
The baseline characteristics of MDCS participants are provided in Tables 1 and 2 stratified according to tertiles of PRS and smoking status, respectively. Similar descriptive data among men and women separately are provided in Tables II through V in the Data Supplement. During a median follow-up period of 19.4 years, 3217 (13.2%) participants developed CHD (21.1% of men and 8.3% of women). We observed a significant association between the PRS and CHD in our study population (odds ratio [OR] per tertile, 1.31; 95% confidence interval [CI], 1.25–1.37). Compared with never smokers, former smokers (OR, 1.19; 95% CI, 1.08–1.31) and current smokers (OR, 1.67; 95% CI, 1.52–1.85) were at increased risk for CHD adjusted for age, sex, total energy intake, alcohol intake, physical activity, and education.
Table 1.
Baseline Characteristics of the MDCS (Malmö Diet and Cancer Study) Participants According to Tertiles of the PRS for Coronary Heart Disease
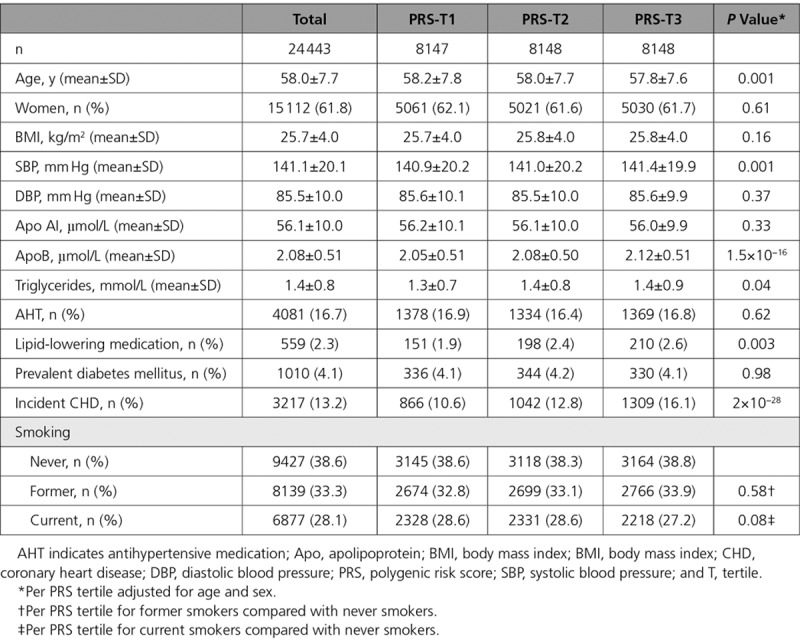
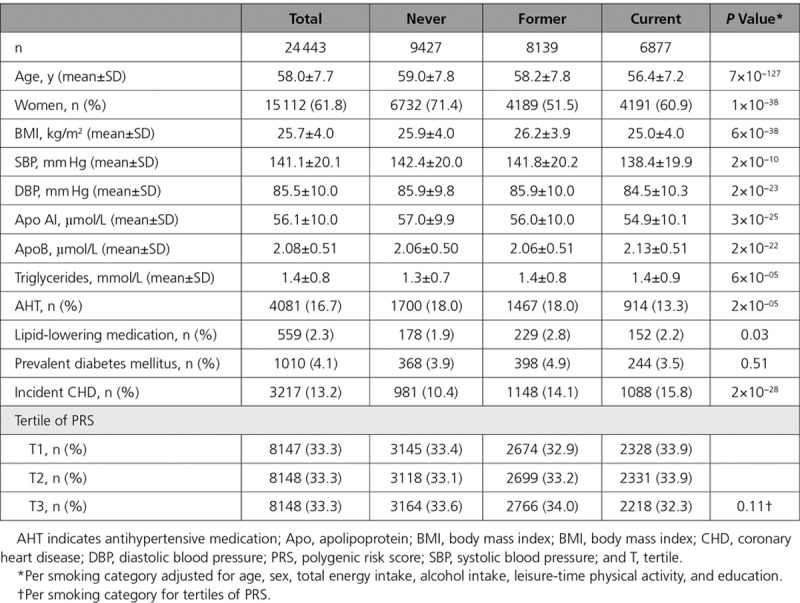
Table 2.
Baseline Characteristics of the MDCS (Malmö Diet and Cancer Study) Participants According to Smoking Status
Interaction Between the PRS and Smoking
The increased risk for CHD conferred by smoking was modified by the PRS (Pinteraction=0.005 and 0.009 for model 1 and 2, respectively). The magnitude of CHD risk elevation by smoking was attenuated among individuals with higher PRS. Compared with never smokers, current smokers in the lowest tertile of PRS had higher CHD risk (OR, 2.01; 95% CI, 1.66–2.42) than current smokers in the highest tertile of PRS (OR, 1.44; CI, 1.23–1.68). A similar but weaker trend was observed among former smokers (Table 3). In a combined analysis of smoking status and PRS, never smokers in the third tertile of PRS were already at a similar risk (2-fold) to current smokers in the first tertile of PRS. The relative increase of CHD risk among current smokers was lower in the highest PRS tertile compared with the lowest, when compared with never smokers (Figure).
Table 3.
Association Between Smoking Status and Coronary Heart Disease in Tertiles of PRS in the MDCS (Malmö Diet and Cancer Study)
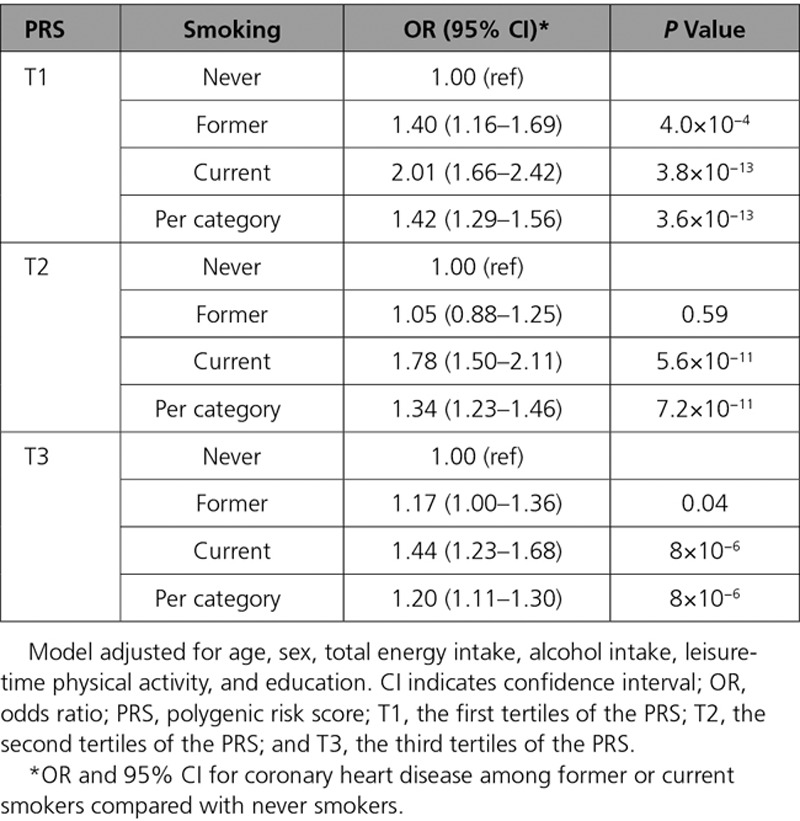
Figure.
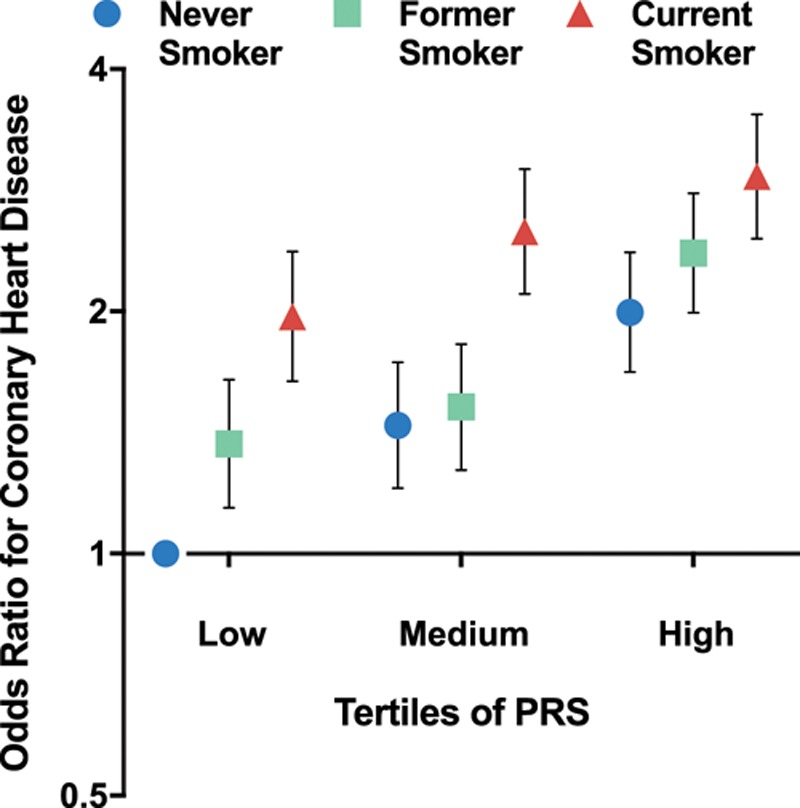
Odds ratio (OR) for coronary heart disease according to tertiles of polygenic risk score (PRS) and smoking status. Individuals who are in the lowest tertile of PRS and are never smokers were considered as a reference group. Smoking was associated with higher risk for coronary heart disease among individuals within each tertile of PRS. The magnitude of relative risk increase by smoking was higher among individuals with low PRS (OR, 1.42; 95% confidence interval [CI], 1.29–1.56) compared with individuals with high PRS (OR, 1.20; 195% CI,.11–1.30). Never smokers in the highest tertile of PRS had a similar risk (OR, 1.97; 95% CI, 1.64–2.37) to current smokers (OR, 1.99; 95% CI, 1.68–2.37) in the lowest tertile of PRS compared with the reference group.
The interaction was observed to be stronger among men (Pinteraction=0.001 and 0.001 for model 1 and model 2, respectively) than among women (Pinteraction=0.44 and 0.67 for model 1 and model 2, respectively; Tables VI and VII in the Data Supplement; Figures I and II in the Data Supplement). The interaction remained significant with the PRS of 49 SNPs excluding the strongest CHD-associated SNP rs4977574 at the 9p21 locus (Pinteraction=0.015 and 0.022 for model 1 and model 2, respectively). To investigate whether changes in smoking behavior during the long follow-up could be driving our results, we censored our follow-up at 10 years (nCHD=1862). Censoring did not dramatically change our results (Pinteraction=0.05 and 0.09 for model 1 and model 2, respectively).
To investigate whether the interaction with PRS may reflect interaction with family history for myocardial infarction, we performed interaction analyses between smoking and family history. We observed no effect modification for the smoking association with CHD by family history (Pinteraction=0.93 and 0.95 for model 1 and model 2, respectively). We additionally performed interaction analyses between smoking and PRS separately among individuals with and without family history. We observed similar effect modification by smoking on the PRS association with CHD, among both individuals with or without family history, although the interaction was not significant among those with family history (n=8949), most likely because of a lower power (Pinteraction=0.13 and 0.02 [model 1]; Pinteraction=0.21 and 0.02 [model2]; Tables VIII and IX in the Data Supplement).
Discrimination and Reclassification
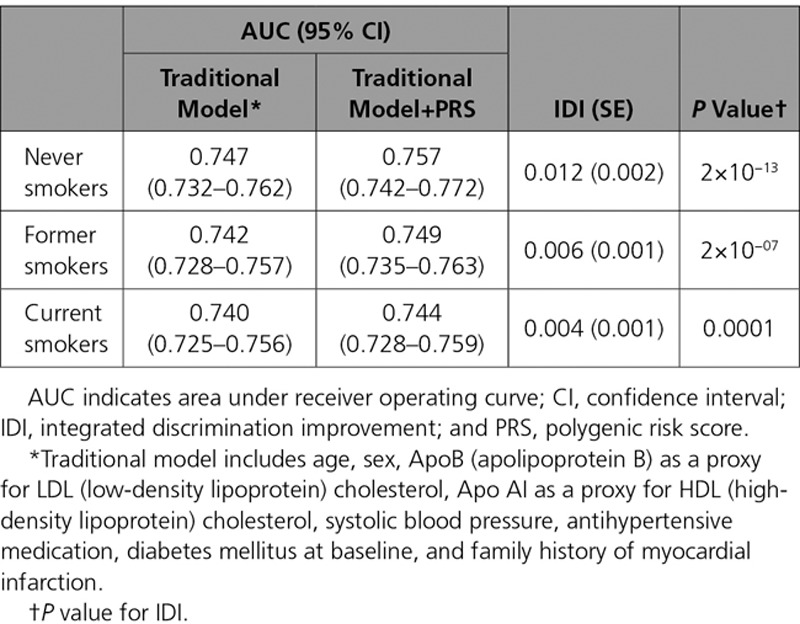
The PRS showed improved discrimination and reclassification of incident CHD on the top of traditional risk factors among never smokers when compared with smokers. In general, the PRS provided modest improvement on the top of traditional risk factors; adding the PRS to a prediction model that included traditional risk factors improved the discrimination of the model (IDI=0.007; P<0.0001). Stratifying by smoking status showed better discrimination by PRS among never smokers (IDI=0.012; P<0.0001) than former smokers (IDI=0.006; P<0.0001) and current smokers (IDI=0.004; P<0.0001; Table 4). This pattern was stronger among men showing even larger discrimination among never smokers (IDI=0.023; P<0.0001) than former smokers (IDI=0.007; P<0.0001) and current smokers (IDI=0.004; P=0.003; Tables X and XI in the Data Supplement).
Table 4.
C Statistics and Discrimination by PRS for Incident Coronary Heart Disease by Smoking Status
Risk reclassification was also improved by adding the PRS to a traditional risk model (NRI=0.18; P<0.0001). The PRS showed better risk reclassification among never smokers (NRI=0.23; P<0.0001) than former smokers (NRI = 0.18, P<0.0001) and current smokers (NRI=0.15; P<0.0001). The magnitude of reclassification by the PRS dependent on smoking status was stronger among men when compared with women, with the highest reclassification observed among never smokers (NRI=0.29; P<0.0001) compared with former smokers (NRI=0.14; P=0.0006) and current smokers (NRI=0.20; P<0.0001). The interaction term provided improved risk reclassification (NRI=0.066; P=0.006) on the top of a model including age, sex, ApoB, Apo AI, systolic blood pressure, antihypertensive medication, diabetes mellitus at baseline, family history of myocardial infarction, smoking, and PRS among current and never smokers.
Individual SNP Interactions With Smoking
To provide information on the contribution of each SNP included in the PRS, we performed interaction analyses between each SNP and smoking on CHD incidence among all individuals and among men and women separately (Figures III through V in the Data Supplement). Among all individuals (n=24 443), none of the interactions passed the Bonferroni-corrected P value of 0.001, whereas 6 SNPs had an interaction P value of <0.05. The strongest interactions were observed with the SNPs in the PPAP2B and HDAC9 loci (Pinteraction=0.003 and 0.009, respectively). In the sex-specific analyses, 4 SNPs in men and 1 in women provided nominally significant interaction P values, and after Bonferroni corrections, 1 SNP in the MRAS locus remained significant in men. Finally, trait-specific PRSs did not show significant interactions with smoking (Pinteraction=0.21, 0.09, and 0.12 for lipids, blood pressure, and type 2 diabetes mellitus PRSs, respectively).
Discussion
In this prospective study, we observed that the elevated risk of CHD incidence by cigarette smoking is modified by genetic risk for CHD assessed as 50 SNPs compiled in a PRS. The relative risk increase of CHD by smoking was significantly higher among individuals with low genetic risk when compared with those with high genetic risk. This interaction was more evident among men than women. No interaction was observed between smoking and family history for CHD. The PRS significantly improved both discrimination risk and net reclassification of a prediction model for CHD based on traditional risk factors. The improvement was significantly better among never smokers than former and current smokers and particularly strong among male never smokers.
Smoking is among the strongest environmental risk factors for CHD, and often it is associated with other unfavorable lifestyle risk factors. Our results were observed after adjusting for other confounders including total energy intake, alcohol consumption, physical activity, and education indicating that the observed interaction is unlikely to be confounded by these factors. In addition, the interaction remained significant after additionally including other possible confounding or mediating risk factors as body mass index, diabetes mellitus, lipid-lowering and antihypertensive medication, ApoB, and systolic blood pressure, suggesting that the observed interaction is unlikely to be confounded or mediated by these factors either. The observed interaction showed a deviation from the independent multiplicative effects of smoking and the PRS on CHD incidence. Individuals within the highest PRS tertile were already at such a high risk of CHD that the additional risk increase by smoking was weaker compared with those within the lowest PRS tertile. In addition, genetic risk was observed to play a more important role among never smokers compared with current smokers. The interaction was stronger and more evident in men, and lost significance when tested among women, although a similar tendency was observed. This could be explained by decreased power among women because of both a markedly lower CHD incidence (8.3% versus 21.1%) and a clearly lower percentage of former smokers (27.7% versus 42.3%) in female when compared with male participants. Our results also indicate that the observed interaction is independent of the family history for myocardial infarction and that stratifying by family history status did not modify the observed interaction with smoking. This is in line with previous evidence showing that a CHD PRS predicts CHD independently from family history.8
In a previous study in MDCS, we reported that the association between smoking and CHD was modified by the strongest CHD-associated variant in the 9p21 locus.9 Carrying the 9p21 risk allele attenuated the relative risk increase by smoking. Because the direction of the 9p21 and smoking interaction was similar to the observed interaction in the current study, we performed a sensitivity analysis removing this variant from the PRS. Our results remained significant, although weaker, after removing the 9p21 variant, indicating that the interaction is partially mediated by the 9p21 variant. This locus has been previously found to interact with dietary factors particularly vegetable intake,28,29 obesity,30 blood pressure,31 and glycemia.32 Gene–smoking interactions for CHD have been extensively studied with a genetic variation in APOE, but a recent meta-analysis found no evidence for such interaction.33 Although interaction between smoking and PRS for CHD has not been studied earlier, a previous study in the Swedish Twin Register suggested that the heritability of CHD is not modified by smoking, a result in line with our results showing no interaction between smoking and family history for myocardial infarction on CHD incidence. However, they reported modification of CHD heritability by body mass index, indicating that genetic factors may play a more important role in CHD in the absence of environmental risk factors.34 This is in line with our observation that the PRS showed a larger effect and a better prediction of CHD among never smokers compared with smokers.
To gain insight on the contribution of the individual 50 SNPs on our interaction results, we performed individual SNP–smoking interaction analyses and observed some nominally significant interactions, of which the strongest were with variants in the PPAP2B and HDAC9 loci. Only 1 SNP, in the MRAS locus, showed significant interaction with smoking after correction for multiple testing, but this was only observed among males. Although our study was not powered for studying interactions with the single SNPs, it may be interesting that the PPAP2B CHD locus has been reported to be a target of oxidized LDL-induced epigenetic regulation with more open chromatin, higher enhancer activity, and increased PPAP2B expression, whereas HDAC9 has been reported be an epigenetic regulator of oxidized LDL-induced atherosclerotic processes.35,36 In addition, smoking has been associated with decreased methylation of CpG sites at the three prime untranslated region MRAS,37 where the rs9818870 SNP is located and reported as a strong expression quantitative trait locus for MRAS expression with the CHD risk T allele associated with increased expression in arterial tissue (www.gtexportal.org).
Our results show that smoking status may provide a valuable stratification tool for prioritization of individuals who would benefit from using a PRS to improve prediction of future CHD. The PRS provided better discrimination on the top of a traditional risk factor model among never smokers compared with smokers. This was more evident among male never smokers where the genetic prediction provided a 2.3% improvement in discrimination. In addition, PRS provided better risk reclassification among never smokers compared with smokers and particularly among male never smokers where adding the PRS provided a total of 29% improved net reclassification.
Our study has some limitations that need to be pointed out. First, our study was conducted in a sample of middle-aged Swedish individuals which may limit the generalizability to other populations. Second, smoking behavior was assessed at baseline, and no record of changes in smoking behavior was available during the follow-up. This could bias our results, especially if some selective smoking cessation has occurred among individuals with high genetic risk for CHD. However, censoring our data at 10 years of follow-up showed similar results but with less significant P values, probably because of decreased statistical power. In addition, we did not have data on baseline LDL and HDL cholesterol levels in the whole MDCS population, and, therefore, ApoB and Apo AI were used as proxies for cholesterol measurements. Further, the 50 SNPs included do not represent all to date identified CHD-associated variants as at least 30 more SNPs have been recently discovered.3,38,39 Finally, we have used logistic regression analyses instead of survival analyses mainly because of violations of the Cox proportional hazards assumptions. However, our results did not change when performed using Cox proportional hazard regressions. The longitudinal design and a low level of measurement errors for our main exposure variables, smoking40 and SNP genotypes, improved the power to detect interactions. Finally, the good ascertainment of CHD cases using multiple registries is another strength of the present study.
In conclusion, our study provides evidence for that smoking may interact with polygenic risk for CHD. Further, the PRS for CHD provided better risk discrimination and reclassification on the top of traditional risk factors among never smokers compared with smokers, and smoking cessation may provide a larger decrease of the relative risk for CHD in individuals with low polygenic risk for CHD.
Acknowledgments
We thank Malin Svensson and Widet Gallo for their excellent technical assistance. We also thank the staff and participants of the MDCS (Malmö Diet and Cancer Study).
Sources of Funding
This work was supported by the European Research Council (Consolidator grant no. 649021, Dr Orho-Melander), the Swedish Research Council, The Swedish Heart and Lung Foundation, the Region Skåne, the Swedish Diabetes Foundation, the Novo Nordisk Foundation, the Albert Påhlsson Foundation, and the Linneus Foundation for the Lund University Diabetes Center.
Disclosures
None.
Supplementary Material
Footnotes
The Data Supplement is available at http://circgenetics.ahajournals.org/lookup/suppl/doi:10.1161/CIRCGEN.117.001856/-/DC1.
An educational video is available at http://circgenetics.ahajournals.org/highwire/filestream/257374/field_highwire_adjunct_files/1/CircGenetics_CIRCCVG-2018-001856_supp3.mp4.
Circ Genom Precis Med is available at http://circgenetics.ahajournals.org.
References
- 1.Mons U, Müezzinler A, Gellert C, Schöttker B, Abnet CC, Bobak M, et al. CHANCES Consortium. Impact of smoking and smoking cessation on cardiovascular events and mortality among older adults: meta-analysis of individual participant data from prospective cohort studies of the CHANCES consortium. BMJ. 2015;350:h1551. doi: 10.1136/bmj.h1551. doi: 10.1136/bmj.h1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thun MJ, Carter BD, Feskanich D, Freedman ND, Prentice R, Lopez AD, et al. 50-year trends in smoking-related mortality in the United States. N Engl J Med. 2013;368:351–364. doi: 10.1056/NEJMsa1211127. doi: 10.1056/NEJMsa1211127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nikpay M, Goel A, Won HH, Hall LM, Willenborg C, Kanoni S, et al. A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet. 2015;47:1121–1130. doi: 10.1038/ng.3396. doi: 10.1038/ng.3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deloukas P, Kanoni S, Willenborg C, Farrall M, Assimes TL, Thompson JR, et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet. 2013;45:25–33. doi: 10.1038/ng.2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ripatti S, Tikkanen E, Orho-Melander M, Havulinna AS, Silander K, Sharma A, et al. A multilocus genetic risk score for coronary heart disease: case-control and prospective cohort analyses. Lancet. 2010;376:1393–1400. doi: 10.1016/S0140-6736(10)61267-6. doi: 10.1016/S0140-6736(10)61267-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ganna A, Magnusson PK, Pedersen NL, de Faire U, Reilly M, Arnlöv J, et al. Multilocus genetic risk scores for coronary heart disease prediction. Arterioscler Thromb Vasc Biol. 2013;33:2267–2272. doi: 10.1161/ATVBAHA.113.301218. doi: 10.1161/ATVBAHA.113.301218. [DOI] [PubMed] [Google Scholar]
- 7.Yiannakouris N, Katsoulis M, Trichopoulou A, Ordovas JM, Trichopoulos D. Additive influence of genetic predisposition and conventional risk factors in the incidence of coronary heart disease: a population-based study in Greece. BMJ Open. 2014;4:e004387. doi: 10.1136/bmjopen-2013-004387. doi: 10.1136/bmjopen-2013-004387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tada H, Melander O, Louie JZ, Catanese JJ, Rowland CM, Devlin JJ, et al. Risk prediction by genetic risk scores for coronary heart disease is independent of self-reported family history. Eur Heart J. 2016;37:561–567. doi: 10.1093/eurheartj/ehv462. doi: 10.1093/eurheartj/ehv462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hamrefors V, Hedblad B, Hindy G, Smith JG, Almgren P, Engström G, et al. Smoking modifies the associated increased risk of future cardiovascular disease by genetic variation on chromosome 9p21. PLoS One. 2014;9:e85893. doi: 10.1371/journal.pone.0085893. doi: 10.1371/journal.pone.0085893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mega JL, Stitziel NO, Smith JG, Chasman DI, Caulfield M, Devlin JJ, et al. Genetic risk, coronary heart disease events, and the clinical benefit of statin therapy: an analysis of primary and secondary prevention trials. Lancet. 2015;385:2264–2271. doi: 10.1016/S0140-6736(14)61730-X. doi: 10.1016/S0140-6736(14)61730-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sotos-Prieto M, Baylin A, Campos H, Qi L, Mattei J. Lifestyle cardiovascular risk score, genetic risk score, and myocardial infarction in Hispanic/Latino adults living in Costa Rica. J Am Heart Assoc. 2016;5:e004067. doi: 10.1161/JAHA.116.004067. doi: 10.1161/JAHA.116.004067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aschard H, Tobin MD, Hancock DB, Skurnik D, Sood A, James A, et al. Understanding Society Scientific Group. Evidence for large-scale gene-by-smoking interaction effects on pulmonary function. Int J Epidemiol. 2017;46:894–904. doi: 10.1093/ije/dyw318. doi: 10.1093/ije/dyw318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khera AV, Emdin CA, Drake I, Natarajan P, Bick AG, Cook NR, et al. Genetic risk, adherence to a healthy lifestyle, and coronary disease. N Engl J Med. 2016;375:2349–2358. doi: 10.1056/NEJMoa1605086. doi: 10.1056/NEJMoa1605086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Callmer E, Riboli E, Saracci R, Akesson B, Lindgärde F. Dietary assessment methods evaluated in the Malmö food study. J Intern Med. 1993;233:53–57. doi: 10.1111/j.1365-2796.1993.tb00648.x. [DOI] [PubMed] [Google Scholar]
- 15.Wirfält E, Mattisson I, Johansson U, Gullberg B, Wallström P, Berglund G. A methodological report from the Malmö Diet and Cancer study: development and evaluation of altered routines in dietary data processing. Nutr J. 2002;1:3. doi: 10.1186/1475-2891-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berglund G, Elmstähl S, Janzon L, Larsson SA. The Malmo Diet and Cancer Study. Design and feasibility. J Intern Med. 1993;233:45–51. doi: 10.1111/j.1365-2796.1993.tb00647.x. [DOI] [PubMed] [Google Scholar]
- 17.Cederholm J, Eeg-Olofsson K, Eliasson B, Zethelius B, Nilsson PM, Gudbjörnsdottir S Swedish National Diabetes Register. Risk prediction of cardiovascular disease in type 2 diabetes: a risk equation from the Swedish National Diabetes Register. Diabetes Care. 2008;31:2038–2043. doi: 10.2337/dc08-0662. doi: 10.2337/dc08-0662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lindholm E, Agardh E, Tuomi T, Groop L, Agardh CD. Classifying diabetes according to the new WHO clinical stages. Eur J Epidemiol. 2001;17:983–989. doi: 10.1023/a:1020036805655. [DOI] [PubMed] [Google Scholar]
- 19.Ludvigsson JF, Andersson E, Ekbom A, Feychting M, Kim JL, Reuterwall C, et al. External review and validation of the Swedish national inpatient register. BMC Public Health. 2011;11:450. doi: 10.1186/1471-2458-11-450. doi: 10.1186/1471-2458-11-450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johansson LA, Westerling R. Comparing Swedish hospital discharge records with death certificates: implications for mortality statistics. Int J Epidemiol. 2000;29:495–502. [PubMed] [Google Scholar]
- 21.Wettermark B, Hammar N, Fored CM, Leimanis A, Otterblad Olausson P, Bergman U. The new Swedish Prescribed Drug Register–opportunities for pharmacoepidemiological research and experience from the first six months. Pharmacoepidemiol Drug Saf. 2007;16:726–735. doi: 10.1002/pds.1294. doi: 10.1002/pds.1294. [DOI] [PubMed] [Google Scholar]
- 22.Lagerqvist B, Swedeheart JS. SCAAR Årsrapport 2008 (In Swedish). 2016. http://www.ucr.uu.se/scaar Accessed August 31, 2016.
- 23.Shiffman D, O’Meara ES, Bare LA, Rowland CM, Louie JZ, Arellano AR, et al. Association of gene variants with incident myocardial infarction in the Cardiovascular Health Study. Arterioscler Thromb Vasc Biol. 2008;28:173–179. doi: 10.1161/ATVBAHA.107.153981. doi: 10.1161/ATVBAHA.107.153981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Erdmann J, Grosshennig A, Braund PS, König IR, Hengstenberg C, Hall AS, et al. Italian Atherosclerosis, Thrombosis, and Vascular Biology Working Group; Myocardial Infarction Genetics Consortium; Wellcome Trust Case Control Consortium; Cardiogenics Consortium. New susceptibility locus for coronary artery disease on chromosome 3q22.3. Nat Genet. 2009;41:280–282. doi: 10.1038/ng.307. doi: 10.1038/ng.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.IBC 50K CAD Consortium. Large-scale gene-centric analysis identifies novel variants for coronary artery disease. PLoS Genet. 2011;7:e1002260. doi: 10.1371/journal.pgen.1002260. doi: 10.1371/journal.pgen.1002260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schunkert H, König IR, Kathiresan S, Reilly MP, Assimes TL, Holm H, et al. Cardiogenics; CARDIoGRAM Consortium. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet. 2011;43:333–338. doi: 10.1038/ng.784. doi: 10.1038/ng.784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Coronary Artery Disease Genetics Consortium. A genome-wide association study in Europeans and South Asians identifies five new loci for coronary artery disease. Nat Genet. 2011;43:339–344. doi: 10.1038/ng.782. doi: 10.1038/ng.782. [DOI] [PubMed] [Google Scholar]
- 28.Hindy G, Ericson U, Hamrefors V, Drake I, Wirfält E, Melander O, et al. The chromosome 9p21 variant interacts with vegetable and wine intake to influence the risk of cardiovascular disease: a population based cohort study. BMC Med Genet. 2014;15:1220. doi: 10.1186/s12881-014-0138-x. doi: 10.1186/s12881-014-0138-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Do R, Xie C, Zhang X, Männistö S, Harald K, Islam S, et al. INTERHEART I; nvestigators. The effect of chromosome 9p21 variants on cardiovascular disease may be modified by dietary intake: evidence from a case/control and a prospective study. PLoS Med. 2011;8:e1001106. doi: 10.1371/journal.pmed.1001106. doi: 10.1371/journal.pmed.1001106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ye S, Willeit J, Kronenberg F, Xu Q, Kiechl S. Association of genetic variation on chromosome 9p21 with susceptibility and progression of atherosclerosis: a population-based, prospective study. J Am Coll Cardiol. 2008;52:378–384. doi: 10.1016/j.jacc.2007.11.087. doi: 10.1016/j.jacc.2007.11.087. [DOI] [PubMed] [Google Scholar]
- 31.Kim DS, Smith JA, Bielak LF, Wu CY, Sun YV, Sheedy PF., II The relationship between diastolic blood pressure and coronary artery calcification is dependent on single nucleotide polymorphisms on chromosome 9p21.3. BMC Med Genet. 2014;15:89. doi: 10.1186/s12881-014-0089-2. doi: 10.1186/s12881-014-0089-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Doria A, Wojcik J, Xu R, Gervino EV, Hauser TH, Johnstone MT, et al. Interaction between poor glycemic control and 9p21 locus on risk of coronary artery disease in type 2 diabetes. JAMA. 2008;300:2389–2397. doi: 10.1001/jama.2008.649. doi: 10.1001/jama.2008.649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Holmes MV, Frikke-Schmidt R, Melis D, Luben R, Asselbergs FW, Boer JM, et al. A systematic review and meta-analysis of 130,000 individuals shows smoking does not modify the association of APOE genotype on risk of coronary heart disease. Atherosclerosis. 2014;237:5–12. doi: 10.1016/j.atherosclerosis.2014.07.038. doi: 10.1016/j.atherosclerosis.2014.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Song C, Chang Z, Magnusson PK, Ingelsson E, Pedersen NL. Genetic factors may play a prominent role in the development of coronary heart disease dependent on important environmental factors. J Intern Med. 2014;275:631–639. doi: 10.1111/joim.12177. doi: 10.1111/joim.12177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reschen ME, Gaulton KJ, Lin D, Soilleux EJ, Morris AJ, Smyth SS, et al. Lipid-induced epigenomic changes in human macrophages identify a coronary artery disease-associated variant that regulates PPAP2B Expression through altered C/EBP-beta binding. PLoS Genet. 2015;11:e1005061. doi: 10.1371/journal.pgen.1005061. doi: 10.1371/journal.pgen.1005061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Han X, Han X, Wang Z, Shen J, Dong Q. HDAC9 regulates ox-LDL-induced endothelial cell apoptosis by participating in inflammatory reactions. Front Biosci (Landmark Ed) 2016;21:907–917. doi: 10.2741/4428. [DOI] [PubMed] [Google Scholar]
- 37.Steenaard RV, Ligthart S, Stolk L, Peters MJ, van Meurs JB, Uitterlinden AG, et al. Tobacco smoking is associated with methylation of genes related to coronary artery disease. Clin Epigenetics. 2015;7:54. doi: 10.1186/s13148-015-0088-y. doi: 10.1186/s13148-015-0088-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Webb TR, Erdmann J, Stirrups KE, Stitziel NO, Masca NG, Jansen H, et al. Wellcome Trust Case Control Consortium; MORGAM Investigators; Myocardial Infarction Genetics and CARDIoGRAM Exome Consortia Investigators. Systematic evaluation of pleiotropy identifies 6 further loci associated with coronary artery disease. J Am Coll Cardiol. 2017;69:823–836. doi: 10.1016/j.jacc.2016.11.056. doi: 10.1016/j.jacc.2016.11.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Klarin D, Zhu QM, Emdin CA, Chaffin M, Horner S, McMillan BJ, et al. CARDIoGRAMplusC4D Consortium. Genetic analysis in UK Biobank links insulin resistance and transendothelial migration pathways to coronary artery disease. Nat Genet. 2017;49:1392–1397. doi: 10.1038/ng.3914. doi: 10.1038/ng.3914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Patrick DL, Cheadle A, Thompson DC, Diehr P, Koepsell T, Kinne S. The validity of self-reported smoking: a review and meta-analysis. Am J Public Health. 1994;84:1086–1093. doi: 10.2105/ajph.84.7.1086. [DOI] [PMC free article] [PubMed] [Google Scholar]