

Abstract
The evolution of the major histocompatibility complex (MHC) is shaped by frequent gene duplications and deletions, which generate extensive variation in the number of loci (gene copies) between different taxa. Here, we collected estimates of copy number at the MHC for over 250 bird species from 68 families. We found contrasting patterns of copy number evolution between MHC class I and class IIB, which encode receptors for intra- and extracellular pathogens, respectively. Across the avian evolutionary tree, there was evidence of accelerated evolution and stabilizing selection acting on copy number at class I, while copy number at class IIB was primarily influenced by fluctuating selection and drift. Reconstruction of MHC copy number variation showed ancestrally low numbers of MHC loci in nonpasserines and evolution toward larger numbers of loci in passerines. Different passerine lineages had the highest duplication rates for MHC class I (Sylvioidea) and class IIB (Muscicapoidea and Passeroidea). We also found support for the correlated evolution of MHC copy number and life-history traits such as lifespan and migratory behavior. These results suggest that MHC copy number evolution in birds has been driven by life histories and differences in exposure to intra- and extracellular pathogens.
Keywords: birds, copy number variation, duplication, evolution, major histocompatibility complex
Introduction
Genes of the major histocompatibility complex (MHC) encode molecules that initiate specific immune responses by binding pathogen-derived antigens and presenting them to T lymphocytes (Klein 1986). These molecules are encoded by two structurally and functionally distinct gene subfamilies, MHC class I and class II, which trigger the immune response against intra- and extracellular pathogens, respectively. Thus, MHC class I and class II genes are considered to play an essential role in the adaptive immunity of vertebrates. As each allele at MHC class I and class II encodes a protein that can respond to a limited number of antigens, high allelic diversity at the MHC may be advantageous because it increases the spectrum of pathogens that an organism can recognize and defend against (Jeffery and Bangham 2000). In fact, the level of within-individual MHC polymorphism has been associated with disease resistance and fitness in fish (Evans and Neff 2009) and all extant tetrapod classes (Madsen and Ujvari 2006; Kloch et al. 2010; Savage and Zamudino 2011; Radwan et al. 2012; Dunn et al. 2013).
The apparent fitness advantage of high MHC heterozygosity at the individual level is reflected in a high allelic diversity of these genes in natural populations. For example, over 3,500 MHC class I alleles were found in a Polish population of sedge warblers Acrocephalus schoenobaenus (Biedrzycka, O’Connor, et al. 2017), while nearly 1,000 class II alleles were recorded in common yellowthroats Geothlypis trichas breeding in Wisconsin (Bollmer et al. 2012). This extreme MHC polymorphism is thought to be maintained primarily via pathogen-mediated balancing selection (Spurgin and Richardson 2010), acting through the mechanisms of overdominance (Hughes and Nei 1988), negative frequency dependence (Takahata and Nei 1990), and fluctuating selection (Hedrick 2002). Although pathogen-mediated selection should act to increase the number of MHC alleles within populations and individuals, the number of MHC variants per individual may be limited by the risk of autoimmune responses (Milinski 2006). Also, T-cell clones that recognize self-peptides bound to MHC molecules are eliminated, which reduces the T-cell clone repertoire that is ultimately available for pathogen recognition (Lawlor et al. 1990; Woelfing et al. 2009). Thus, a balance between the opposing selective forces of pathogen detection versus T-cell repertoire depletion and the risk of autoimmune disease may lead to the evolution of an optimal rather than maximal individual MHC diversity (Nowak et al. 1992).
The high allelic diversity at the MHC is thought to arise from the processes of gene duplication, producing variation in the number of loci (copy number). The evolution of genes in the MHC is generally consistent with a birth-and-death model, in which new genes are created by repeated gene duplication and some duplicate genes are maintained in the genome for long evolutionary times, whereas other genes are deleted or become inactivated through deleterious mutations (Nei et al. 1997). It has been proposed that birth-and-death processes at the MHC may be primarily driven by evolutionary forces such as exposure to different abundances and diversities of pathogens, rather than simply reflecting phylogenetic constraints (Woelfing et al. 2009). This, in turn, can create extensive variation in the number of MHC gene copies and large differences in the organization of the MHC region between different taxa. For example, 7 to ∼100 classical (highly expressed and polymorphic) MHC class I genes were found in teleost fish (Grimholt 2016) and the number of classical MHC class II genes varies from zero in the Atlantic cod Gadus morhua (Star et al. 2011) up to 33 genes in tilapia (Sato et al. 2012). The organization of the mammalian MHC shows similar levels of diversity. In contrast to eutherians, marsupials have class I and class II genes interspersed within one region (Belov 2006), and classical class IIB genes (DAB, DBB, and DCB) are not orthologous to eutherian genes (Belov et al. 2004). However, MHC class I loci appear to experience a much faster rate of birth-and-death evolution than class II loci in eutherian mammals, and, thus, there seem to be no orthologous relationships of different class I loci among different mammalian orders (Hughes and Nei 1989). There is robust support for birth-and-death evolution of MHC class I loci in primates, as inferred from frequent gene duplications and deletions, as well as from the presence of multiple pseudogenes (Piontkivska and Nei 2003). For example, the MHC class I supercluster in humans comprises 19 loci, out of which only three are classical (HLA-A, -B, and -C), while 12 are recognized as pseudogenes (Horton et al. 2004). So far, mammals (including humans) have been subject to the most rigorous studies on the evolution and architecture of the MHC (Bernatchez and Landry 2003), while evolutionary mechanisms that generate and maintain MHC copy number variation are much less well understood in other vertebrate groups, such as birds.
The aim of this study was to assess historical patterns and mechanisms of gene copy number evolution at the avian MHC. The genetic architecture of the MHC varies substantially between species of birds. The number of loci range from a single dominantly expressed gene at both class I and class II in galliforms, birds of prey and penguins (so called “minimal essential MHC”; Kaufman et al. 1999) up to tens of putatively transcribed loci in some passerines (Bollmer et al. 2012; Sepil et al. 2012; Biedrzycka, O’Connor, et al. 2017). A recent molecular study showed large variation in copy number of MHC class I genes within 12 passerine species and provided evidence for the primary roles of genetic drift and fluctuating selection (rather than selection toward a single optimal value or evolutionary bursts) in the evolution of MHC class I (O’Connor et al. 2016). Here, we used all available estimates of MHC copy number in birds to analyze the macroevolutionary history of MHC class I and class II across the entire avian tree. We also used a phylogenetically informed comparative analysis to test for correlated evolution between the number of MHC gene copies and life-history traits in birds. We have recently shown that the strength of balancing selection (as measured with nucleotide substitution rates) on the avian MHC increased with life-history traits, migration and sociality, possibly due to an exposure to more diverse pathogen faunas (migratory species) and an elevated transmission rate of pathogens (colonial species) (Minias et al. 2017). In this study, we hypothesized that similar life-history traits that increase exposure to pathogens will also be associated with a larger number of MHC gene copies in birds.
Materials and Methods
Gene Copy Number Estimates
Data on gene copy number at MHC class I and II of birds were collected from the literature. For this purpose, we checked published sources for all avian MHC sequences deposited in GenBank. First, we used a combination of search terms “MHC,” “aves,” and “class I” or “class II” to gather information on 9,212 MHC class I and 7,556 MHC class II sequences of birds from GenBank, as accessed on September 1, 2017. Next, we examined references provided for these sequences (n = 135 studies) to retrieve estimates of the number of classical MHC loci. All MHC loci identified as nonclassical (e.g., MHC-Y genes in Phasianidae) or nonfunctional (pseudogenes) were excluded from our data. At MHC class II we focused exclusively on B genes, as there were little data on the number of MHC class IIA copies in birds. In total, we compiled 101 and 279 different estimates of gene copy number at MHC class I and class IIB, respectively. Data for MHC class I and class IIB were collected for 78 species from 38 families and 221 species from 58 families, respectively. Distribution of species within families was relatively uneven, ranging from 1 to 29 species per family, with a mean of 2.05 ± 0.33 (SE) and 3.81 ± 0.69 (SE) species per family for MHC class I and class IIB, respectively. Our entire data set represented 64% of all extant avian orders and 28% of all extant avian families, according to the classification proposed by Winkler et al. (2015).
Most estimates of gene copy number (92% and 97% for MHC class I and class IIB, respectively) were obtained by dividing the maximum number of putatively functional (e.g., lacking stop codons) MHC alleles detected in any individual from a given species by 2 (i.e., assuming heterozygosity at each locus). This is a common practice, as due to its high complexity, the genetic architecture of the MHC region has been resolved for few (n = 9 species within our data set) avian species, mostly nonpasserines with a small number of loci. Here, we explicitly acknowledge that indirect estimates of MHC copy number can be imprecise and prone to certain biases. First, the estimates of the maximum number of alleles per individual can vary between genotyping techniques. To test this prediction, we collected data on the genotyping methods used in each of the studies (see Supplementary Material online for details) and showed that 454 pyrosequencing and other next-generation sequencing methods yielded higher estimates of gene copy numbers than cloning (supplementary table S1, Supplementary Material online). Thus, we carefully controlled for genotyping methods in all further analyses. Although the number of MHC alleles recorded per individual could also vary with the methods used to process the data, it has recently been shown that allele calling had high (>90%) agreement between the most common methods of data processing (Biedrzycka, Sebastian, et al. 2017), so we assumed that this variation should not introduce any major bias in the data. Second, a different number of alleles per individual can be obtained using different pairs of primers. Although we could not directly control for the identity of primers used in different studies, previous research on passerines showed that interspecific variation in the number of MHC alleles is highly repeatable with different pairs of primers (O’Connor et al. 2016). Also, we showed that MHC gene number estimates, measured independently with the same genotyping technique in the same species, were highly repeatable (R = 0.92, 95% CI: 0.82–0.97; n = 22, P < 0.001; see Supplementary Material online for details), irrespective of the primers used for genotyping. Further, the estimates of gene copy number should depend on the number of genotyped individuals, as the probability of genotyping an individual having different alleles at each locus increases with sample size. Consistent with this prediction, the estimates of gene copy number within our data set were significantly or nearly significantly correlated with the number of individuals genotyped (supplementary table S1, Supplementary Material online) and, thus, we controlled for the sample size in all our analyses. Finally, we assumed that genotyping errors did not constitute a major bias in our database, as a majority (>85%) of studies from which we collected data were conducted within the last decade, when stringent quality control procedures (e.g., Burri et al. 2008; Zagalska-Neubauer et al. 2010) were put in place to differentiate putative functional MHC alleles from artifacts. Finally, it is important to note that although copy number estimates collected for this study are likely to have biases, we do not expect them to vary systematically in relation to our hypothesized relationships with life history or other variables and, consequently, we do not expect them to confound our results.
Inferring Historical Patterns of Evolution
The evolutionary history of copy number at the avian MHC was analyzed in three major steps. First, we fitted six macroevolutionary models to our data. These included three general models: 1) Brownian motion (BM), 2) BM adjusted for the phylogenetic scaling parameter λ, 3) Ornstein–Uhlenbeck (OU), and three models with time dependence in the rate of evolution: 4) early versus late differences in evolution rate, 5) linear trend in evolution rate, and 6) exponential trend in evolution rate. BM constitutes a basic model of evolution that describes the evolutionary dynamics of a trait that changes randomly in direction and distance over any time interval. The BM model is consistent with purely neutral evolution (trait changes only due to genetic drift), but it can also reflect fluctuating selection toward a moving optimum (Hansen and Martins 1996). The standard BM model is associated with a constant-rate process of evolution across a phylogenetic tree, but this assumption can be weakened by the phylogenetic scaling parameter λ, which introduces a transformation of the phylogenetic variance–covariance matrix and effectively changes branch lengths of the phylogeny (Pagel 1999). In general, λ varies between 0 and 1, where 0 indicates phylogenetic independence (producing a single polytomy where all species are equally related), and a value of 1 retains the original tree and indicates that trait evolution corresponds to the standard BM model (Freckleton et al. 2002). With the decreasing of λ from 1 to 0, internal nodes are pushed toward the root, which is consistent with stronger transformation of phylogenetic variance–covariance matrix. Low λ values can be caused by fluctuating selection with a low rate of fluctuation or by genetic drift that was initially low, but increased over time (Revell et al. 2008). In contrast, high λ values may be indicative of constant-rate genetic drift and fluctuating selection with a high fluctuation rate (Revell et al. 2008). All BM models assume that the correlation structure among trait values is proportional to the extent of shared ancestry for pairs of species (Felsenstein 1973) and the variance grows with time in an unbounded fashion, in contrast to stabilizing selection (Butler and King 2004). The OU model has been introduced to describe evolution of traits under stabilizing selection with a constant optimum or optima (Hansen 1997; Butler and King 2004). Finally, models with time dependence in evolutionary rates either fit relative contributions of early versus late evolution in the tree to the covariance of species trait values (Pagel 1999) or assume a linear or exponential trend in rates through time (Blomberg et al. 2003; Harmon et al. 2010). All of our models were fit with the fitContinuous function in the geiger R package (Harmon et al. 2008) using specifications of the following models: BM (Brownian motion), lambda (Brownian motion adjusted for λ), OU (Ornstein–Uhlenbeck), delta (differences in early vs. late evolution rate), trend (linear change in evolution rate), and EB (exponential change in evolutionary rate). Relative fit of all models was compared using differences in the Akaike information criterion corrected for small sample sizes (ΔAICC) and Akaike weights (ωi).
Second, we estimated phylogenetic signal and phylogenetic autocorrelation in the data. Phylogenetic signal (the tendency of related species to resemble each other) was estimated with the Pagel’s (1999) scaling parameter λ (the one used in lambda model from geiger package) and Blomberg’s K (Blomberg et al. 2003), as λ performs better for discriminating random and BM patterns of trait distribution in the phylogeny, while Blomberg’s K is most suitable to capture the effects of changing evolutionary rates (Münkemüller et al. 2012). Both measures of phylogenetic signal were estimated using the phylosig function in the phytools (Revell 2012) R package. Statistical significance of phylogenetic signal was determined using the randomization test of Blomberg et al. (2003) for K and a likelihood ratio test for λ. Phylogenetic autocorrelation was estimated using spatial autocorrelation statistic Moran’s I (Gittleman and Kot 1990). Phylogenetic correlograms of normalized Moran’s I (I/Imax) were used to assess the strength of autocorrelation in gene copy numbers at different taxonomic levels (genus, family, and order). The analysis of Moran’s I was conducted using the ape (Paradis et al. 2004) package in R.
Third, we reconstructed ancestral states for the number of MHC class I and class IIB gene copies using the maximum likelihood approach under the assumptions of BM processes, as implemented in fastAnc function in the phytools R package. For MHC class I we also estimated ancestral states using the anc.ML function in phytools, which uses the OU model of evolution (it provided better fit to the MHC class I data than the BM model; see Results for details). As both approaches (fastAnc and anc.ML) produced almost identical results (between-method repeatability: R = 1, P < 0.001), only fastAnc point estimates and corresponding 95% confidence intervals were presented in the text (anc.ML does not compute variances and confidence intervals). The estimated ancestral states (fastAnc function) were mapped on the phylogeny using the contMap function in phytools, while the 95% confidence intervals for point estimates of ancestral states were plotted on the traitgrams using the fancyTree function from the same package.
Life-History Variables
For all species we collected data on the following life-history variables: clutch size, incubation period, body mass, sociality, migratory behavior, and lifespan. Data on clutch size, incubation period, body mass, and sociality were compiled from standard references (del Hoyo et al. 1992–2011; Snow and Perrins 1998). Body mass was log-transformed prior to analyses. Sociality was coded as a categorical variable with three levels: 1) solitary species (n = 163), 2) semisocial (group-living and semicolonial) species (n = 39), and 3) predominantly colonial species (n = 50). Migratory behavior was assessed based on the total migration distance (TMD), which was coded as a categorical variable with three levels: 1) resident (TMD = 0 km, n = 112), 2) short-distance migrants (0 > TMD < 2,000 km, n = 75), and 3) long-distance migrants (TMD ≥ 2,000 km, n = 65) (see Supplementary Material online for details). Lifespan was adjusted for allometry, sampling effort and source of data (see Supplementary Material online for details), and is henceforth referred to as residual lifespan. The distribution of species with different life histories was relatively homogeneous across different methods of MHC genotyping (supplementary table S2, Supplementary Material online).
Comparative Analyses
Bayesian phylogenetic mixed models (Hadfield and Nakagawa 2010), as implemented in the MCMCGlmm (Hadfield 2010) package developed for R statistical environment (R Development Core Team 2013), assume the BM model of evolution and, thus, might be inappropriate to model traits that evolved under different evolutionary scenarios (Hadfield 2015). In our study, the BM model provided a poor fit to data on copy number for MHC class I (see Results for details; table 1). Consequently, we used the phylogenetic generalized least squares (PGLS) method (Martins and Hansen 1997) to test for relationships between copy number at the MHC class I and life-history variables. In general, PGLS incorporates a matrix of the covariances among species into a model fitted by generalized least squares and alters the correlation between error terms to reflect the degree of phylogenetic relatedness among the species. In the PGLS analysis of MHC class I copy number, we specified the OU model of evolution, which provided a better fit to the data than BM (table 1). In contrast, the evolution of copy number at the MHC class IIB was best described by the BM model adjusted for λ (table 1) and, thus, we used both PGLS and BPMM models to test for the relationships of this trait with life-history characteristics. The phylogenetic scaling parameter λ was set to its maximum likelihood estimate in all BM-based PGLS models.
Table 1.
Relative Fit (ΔAICC) of Six Evolutionary Models Describing Gene Copy Number Variation at MHC Class I and Class II of Birds
| MHC | Evolution Model | AICC | ΔAICC | Relative Importance (ωi) |
|---|---|---|---|---|
| Class I | Time-dependent (early vs. late evolution) | 462.3 | 0.0 | 0.662 |
| Ornstein–Uhlenbeck | 464.2 | 1.9 | 0.301 | |
| Time-dependent (linear) | 468.5 | 6.2 | 0.036 | |
| Brownian motion adjusted for λ | 477.1 | 14.8 | <0.001 | |
| Brownian motion | 478.4 | 16.1 | <0.001 | |
| Time-dependent (exponential) | 480.6 | 18.3 | <0.001 | |
| Class II | Brownian motion adjusted for λ | 1,010.3 | 0.0 | 0.999 |
| Ornstein–Uhlenbeck | 1,038.8 | 28.5 | <0.001 | |
| Time-dependent (early vs. late evolution) | 1,094.6 | 84.3 | <0.001 | |
| Time-dependent (linear) | 1,120.1 | 109.8 | <0.001 | |
| Brownian motion | 1,150.1 | 139.8 | <0.001 | |
| Time-dependent (exponential) | 1,152.1 | 141.8 | <0.001 |
Note.—Relative importance of each model is assessed with Akaike weight (ωi).
The BPMM model for MHC class IIB was run according to the methodology described in the Supplementary Material online. While BPMM allowed us to incorporate within-species variation in the response variable, PGLS models required only one estimate of copy number per species. Thus, for species with multiple estimates of gene copy number we used the highest available estimate, as some measures of MHC copy number are likely to be underestimated (e.g., because of ineffective amplification of all loci by the primers or limited probability of genotyping an individual that would be heterozygous at all loci). All PGLS models were fitted using gls function in the ape R package (see Supplementary Material online for details). In both PGLS (MHC class I and class IIB) and BPMM (only MHC class IIB) models, we entered log-transformed copy number as the response variable. Both the BPMM and PGLS models were run with phylogenies extracted from the BirdTree database (see Supplementary Material online for more details). Clutch size, incubation period, residual lifespan, log-transformed body mass, and genotyping effort (log-transformed number of genotyped individuals) were entered as covariates, while migratory behavior, sociality, and genotyping method were included as fixed factors in each model. To obtain more parsimonious reduced models, highly nonsignificant (P > 0.10) traits were removed from the initial full models. Statistical significance was inferred at P < 0.05.
Results
Patterns of Evolution and Phylogenetic Autocorrelation
We found that evolutionary patterns of gene copy number differed between MHC class I and class IIB. The time-dependent model of evolution best fit the copy number variation at MHC class I (table 1), providing evidence for relatively fast (accelerated) recent evolution (δ > 0). Evolution of MHC class I copy number was also well explained by the OU process, consistent with the action of stabilizing selection. In contrast, we found no evidence for time-dependent changes in the rate of evolution at MHC class IIB. The BM model adjusted with the phylogenetic scaling parameter λ provided the best fit with copy number variation at MHC class IIB (λ = 0.39; table 1). Both λ and Blomberg’s K indicated moderate, but significant phylogenetic signal in the number of gene copies at MHC class I (λ = 0.57, P < 0.001; K = 0.38, P = 0.011) and class IIB (λ = 0.39, P < 0.001; K = 0.15, P = 0.007). The measure of phylogenetic autocorrelation (Moran’s I) in copy number at the MHC was highest at the level of genera (I = 0.63, P < 0.001 for class I; I = 0.42, P < 0.001 for class IIB; supplementary fig. S1, Supplementary Material online). Much weaker phylogenetic autocorrelation was recorded at the level of families and orders (supplementary fig. S1, Supplementary Material online), suggesting that most diversification in MHC class I and class IIB copy numbers occurred relatively late in avian radiation.
Ancestral State Reconstruction
Ancestral character estimates of the number of gene copies at the root nodes of avian trees were 3.34 (95% CI: −5.05 to 11.74) and 2.38 (95% CI: −3.75 to 8.51) for MHC class I and class IIB, respectively. In general, nonpasserines showed a consistently lower number of MHC class I and class IIB copies than passerines. In nonpasserines, the mean number of copies was 2.60 ± 0.29 (SE) (n = 43) at class I and 2.04 ± 0.08 (SE) (n = 142) at class IIB, while passerines had, on average, 7.66 ± 1.23 (SE) copies at class I (n = 35) and 4.97 ± 0.41 (SE) copies at class IIB (n = 79). Some nonpasserine taxa have evolved moderately high numbers of MHC gene copies with up to eight class I copies in the blue petrel Halobaena caerulea (Procellariformes) and eight class IIB copies in Blakiston’s fish owl Ketupa blakistoni (Strigiformes). However, these relatively high numbers of MHC gene copies for nonpasserines were most likely very recent, as in most cases they were not shared with closely related taxa (fig. 1). Ancestral character estimates for the basal node of passerines were 6.44 (95% CI: 1.02–11.88) copies at class I and 4.41 (95% CI: −0.43 to 7.80) copies at class IIB. Within passerines, the highest diversification of gene copy numbers at MHC class I was recorded in the superfamily Sylvioidea (fig. 2A). An ancestral character estimate for the basal node of this clade was 5.26 (95% CI: 4.30–13.29) copies of MHC class I, but over 30 copies were recorded at some terminal branches (32 and 33 copies in the great tit Parus major and sedge warbler, respectively). The number of MHC class I copies was ≤10 in other passerine superfamilies. In contrast, the highest numbers (>10) of MHC class IIB copies were recorded in the Muscicapoidea and Passeroidea superfamilies, and not in Sylvioidea (fig. 2B), suggesting that the evolution of MHC architecture in passerines differed between class I and class IIB genes. Some recently diverged passerine families (Cardinalidae, Icteridae, and Thraupidae) had low numbers (≤5) of MHC class I genes, despite originating from ancestors with a higher number of MHC class I copies (fig. 1A). Similarly, at MHC class IIB we recorded several transitions from high to low number of gene copies (fig. 1B), consistent with a birth-and-death model of MHC evolution.
Fig. 1.

—Ancestral character estimation of gene copy number at MHC class I (A) and class II (B) along the branches and nodes of avian phylogeny. Bars associated with each terminal node indicate raw number of gene copies.
Fig. 2.—

Traitgrams for gene copy number at MHC class I (A) and class II (B) of birds. The vertical position of nodes shows reconstructed ancestral states for the number of MHC gene copies, while the horizontal position gives time from the root. Uncertainty is shown via increasing transparency of blue lines plotted along branches and at nodes which indicate 95% confidence intervals point estimates.
Fig. 1.

—Continued
Life-History Correlates
After controlling for genotyping effort and method, we found that copy number variation at MHC class I and class IIB correlated with different life-history variables. The only variable that correlated with copy number at both MHC class I and IIB was body mass, as heavier species had a lower number of MHC genes (supplementary tables S3 and S4 and fig. S2, Supplementary Material online). PGLS models indicated that the number of MHC class I copies also correlated positively with residual lifespan (supplementary tables S3 and S4, Supplementary Material online, and fig. 3). In contrast, migratory behavior was identified as a significant predictor of copy number at MHC class IIB (supplementary table S3, Supplementary Material online); here both short- and long-distance migrants had a higher number of MHC class IIB copies than resident species (supplementary table S4, Supplementary Material online, and fig. 4). The effect of migratory behavior on MHC class IIB copy number was supported by both PGLS and BPMM analyses, the latter of which incorporated intraspecific variation in copy number (supplementary table S5, Supplementary Material online). None of the models provided support for the effect of sociality, clutch size, or incubation period on the number of MHC class I or IIB genes (supplementary table S2, Supplementary Material online).
Fig. 3.—
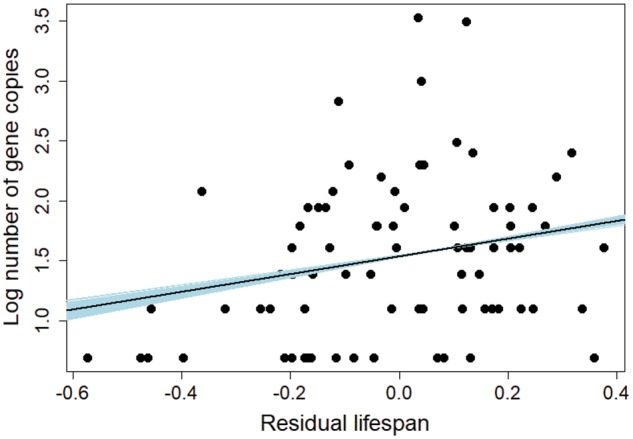
Relationship of gene copy number at MHC class I with residual lifespan of birds. Regression lines for 1,000 random phylogenies are marked with blue, and regression line averaged across these phylogenies is marked with black.
Fig. 4.—

Relationship of gene copy number at MHC class II with migratory behavior of birds. Mean ± SE estimates for 1,000 random phylogenies are shown for resident, short-distance migratory, and long-distance migratory species.
Discussion
Our study provides a taxonomically broad assessment of gene copy number evolution at the avian MHC. Using data for over 250 species from 68 families, we showed that copy number variation at MHC class I and IIB has different macroevolutionary patterns and is likely shaped by different selective forces. First, we found that variation in MHC class I copy numbers was shaped primarily by accelerated evolution and stabilizing selection (the time-dependent and OU models provided the best fit), while the evolution of copy numbers at MHC class IIB was more likely governed by fluctuating selection and drift (BM model adjusted for λ provided the best fit). Second, a low number of MHC loci was identified as the ancestral state in birds, and we showed that the highest diversification in copy numbers occurred relatively late in avian radiation, mostly in passerines. However, the higher duplication rate of MHC class I and IIB in passerines occurred in different lineages, suggesting that different passerine clades may have been under varying selective pressure by intra- and extracellular pathogens and parasites, although we cannot exclude the possibility that duplication processes in some taxa could be due to neutral evolution. Finally, we found evidence for the correlated evolution of MHC copy number and life-history traits that may be associated with pathogen exposure.
Direct comparisons of MHC copy number across species have been largely hindered by inconsistent methodological framework used for MHC genotyping in different taxa (O’Connor et al. 2016). One solution to this problem would be to estimate copy numbers with a uniform genotyping methodology in multiple species, but such an approach is limited by the immense genotyping effort and costs involved in studying a wide range of species. O’Connor et al. (2016) used this method to investigate copy number variation at MHC class I, but only 12 passerine species were genotyped and sample sizes were small (2–4 individuals genotyped per species). In contrast, our study aimed to maximize taxonomic breadth while at the same time taking care to exclude or control for possible methodological bias in the data. In particular, we controlled for differences in gene copy number associated with different genotyping techniques and different sample sizes. We also showed that MHC gene number estimates, measured independently with the same genotyping technique in the same species, were highly repeatable (R = 0.92), irrespective of the primers used for genotyping. Finally, we also showed that the estimates were not affected by the coverage of the most common genotyping techniques, that is, by the number of clones genotyped per individual or the average number of reads per individual in the next-generation sequencing methods. We acknowledge that MHC copy numbers might have been underestimated in some taxa (e.g., because of ineffective amplification of all loci by the primers or limited probability of genotyping an individual that would be heterozygous at all loci). In an attempt to deal with this problem, we ran phylogenetically informed comparative models that either incorporated within-species variation in the data (BPMM) or were based on the highest estimates of gene copy number available for each species (PGLS). Finally, we acknowledge that the estimates of MHC gene copy number may, to some extent, reflect variation in the rates of concerted evolution among species, as species with high rates of concerted evolution may exhibit more allele sharing between loci, and this may lead to an underestimation of the number of paralogs. For example, it has been shown that elevated rates of concerted evolution may mask the presence of two ancient MHC IIB lineages in birds (Goebel et al. 2017). We assume that any possible biases in our data are not taxonomically skewed because, if present, they would be expected to obscure rather than produce the patterns described in our study. The high repeatabilities of genotyping techniques and our conservative analyses with controls for differences in methods and sample sizes provide the most reliable information to date on the evolution of MHC copy number across the avian tree of life.
One of the major aims of our study was to reconstruct the patterns of copy number evolution at the MHC class I and IIB of birds. Our ancestral state reconstruction provided qualitative evidence for the ancestrally low number of MHC genes in nonpasserines and elevated gene duplication rate in passerines. Although it has been recognized within the last decade that some passerine species have more MHC loci than nonpasserines (e.g., Bollmer et al. 2010), this pattern has never been investigated at a broad taxonomical scale. Here, we showed that most diversification in MHC copy number occurred relatively late in avian evolution and the strongest phylogenetic autocorrelation in this trait was recorded at the level of genus. Although previous research indicated that some diversification in MHC copy numbers occurred prior to or early in the passerine radiation (Balasubramaniam 2016; Eimes et al. 2016), our results suggest that MHC architecture continued to diversify during passerine evolution, producing great variation in MHC copy numbers within this group. Based on the time-dependent model, we also provided support for an accelerated rate of MHC class I evolution in birds, in contrast to the previous study by O’Connor et al. (2016) which found no evidence for an evolutionary burst in the copy number of MHC class I within Passerida. Evolution of MHC class I copy number was also well explained by the OU process with a central tendency, which is consistent with stabilizing selection (Hansen 1997; Butler and King 2004). In contrast, gene copy numbers at the MHC class IIB evolved according to the BM model adjusted for λ, which is consistent with fluctuating selection toward a moving optimum and genetic drift. The value of λ was moderately low (though significant), which could be caused by fluctuating selection with a low rate of fluctuation or by genetic drift that was initially low, but increased over time (Revell et al. 2008). Although we showed that different modes of selection were primarily responsible for the evolution of copy numbers at the MHC class I versus class II, we cannot exclude, based on our analyses, that drift also played some role in the evolution of MHC class I. However, consistently with the conclusions by O’Connor et al. (2016), copy number variation at the MHC is unlikely to have arisen solely through genetic drift, as this region is generally under strong positive selection (Minias et al. 2018).
We also demonstrated that nonpasserines showed consistently lower numbers of MHC class I and class IIB copies than passerines and that the duplication rate of MHC class I and class IIB varied between passerine lineages. While the low number of MHC loci during early avian radiation was probably constrained by evolutionary history (i.e., low number of loci in the common ancestor of birds), it remains unresolved why duplication rate did not accelerate during the evolution of old avian lineages (nonpasserines). This could reflect lower overall rates of evolution in nonpasserines than passerines resulting from their larger body size and longer generation time (Nam et al. 2010). Alternatively, a low number of MHC copies in nonpasserines could be associated with the evolution of some compensatory immunological mechanisms (Acevedo-Whitehouse and Cunningham 2006). For example, pathogen recognition in nonpasserines could be primarily governed by innate immune genes, such as toll-like receptors. Nonpasserines could also invest more in other innate defenses, overriding the need to trigger an adaptive response that may require additional metabolic costs (Lochmiller and Deerenberg 2000). These mechanisms are consistent with the hypothesis of an evolutionary trade-off between innate versus adaptive immunity (Wegner et al. 2007) and have already been employed to explain extraordinarily low levels of polymorphism at the MHC of some nonpasserine lineages, such as falcons (Gangoso et al. 2012). Within passerines, most MHC class I gene copies were recorded in the members of Sylvioidea superfamily, which contains 23 avian families, including tits and chickadees, swallows, and warblers (Winkler et al. 2015). In contrast, the highest number of MHC class IIB copies was found in the Passeroidea and Muscicapoidea superfamilies. These differences suggest that different passerine lineages may have been subject to a varying selective pressure by different groups of pathogens or parasites. In general, selective forces exerted by intra- and extracellular pathogens should lead to increasing polymorphism (e.g., via loci duplication) of MHC class I and class II, respectively. Thus, it is possible that differences in exposure to intra- and extracellular pathogens could be associated with variation in MHC copy number between both nonpasserines and passerines, as well as between different passerine clades.
Our study provides evidence that gene copy number at the avian MHC might have evolved in response to basic life-history traits. Specifically, we found that MHC class I and class II copy numbers correlated negatively with average body mass. Also, we found significant positive relationships between gene copy number and both residual lifespan (MHC class I) and migratory behavior (MHC class IIB). However, contrary to our previous comparative study of diversifying selection at the MHC class IIB in nonpasserine birds (Minias et al. 2017), we found no effect of sociality on gene copy number at the avian MHC. The relationship with body mass likely reflects historical patterns in copy number evolution in birds, changing from ancestrally few loci in nonpasserines toward larger numbers of loci in passerines. On average, nonpasserines have much greater body masses than passerines (McKechnie et al. 2006), and this general pattern was apparent within our data set (3,263 ± 849 g in nonpasserines vs. 45.5 ± 8.6 g in passerines; P < 0.001). Thus, a correlation between body mass and MHC copy number could be a mere byproduct of avian evolutionary history. In contrast, there were no statistical differences in residual lifespan or migratory distance (all P > 0.1) between passerines and nonpasserines within our data set. Residual lifespan and migratory behavior can be associated with the level of exposure to pathogens or parasites and, thus, could possibly exert direct selective pressure on the MHC (Minias et al. 2017). First, as the lifespan increases, an average individual is exposed to pathogens longer and, thus, might interact with greater number of pathogen antigenic types during lifetime. Also, reemergence of certain antigenic types of pathogens is much more frequent in short-lived species and decreases with lifespan, meaning that populations of long-lived species are unlikely to be exposed to the same dominant strain in the successive years (Wikramaratna et al. 2014). In fact, a comparative study of Neotropical birds showed that slow-living species (longer development and lifespan) invest more in adaptive immunity than fast-living species (Lee et al. 2008). Similarly, long-distance migrants are usually exposed to more diverse, geographically distinct faunas of pathogens during different stages of their annual cycle (Figuerola and Green 2000; Altizer et al. 2011). Thus, residual lifespan and migratory behavior could both act to increase within-individual diversity of MHC via locus duplication, which would enhance the spectrum of pathogens recognized by an organism and, thus, could counteract detrimental effects of elevated pathogen exposure. These results are consistent with our previous study of nonpasserines, in which migratory behavior was identified as a key correlate of diversifying selection acting on the peptide-binding residues of MHC class IIB (Minias et al. 2017). On the other hand, long-distance migrant songbirds had lower or similar MHC class I diversity to African and Palearctic resident species (O’Connor et al. 2018), which combined with our results (no effect of migratory behavior on MHC class I copy numbers) suggests that MHC class I may be less important than MHC class II for migratory birds. While this hypothesis needs further testing, we also have to acknowledge that the link between migration and the mechanisms shaping genetic variation at the two MHC classes may differ between species. For example, a recent study by Whittingham et al. (2018) revealed higher MHC class I, but not class II, diversity in migratory than resident populations of the common yellowthroat. As expected for class I, the migratory individuals also had higher levels of infection by intracellular hemosporidians.
In conclusion, our study provides novel insights into the history and contrasting mechanisms of copy number evolution at the avian MHC class I and class IIB, as assessed in a broad phylogenetic context (over 250 bird species from 68 families). We found contrasting patterns of copy number evolution between MHC class I (accelerated evolution and stabilizing selection) and class IIB (fluctuating selection and drift). Ancestral reconstruction of MHC copy number also provided qualitative evidence for low numbers of MHC loci in nonpasserines and for different rates of MHC class I and IIB duplication in different passerine lineages. Finally, we provided evidence for the role of life-history traits in the evolution of copy numbers at the avian MHC and suggest that future studies of different types of pathogens (intra- and extracellular) may expand our understanding of macroevolution of the MHC in different lineages of birds.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
We thank Emily O’Connor and an anonymous reviewer for constructive comments on an earlier draft of the article. The study was financially supported by the research grant of the National Science Centre in Poland (2015/19/D/NZ8/01310).
Literature Cited
- Acevedo-Whitehouse K, Cunningham AA.. 2006. Is MHC enough for understanding wildlife immunogenetics? Trends Ecol Evol. 21(8):433–438. [DOI] [PubMed] [Google Scholar]
- Altizer S, Bartel R, Han BA.. 2011. Animal migration and infectious disease risk. Science 331(6015):296–302. [DOI] [PubMed] [Google Scholar]
- Balasubramaniam S. 2016. New data from basal Australian songbird lineages show that complex structure of MHC class II β genes has early evolutionary origins within passerines. BMC Evol Biol. 16(1):112.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belov K. 2006. Reconstructing an ancestral mammalian immune supercomplex from a marsupial major histocompatibility complex. PLoS Biol. 4(3):e46.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belov K, Lam MP, Colgan DJ.. 2004. Marsupial MHC class II β genes are not orthologous to the eutherian β gene families. J Hered. 95(4):338–345. [DOI] [PubMed] [Google Scholar]
- Bernatchez L, Landry C.. 2003. MHC studies in nonmodel vertebrates: what have we learned about natural selection in 15 years? J Evol Biol. 16(3):363–377. [DOI] [PubMed] [Google Scholar]
- Biedrzycka A, O’Connor E et al. 2017. Extreme MHC class I diversity in the sedge warbler (Acrocephalus schoenobaenus); selection patterns and allelic divergence suggest that different genes have different functions. BMC Evol Biol. 17:159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biedrzycka A, Sebastian A, Migalska M, Westerdahl H, Radwan J.. 2017. Testing genotyping strategies for ultra‐deep sequencing of a co‐amplifying gene family: MHC class I in a passerine bird. Mol Ecol Res. 17(4):642–655. [DOI] [PubMed] [Google Scholar]
- Blomberg SP, Garland T, Ives AR.. 2003. Testing for phylogenetic signal in comparative data: behavioral traits are more labile. Evolution 57(4):717–745. [DOI] [PubMed] [Google Scholar]
- Bollmer JL, Dunn PO, Freeman-Gallant CR, Whittingham LA.. 2012. Social and extra-pair mating in relation to major histocompatibility complex variation in common yellowthroat. Proc R Soc B. 279:4478–4785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollmer JL, Dunn PO, Whittingham LA, Wimpee C.. 2010. Extensive MHC class II B gene duplication in a passerine, the common yellowthroat. J Hered. 101(4):448–460. [DOI] [PubMed] [Google Scholar]
- Burri R, Hirzel HN, Salamin N, Roulin A, Fumagalli L.. 2008. Evolutionary patterns of MHC class II B in owls and their implications for the understanding of avian MHC evolution. Mol Biol Evol. 25(6):1180–1191. [DOI] [PubMed] [Google Scholar]
- Butler MA, King AA.. 2004. Phylogenetic comparative analysis: a modeling approach for adaptive evolution. Am Nat. 164(6):683–695. [DOI] [PubMed] [Google Scholar]
- del Hoyo J, Elliott A, Sargatal J.. 1992. –2011. Handbook of the birds of the world. Vol. 1–16 Barcelona (Spain): Lynx Edicions. [Google Scholar]
- Dunn PO, Bollmer JL, Freeman-Gallant CR, Whittingham LA.. 2013. MHC variation is related to a sexually selected ornament, survival and parasite resistance in the common yellowthroat. Evolution 67(3):679–687. [DOI] [PubMed] [Google Scholar]
- Eimes JA, et al. 2016. Early duplication of a single MHC IIB locus prior to the passerine radiations. PLoS One 11(9):e0163456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans ML, Neff BD.. 2009. Major histocompatibility complex heterozygote advantage and widespread bacterial infections in populations of Chinook salmon (Oncorhynchus tshawytscha). Mol Ecol. 18(22):4716–4729. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. 1973. Maximum likelihood estimation of evolutionary trees from continuous characters. Am J Hum Genet. 25(5):471–492. [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. 1985. Phylogenies and the comparative method. Am Nat. 125(1):1–15. [Google Scholar]
- Figuerola J, Green AJ.. 2000. Haematozoan parasites and migratory behaviour in waterfowl. Evol Ecol. 14(2):143–153. [Google Scholar]
- Freckleton RP, Harvey PH, Pagel M.. 2002. Phylogenetic analysis and comparative data: a test and review of evidence. Am Nat. 160(6):712–716. [DOI] [PubMed] [Google Scholar]
- Gangoso L, et al. 2012. Colonizing the world in spite of reduced MHC variation. J Evol Biol. 25(7):1438–1447. [DOI] [PubMed] [Google Scholar]
- Gittleman JL, Kot M.. 1990. Adaptations: statistics and null model for estimating phylogenetic effects. Syst Zool. 39(3):227–241. [Google Scholar]
- Goebel J, et al. 2017. 100 million years of multigene family evolution: origin and evolution of the avian MHC class IIB. BMC Genomics 18(1):460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimholt U. 2016. MHC and evolution in teleosts. Biology 5(1):6.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadfield JD. 2010. MCMC methods for multi-response generalized linear mixed models: the MCMCglmm R package. J Stat Soft. 33:1–22. [Google Scholar]
- Hadfield JD. 2015. Increasing the efficiency of MMC for hierarchical phylogenetic models of categorical traits using reduced mixed models. Methods Ecol Evol. 6(6):706–714. [Google Scholar]
- Hadfield JD, Nakagawa S.. 2010. General quantitative genetic methods for comparative biology: phylogenies, taxonomies, and multi-trait models for continuous and categorical characters. J Evol Biol. 23(3):494–508. [DOI] [PubMed] [Google Scholar]
- Hansen TF. 1997. Stabilizing selection and the comparative analysis of adaptation. Evolution 51(5):1341–1351. [DOI] [PubMed] [Google Scholar]
- Hansen TF, Martins EP.. 1996. Translating between microevolutionary process and macroevolutionary patterns: the correlation structure of interspecific data. Evolution 50(4):1404–1417. [DOI] [PubMed] [Google Scholar]
- Harmon LJ, et al. 2010. Early bursts of body size and shape evolution are rare in comparative data. Evolution 64(8):2385–2396. [DOI] [PubMed] [Google Scholar]
- Harmon LJ, Weir J, Brock C, Glor RE, Challenger W.. 2008. GEIGER: investigating evolutionary radiations. Bioinformatics 24(1):129–131. [DOI] [PubMed] [Google Scholar]
- Hedrick PW. 2002. Pathogen resistance and genetic variation at MHC loci. Evolution 56(10):1902–1908. [DOI] [PubMed] [Google Scholar]
- Horton R, et al. 2004. Gene map of the extended human MHC. Nat Rev Genet. 5(12):889–899. [DOI] [PubMed] [Google Scholar]
- Hughes AL, Nei M.. 1988. Pattern of nucleotide substitution at major histocompatibility complex class I loci reveals overdominant selection. Nature 335(6186):167–170. [DOI] [PubMed] [Google Scholar]
- Hughes AL, Nei M.. 1989. Evolution of the major histocompatibility complex: independent origin of nonclassical class I genes in different groups of mammals. Mol Biol Evol. 6:559–579. [DOI] [PubMed] [Google Scholar]
- Jeffery KJ, Bangham CR.. 2000. Do infectious diseases drive MHC diversity? Microbes Infect. 2(11):1335–1341. [DOI] [PubMed] [Google Scholar]
- Kaufman J, et al. 1999. The chicken B locus is a minimal essential major histocompatibility complex. Nature 401(6756):923–925. [DOI] [PubMed] [Google Scholar]
- Klein J. 1986. Natural history of the major histocompatibility complex. New York: Wiley. [Google Scholar]
- Kloch A, Babik W, Bajer A, Siński E, Radwan J.. 2010. Effects of an MHC-DRB genotype and allele number on the load of gut parasites in the bank vole Myodes glareolus. Mol Ecol. 19:255–265. [DOI] [PubMed] [Google Scholar]
- Lawlor DA, Zemmour J, Ennis PD, Parham P.. 1990. Evolution of class-I MHC genes and proteins: from natural selection to thymic selection. Ann Rev Immunol. 8:23–63. [DOI] [PubMed] [Google Scholar]
- Lee KA, Wikelski M, Robinson WD, Robinson TR, Klasing KC.. 2008. Constitutive immune defences correlate with life-history variables in tropical birds. J Anim Ecol. 77(2):356–363. [DOI] [PubMed] [Google Scholar]
- Lochmiller RL, Deerenberg C.. 2000. Trade‐offs in evolutionary immunology: just what is the cost of immunity? Oikos 88(1):87–98. [Google Scholar]
- Madsen T, Ujvari B.. 2006. MHC class I variation associates with parasite resistance and longevity in tropical pythons. J Evol Biol. 19(6):1973–1978. [DOI] [PubMed] [Google Scholar]
- Martins EP, Hansen TF.. 1997. Phylogenies and the comparative method: a general approach to incorporating phylogenetic information into the analysis of interspecific data. Am Nat. 149(4):646–667. [Google Scholar]
- McKechnie AE, Freckleton RP, Jetz W.. 2006. Phenotypic plasticity in the scaling of avian basal metabolic rate. Proc R Soc B. 273(1589):931–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milinski M. 2006. The major histocompatibility complex, sexual selection, and mate choice. Ann Rev Ecol Evol Syst. 37(1):159–186. [Google Scholar]
- Minias P, Pikus E, Whittingham LA, Dunn PO.. 2018. A global analysis of selection at the avian MHC. Evolution 72(6):1278–1293. [DOI] [PubMed] [Google Scholar]
- Minias P, Whittingham LA, Dunn PO.. 2017. Coloniality and migration are related to selection on MHC genes in birds. Evolution 71(2):432–441. [DOI] [PubMed] [Google Scholar]
- Münkemüller T, et al. 2012. How to measure and test phylogenetic signal. Methods Ecol Evol. 3(4):743–756. [Google Scholar]
- Nam K, et al. 2010. Molecular evolution of genes in avian genomes. Genome Biol. 11(6):R68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M, Gu X, Sitnikova T.. 1997. Evolution by the birth-and-death process in multigene families of the vertebrate immune system. Proc Natl Acad Sci U S A. 94(15):7799–7806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak MA, Tarczy-Hornoch K, Austyn JM.. 1992. The optimal number of major histocompatibility complex molecules in an individual. Proc Natl Acad Sci U S A. 89(22):10896–10899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor EA, Cornwallis CK, Hasselquist D, Nilsson J-Å, Westerdahl H.. 2018. The evolution of immunity in relation to colonization and migration. Nat Ecol Evol. 2(5):841.. [DOI] [PubMed] [Google Scholar]
- O’Connor EA, Strandh M, Hasselquist D, Nilsson J-Å, Westerdahl H.. 2016. The evolution of highly variable immunity genes across a passerine bird radiation. Mol Ecol. 25(4):977–989. [DOI] [PubMed] [Google Scholar]
- Pagel M. 1999. Inferring the historical patterns of biological evolution. Nature 401(6756):877–884. [DOI] [PubMed] [Google Scholar]
- Paradis E, Claude J, Strimmer K.. 2004. APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20(2):289–290. [DOI] [PubMed] [Google Scholar]
- Piontkivska H, Nei M.. 2003. Birth-and-death evolution in primate MHC class I genes: divergence time estimates. Mol Biol Evol. 20(4):601–609. [DOI] [PubMed] [Google Scholar]
- R. Development Core Team. 2013. R: a language and environment for statistical computing. Vienna (Austria: ): R Foundation for Statistical Computing. [Google Scholar]
- Radwan J, et al. 2012. MHC diversity, malaria and lifetime reproductive success in collared flycatchers. Mol Ecol. 21(10):2469–2479. [DOI] [PubMed] [Google Scholar]
- Revell LJ. 2012. phytools: an R package for phylogenetic comparative biology (and other things). Methods Ecol Evol. 3(2):217–223. [Google Scholar]
- Revell LJ, Harmon LJ, Collar DC.. 2008. Phylogenetic signal, evolutionary process, and rate. Syst Biol. 57(4):591–601. [DOI] [PubMed] [Google Scholar]
- Sato A, Dongak R, Hao L, Shintani S, Sato T.. 2012. Organization of MHC class II A and B genes in the tilapiine fish Oreochromis. Immunogenetics 64(9):679–690. [DOI] [PubMed] [Google Scholar]
- Savage AE, Zamudino KR.. 2011. MHC genotypes associate with resistance to a frog-killing fungus. Proc Natl Acad Sci U S A. 108(40):16705–16710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepil I, Moghadam HK, Hucard E, Sheldon BC.. 2012. Characterization and 454 pyrosequencing of Major Histocompatibility Complex class I genes in the great tit reveal complexity in a passerine system. BMC Evol Biol. 12(1):68.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snow DW, Perrins CM.. 1998. The birds of the Western Palearctic (concise edition). Vol. 1–2 Oxford: Oxford University Press. [Google Scholar]
- Spurgin LG, Richardson DS.. 2010. How pathogens drive genetic diversity: MHC, mechanisms and misunderstandings. Proc R Soc B. 277(1684):979–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Star B, et al. 2011. The genome sequence of Atlantic cod reveals a unique immune system. Nature 477(7363):207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahata N, Nei M.. 1990. Allelic genealogy under overdominant and frequency-dependent selection and polymorphism of major histocompatibility complex loci. Genetics 124(4):967–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegner KM, Kalbe M, Reusch TBH.. 2007. Innate versus adaptive immunity in sticklebacks: evidence for trade-offs from a selection experiment. Evol Ecol. 21(4):473–483. [Google Scholar]
- Whittingham LA, Dunn PO, Freeman‐Gallant CR, Taff CC, Johnson JA.. 2018. Major histocompatibility complex variation and blood parasites in resident and migratory populations of the common yellowthroat. J Evol Biol. 31(10):1544–1557. [DOI] [PubMed] [Google Scholar]
- Wikramaratna PS, Pybus OG, Gupta S.. 2014. Contact between bird species of different lifespans can promote the emergence of highly pathogenic avian influenza strains. Proc Natl Acad Sci U S A. 111(29):10767–10772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler DW, Billerman SM, Lovette IJ.. 2015. Bird families of the world: an invitation to the spectacular diversity of birds. Barcelona (Spain): Lynx Edicions. [Google Scholar]
- Woelfing B, Traulsen A, Milinski M, Boehm T.. 2009. Does intra-individual major histocompatibility complex diversity keep a golden mean? Philos Trans R Soc B. 364(1513):117–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagalska-Neubauer M, et al. 2010. 454 sequencing reveals extreme complexity of the class II major histocompatibility complex in the collared flycatcher. BMC Evol Biol. 10:395.. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.