

Abstract
GIPR signaling in adipose tissue plays an important role in HFD‐induced insulin resistance and hepatic steatosis in vivo, with no direct effect on fat accumulation, through IL‐6 signaling
![]()
Introduction
Obesity has been recognized as a worldwide problem, especially for developing insulin resistance and increasing the risk of type 2 diabetes. The increased adipose tissue associated with obesity secretes various kinds of cytokines, adipokines and lipokines, and triggers systemic inflammatory response, resulting in the disturbance of metabolic homeostasis.
Gastric inhibitory polypeptide/glucose‐dependent insulinotropic polypeptide (GIP) is an incretin secreted from enteroendocrine K cells in response to meal ingestion1, and potentiates insulin secretion through the GIP receptor (GIPR) expressed in pancreatic β‐cells2. Dietary lipid is a very strong stimulant of GIP secretion. For example, the peak value of GIP increase in response to a high‐fat meal (450 kcal containing 33.3% of fat) is threefold higher than that in the 75‐g oral glucose tolerance test in human subjects, suggesting that fat content in a mixed meal strongly stimulates GIP secretion1. Chronic high‐fat consumption induces hypersecretion of GIP from K cells3, and insulin secretion in response to GIP is enhanced in high‐fat diet (HFD)‐fed obese mice compared with that in lean mice fed a normal‐fat diet4, indicating that GIPR signaling plays a critical role in postprandial hyperinsulinemia associated with chronic HFD feeding and the subsequent increase in lipoprotein lipase activity, which results in HFD‐induced fat accumulation (Figure 1). Actually, reduction in GIPR signaling using systemic GIPR knockout (GIPR−/−) mice5, a GIP antagonist6 and GIP immunoneutralization ameliorates obesity under HFD‐fed conditions. In addition, we have reported that genetic deletion of GIP alleviates obesity and lessens the degree of insulin resistance in HFD‐fed mice7. In contrast, given that functional GIPR is expressed in rodent and human adipocytes, it has been assumed that GIP also has direct effects on adipose tissue and plays some roles in lipid metabolism under HFD‐fed conditions.
Figure 1.
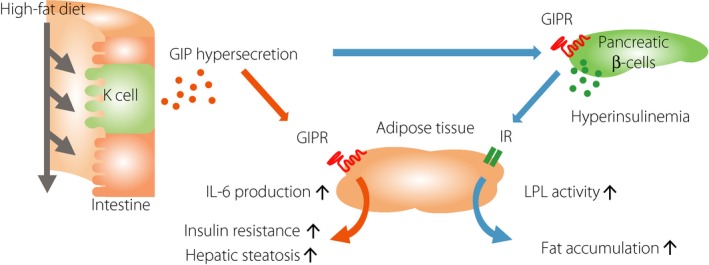
Schematic representation illustrating potential mechanisms by which GIP hypersecretion in high‐fat diet conditions induces insulin resistance and hepatic steatosis. GIP, gastric inhibitory polypeptide/glucose‐dependent insulinotropic polypeptide; GIPR, gastric inhibitory polypeptide/glucose‐dependent insulinotropic polypeptide receptor; IR, insulin receptor; IL‐6, interleukin‐6; LPL, lipoprotein lipase.
GIP Signaling in Adipose Tissue
In vitro studies have shown that GIP directly induces fat accumulation in adipose tissue by increasing lipoprotein lipase expression through the cyclic adenosine monophosphate response element‐binding protein (CREB)‐regulated transcription coactivator 2‐mediated pathway, and by increasing lipoprotein lipase enzyme activity and plasma membrane glucose transporter 4 expression through the protein kinase B (PKB)‐mediated pathway8. However, the physiological significance of GIPR signaling in adipocytes, namely, whether GIPR expressed in adipose tissue plays some roles in HFD‐induced obesity and insulin resistance in vivo, has remained unproven.
To address this problem, we generated adipose tissue‐specific GIPR knockout (GIPRadipo−/−) mice9. Under HFD‐fed conditions, GIPRadipo−/− mice had significantly lower bodyweight and lean body mass compared with those in floxed GIPR (GIPRfl/fl) mice, although the fat volume was not significantly different between the two groups. Interestingly, insulin resistance, liver weight and hepatic steatosis were reduced in HFD‐fed GIPRadipo−/− mice compared with those in GIPRfl/fl mice. Microarray analysis of adipose tissues showed that expression of interleukin‐6 (IL‐6), a pro‐inflammatory cytokine that induces insulin resistance and hepatic steatosis10, was significantly decreased in adipose tissue of GIPRadipo−/− mice, and plasma levels of IL‐6 were also reduced in HFD‐fed GIPRadipo−/− mice compared with those in HFD‐fed GIPRfl/fl mice.
Previous in vitro studies have shown that GIP increases IL‐6 expression levels in adipose tissue11. In astrocytes, CREB activated by β‐adrenergic receptor enhances IL‐6 expression induced by tumor necrosis factor (TNF)‐α receptor signaling12. It has been reported that GIP activates CREB in adipose tissue13. In our study using differentiated 3T3‐L1 adipocytes, IL‐6 expression was significantly augmented by GIP in the presence of TNF‐α, suggesting that GIP enhances IL‐6 messenger ribonucleic acid (mRNA) expression induced by TNF‐α in adipose tissue, possibly through CREB activation.
Suppressor of cytokine signaling (SOCS) protein is induced by the activation of cytokine receptors, including the IL‐6 receptor, TNF‐α receptor and Toll‐like receptors, and attenuates cytokine signal transduction14. Seven SOCS proteins have been identified, and SOCS1 and SOCS3 were recently reported to negatively regulate insulin signaling by competitively interfering with insulin receptor substrate binding to the insulin receptor15, by inhibiting insulin receptor autophosphorylation16 and by promoting proteasomal degradation of insulin receptor substrate17. In particular, SOCS3 signaling is located downstream of the IL‐6 receptor14, and is associated with insulin resistance and hepatic steatosis18. Our in vitro experiments showed that GIP induced mRNA expression of IL‐6 and SOCS3 in the presence of TNF‐α in differentiated 3T3‐L1 adipocytes. In addition, expression levels of SOCS3 mRNA were decreased in adipose and liver tissues of GIPRadipo−/− mice. These results suggest that a decrease in SOCS3 expression by GIPR deficiency might enhance insulin sensitivity and potentiate PKB phosphorylation in adipose and liver tissues. However, SOCS3 mRNA expression in skeletal muscle was not different between the two groups of mice, although PKB phosphorylation was increased in the skeletal muscle of GIPRadipo−/− mice. Further study is required to clarify the mechanism of the increase in insulin‐induced PKB phosphorylation in the skeletal muscle of GIPR adipo−/− mice.
It has also been reported that SOCS3 binds to the gene promoter of sterol regulatory element‐binding protein‐1c (SREBP‐1c), which has critical roles in fat synthesis in the liver, and increases SREBP‐1c mRNA expression18. Suppression of SOCS3 expression in db/db mice improves the hepatic steatosis through downregulation of SREBP‐1c19. In our study, SREBP‐1c and SOCS3 mRNA expression were decreased in the liver of HFD‐fed GIPR adipo−/− mice, indicating that deletion of GIPR signaling in adipocytes results in the decrease in IL‐6 production from adipose tissue and subsequent fat synthesis in the liver through the IL‐6–SOCS3–SREBP‐1c pathway.
Although GIPR function in human adipose tissue is still unclear in vivo, in vitro study shows that GIP induces IL‐6 mRNA expression and potentiates IL‐6 secretion in the presence of TNF‐α in human adipocytes11, which is consistent with our data showing that IL‐6 mRNA expression is decreased in the adipocytes of HFD‐fed GIPRadipo−/− mice.
Based on the phenotypes of GIPRadipo−/− mice, it can be concluded that GIPR signaling in adipose tissue plays an important role in HFD‐induced insulin resistance and hepatic steatosis in vivo, with no direct effect on fat accumulation, through IL‐6 signaling (Figure 1).
Future Perspective
As shown above, there is sufficient evidence to confirm that GIP is an obesity‐promoting factor in HFD conditions and that deletion of GIPR signaling causes resistance to diet‐induced obesity. Additionally, we have reported that partial reduction of GIP alleviates obesity and lessens the degree of insulin resistance under HFD conditions7. These findings suggest that regulation of GIPR signaling, especially after fat intake, is a promising therapeutic approach to obesity and type 2 diabetes. GIP antagonism is one of the candidates for GIP‐based treatment for diabetes and obesity. However, the results in mice are not necessarily recapitulated in humans. For example, previously developed (Pro3) GIP as a potent antagonist of GIPR in mice was shown to stimulate GIPR in humans20. In addition, given that GIPR is also expressed in various tissues, except for pancreatic β‐cells and adipocytes, including the gastrointestinal tract, heart, inner layers of the adrenal cortex and several brain regions, further investigation is required to establish the systemic biological action of GIP before the implementation of GIPR antagonism as an anti‐obesity/diabetes therapy to minimize unexpected adverse events.
Disclosure
The authors declare no conflict of interest.
Acknowledgments
This study was supported by grants from the Ministry of Education, Culture, Sports, Science and Technology (MEXT); Japan Society for the Promotion of Science; Ministry of Health, Labor and Welfare; Ministry of Agriculture, Forestry and Fisheries; Japan Diabetes Foundation; Japan Association for Diabetes Education and Care; Merck Sharp & Dohme (MSD); Novo Nordisk Pharma; and Banyu Life Science Foundation International.
References
- 1. Yamane S, Harada N, Hamasaki A, et al Effects of glucose and meal ingestion on incretin secretion in Japanese subjects with normal glucose tolerance. J Diabetes Investig 2012; 3: 80–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Seino Y, Yabe D. Glucose‐dependent insulinotropic polypeptide and glucagon‐like peptide‐1: incretin actions beyond the pancreas. J Diabetes Investig 2013; 4: 108–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Suzuki K, Harada N, Yamane S, et al Transcriptional regulatory factor X6 (Rfx6) increases gastric inhibitory polypeptide (GIP) expression in enteroendocrine K‐cells and is involved in GIP hypersecretion in high‐fat diet‐induced obesity. J Biol Chem 2013; 288: 1929–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Harada N, Yamada Y, Tsukiyama K, et al A novel GIP receptor splice variant influences GIP sensitivity of pancreatic beta‐cells in obese mice. Am J Physiol Endocrinol Metab 2008; 294: E61–E68. [DOI] [PubMed] [Google Scholar]
- 5. Miyawaki K, Yamada Y, Ban N, et al Inhibition of gastric inhibitory polypeptide signaling prevents obesity. Nat Med 2002; 8: 738–742. [DOI] [PubMed] [Google Scholar]
- 6. McClean PL, Irwin N, Cassidy RS, et al GIP receptor antagonism reverses obesity, insulin resistance, and associated metabolic disturbances induced in mice by prolonged consumption of high‐fat diet. Am J Physiol Endocrinol Metab 2007; 293: E1746–E1755. [DOI] [PubMed] [Google Scholar]
- 7. Nasteska D, Harada N, Suzuki K, et al Chronic reduction of GIP secretion alleviates obesity and insulin resistance under high‐fat diet conditions. Diabetes 2014; 63: 2332–2343. [DOI] [PubMed] [Google Scholar]
- 8. Kim SJ, Nian C, McIntosh CH. GIP increases human adipocyte LPL expression through CREB and TORC2‐mediated trans‐activation of the LPL gene. J Lipid Res 2010; 51: 3145–3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Joo E, Harada N, Yamane S, et al Inhibition of gastric inhibitory polypeptide receptor signaling in adipose tissue reduces insulin resistance and hepatic steatosis in high‐fat diet‐fed mice. Diabetes 2017; 66: 868–879. [DOI] [PubMed] [Google Scholar]
- 10. Sabio G, Das M, Mora A, et al A stress signaling pathway in adipose tissue regulates hepatic insulin resistance. Science 2008; 322: 1539–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen S, Okahara F, Osaki N, et al Increased GIP signaling induces adipose inflammation via a HIF‐1α‐dependent pathway and impairs insulin sensitivity in mice. Am J Physiol Endocrinol Metab 2015; 308: E414–E425. [DOI] [PubMed] [Google Scholar]
- 12. Spooren A, Kooijman R, Lintermans B, et al Cooperation of NFκB and CREB to induce synergistic IL‐6 expression in astrocytes. Cell Signal 2010; 22: 871–881. [DOI] [PubMed] [Google Scholar]
- 13. Mohammad S, Ramos LS, Buck J, et al Gastric inhibitory peptide controls adipose insulin sensitivity via activation of cAMP response element‐binding protein and p110β isoform of phosphatidylinositol 3‐kinase. J Biol Chem 2011; 286: 43062–43070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Johnston JA, O'Shea JJ. Matching SOCS with function. Nat Immunol 2003; 4: 507–509. [DOI] [PubMed] [Google Scholar]
- 15. Galic S, Sachithanandan N, Kay TW, et al Suppressor of cytokine signalling (SOCS) proteins as guardians of inflammatory responses critical for regulating insulin sensitivity. Biochem J 2014; 461: 177–188. [DOI] [PubMed] [Google Scholar]
- 16. Trengove MC, Ward AC. SOCS proteins in development and disease. Am J Clin Exp Immunol 2013; 2: 1–29. [PMC free article] [PubMed] [Google Scholar]
- 17. Ueki K, Kondo T, Kahn CR. Suppressor of cytokine signaling 1 (SOCS‐1) and SOCS‐3 cause insulin resistance through inhibition of tyrosine phosphorylation of insulin receptor substrate proteins by discrete mechanisms. Mol Cell Biol 2004; 24: 5434–5446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Xu X, So JS, Park JG, et al Transcriptional control of hepatic lipid metabolism by SREBP and ChREBP. Semin Liver Dis 2013; 33: 301–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ueki K, Kondo T, Tseng Y‐H, et al Central role of suppressors of cytokine signaling proteins in hepatic steatosis, insulin resistance, and the metabolic syndrome in the mouse. Proc Natl Acad Sci USA 2004; 101: 10422–10427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sparre‐Ulrich AH, Hansen LS, Svendsen B, et al Species‐specific action of (Pro3)GIP ‐ A full agonist at human GIP receptors, but a partial agonist and competitive antagonist at rat and mouse GIP receptors. Br J Pharmacol 2016; 173: 27–38. [DOI] [PMC free article] [PubMed] [Google Scholar]