

Abstract
The alpha/beta‐hydrolases (ABH) are among the largest structural families of proteins that are found in nature. Although they vary in their sequence and function, the ABH enzymes use a similar acid–base‐nucleophile catalytic mechanism to catalyze reactions on different substrates. Because ABH enzymes are biocatalysts with a wide range of potential applications, protein engineering has taken advantage of their catalytic versatility to develop enzymes with industrial applications. This study is a comprehensive analysis of 40 ABH enzyme families focusing on two identified substructures: the nucleophile zone and the oxyanion zone, which co‐ordinate the catalytic nucleophile and the residues of the oxyanion hole, and independently reported as critical for the enzymatic activity. We also frequently observed an aromatic cluster near the nucleophile and oxyanion zones, and opposite the ligand‐binding site. The nucleophile zone, the oxyanion zone and the residue cluster enriched in aromatic side chains comprise a three‐dimensional structural organization that shapes the active site of ABH enzymes and plays an important role in the enzymatic function by structurally stabilizing the catalytic nucleophile and the residues of the oxyanion hole. The structural data support the notion that the aromatic cluster can participate in co‐ordination of the catalytic histidine loop, and properly place the catalytic histidine next to the catalytic nucleophile.
Keywords: alpha/beta‐hydrolases, catalytic triad, structural motif, carboxylesterase; structural framework
Abbreviations
- ABH
structural family of alpha/beta‐hydrolases
- EstFa_R
recombinant carboxylesterase from Ferroplasma acidiphilum
- Nuc
catalytic nucleophile residue
- PDB
Protein Data Bank
- SCOP database
Structural Classification of Proteins database
- Smnuc‐2
small residue located two positions before the catalytic nucleophile residue
- Xnuc‐1
residue located adjacent (N‐terminal) to the catalytic nucleophile residue
- XoxyI
residues of the oxyanion hole, first position
- XoxyII
residues of the oxyanion hole, second position
- XozI
residue at position “Oxyanion zone I”
- XozII
residue at position “Oxyanion zone II”
Introduction
The alpha/beta‐hydrolases (ABH) are a structural family of enzymes that share a common fold structure and a catalytic mechanism, while differing in their degree of sequence similarity and function.1, 2, 3, 4 The ABH enzymes catalyze reactions on different substrates, but some ABH enzymes can also act as transporters, receptors, or have other ancillary functions.5 The ABH‐fold enzymes are structurally stable, and thus have been extensively used in protein engineering.6, 7
The ABH family members maintain a half‐barrel structure shaped by eight, mostly parallel β‐strands (the strand β2 is antiparallel) and six α‐helices (αA‐αF) flanking the β‐sheet. The β‐strands and the α‐helices are organized in an α‐turn‐β supersecondary structure geometry that starts with the αA‐helix followed by strand β4. The catalytic machinery of all ABH enzymes is similar, with the catalytic triad consisting of an acid: aspartic or glutamic acid; a base: histidine; and a nucleophile: serine, cysteine or aspartate. This catalytic triad is found at identical locations within the ABH fold (Fig. 1). In particular, the catalytic acid residue is located on a reverse turn after strand β7 or on a tight turn after strand β6; and the catalytic base is a histidine located on the loop following strand β8.1
Figure 1.
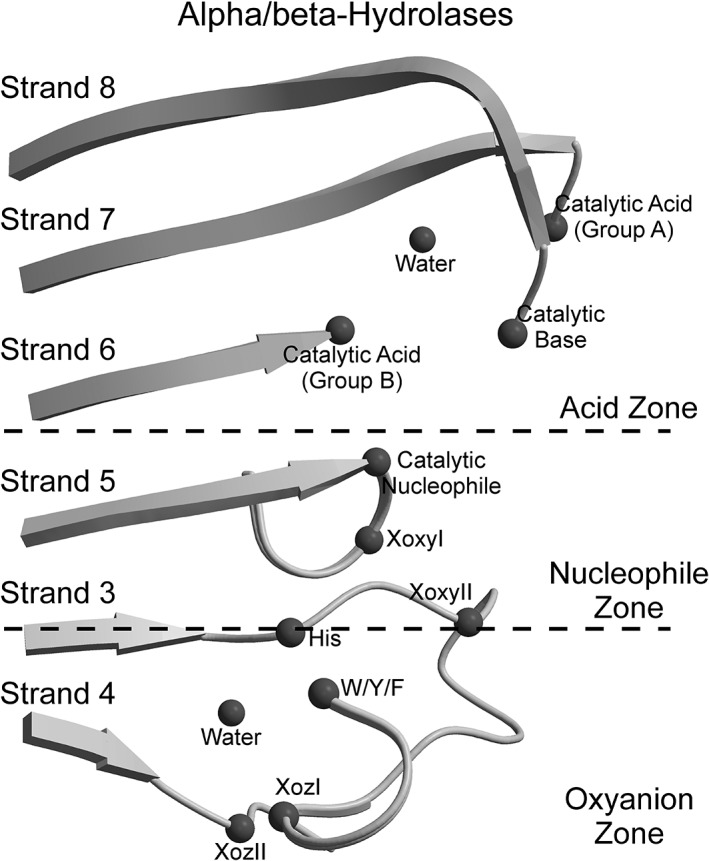
The active site of the ABH‐fold enzymes. Elements of the catalytic machinery are arranged as follows: the catalytic acid (“Catalytic Acid”) is located either after strand β7 (Group A) or strand β6 (Group B); the catalytic base (“Catalytic Base”); and the catalytic nucleophile (“Catalytic Nucleophile”). The oxyanion hole is usually formed by two residues: the Oxyanion I residue (“XoxyI”), adjacent to the catalytic nucleophile, and the Oxyanion II residue (“XoxyII”) in the loop following strand β3. The “Acid zone”, the “Nucleophile zone” and the “Oxyanion zone” designate the positions of three clusters of conserved residues that are linked by hydrogen bonds around the catalytic acid, the catalytic nucleophile and the residues of the oxyanion hole, respectively; the boundaries of each zone are marked by dashed lines. The positions of conserved elements of the zones are shown as: “Water” for structural water molecules; “His” for the conserved histidine of the Oxyanion zone; and “W/Y/F” for the aromatic residues tryptophan, tyrosine or phenylalanine from the aromatic cluster. The positions “Oxyanion zone I” (“XozI”) and “Oxyanion zone II” (“XozII”) are part of the Oxyanion zone
The catalytic nucleophile is the third key player of the charge‐relay system that enables the covalent catalysis by nucleophilic attack. The nucleophile is located at the apex of a sharp turn after strand β5, known as the “nucleophile elbow”, identified by the sequence motif Smnuc‐2–Xnuc‐1–Nuc–Xnuc + 1–Smnuc + 2 (Sm, small residue; X, any residue; Nuc, nucleophile). The dipeptide Smnuc‐2–Xnuc‐1 is located at the end of strand β5 prior to the nucleophile and followed by the dipeptide Xnuc + 1–Smnuc + 2 located at the N‐terminal end of the αC‐helix.
The helical axis of the αC‐helix forms an acute angle with respect to strand β5 and the helical N‐terminus is placed close to the C‐terminus of strand β3; this architecture leads to the arrangement of the “oxyanion hole” that stabilizes the tetrahedral intermediate during catalysis.1 The oxyanion hole is situated adjacent to the nucleophile and is mainly shaped by the main‐chain nitrogen atoms of two residues that we term here oxyanion I (XoxyI) and oxyanion II (XoxyII). Residue XoxyI is always located after the nucleophile, at position Xnuc + 1 of the nucleophile elbow.1 Residue XoxyII is located after the start of loopβ3‐αA, from which the main‐chain nitrogen atom helps form the oxyanion hole in the majority of ABH enzymes. Sometimes, residue XoxyII is located at an alternate position and the oxyanion hole is formed by a side chain. For example, residue XoxyII can be a tyrosine at the start of loopβ3‐αA,8, 9 an aspartic acid after the start of loopβ3‐αA,10 or an arginine emerging from an α‐helix that is located opposite the active site.11 For some ABH enzymes, the oxyanion hole is formed by the main‐chain amide NH group from three residues.12, 13, 14
The nucleophile and the residues forming the oxyanion hole are essential for the enzymatic activity of ABH enzymes. Site‐directed mutagenesis of the nucleophile and residues forming the oxyanion hole results in the loss of enzymatic function, usually with little change in the local arrangement of the catalytic site and without interfering with the binding of the ligand8, 14, 15, 16, 17; however, it is possible that the mutation of residues of the oxyanion hole radically distorts the shape of loopβ3‐αA and hence inactivate the enzyme.18
In addition to the residues of the charge‐relay network, other residues around the active site have been shown to be essential for enzymatic activity. For example, Kiss et al.19 have shown experimentally that a residue located in the vicinity of the oxyanion hole affects the activity of the enzyme acylaminoacyl‐peptidase from Aeropyrum pernix. Specifically, the site‐directed mutation of His367 to alanine, located two residue‐positions before Gly369 of the Oxyanion II, distorted the shape of the oxyanion hole and consequently, peptidase activity was lost.
In our previous work, we studied the structure surrounding the catalytic acid and identified common structural elements that comprise the catalytic acid zone in ABH enzymes.20 We have shown that the catalytic acid zone co‐ordinates the position of the catalytic histidine loop relative to the catalytic acid and histidine residues. We have also shown that the catalytic acid zone is sometimes found at domain‐domain interfaces and residues of the acid zone are directly involved in the formation of multi‐subunit complexes and in protein–protein interactions.
Both the experimental study from Kiss et al.19 and our work on the catalytic acid zone20 suggested that there were additional elements important for the enzymatic activity in the structure around the active site. This motivated us to focus our attention on the architecture around the nucleophile and the oxyanion hole and describe their structural frameworks. In particular, we examined structures for the presence of common structural elements around each key part of the catalytic machinery of the charge‐relay system. Herein, we report the occurrence of unique substructures, which we refer to as the nucleophile zone and oxyanion zone, and discuss their role as supporting structural scaffolds for the enzymatic activity of ABH enzymes.
Results
From the structural point of view, ABH‐fold enzymes maintain a catalytic mechanism, which consists of an acid–base‐nucleophile triad and the oxyanion hole. Here, we have extensively studied all residues that directly surround key elements of the catalytic machinery; specifically, those residues adjacent to the nucleophile and the oxyanion hole, aiming to understand their influence on the activity of the ABH‐fold enzymes. We compared 41 structures, obtained from the Protein Data Bank21 (PDB), representing each of the 40 ABH enzyme families described in the SCOP database.22
This comprehensive analysis revealed structural conservation in three regions that surround the catalytic site: the nucleophile zone near the catalytic nucleophile and Oxyanion I residue XoxyI; the oxyanion zone near Oxyanion II residue XoxyII; and a cluster of aromatic residues that often surrounds both zones.
The nucleophile zone
The nucleophile zone is a planar structural organization that is found repeatedly in the ABH enzymes (Table 1). It co‐ordinates two key units of the catalytic mechanism: the catalytic nucleophile and the following residue, which is XoxyI of the oxyanion hole. Structurally, the nucleophile zone is formed by residues that belong to the nucleophile elbow and the loopβ3‐αA, and its hydrogen‐bond network consists of two conserved, weak hydrogen bonds that connect residues between the nucleophile elbow and the loopβ3‐αA [for the definition of weak hydrogen bonds, see Materials and methods].
Table 1.
Inventory of Interactions within the Nucleophile Zone in 36 ABH‐Fold Families
| N | SCOP Family #/ SCOP family name |
PDB ID resolution |
S nuc | O/Xnuc‐1 – CA/GoxyII‐1 | CA/Gnuc + 2 – O/HoxyII‐2 | N/XoxyI | N/XoxyII |
|---|---|---|---|---|---|---|---|
| A | 2. Carboxylesterase | 3WJ2_A23
1.61 Å |
S156 | O/D155‐CA/G84 3.1 (2.7) |
CA/G158‐O/H83 3.3 (2.7) |
N/A157 | N/G85 |
| 1 | 1. Acetylcholinesterase‐like | 1QE3_A75
1.50 Å |
S189 | O/E188‐CA/G105 3.2 (2.5) |
CA/G191‐O/H104 3.6 (3.2) |
N/A190 | N/G106 |
| 2 | 2. Carboxylesterase | 1LZL_A76
1.30 Å |
S160 | O/Q159‐CA/G87 3.1 (2.7) |
CA/G162‐O/H86 3.2 (2.6) |
N/A161 | N/G88 |
| 3 | 3. Mycobacterial antigens | 1DQZ_A47
1.50 Å |
S124 | O/L123‐CA/G39 4.1 (3.1) |
OG/S126‐O/D38 2.7 |
N/M125 | N/L40 |
| 4 | 4. Hypothetical protein TT1662 | 1UFO_A77
1.60 Å |
S113 | O/G112‐CA/G32 3.2 (2.9) |
CA/G115‐O/H31 3.4 (2.9) |
N/L114 | N/L33 |
| 5 | 5. PepX catalytic domain‐like | 3PUI_A24
1.53 Å |
S117 | ‐ | ‐ | N/Y118 | OH/Y44 |
| 6 | 6. Prolyl oligopeptidase, C‐terminal domain | 1H2W_A25
1.39 Å |
S554 | ‐ | ‐ | N/N555 | OH/Y473 |
| 7 | 7. DPP4 catalytic domain‐like | 1ORV_A26
1.80 Å |
S630 | ‐ | ‐ | N/Y631 | OH/Y547 |
| 8 | 8. Serine carboxypeptidase‐like | 3SC2_A48
2.20 Å |
S146 | O/E145‐CA/G52 3.8 (3.2) |
CB/A148‐O/N51 4.0 (2.9) |
N/Y147 | N/G53 |
| 9 | 9. Gastric lipase | 1HLG_A46
3.00 Å |
S153 | O/H152‐CA/G66 3.1 (2.7) |
CA/G155‐O/H65 3.3 (2.8) |
N/Q154 | N/L67 |
| 10 | 10. Proline iminopeptidase‐like | 1MTZ_A78
1.80 Å |
S105 | O/S104‐CA/G36 3.2 (2.8) |
CA/G107‐O/H35 3.3 (2.7) |
N/Y106 | N/G37 |
| 11 | 11. Acetyl xylan esterase‐like | 1L7A_A79
1.50 Å |
S181 | O/G180‐CA/G90 4.0 (3.3) |
CA/G183‐O/H89 3.6 (3.4) |
N/Q182 | N/Y91 |
| 12 | 12. Haloalkane dehalogenase | 1MJ5_A80
0.95 Å |
D108 | O/H107‐CA/G37 4.2 (3.6) |
CA/G110‐O/H36 3.4 (3.0) |
N/W109 | N/N38 |
| 13 | 13. Dienelactone hydrolase | 1ZI9_A27
1.50 Å |
S123 | O/Y122‐CA/D36 3.1 (2.3) |
CA/G125‐O/Q35 3.3 (2.8) |
N/L124 | N/I37 |
| 14 | 14. Carbon–carbon bond hydrolase | 2OG1_A81
1.60 Å |
S112 | O/N111‐CA/G41 3.3 (2.4) |
CA/G114‐O/H40 3.2 (2.8) |
N/M113 | N/G42 |
| 15 | 15. Biotin biosynthesis protein BioH | 4ETW_A82
2.05 Å |
A82a | O/W81‐CA/G21 3.3 (2.7) |
CA/G84‐O/H20 3.3 (2.7) |
N/L83 | N/W22 |
| 16 | 16. Aclacinomycin methylesterase RdmC | 1Q0R_A28
1.45 Å |
S102 | O/L101‐CA/G31 3.3 (2.7) |
CA/G104‐O/M30 3.2 (2.7) |
N/M103 | N/G32 |
| 17 | 17. Carboxylesterase/lipase | 4DIU_A83
2.00 Å |
S93 | O/L92‐CA/G23 3.3 (2.7) |
CA/G95‐O/H22 3.5 (3.0) |
N/L94 | N/F24 |
| 18 | 18. Epoxide hydrolase | 1QO7_A84
1.80 Å |
D192 | O/G191‐CA/G116 4.4 (3.7) |
CA/G194‐O/H115 3.4 (2.9) |
N/I193 | N/W117 |
| 19 | 19. Haloperoxidase | 1BRT_A85
1.50 Å |
S98 | O/F97‐CA/G31 3.7 (3.1) |
CA/G100‐O/H30 3.3 (2.4) |
N/T99 | N/F32 |
| 20 | 20. Thioesterases | 1EI9_A86
2.25 Å |
S115 | O/F114‐CA/G40 3.2 (2.8) |
CA/G117‐O/H39 3.8 (3.2) |
N/Q116 | N/M41 |
| 21 | 21. Carboxylesterase/thioesterase 1 | 1FJ2_A87
1.50 Å |
S114 | O/F113‐CA/G24 3.3 (2.9) |
CA/G116‐O/H23 3.4 (2.6) |
N/Q115 | N/L25 |
| 22 | 22. Ccg1/TafII250‐interacting factor B (Cib) | 1IMJ_A88
2.20 Å |
S111 | O/P110‐CA/G40 4.2 (3.4) |
CB/S113‐O/H39 3.6 (2.6) |
N/L112 | N/I41 |
| 23 | 23. A novel bacterial esterase | 1QLW_A89
1.09 Å |
S206 | O/H205‐CA/G70 3.3 (2.7) |
OG/S208‐O/H69 2.9 |
N/Q207 | N/C71 |
| 24 | 24. Lipase | 1JFR_A30
1.90 Å |
S131 | O/H130‐CA/G62 3.3 (3.0) |
CA/G133‐O/P61 3.4 (2.7) |
N/M132 | N/F63 |
| 25 | 25. Fungal lipases | 1TCA_A31
1.55 Å |
S105 | O/W104‐CA/G39 3.5 (2.8) |
CA/G107‐O/P38 3.5 (3.1) |
N/Q106 | N/T40 |
| 26 | 26. Bacterial lipase | 1ISP_A90
1.30 Å |
S77 | O/H76‐CA/G11 3.6 (3.3) |
CA/G79‐O/H10 3.4 (2.9) |
N/M78 | N/I12 |
| 27 | 27. Pancreatic lipase, N‐terminal domain | 1BU8_A91
1.80 Å |
S152 | O/H151‐CA/G76 3.6 (2.8) |
CA/G154‐O/H75 3.3 (2.7) |
N/L153 | N/F77 |
| 28 | 28. Hydroxynitrile lyase‐like | 3C6X_A92
1.05 Å |
S80 | O/E79‐CB/T11 3.5 (2.7) |
CA/G82‐O/H10 4.1 (3.7) |
N/C81 | N/I12 |
| 29 | 29. Thioesterase domain of polypeptide, polyketide and fatty acid synthases | 1JMK_C32
1.71 Å |
S80 | O/Y79‐CA/P26 3.3 (2.3) |
CA/G82‐O/P25 3.0 (2.5) |
N/A81 | N/V27 |
| 30 | 30. Cutinase‐like | 1BS9_A29
1.10 Å |
S90 | O/Y89‐CA/E12 3.4 (2.8) |
CA/G92‐O/R11 3.6 (3.1) |
N/Q91 | N/T13 |
| 31 | 31. YdeN‐like | 1UXO_A93
1.80 Å |
S71 | O/H70‐CA/G10 3.4 (3.1) |
CA/G73‐O/H9 3.5 (2.9) |
N/L72 | N/Y11 |
| 32 | 32. Putative serine hydrolase Ydr428c | 1VKH_A94
1.85 Å |
S110 | O/H109‐CA/G37 3.2 (2.5) |
CA/G112‐O/H36 3.5 (3.0) |
N/V111 | N/G38 |
| 33 | 33. Acylamino‐acid‐releasing enzyme, C‐terminal domain | 1VE6_A95
2.10 Å |
S445 | O/Y444‐CA/G368 3.7 (3.3) |
CA/G447‐O/H367 3.5 (3.2) |
N/Y446 | N/G369 |
| 34 | 34. Hypothetical esterase YJL068C | 1PV1_A96
2.30 Å |
S161 | O/H160‐CA/G57 3.6 (3.2) |
CA/G163‐O/S56 3.4 (3.0) |
N/M162 | N/L58 |
| 35 | 35. Hypothetical protein VC1974 | 1R3D_A97
1.90 Å |
S91 | O/Y90‐CA/G23 3.4 (3.0) |
CA/G93‐O/H22 3.3 (2.6) |
N/L92 | N/L24 |
| 36 | 36. Atu1826‐like | 2I3D_A98
1.50 Å |
S108 | O/Y107‐CA/P33 3.5 (2.8) |
CA/G110‐O/H32 3.3 (2.9) |
N/F109 | N/H34 |
| 37 | 37. PHB depolymerase‐like | 2D80_A35
1.70 Å |
S39 | O/L38‐CA/G249 3.1 (2.5) |
CA/G41‐O/H248 3.4 (2.6) |
N/S40 | N/C250 |
| 38 | 38. IroE‐like | 2GZS_A11
1.40 Å |
S189 | ‐ | ‐ | N/Y190 | NH1/R130 |
| 39 | 40. O‐acetyltransferase | 2B61_A99
1.65 Å |
S143 | O/G142‐CB/A48 3.7 (2.8) |
CA/G145‐O/H47 3.3 (2.9) |
N/F144 | N/L49 |
| 40 | 41. 2,6‐dihydropseudo‐oxynicotine hydrolase‐like | 2JBW_A49
2.10 Å |
S217 | O/R216‐CA/G146 3.6 (3.1) |
CA/G219‐O/G145 3.0 (2.7) |
N/L218 | N/L147 |
The nucleophile zone coordinates the catalytic nucleophile (Column Snuc) and the Oxyanion I residue (Column N/XoxyI), and supports the formation of the oxyanion hole by fixing their relative conformation near the Oxyanion II residue (Column N/XoxyII). The two weak hydrogen bonds of the nucleophile zone (Columns O/Xnuc‐1–CA/GoxyII‐1 and CA/Gnuc + 2–O/HoxyII‐2) occur in 36 ABH families, including the carboxylesterase EstFa_R (PDB ID: 3WJ2), which we use as the reference structure (row A). In four families (Rows 5, 6, 7, and 38), the contact network of the nucleophile zone is disrupted. The “‐” designates that the respective hydrogen bond is not formed; values in parentheses show distances to the hydrogen atom of the corresponding hydrogen bond.
In biotin biosynthesis protein BioH (SCOP Family 15), the nucleophile is serine,100, 101 and not alanine as in the mutant with the PDB ID: 4ETW.82 However, the nucleophile side chain is not involved in the formation of the nucleophile zone. It points outwards, and does not interfere with the network of contacts within the zone.
For example, in the carboxylesterase EstFa_R from Ferroplasma acidiphilum 23 (PDB ID: 3WJ2), the shape of the nucleophile zone is defined by residues of the nucleophile elbow Asp155–Ser156–Ala157–Gly158 and by the dipeptide His83–Gly84 at the start of loopβ3‐αA [Fig. 2(A)]. One of the two connecting weak hydrogen bonds is formed between O/Asp155 at position Xnuc‐1 and CA/Gly84 that precedes Gly85 at XoxyII. The second weak hydrogen bond is formed between CA/Gly158nuc + 2 and O/His83oxyII‐2; this contact anchors the αC‐helix to loopβ3‐αA.
Figure 2.
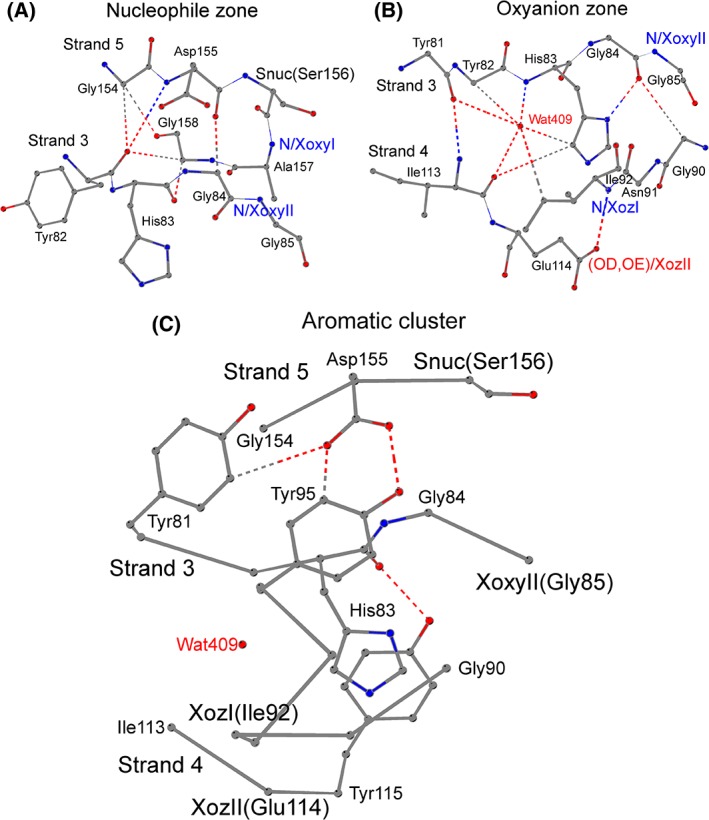
The nucleophile zone (A), the oxyanion zone (B), and the aromatic cluster (C) in the active site of ABH enzymes. (A) The nucleophile zone in the structure of the carboxylesterase EstFa_R from Ferroplasma acidiphilum (PDB ID: 3WJ2). The zone is shaped by the residues Asp155–Ser156–Ala157–Gly158 of the nucleophile elbow and His83–Gly84 at the start of loopβ3‐αA, and involves two, weak hydrogen bonds, O/Asp155–CA/Gly84 and CA/Gly158–O/His83. The residues Tyr82, Gly85, and Gly154 occupy semi‐conserved, peripheral positions and form four, supplementary hydrogen bonds to the nucleophile zone. “Snuc” designates the catalytic nucleophile residue, which is a serine in the majority of ABH enzymes; main‐chain nitrogen atoms at the oxyanion I (“N/XoxyI” – from Ala157 in EstFa_R) and Oxyanion II (“N/XoxyII” – from Gly85 in EstFa_R) positions form the oxyanion hole. (B) The oxyanion zone in the structure of the carboxylesterase EstFa_R from Ferroplasma acidiphilum (PDB ID: 3WJ2). The zone is formed by: the residues Tyr81–Tyr82–His83–Gly84 at the end of strand β3 and the start of loopβ3‐αA; the Gly90–Asn91–Ile92 at the end of loopβ3‐αA; and Ile113–Glu114 from strand β4. All amino acids except XoxyII, and all contacts except the structural β‐sheet hydrogen bond between Tyr81 and Ile113, belong to the oxyanion zone.“N/XoxyII” designates the main‐chain nitrogen atom of oxyanion II residue Gly85 in EstFa_R; and “Wat409” designates the conserved structural water molecule forming multiple hydrogen bonds in the oxyanion zone of the ABH enzymes. A hydrogen bond is formed between the main‐chain nitrogen atom of the residue at “oxyanion zone I” (“N/XozI”, Ile92 in EstFa_R) and the residue at “Oxyanion zone II” (“OD,OE/XozII”, side‐chain oxygen atom of Glu114 in EstFa_R). (C) Conserved residues at the aromatic cluster near the active site of the carboxylesterase EstFa_R from Ferroplasma acidiphilum (PDB ID: 3WJ2). The residues Tyr81, Tyr95, and Asp155 are located at positions where aromatic amino acids (as well as hydrophobic residues) are frequently found in all ABH enzymes; the side‐chain interactions of the residues at these positions form a “roof”‐like structural arrangement over the β‐sheet. Conservation of an aromatic residue is also observed before or after the residue at the position “oxyanion zone II” (“XozII”, Glu114 in EstFa_R); in EstFa_R, the side‐chain hydroxyl of Tyr115 interacts with the main‐chain oxygen of His83, and the two residues form an aromatic pair below the β‐sheet.“Snuc” and “XoxyII” designate the catalytic nucleophile and oxyanion II residue of the catalytic machinery, and “Wat409” and “XozI” designate the conserved structural water molecule and the residue at position “oxyanion zone I” of the Oxyanion zone.
Besides the residues of the nucleophile zone, there are also flanking residues, which in EstFa_R are Tyr82oxyII‐3, Gly85oxyII and Gly154nuc‐2. XoxyII‐3, XoxyII and Smnuc‐2 interact with the nucleophile zone and with each other [Fig. 2(A)], and contribute to the local stability. The distances of these peripheral contacts are detailed in the Supplementary material (Table S1).
Unlike most ABH families that have the main and supplementary contacts of the nucleophile zone, four structural representatives are exceptions (PDB IDs: 3PUI,24 1H2W,25 1ORV,26 and 2GZS11 in Table 2), where the entire contact network of the nucleophile zone is disrupted when residue XoxyII is located at an alternate position in the fold. In these cases, the nucleophile zone is not formed (see Discussion).
Table 2.
Inventory of Interactions within “the Oxyanion Zone” in 29 ABH‐Fold Families
| N | SCOP Family #/ SCOP family name |
PDB ID resolution |
(ND1, CD2)/HoxyII‐2 – O/XoxyII‐1 | (ND1, CD2)/HoxyII‐2
– O/XozII‐1 |
N/XozI‐2 – O/XoxyII‐1 | N/XozI – OD/XozII |
|---|---|---|---|---|---|---|
| A | 2. Carboxylesterase | 3WJ2_A23
1.61 Å |
ND1/H83‐O/G84 2.6 |
CD2/H83‐O/I113 3.9 (3.0) |
CA/G90‐O/G84 3.9 (2.9) |
N/I92‐OE2/E114 2.8 |
| 1 | 1. Acetylcholinesterase‐like | 1QE3_A75
1.50 Å |
ND1/H104‐O/G105 2.7 |
CD2/H104‐O/L134 3.8 (2.7) |
CA/G111‐O/G105 4.1 (3.1) |
N/G113‐OD1/N135 2.9 |
| 2 | 2. Carboxylesterase | 1LZL_A76
1.30 Å |
ND1/H86‐O/G87 2.7 |
CD2/H86‐O/V116 4.0 (3.1) |
CA/G93‐O/G87 4.2 (3.1) |
N/A95‐OE1/E117 2.8 |
| 3 | 4. Hypothetical protein TT1662 | 1UFO_A77
1.60 Å |
ND1/H31‐O/G32 2.8 |
CD2/H31‐O/F57 3.5 (2.7) |
N/G35‐O/G32 2.9 |
N/K37‐OD2/D58 2.9 |
| 4 | 9. Gastric lipase | 1HLG_A46
3.00 Å |
CD2/H65‐O/G66 3.0 (1.9) |
ND1/H65‐O/G97 4.2 |
N/A69‐O/G66 3.1 |
CB/A71‐O/G97 3.3 (2.5) |
| 5 | 10. Proline iminopeptidase‐like | 1MTZ_A78
1.80 Å |
CD2/H35‐O/G36 2.9 (1.9) |
ND1/H35‐O/Y61 3.3 |
N/M40‐O/G36 3.3 |
N/H42‐OD2/D62 2.9 |
| 6 | 11. Acetyl xylan esterase‐like | 1L7A_A79
1.50 Å |
ND1/H89‐O/G90 2.7 |
CD2/H89‐O/M115 3.4 (2.5) |
N/A93‐O/G90 3.1 |
OG/S94‐CD2/L116 3.7 (2.8) |
| 7 | 12. Haloalkane dehalogenase | 1MJ5_A80
0.95 Å |
ND1/H36‐O/G37 2.8 |
CD2/H36‐O/C61 3.2 (2.4) |
N/T40‐O/G37 3.0 |
N/S42‐OD1/D62 2.9 |
| 8 | 13. Dienelactone hydrolase | 1ZI9_A27
1.50 Å |
NE2/Q35‐O/D36 3.3 |
OE1/Q35‐N/L63 3.1 |
N/G39‐O/D36 3.6 |
CG2/V40‐OD1/D62 4.3 (3.5) |
| 9 | 14. Carbon–carbon bond hydrolase | 2OG1_A81
1.60 Å |
ND1/H40‐O/G41 2.8 |
CD2/H40‐O/K69 3.1 (2.3) |
CB/A46‐O/G41 3.4 (3.0) |
N/G48‐OD1/D70 2.8 |
| 10 | 15. Biotin biosynthesis protein BioH | 4ETW_A82
2.05 Å |
ND1/H20‐O/G21 2.8 |
CD2/H20‐O/V45 3.1 (2.2) |
N/L24‐O/G21 3.4 |
N/A26‐OD1/D46 2.9 |
| 11 | 16. Aclacinomycin methylesterase RdmC | 1Q0R_A28
1.45 Å |
CB/M30‐O/G31 3.9 (2.9) |
‐ | N/L34‐O/G31 3.4 |
N/A36‐OD1/D58 2.8 |
| 12 | 17. Carboxylesterase/ lipase |
4DIU_A83
2.00 Å |
ND1/H22‐O/G23 2.8 |
CD2/H22‐O/P48 3.4 (2.4) |
N/G26‐O/G23 2.9 |
OG/S28‐CD1/I49 3.1 (2.0) |
| 13 | 18. Epoxide hydrolase | 1QO7_A84
1.80 Å |
CD2/H115‐O/G116 3.0 (1.9) |
ND1/H115‐O/P147 3.4 |
N/G119‐O/G116 3.0 |
CD1/F121‐OG/S148 3.3 (2.7) |
| 14 | 19. Haloperoxidase | 1BRT_A85
1.50 Å |
ND1/H30‐O/G31 2.8 |
CD2/H30‐O/Y56 3.3 (2.3) |
N/L34‐O/G31 3.0 |
N/G36‐OD1/D57 2.8 |
| 15 | 20. Thioesterases | 1EI9_A86
2.25 Å |
CD2/H39‐O/G40 3.0 (2.0) |
ND1/H39‐O/L70 3.7 |
N/D43‐O/G40 3.4 |
N/C45‐OD2/D79 3.0 |
| 16 | 21. Carboxylesterase/ thioesterase 1 |
1FJ2_A87
1.50 Å |
ND1/H23‐O/G24 2.7 |
CD2/H23‐O/P49 3.6 (2.8) |
N/D27‐O/G24 3.5 |
N/H30‐Wat859‐ ‐CE1/H50 2.8; 3.4 (2.6) |
| 17 | 22. Ccg1/TafII250‐interacting factor B (Cib) | 1IMJ_A88
2.20 Å |
ND1/H39‐O/G40 2.7 |
CD2/H39‐O/I67 3.3 (2.3) |
N/F43‐O/G40 3.1 |
N/S45‐OD1/D68 2.9 |
| 18 | 23. A novel bacterial esterase | 1QLW_A89
1.09 Å |
ND1/H69‐O/G70 2.8 |
CD2/H69‐O/I102 3.4 (2.4) |
N/L73‐O/G70 4.0 |
N/G75‐OD1/D103 2.8 |
| 19 | 26. Bacterial lipase | 1ISP_A90
1.30 Å |
ND1/H10‐O/G11 2.8 |
CD2/H10‐O/V39 3.3 (2.3) |
N/G14‐O/G11 2.9 |
N/S16‐OD1/D40 2.9 |
| 20 | 27. Pancreatic lipase, N‐terminal domain | 1BU8_A91
1.80 Å |
CD2/H75‐O/G76 3.2 (2.2) |
ND1/H75‐O/V104 3.3 |
CG1/I78‐O/G76 3.3 (2.3) |
CD1/I78‐(CG,CD)/R107 4.0; 4.0 |
| 21 | 28. Hydroxynitrile lyase‐like | 3C6X_A92
1.05 Å |
ND1/H10‐O/T11 2.7 |
CD2/H10‐O/L36 3.2 (2.4) |
N/H14‐O/T11 3.1 |
N/A16‐OD1/D37 2.8 |
| 22 | 30. Cutinase‐like | 1BS9_A32
1.10 Å |
CG/R11‐O/E12 3.6 (2.6) |
NH1/R11‐O/I41 3.7 |
N/A15‐O/E12 3.2 |
N/G18‐Wat320‐ ND2/N42 3.0; 3.5 |
| 23 | 31. YdeN‐like | 1UXO_A29
1.80 Å |
ND1/H9‐O/G10 2.7 |
CD2/H9‐O/L37 3.5 (2.8) |
N/A13‐O/G10 3.5 |
N/S15‐OD1/N38 2.8 |
| 24 | 32. Putative serine hydrolase Ydr428c | 1VKH_A94
1.85 Å |
ND1/H36‐O/G37 2.8 |
CD2/H36‐O/I71 3.5 (2.7) |
CB/N45‐O/G37 3.0 (2.5) |
CD/P47‐OE2/E72 3.2 (2.2) |
| 25 | 33. Acylamino‐acid‐releasing enzyme, C‐terminal domain | 1VE6_A95
2.10 Å |
CD2/H367‐O/G368 2.6 (1.6) |
ND1/H367‐O/P395 3.9 |
CB/A372‐O/G368 3.5 (2.6) |
N/D374‐OD1/N396 2.7 |
| 26 | 35. Hypothetical protein VC1974 | 1R3D_A97
1.90 Å |
CD2/H22‐O/G23 2.8 (1.7) |
ND1/H22‐O/L48 3.3 |
N/G26‐O/G23 3.0 |
N/G28‐OD1/D49 2.8 |
| 27 | 36. Atu1826‐like | 2I3D_A98
1.50 Å |
ND1/H32‐O/P33 2.7 |
CD2/H32‐O/F63 3.3 (2.3) |
CA/G39‐O/P33 3.3 (2.3) |
N/M41‐OD1/N64 2.7 |
| 28 | 37. PHB depolymerase‐like | 2D80_A35
1.70 Å |
ND1/H248‐O/G249 2.8 |
CD2/H248‐O/P281 3.6 (2.7) |
N/Q252‐O/G249 3.1 |
N/Y254‐NE2/Q282 3.0 |
| 29 | 40. O‐acetyltransferase | 2B61_A99
1.65 Å |
ND1/H47‐O/A48 2.7 |
CD2/H47‐O/S85 3.2 (2.4) |
N/G51‐O/A48 3.0 |
CA/D52‐OD1/N86 3.9 (2.9) |
Contacts between residues at Positions HoxyII‐2 and XoxyII‐1, HoxyII‐2 and XozII‐1, XozI‐2 and XoxyII‐1, XozI and XozII, are shown. The ABH families that partially or entirely lack the contact network of the oxyanion zone are not shown (SCOP ABH Families 3, 5, 6, 7, 8, 24, 25, 29, 34, 38. and 41). Each “‐” sign designates that the respective hydrogen bond is not formed, while values in parentheses show distances to the hydrogen atom of the corresponding hydrogen bond.
The nucleophile zone and its adjacent structural elements that have been described, altogether serve to optimally arrange the oxyanion hole for the 36 of 40 ABH enzyme families. The hydrogen‐bonding network of the nucleophile zone, together with the peripheral interactions, fix the conformation of the catalytic nucleophile relative to the residues forming the oxyanion hole; the co‐ordination of the second residue of the oxyanion hole also depends on the oxyanion zone that surrounds loopβ3‐αA, as is described in the following section.
The oxyanion zone
Adjacent to the nucleophile zone, there is a planar structural organization that we call the oxyanion zone. The oxyanion zone stabilizes the shape of loopβ3‐αA and co‐ordinates residue XoxyII. With regard to the structure of the active site, the role of the oxyanion zone is complementary to that of the nucleophile zone with which it overlaps through the dipeptide XoxyII‐2–XoxyII‐1. Structurally, the oxyanion zone is formed by residues that belong to strand β3, loopβ3‐αA and strand β4, and it is mainly organized around a conserved histidine residue at the start of loopβ3‐αA; histidine is located two residues before XoxyII and the imidazole side chain faces the interior of the protein in 26 of 40 ABH families (Columns 4 and 5, Table 2). The hydrogen‐bond network of the oxyanion zone consists of four contacts: two contacts link the imidazole ring with neighboring residues and two contacts are formed among other residues of that zone. In 3 of the 40 ABH families, alternative residues are present having the same conformation as the histidine and the zone is formed; whereas, in 11 of 40 ABH families, the oxyanion zone is not present because the hydrogen‐bonding network is not properly formed due to short side chains or a side chains having a different conformation from histidine.
In EstFa_R23, for example, the oxyanion zone is shaped by three elements: (i) the tetrapeptide Tyr81–Tyr82–His83–Gly84 at the C‐terminus of strand β3 and the start of loopβ3‐αA; (ii) the tripeptide Gly90–Asn91–Ile92 at the C‐terminus of loopβ3‐αA; and (iii) the dipeptide Ile113–Glu114 at strand β4 [Fig. 2(B)]. The conserved histidine at the start of loopβ3‐αA (His83 in EstFa_R) forms two conserved contacts: a hydrogen bond with the adjacent Gly84 (ND1/His83–O/Gly84), and a weak hydrogen bond with Ile113 at strand β4 (CD2/His83–O/Ile113). The Residues Gly84 and Gly90, located at each side of loopβ3‐αA, are linked by a weak hydrogen bond (O/Gly84–CA/Gly90). Lastly, a hydrogen bond N/Ile92–OE2/Glu114 connects the end of loopβ3‐αA with strand β4 at positions that we refer to as “Oxyanion zone I” (XozI; Ile92 in EstFa_R) and “Oxyanion zone II” (XozII; Glu114 in EstFa_R).
In addition to the geometry described above, we frequently observed a conserved structural water molecule positioned near the side chain of the residue at the start of loopβ3‐αA, and bound by multiple interactions [Fig. 2(B)]. For example, in EstFa_R, the water Wat409 interacts with His83 through a weak hydrogen bond (Wat409–CD2/His83), and with the other residues of the oxyanion zone through standard (Tyr81, His83, Ile113) and weak (Tyr82, Ile92) hydrogen bonds. The interaction distances of the water molecule‐based contact network can be found in the Supplementary material (Table S2).
Besides the conserved histidine at the start of loopβ3‐αA, which is critical for maintaining the shape of the loop, two key residues are highly conserved within the oxyanion zone. Gly84 (GlyoxyII‐1) in EstFa_R is located between the conserved histidine and XoxyII. Like histidine, GlyoxyII‐1 is a structural element shared between the nucleophile and oxyanion zones present in 30 of the 40 ABH families (Table 1). The second conserved residue of the oxyanion zone is an acidic amino acid – aspartate in 13 families and glutamate in 3 families – located at strand β4 at Position XozII in 16 of the 29 ABH families having the oxyanion zone (Table 2). Alternatively, position XozII can be occupied by an amide – asparagine in 6 families and glutamine in 1 family and they play a similar role in 7 of the 29 ABH families. In contrast to XozII, the type of amino acid that occupies position XozI is variable since it is the main‐chain nitrogen atom that participates in the conserved interaction between residues XozI and XozII (Column N/XozI–OD/XozII in Table 2); this interaction likely offers additional stabilization of the geometry of loopβ3‐αA.
Thus, the oxyanion zone is formed by seven basic elements: (i) a tetrapeptide at the end of strand β3 and the start of loopβ3‐αA; (ii) a tripeptide at the end of loopβ3‐αA; (iii) a dipeptide at strand β4; (iv) a conserved histidine at position XoxyII‐2; (v) a conserved glycine at position XoxyII‐1; (vi) a conserved acidic residue at position XozII; and (vii) a conserved structural water molecule that is positioned at the center of the zone, near the imidazole ring of the conserved histidine at XoxyII‐2 [Fig. 2(B)].
All in all, the oxyanion zone, the nucleophile zone and the previously described catalytic acid zone20 are three common structural organizations of the active site of ABH enzymes that lie on the same plane within the β‐sheet and co‐ordinate the residues of the oxyanion hole, the catalytic nucleophile and the catalytic acid residue. The catalytic acid zone interacts with the catalytic histidine loop and co‐ordinates the catalytic histidine next to the catalytic acid residue.20 The results of that study showed that the conservation of the structure of the active site is extended beyond the plane of the β‐sheet. This finding appears to be valid also for the space that surrounds the nucleophile and the oxyanion zones, as described below.
Aromatic residues surround the catalytic nucleophile and residues of the oxyanion hole
The planar structural organization of both the nucleophile zone and the oxyanion zone primarily involve main‐chain interactions, and thus, the overall shapes of the zones are based on the protein fold. Indeed, apart from the residues at positions XoxyII‐2 and XozII, the side chains of other residues of the nucleophile and oxyanion zones are not involved in the formation of the geometry of the two zones. Within the nucleophile and oxyanion zones, or located nearby, there are, however, conserved residues whose side chains interact outside of the plane of the β‐sheet.
For example, in the carboxylesterase EstFa_R23 the Residues Xnuc‐1 (Asp155) from the nucleophile zone and XoxyII‐4 (Tyr81) from the oxyanion zone interact through a weak hydrogen bond [OD1/Asp155–CE2/Tyr81 in Fig. 2(C)], and their side‐chains lie parallel and above these two zones. A nearby residue, Tyr95, which is located after position XozI (Ile92ozI), has a similar side‐chain conformation and interacts with both Xnuc‐1 (OD1/Asp155–CE2/Tyr95) and XoxyII‐4 (OD2/Asp155–OH/Tyr95) through weak and conventional hydrogen bonds; the aromatic ring of Tyr95 also forms a CH‐π interaction with the side‐chain CH2 group of His83, and seems to play a role in stabilizing and directing the side chain of His83 toward the protein interior. Thus, the side chains of all three residues are linked outside the plane of the nucleophile and the oxyanion zones.
As a unit, the hydrogen‐bond network among these three residues (Asp155, Tyr81, and Tyr95 in EstFa_R) creates a “roof”‐like structural arrangement, positioned over the plane of the nucleophile and oxyanion zones. In order to establish whether this is a common aspect among the ABH families, we have compared their equivalent positions with the structure of carboxylesterase EstFa_R: namely, (i) the residue at Position Xnuc‐1 (Asp155), (ii) the residue at Position XoxyII‐4 (Tyr81), and (iii) the residue (Tyr95) that protrudes from the start of the αA‐helix or the end of loopβ3‐αA and lies above the β‐sheet and within an interaction distance of Xnuc‐1 and XoxyII‐4 – two positions of the nucleophile and oxyanion zones.
By comparing the active sites of the 40 ABH family representatives, we observed aromatic residues (Trp, Tyr, Phe, His; Table 3): in 22 of 40 ABH families at position Xnuc‐1, in 8 of 40 ABH families at XoxyII‐4, and in 24 of 40 ABH families at the position equivalent to Tyr95 in EstFa_R. This latter position is occupied by tryptophan in 14 of the 40 ABH families and its side chain has a characteristic perpendicular conformation over the β‐sheet (Column W/Y/F in Table 3). The amino‐acid type at position Xnuc‐1 is more variable than for the other two locations: it is the only position that is occupied by a histidine residue in addition to tryptophan, tyrosine, and phenylalanine (Column Xnuc‐1 in Table 3). Residue XoxyII‐4 has a lower occupancy of aromatic residues but larger hydrophobic amino acids (leucine, isoleucine, methionine) are present in the “aromatic cluster” in 24 of the 40 ABH families (Column XoxyII‐4 of Table 3). Together, we observed that the residues from these three positions often cluster – forming a “roof”‐like structural arrangement – frequently enriched in aromatic side chains positioned over the plane of nucleophile and oxyanion zones: 13 ABH families have aromatic pairs and 3 ABH families have aromatic triads but only 5 ABH families do not have any aromatic residue at one of these three positions.
Table 3.
Amino Acid Sets that Form Aromatic Clusters Near the Active Site
| N | SCOP Family #/ SCOP family name |
PDB ID Resolution |
XoxyII | XoxyII‐4 | W/Y/F | Xnuc‐1 | Snuc | SC/XozII ± 1 – O/HoxyII‐2 |
|---|---|---|---|---|---|---|---|---|
| A | 2. Carboxylesterase | 3WJ2_A23
1.61 Å |
G85 | Y81 | Y95 | D155 | S156 | OH/Y115‐O/H83; 2.6 |
| 1 | 1. Acetylcholinesterase‐like | 1QE3_A75
1.50 Å |
G106 | W102 | Y118 | E188 | S189 | OH/Y136‐O/H104; 2.7 |
| 2 | 2. Carboxylesterase | 1LZL_A76
1.30 Å |
G88 | W84 | S98 | Q159 | S160 | OH/Y118‐O/H86; 2.7 |
| 3 | 3. Mycobacterial antigens | 1DQZ_A47
1.50 Å |
L40 | L36 | W49 | L123 | S124 | O/G71‐OH/Y77; 4.0 CE1/Y77‐O/D38; 3.6 |
| 4 | 4. Hypothetical protein TT1662 | 1UFO_A77
1.60 Å |
L33 | A29 | I40 | G112 | S113 | CB/A59‐π/F88; 3.4 CE2/F88‐O/H31; 3.5 |
| 5 | 5. PepX catalytic domain‐like | 3PUI_A24
1.53 Å |
OH/Y44 | V40 | W52 | V116 | S117 | ‐ |
| 6 | 6. Prolyl oligopeptidase, C‐terminal domain | 1H2W_A25
1.39 Å |
OH/Y473 | Y471 | I480 | G553 | S554 | ‐ |
| 7 | 7. DPP4 catalytic domain‐like | 1ORV_A26
1.80 Å |
OH/Y547 | E545 | K554 | W629 | S630 | ‐ |
| 8 | 8. Serine carboxypeptidase‐like | 3SC2_A48
2.20 Å |
G53 | W49 | S58 | E145 | S146 | O/S95‐OH/Y151; 4.4 CE2/Y151‐O/N51; 3.1 |
| 9 | 9. Gastric lipase | 1HLG_A46
3.00 Å |
L67 | L63 | W74 | H152 | S153 | OG/S99‐OE1/Q64; 3.7 NE2/Q64‐O/H65; 3.3 |
| 10 | 10. Proline iminopeptidase‐like | 1MTZ_A78
1.80 Å |
G37 | T33 | L45 | S104 | S105 | OE1/Q63‐O/H35; 3.0 |
| 11 | 11. Acetyl xylan esterase‐like | 1L7A_A79
1.50 Å |
Y91 | K87 | E98 | G180 | S181 | CG2/V117‐NE2/Q182;3.9 NE2/Q182‐O/H89; 2.8 |
| 12 | 12. Haloalkane dehalogenase | 1MJ5_A80
0.95 Å |
N38 | F34 | W45 | H107 | D108 | CD1/L63‐O/H36; 3.8 |
| 13 | 13. Dienelactone hydrolase | 1ZI9_A27
1.50 Å |
I37 | I33 | M44 | Y122 | S123 | CD1/L63‐CD1/L128; 3.7 CD1/L128‐O/Q35; 5.4 |
| 14 | 14. Carbon–carbon bond hydrolase | 2OG1_A81
1.60 Å |
G42 | M38 | Y52 | N111 | S112 | CB/S71‐CE/M113; 4.0 CE/M113‐O/H40; 4.0 |
| 15 | 15. Biotin biosynthesis protein BioH | 4ETW_A82
2.05 Å |
W22 | L18 | W29 | W81 | A82a | CD1/L47‐O/H20; 3.5 |
| 16 | 16. Aclacinomycin methylesterase RdmC | 1Q0R_A28
1.45 Å |
G32 | L28 | W39 | L101 | S102 | CE1/H59‐O/M30; 2.9 |
| 17 | 17. Carboxylesterase/lipase | 4DIU_A83
2.00 Å |
F24 | L20 | V31 | L92 | S93 | OH/Y50‐O/H22; 3.0 |
| 18 | 18. Epoxide hydrolase | 1QO7_A84
1.80 Å |
W117 | L113 | F124 | G191 | D192 | CD1/L149‐O/H115; 3.6 |
| 19 | 19. Haloperoxidase | 1BRT_A85
1.50 Å |
F32 | L28 | W39 | F97 | S98 | CD/R58‐O/H30; 3.2 |
| 20 | 20. Thioesterases | 1EI9_A86
2.25 Å |
M41 | I37 | M51 | F114 | S115 | CD1/I72‐CZ2/W38; 4.3 CD1/W38‐O/H39; 3.2 |
| 21 | 21. Carboxylesterase/ thioesterase 1 |
1FJ2_A87
1.50 Å |
L25 | F21 | W32 | F113 | S114 | CB/A51‐π/W67; 3.5 NE1/W67‐O/H23; 2.7 |
| 22 | 22. Ccg1/TafII250‐interacting factor B (Cib) | 1IMJ_A88
2.20 Å |
I41 | L37 | W48 | P110 | S111 | CD2/L69‐O/H39; 4.4 |
| 23 | 23. A novel bacterial esterase | 1QLW_A89
1.09 Å |
C71 | L67 | W78 | H205 | S206 | NE2/Q104‐O/H69; 3.0 |
| 24 | 24. Lipase | 1JFR_A30
1.90 Å |
F63 | I59 | I70 | H130 | S131 | OG1/T89‐NH1/R99; 3.9 NH1/R99‐O/P61; 2.8 |
| 25 | 25. Fungal lipases | 1TCA_A31
1.55 Å |
T40 | L36 | F48 | W104 | S105 | CG/P68‐SD/M72; 3.7 CE/M72‐O/P38; 3.2 |
| 26 | 26. Bacterial lipase | 1ISP_A90
1.30 Å |
I12 | M8 | F19 | H76 | S77 | CE2/F41‐O/H10; 3.7 |
| 27 | 27. Pancreatic lipase, N‐terminal domain | 1BU8_A91
1.80 Å |
F77 | I73 | W85 | H151 | S152 | NE1/W106‐O/H75; 3.0 |
| 28 | 28. Hydroxynitrile lyase‐like | 3C6X_A92
1.05 Å |
I12 | L8 | W19 | E79 | S80 | CD1/L38‐O/H10; 3.9 |
| 29 | 29. Thioesterase domain of polypeptide, polyketide and fatty acid synthases | 1JMK_C32
1.71 Å |
V27 | A23 | Y34 | Y79 | S80 | CB/F51‐π/F155; 4.3 CE1/F155‐O/P25; 3.3 |
| 30 | 30. Cutinase‐like | 1BS9_A29
1.10 Å |
T13 | G9 | S22 | Y89 | S90 | OH/Y43‐O/R11; 2.7 |
| 31 | 31. YdeN‐like | 1UXO_A93
1.80 Å |
Y11 | I7 | F20 | H70 | S71 | CG/M39‐π/W50; 3.5 CZ3/W50‐O/H9; 3.5 |
| 32 | 32. Putative serine hydrolase Ydr428c | 1VKH_A94
1.85 Å |
G38 | Y34 | F50 | H109 | S110 | OH/Y73‐O/H36; 2.7 |
| 33 | 33. Acylamino‐acid‐releasing enzyme, C‐terminal domain | 1VE6_A95
2.10 Å |
G369 | L365 | F381 | Y444 | S445 | OH/Y397‐O/H367; 2.8 |
| 34 | 34. Hypothetical esterase YJL068C | 1PV1_A96
2.30 Å |
L58 | Y54 | A65 | H160 | S161 | CG2/T87‐O/S56; 3.7 |
| 35 | 35. Hypothetical protein VC1974 | 1R3D_A97
1.90 Å |
L24 | L20 | W31 | Y90 | S91 | CD1/L50‐O/H22; 3.5 |
| 36 | 36. Atu1826‐like | 2I3D_A98
1.50 Å |
H34 | I30 | V46 | Y107 | S108 | CE2/F65‐O/H32; 3.4 |
| 37 | 37. PHB depolymerase‐like | 2D80_A35
1.70 Å |
C250 | A246 | F261 | L38 | S39 | CB/A283‐π/W305; 4.0 NE1/W305‐O/H248; 2.9 |
| 38 | 38. IroE‐like | 2GZS_A11
1.40 Å |
NH1/R130 | M88 | V94 | H188 | S189 | ‐ |
| 40 | 39. O‐acetyltransferase | 2B61_A99
1.65 Å |
L49 | I45 | W65 | G142 | S143 | CG1/V87‐OE1/Q148; 3.4 NE2/Q148‐O/H47; 2.9 |
| 41 | 40. 2,6‐dihydropseudo‐ oxynicotine hydrolase‐like |
2JBW_A49
2.10 Å |
L147 | M143 | S154 | R216 | S217 | N/G173‐OE1/Q176; 2.8 NE2/Q176‐O/G145; 3.0 |
The plane of nucleophile and oxyanion zones in ABH families is sandwiched between aromatic residues. Three specific positions (Columns XoxyII‐4, W/Y/F, Xnuc‐1), which lie close to the catalytic nucleophile (Column Snuc), and the Oxyanion I residue (Column XoxyII), are frequently occupied by aromatic amino acids. An aromatic pair is often formed between the residue side chain (SC) that precedes or follows the position “Oxyanion zone II” and the conserved histidine of the oxyanion zone (Column SC/XozII ± 1–O/HoxyII‐2). The “‐” sign designates exceptions, where the respective bond is not formed, or where there is no residue at that position that can interact with other residues of the aromatic cluster.
On the opposite side of the β‐sheet we also observed an aromatic residue packing against the imidazole ring of the conserved histidine (His83 in EstFa_R) in 13 of 26 ABH families. The aromatic residue [Tyr115ozII ± 1 in EstFa_R, in Fig. 2(C)] is typically located either before or after Residue XozII (interaction distances: see Column SC/XozII ± 1–O/HoxyII‐2 in Table 3).
In summary, the arrangement of the nucleophile and oxyanion zones and the conserved residues that interact and are located outside of the β‐sheet is such that an aromatic cluster can occur above the two planar zones and directly over the side chain of the conserved histidine (or the equivalent residue) of the oxyanion zone; an aromatic residue also tends to occur “below” the imidazole ring, opposite to and symmetrically to the aromatic cluster. The conservation of these residues clearly shows that the active site structure is extended outside the plane of the β‐sheet, where main elements of the catalytic mechanism are situated.
Discussion
The nucleophile zone and the oxyanion zone are planar, structural organizations that co‐ordinate the catalytic nucleophile and the two residues that form the oxyanion hole. Both zones are located at the catalytic site in the majority of ABH enzymes, while aromatic residues frequently surround the β‐sheet region where the key units of the catalytic machinery are situated. Here, we discuss residues of the zones that have structural and functional meaning for the ABH enzymes.
The nucleophile zone co‐ordinates the catalytic nucleophile and the oxyanion I residue, and aids the optimal arrangement of the oxyanion hole in most ABH enzymes
The nucleophile zone is situated in the core of the active site of ABH enzymes of the overwhelming majority of ABH enzymes, where it directly co‐ordinates the catalytic nucleophile residue and the Oxyanion I residue. Besides the co‐ordination of these two key units of the catalytic machinery, the weak hydrogen bonds of the zone (between Asp155 and Gly84; Gly158 and His83) bind the nucleophile elbow and the loopβ3‐αA. Consequently, it seems likely that the nucleophile zone is required for the optimal arrangement of the oxyanion hole adjacent to the catalytic nucleophile. This optimal arrangement is achieved by placement of the catalytic nucleophile and the Oxyanion I residues of the nucleophile elbow close to the Oxyanion II residue that follows the His–Gly dipeptide on loopβ3‐αA.
The nucleophile zone is only formed when the Oxyanion II residue is located two positions after the start of loopβ3‐αA. The importance of the nucleophile zone for the optimal geometry of the oxyanion hole is further confirmed in that only four ABH families lack important features of the nucleophile zone. In these four ABH families, the Oxyanion II residue is located at an alternate position and employs its side chain to assemble the oxyanion hole. The weak hydrogen bonds of the nucleophile zone are apparently not required for the formation of the oxyanion hole in these cases.
While analyzing structural consequences in the four ABH families that lack the nucleophile zone, we deduced that the residue at the start of loopβ3‐αA – equivalent to His83 in EstFa_R – is a determinant of the local geometry, and the type of amino acid and the side‐chain conformation of the residue at the start of loopβ3‐αA correlates with the existence of the nucleophile zone in the active site of ABH enzymes. The four exceptions fall into two separate cases. In the first case (PDB IDs: 3PUI,24 1H2W,25 1ORV26), the Oxyanion II residue is a tyrosine, which emerges from the start of loopβ3‐αA and provides its hydroxyl group to form the oxyanion hole. This tyrosine occupies the sequence and main‐chain structural position of the conserved histidine (His83 in EstFa_R). In the second case (PDB ID: 2GZS11), the residue that occurs at the start of loopβ3‐αA is an aspartic acid. The aspartic acid at this position helps form the oxyanion hole through a salt bridge with arginine – the oxyanion II residue – which is located on an α‐helix opposite to the active site and distant from the β‐sheet. Thus, in both cases where the nucleophile zone is not formed, the residue at the start of loopβ3‐αA is either the Oxyanion II residue or a residue that forms a salt bridge with it. However, in the remaining 36 ABH enzymes that have the nucleophile zone, the residue at the start of loopβ3‐αA is mostly a histidine, which is critical for the formation of the oxyanion hole, as we describe below.
In addition to the two weak hydrogen bonds important for the nucleophile zone, there are flanking residues (Gly154, Tyr82, an dGly85 in EstFa_R) that structurally support the rigidity of the nucleophile zone by interacting with residues of the zone and with each other. Both the principal and the peripheral contacts of the nucleophile zone are located in the area of the active site of the ABH enzymes and are shown in Figure 2(A). However, their logical division into main and flanking types is based on the role of the nucleophile zone. We consider that the two conserved weak hydrogen bonds of the nucleophile zone actively co‐ordinate the catalytic nucleophile and oxyanion I residues. In contrast, the other contacts, which are shown in Figure 2(A), are situated further away from the catalytic site. They seem to play supportive roles for the local supersecondary structure rather than a straightforward role in the co‐ordination of the key units of the catalytic machinery and the formation of the oxyanion hole. Our assumption for the supplementary structural role of the flanking elements is also supported by the high conservation of glycines at positions Smnuc‐2 and Smnuc + 2. Because of the lack of steric clashes and the existence of the two extra weak hydrogen bonds that can be formed by the CA atoms of both glycine residues [Fig. 2(A)], these residues may help enhance the local stability. With the β‐sheet contact N/Asp155–O/Tyr82 creating an imaginary line of separation, this sets a boundary between the elements of function [nucleophile zone; Fig. 2(A), right] and the flanking elements [Fig. 2(A), left].
The oxyanion zone is organized around a conserved histidine residue, which is relatively remote from the active site, but structurally important for the enzymatic function
In most ABH enzymes, the oxyanion zone of the active site is situated right next to the nucleophile zone. It includes seven structural elements, which are connected through an extended hydrogen‐bonding network that functions to stabilize elements of the catalytic machinery [Fig. 2(B)]. Among the elements of the oxyanion zone, we consider the conserved Residues HisoxyII‐2, GlyoxyII‐1, and the acidic Residue XozII to be critical for the stability of the overall shape of loopβ3‐αA, that eventually leads to the optimal arrangement of the oxyanion hole. By being critical for the stability of loopβ3‐αA, we mean that the hydrogen‐bonding network of the oxyanion zone is committed to co‐ordinating the Oxyanion II residue next to Oxyanion I residue and the catalytic nucleophile, where the latter two residues are co‐ordinated by the nucleophile zone, as we discussed earlier. The structures of the nucleophile and oxyanion zones are linked through the shared dipeptide HisoxyII‐2–GlyoxyII‐1 that participates in the contact networks of both zones. The association of the two zones through this connecting segment shows that the nucleophile and oxyanion zones have complementary roles in the overall arrangement of the structure of the catalytic site. However, their clear structural separation results in different levels of structural variability, where the nucleophile zone appears to be much more conserved than the oxyanion zone, that may lead to diverse catalytic activities among the ABH families.
HisoxyII‐2 appears to be the central organizing unit of the oxyanion zone. Half of the contacts of the zone are formed around its side chain. The imidazole ring is placed centrally in the area defined by loopβ3‐αA and interacts with the adjacent Residue XoxyII‐1 and a neighboring residue from Strand β4. Other residues than histidine at the Position XoxyII‐2 can play a similar structural role when their side‐chain points toward the center of loopβ3‐αA. For example, in our dataset, we have found that the hydrogen‐bonding network of the oxyanion zone is fully maintained when glutamine (PDB ID: 1ZI927), methionine (PDB ID: 1Q0R28), or arginine (PDB ID: 1BS929) appear at the start of loopβ3‐αA (Columns 4 and 5 in Table 2). A proline (PDB IDs: 1JFR,30 1TCA,31 1JMK32) at the same position partially maintains the bonds of the oxyanion zone. Outside the dataset, we observed another variation at this position, in the structure of cholesterol esterase from Candida cylindracea (PDB ID: 1LLF33, fungal lipases family): a phenylalanine occurs in the position of the conserved histidine and maintains all of the contacts of the oxyanion zone.
The conserved histidine at position XoxyII‐2 was previously suggested to be part of the oxyanion hole, with the dipeptide HisoxyII‐2–GlyoxyII‐1 termed the “oxyanion pocket”.34 This suggestion was later withdrawn because the histidine does not directly participate in catalysis.35, 36 Here, we show that the conserved histidine (or the equivalent residue) in the middle of oxyanion zone is indirectly involved in the ideal positioning of Oxyanion II to complete the shape of the oxyanion hole, due to the hydrogen bond with the adjacent residue that appears to stabilize the position of the Oxyanion II backbone amide.
The first four residues of loopβ3‐αA – the conserved histidine being the first residue – have been previously mentioned as a possible sequence identifier of different ABH families.37, 38, 39 The four‐residue loop segment is suggested to be involved in substrate binding40 and in the stabilization of the position of the putative hydrolytic water molecule.37 This indicates that the area around the oxyanion zone is actively involved in several aspects of catalytic activity, as mentioned above.
Multiple experimental studies confirm the importance of the conserved histidine near the oxyanion hole, and thus, the oxyanion zone in general. Site‐directed mutagenesis of histidine to alanine resulted in the complete loss of enzymatic function.19, 34, 41, 42 It is suggested that this mutation is responsible for the distortion of the shape of loopβ3‐αA and thus, the oxyanion hole fails to be formed.19, 34, 42 The mutation of histidine to leucine and asparagine had a similar adverse effect on enzymatic activity.43 Experimental studies have also been applied to those residues that have a similar side‐chain conformation as the conserved histidine. The mutation of glutamine to histidine improved the catalytic activity,44 while the mutation of arginine to alanine and lysine again resulted in a loss of function.45 Notably, site‐directed mutagenesis of the residue XoxyII‐1 that follows the conserved histidine also leads to a loss of activity, for example, mutation of the glycine12, 46 or aspartate at this position.27 Taken together, these mutagenesis studies show that the Position XoxyII‐2 should preferably be occupied by a histidine, since in many structures, mutating histidine leads to loss of function as does the adjacent residue; in contrast, mutating other residues to histidine can improve the enzymatic activity.
Thus, for the oxyanion zone, the residue at Position XoxyII‐2 (His83 in EstFa_R) is critical for the geometry of loopβ3‐αA. In those ABH families that do not have the oxyanion zone, the type of amino acid at that position can be part of large‐scale variations of the structure of the active site that serve the enzymatic function,47, 48, 49 or its specific occurrence can contribute to the substrate binding,26 the regulation of pH optimum50 or the formation of the oxyanion hole.8, 9, 11, 51 Nonetheless, regardless the amino acid variation, the residue at Position XoxyII‐2 also affects the enzymatic activity in the ABH enzymes without the oxyanion zone. Indeed, experimental studies have shown that site‐directed mutation of the residue at the start of loopβ3‐αA can decrease9 or eliminate the enzymatic activity entirely.8, 51
Residue XozII enhances the stability of the oxyanion zone
The acidic side chain of XozII faces the exterior of the protein, opposite to the conserved histidine that points toward the core of the protein. In many cases, Residue XozII is an aspartic acid, and its side‐chain oxygen atom forms a hydrogen bond with the amide of Residue XozI located near the end of loopβ3‐αA. This contact seems to stabilize the shape of loopβ3‐αA in the ABH enzymes with the oxyanion zone. Although Position XozII is located distantly from the catalytic site, site‐directed mutagenesis of the aspartic acid results in detrimental loss of activity, which suggests that this amino acid plays an important role in maintaining local folding and stability of the enzyme structure.41 A second mutational study showed that this conserved aspartic acid is actively involved in the formation of the protein–protein complex in the case of acyl esterase Aes from Escherichia coli.52
The aromatic residues around the nucleophile and oxyanion zones can affect the stability of the catalytic site
Besides the conservation of the nucleophile and oxyanion zones, we showed that aromatic residues regularly occur around both structural organizations. Particularly, we showed that aromatic residues often cluster over the nucleophile and oxyanion zones [Fig. 2(C)], while aromatic pairs are formed by the conserved histidine and a residue whose side chain lies opposite to the aromatic cluster with respect to the β‐sheet.
The conservation of aromatic residues in the periphery of the catalytic site can prove beneficial for the local structural stability. Several studies have previously analyzed the importance of aromatic‐aromatic interactions around the active site and shown their influence on the enzymatic42, 53, 54, 55, 56, 57 and the thermal58 stability of the protein, on the lining of the binding pocket25 and on the non‐local stabilization of the protein tertiary structure.59
We observed aromatic residues at three specific positions forming the aromatic cluster: the Residue Xnuc‐1 at the C‐terminus of strand β5, the Residue XoxyII‐4 at the C‐terminus of strand β3 and a residue at the end of loopβ3‐αA or the start of αA‐helix. Our results indicate that the Residue Xnuc‐1, located before the nucleophile, is not a randomly occurring amino acid, but it is very often an aromatic residue. Experimental studies have shown that site‐directed mutation of Residue Xnuc‐1 results in the reduction60 or the elimination of the catalytic efficiency of the enzyme.61 It is suggested that the Residue Xnuc‐1 plays a structural role in the correct geometry of the catalytic site.60, 61, 62 Residue XoxyII‐4 appears to be the most conserved of the aromatic cluster residues, as it is mostly a hydrophobic or else an aromatic residue.
The third residue of the aromatic cluster does not have a defined sequence position, because its position heavily depends on the length of loopβ3‐αA. Nonetheless, it is always linked with the Residues Xnuc‐1 and XoxyII‐4. In the majority of ABH families, this position is occupied by an aromatic amino acid, quite often by a tryptophan residue, whose side chain has a characteristic, nearly perpendicular conformation over the β‐sheet, with the indole ring landing opposite to residue Smnuc‐2 of the nucleophile elbow. Interestingly enough, this structural phenomenon has been previously observed63 in a structurally similar part of the Clostridium MP flavodoxin64 structure, which also belongs to α/β‐fold class. Experimental studies, that refer to the third residue of the aromatic cluster, have showed that its site‐directed mutation can lead to decreased catalytic activity60, 65, 66 and affect the thermostability of the enzyme.66
Finally, the location of the aromatic cluster suggests that it would be involved in the co‐ordination of the catalytic histidine loop. Two separate studies have mentioned that Residue Xnuc‐1 62 and the third residue of the cluster42 form contacts with aromatic residues that occur at the loop fragment after the catalytic histidine, and thus participate in extended aromatic interactions near the catalytic histidine. This fact is rather interesting, because in our previous study20 we have shown that the catalytic acid zone residues co‐ordinate the loop fragment before the catalytic histidine.
Key residues surrounding the catalytic nucleophile retain their amino‐acid conservation, even in the absence of a canonical catalytic triad
Recently, the crystallographic structures of two ABH proteins were resolved, the Methylketone Synthase1 (MKS1) from Solanum habrochaites 67 and the aminoacrylate hydrolase RutD from Escherichia coli, 68 which have enzymatic activity and use a non‐canonical catalytic triad. MKS1 has an Ala–His–Asn triad in the place of the canonical catalytic triad and the residue Thr18 at the canonical Position XoxyII‐1 is proposed to play the role of the putative, alternative nucleophile. RutD has a non‐canonical catalytic triad His–His–Asp, with its putative, alternative nucleophile His87 situated at the canonical Position Xnuc‐1. Given the exceptional catalytic mechanisms of MKS1 and RutD, we would like to examine the existence and conservation of the nucleophile zone, the oxyanion zone and the aromatic cluster in ABH enzymes with non‐canonical triads. We were able to identify eight ABH proteins (Entries B‐I in Table 4) that lack a canonical catalytic triad, two of which have a putative, alternative nucleophile (ABH enzymes MKS1 and RutD, Entries B and E respectively), while the rest lack some or all the catalytic residues (Entries C, D, F, G, H, I). These eight new ABH proteins have different degrees of activity, spanning from catalytically active to non‐enzymatic ABH proteins.
Table 4.
Relatedness of ABH Proteins Based on the Amino‐Acid Conservation of Five Key Positions of the Catalytic Site, Surrounding the Catalytic Nucleophile
| N | SCOP Family #/ SCOP family name | PDB ID | XoxyII‐2 | XoxyII‐1 | W/Y/F | XozII | Xnuc‐1 |
|---|---|---|---|---|---|---|---|
| I. “+++++” group | |||||||
| 1 | 9. Gastric lipase | 1HLG_A | H65 | G66 | W74 | N98 | H152 |
| 2 | 12. Haloalkane dehalogenase | 1MJ5_A | H36 | G37 | W45 | D62 | H107 |
| 3 | 23. A novel bacterial esterase | 1QLW_A | H69 | G70 | W78 | D103 | H205 |
| 4 | 26. Bacterial lipase | 1ISP_A | H10 | G11 | F19 | D40 | H76 |
| 5 | 27. Pancreatic lipase, N‐terminal domain | 1BU8_A | H75 | G76 | W85 | D105 | H151 |
| 6 | 31. YdeN‐like | 1UXO_A | H9 | G10 | F20 | N38 | H70 |
| 7 | 32. Putative serine hydrolase Ydr428c | 1VKH_A | H36 | G37 | F50 | E72 | H109 |
| 8 | 15. Biotin biosynthesis protein BioH | 4ETW_A | H20 | G21 | W29 | D46 | W81 |
| 9 | 19. Haloperoxidase | 1BRT_A | H30 | G31 | W39 | D57 | F97 |
| 10 | 21. Carboxylesterase/thioesterase 1 | 1FJ2_A | H23 | G24 | W32 | H50 | F113 |
| 11 | 33. Acylamino‐acid‐releasing enzyme, C‐terminal domain | 1VE6_A | H367 | G368 | F381 | N396 | Y444 |
| 12 | 35. Hypothetical protein VC1974 3BE8 | 1R3D_A | H22 | G23 | W31 | D49 | Y90 |
| II. “+ − +++” group | |||||||
| B | Methylketone synthase 1 | 3STT_A67 | H17 | T18 | W26 | D44 | H86 |
| III. “++++−” group | |||||||
| A | 2. Carboxylesterase | 3WJ2_A | H83 | G84 | Y95 | E114 | D155 |
| 13 | 1. Acetylcholinesterase‐like | 1QE3_A | H104 | G105 | Y118 | N135 | E188 |
| 14 | 14. Carbon–carbon bond hydrolase | 2OG1_A | H40 | G41 | Y52 | D70 | N111 |
| 15 | 16. Aclacinomycin methylesterase RdmC | 1Q0R_A | M30 | G31 | W39 | D58 | L101 |
| 16 | 37. PHB depolymerase‐like | 2D80_A | H248 | G249 | F261 | Q282 | L38 |
| 17 | 22. Ccg1/TafII250‐interacting factor B (Cib) | 1IMJ_A | H39 | G40 | W48 | D68 | P110 |
| 18 | 18. Epoxide hydrolase | 1QO7_A | H115 | G116 | F124 | S148 | G191 |
| IV. “+ − ++−” group | |||||||
| 19 | 28. Hydroxynitrile lyase‐like^ | 3C6X_A | H10 | T11 | W19 | D37 | E79 |
| 20 | 40. O‐acetyltransferase | 2B61_A | H47 | A48 | W65 | N86 | G142 |
| C | Protein NDRG2 | 2QMQ_A102 | H69 | D70 | F79 | D101 | V145 |
| V. “++ − ++” group | |||||||
| 21 | 20. Thioesterases | 1EI9_A | H39 | G40 | M51 | D79 | F114 |
| VI. “+−−++” group | |||||||
| 22 | 13. Dienelactone hydrolase | 1ZI9_A | Q35 | D36 | M44 | D62 | Y122 |
| 23 | 30. Cutinase‐like | 1BS9_A | R11 | E12 | S22 | N42 | Y89 |
| 24 | 36. Atu1826‐like | 2I3D_A | H32 | P33 | V46 | N64 | Y107 |
| VII. “++ − +−” group | |||||||
| 25 | 2. Carboxylesterase | 1LZL_A | H86 | G87 | S98 | E117 | Q159 |
| 26 | 10. Proline iminopeptidase‐like | 1MTZ_A | H35 | G36 | L45 | D62 | S104 |
| 27 | 4. Hypothetical protein TT1662 | 1UFO_A | H31 | G32 | I40 | D58 | G112 |
| D | Neuroligin‐4, X‐linked | 3BE8_A103 | H173 | G174 | I185 | N202 | S253 |
| VIII. “++ − ‐‐” group | |||||||
| 28 | 11. Acetyl xylan esterase‐like | 1L7A_A | H89 | G90 | E98 | L116 | G180 |
| 29 | 17. Carboxylesterase/lipase | 4DIU_A | H22 | G23 | V31 | I49 | L92 |
| IX. “−++++” group | |||||||
| E | Putative aminoacrylate hydrolase RutD | 3V48_A68 | S20 | G21 | W29 | D46 | H87 |
| 30 | 25. Fungal lipases | 1TCA_A | P38 | G39 | F48 | S67 | W104 |
| X. “‐ − +++” group | |||||||
| 31 | 29. Thioesterase domain of polypeptide, polyketide and fatty acid synthases | 1JMK_C | P25 | P26 | Y34 | D50 | Y79 |
| XI. “‐ − ++−” group | |||||||
| 32 | 5. PepX catalytic domain‐like | 3PUI_A | N42 | P43 | W52 | D73 | V116 |
| F | 39. TTHA1544‐like | 2DST_A70 | A29 | E30 | W35 | D49 | A87 |
| XII. “− + −++” group | |||||||
| 33 | 34. Hypothetical esterase YJL068C | 1PV1_A | S56 | G57 | A65 | D86 | H160 |
| 34 | 24. Lipase | 1JFR_A | P61 | G62 | I70 | D88 | H130 |
| G | UPF0255 protein FrsA | 4I4C_A104 | A200 | G201 | M209 | D228 | F271 |
| XIII. “‐‐ − ++” group | |||||||
| 35 | 7. DPP4 catalytic domain‐like | 1ORV_A | Y547 | A548 | K554 | D579 | W629 |
| XIV. “‐‐ − +−” group | |||||||
| H | Inactive dipeptidyl peptidase 10 (DPP10) | 4WJL_A69 | D568 | E569 | L575 | D600 | K650 |
| XV. “− + − + −” group | |||||||
| 36 | 6. Prolyl oligopeptidase, C‐terminal domain | 1H2W_A | Y473 | G474 | I480 | N503 | G553 |
| 37 | 8. Serine carboxypeptidase‐like | 3SC2_A | N51 | G52 | S58 | D94 | E145 |
| 38 | 41. 2,6‐dihydropseudo‐oxynicotine hydrolase‐like | 2JBW_A | G145 | G146 | S154 | D172 | R216 |
| XVI. “−++ − ‐” group | |||||||
| 39 | 3. Mycobacterial antigens | 1DQZ_A | D38 | G39 | W49 | V70 | L123 |
| XVII. “‐ − + − ‐” group | |||||||
| I | MPT51/MPB51 antigen | 1R88_A105 | D66 | A67 | W77 | A98 | A144 |
| XVIII. “‐‐‐ − +” group | |||||||
| 40 | 38. IroE‐like | 2GZS_A | ‐ | ‐ | V94 | G118 | H188 |
The active site of ABH proteins is surrounded by five key residues from three structural organizations: the nucleophile zone (Columns Xnuc‐1, XoxyII‐2, and XoxyII‐1), the oxyanion zone (Columns XoxyII‐2, XoxyII‐1, and XozII), and the aromatic cluster (Columns W/Y/F and Xnuc‐1). ABH proteins with canonical catalytic triads (Entries 1–40, A) and ABH proteins with non‐canonical catalytic triads (Entries B‐I) are classified in 18 groups (Groups I–XVIII), based on the amino‐acid type present at the five key positions. Every group is labeled by a ± five‐character sequence, which corresponds to the sequence XoxyII‐2 − XoxyII‐1− W/Y/F − XozII − Xnuc‐1. Each character of the sequence is marked with “+”, if the corresponding position of ABH protein is occupied by the most conserved amino‐acid according to the scheme (±H)oxyII‐2 − GoxyII‐1 − (±Φ)W/Y/F−(±D/E/N/Q)ozII−(±H/Φ)nuc‐1 (“Φ” designates the presence of an aromatic amino acid). Alternatively, the character is marked with “‐”, if this position is occupied by a non‐conserved residue. Thus, Group I (“+++++”) gathers all those ABH proteins that retain the amino‐acid conservation in all five key residues, while Group XIV (“‐‐ − +−”), Group XVII (“‐ − + − ‐”), and Group XVIII (“‐‐‐ − +”) retain the amino‐acid conservation in none‐but‐one position. In the five columns that contain the amino‐acid sets, the “‐” sign designates that the respective position is not occupied by any residue.
To compare the ABH enzymes that have a canonical catalytic triad and the eight exceptional cases that have a non‐canonical catalytic triad with respect to our findings, we have singled‐out five key residues from the nucleophile zone, the oxyanion zone and the aromatic cluster in order to assess how similar or different is the structural environment around the catalytic nucleophile in canonical and non‐canonical settings. We have selected the nucleophile zone Residues Xnuc‐1, XoxyII‐2, XoxyII‐1 (from which the first residue is shared with the aromatic cluster, and the latter two are shared with the oxyanion zone), the oxyanion zone Residue XozII and the third residue of the aromatic cluster, based on their proximity to the catalytic nucleophile, the experimental data that show their effect in the enzymatic activity, and their high conservation rate. These five residues constitute the structural core around the active site of the ABH enzymes. As we have shown in the Results section, each of these five positions is occupied by conserved and specific amino‐acids: Residue Xnuc‐1 is often aromatic (Trp, Tyr, Phe, and His); Residue XoxyII‐2 is usually a histidine or an amino‐acid with a similar side‐chain conformation; Residue XoxyII‐1 is mostly a glycine; Residue XozII has an acidic (Asp, Glu) or an amidic side chain (Asn, Gln); and the third residue of the aromatic cluster is clearly aromatic (Trp, Tyr, Phe).
Using the five key positions shown above, we have analyzed the relatedness of ABH families that have canonical catalytic triads (SCOP Families #1–38, 40, 41 in Table 4) with those that have non‐canonical catalytic triads (Entries B‐I in Table 4). It appears that the majority of ABH families that have both canonical and non‐canonical catalytic triads retain the amino‐acid conservation in all (Group I in Table 4) or all‐but‐one (Groups III, V, and IX in Table IV) five key positions; Groups I and III are the largest categories, with the ABH enzymes of the two groups differing in the amino‐acid occupancy of Position Xnuc‐1. The groups that retain the amino‐acid conservation in two or three positions are generally poorly populated (Groups IV, VI, VII, VIII, X, XI, XII, XIII, XV, and XVI in Table 4).
Based on the classification that arose from the comparison of ABH enzymes with a canonical catalytic triad, five of the eight ABH proteins with non‐canonical catalytic triad (Entries C, D, E, F, and G in Table 4) fall in the same categories with ABH enzymes that have a canonical catalytic triad (Groups IV, VII, IX, XI, and XII in Table 4, respectively). Three of the eight additional ABH proteins (Entries B, H, and I) make up new, single‐entry categories (Groups II, XIV, and XVII, respectively). Group II is similar to Group I, while Groups XIV and XVII are very different from Group I with respect to the amino acids that occupy the five key positions. Both Entries H and I, however, are enzymatically inactive proteins that contain incomplete catalytic triads (Table 4). In the case of Entry H, the catalytic triad appears almost intact, except there is a glycine, which occupies the canonical position of the catalytic nucleophile.69 Nonetheless, the mutation of this glycine to serine failed to induce any enzymatic activity, probably due the long side‐chain amino acids (Asp568 and Glu569) at the key Positions XoxyII‐2 and XoxyII‐1, that apparently hinder both the ligand binding and optimal formation of the oxyanion hole.69
Thus, while there are ABH fold proteins that have non‐conventional catalytic triads, with some of them being enzymatically active (Entry B in Table 4) and others lacking any catalytic activity (Entries H and I in Table 4), then based on our analysis and grouping, we can conclude that (1) these non‐canonical proteins still have largely the same structural cores as the ABH enzymes with the canonical catalytic triads; and (2) the more the structural core of a protein with a non‐canonical catalytic triad differs from that of Group I (Table 4), the less likely it will retain any catalytic activity.
Conclusions
In this study, we analyzed the structure around the nucleophile elbow and the oxyanion hole in representative structures from 40 ABH families. We have shown that the nucleophile zone, the oxyanion zone and the aromatic cluster comprise a three‐dimensional structural organization that shapes the active site of ABH enzymes and plays an important role in the enzymatic function by structurally stabilizing the catalytic nucleophile and the residues of the oxyanion hole. This finding is complementary to the result from our previous study, where we showed that there is a common structural organization that co‐ordinates the catalytic acid residue and links it with the catalytic histidine. Overall, we were able to confirm that there is a common supporting architecture around each key unit of the catalytic machinery.
Undeniably, Position XoxyII‐2 at the start of loopβ3‐αA is paramount for the local structure of the catalytic site and the activity of ABH enzymes, and is shared by both the nucleophile and the oxyanion zones. Regardless the type of amino acid, position XoxyII‐2 is typically occupied by a residue that is suitable for the optimal geometry of the oxyanion hole, either indirectly by stabilizing the arrangement of the oxyanion hole, or directly by providing a side‐chain group to form the oxyanion hole. In addition to that, a conserved acidic residue at Position XozII of the oxyanion zone proves to be essential for the formation of the oxyanion hole, although it is located remote from the active site.
Finally, we showed that aromatic residues tend to appear around the nucleophile and the oxyanion zones in most ABH families, often forming an aromatic cluster over and an aromatic pair below the plane of the nucleophile and oxyanion zones. Aromatic residues are shown to enhance the stability of the active site, and thus, we consider that they also affect the stability of the ABH enzymes. The aromatic cluster, which is located close to the active site, connects the nucleophile and the oxyanion zones through interactions between residues that belong to both zones. In addition, there is evidence that the aromatic cluster can be involved in the co‐ordination of the catalytic histidine loop. Further experimental studies on the conserved aromatic residues may provide valuable insight about their role in the enzymatic activity.
Materials and Methods
We have used the Structural Classification Of Proteins (SCOP) database22 to choose representative structures for our dataset. There are 41 ABH families according to SCOP classification, which contain 128 different entries of proteins with the ABH fold. The single entry from the TTHA1544‐like family (SCOP Family #39) is not included in our dataset because it is not a hydrolase.70 From the 127 entries, we have selected 40 representative structures ensuring that they represent each listed ABH family, on the condition that they are mainly unligated and have the highest resolution. In total, we have formed a dataset, which contains 40 structures plus the carboxylestarase EstFa_R from the study of Ohara et al.23 that we use as the reference structure throughout this study. We have obtained all protein structures from Protein Data Bank (PDB).21
The choice of structures to be analyzed: The results of this study are presented in four tables. Each table has a different number of entries, depending on the subset of representative structures that have each conserved structural motif. Specifically, Table 1 describes 40 representative structures from 40 ABH enzyme families (Rows 1–40) plus the carboxylesterase EstFa_R (Row A), with the exception of the single‐entry protein from the TTHA1544‐like family. In Table 1, we show not only the 36 ABH structures and EstFa_R that maintain the contact network of the nucleophile zone but also the four ABH structures that lack all interactions of the nucleophile zone. Table 2 only includes those structures that maintain the contact network of the oxyanion zone and specifically, 29 representative structures from the 40 ABH enzyme families (Rows 1–29) plus the carboxylesterase EstFa_R (Row A). The protein from SCOP Family TTHA1544‐like is not taken into account here as well. Table 3 includes all 40 representative structures (Rows 1–40) from the 40 ABH families (except the TTHA1544‐like family) and the carboxylesterase EstFa_R (Row A).
“Conserved amino acid residues” and “conserved interactions and bonds”: Throughout the text we use this terminology to show amino acids and bonds, which are repeatedly found at the same structural positions and places among many families of ABH proteins. For example, structurally, the oxyanion zone is formed around a conserved histidine residue at the start of loopβ3‐αA, and exactly two residues before XoxyII in 26 of 40 ABH fold families (Columns 4 and 5, Table 2). If two structurally conserved residues or water molecules make an interaction or form a bond, which is also observed in many different ABH families, we call it a conserved bond or interaction. A Structural Motif, common to many ABH protein families, is essentially a set of structurally conserved amino acids joined by similar interactions in a similar way.
For all structural analysis, such as hydrogen bonds, hydrophobic or other types of weak interactions, we have used the BIOVIA (Accelrys) Discovery Studio71 (http://accelrys.com/products/collaborativescience/bioviadiscoverystudio/). We have identified the weak hydrogen bonds from CHO contacts in structures with at least 2.0 Å resolution based on geometrical criteria given in (72) and with distances C…O ≤ 4.1 Å and H…O ≤ 3.0 Å, and the weak hydrogen bonds from CH‐π contacts with distance between 3.4 Å and 6 Å.57
Lastly, for the visualization and analysis of the structural data, we have used Discovery Studio71 and Bodil.73 Figures are drawn with MolScript.74
Supporting information
Appendix S1: Supporting information
Acknowledgments
This work is supported by a grant from the Sigrid Juselius Foundation and Joe, Pentti, and Tor Borg Memorial Fund. We thank the Biocenter Finland Bioinformatics Network (Dr. Jukka Lehtonen) and CSC IT Center for Science for computational support for the project. P. S. D. is funded by the Åbo Akademi Doctoral Network of Informational and Structural Biology.
For the catalytic acid‐base‐nucleophile machinery of alpha/beta‐hydrolases, unique structural motifs—termed catalytic zones—were identified that serve to properly position the catalytic machinery and co‐ordinate function. Understanding the role of these structural features can contribute to engineering this large family of enzymes for novel applications and industrial processes.
REFERENCES
- 1. Nardini M, Dijkstra BW (1999) α/β‐Hydrolase fold enzymes: the family keeps growing. Curr Opin Struct Biol 9:732–737. [DOI] [PubMed] [Google Scholar]
- 2. Mindrebo JT, Nartey CM, Seto Y, Burkart MK, Noel JP (2016) Unveiling the functional diversity of the alpha/beta‐hydrolase superfamily in the plant kingdom. Curr Opin Struct Biol 41:233–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lenfant N, Hotelier T, Velluet E, Bourne Y, Marchot P, Chatonnet A (2013) ESTHER, the database of the α/β‐hydrolase fold superfamily of proteins: tools to explore diversity of functions. Nucleic Acids Res 41:D423–D429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rauwerdink A, Kazlauskas RJ (2015) How the same core catalytic machinery catalyzes 17 different reactions: the serine–histidine–aspartate catalytic triad of α/β‐hydrolase fold enzymes. ACS Catal 5:6153–6176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Marchot P, Chatonnet A (2012) Enzymatic activity and protein interactions in alpha/beta‐hydrolase fold proteins: moonlighting versus promiscuity. Protein Pept Lett 19:132–143. [DOI] [PubMed] [Google Scholar]
- 6. Jochens H, Hesseler M, Stiba K, Padhi SK, Kazlauskas RJ, Bornscheuer UT (2011) Protein engineering of α/β‐hydrolase fold enzymes. ChemBioChem 12:1508–1517. [DOI] [PubMed] [Google Scholar]
- 7. Suplatov DA, Besenmatter W, Svedas VK, Svendsen A (2012) Bioinformatic analysis of alpha/beta‐hydrolase fold enzymes reveals subfamily‐specific positions responsible for discrimination of amidase and lipase activities. Protein Eng Des Sel 25:689–697. [DOI] [PubMed] [Google Scholar]
- 8. Turner JM, Larsen NA, Basran A, Barbas CF, Bruce NC, Wilson IA, Lerner RA (2002) Biochemical characterization and structural analysis of a highly proficient cocaine esterase. Biochemistry 41:12297–12307. [DOI] [PubMed] [Google Scholar]
- 9. Szeltner Z, Renner V, Polgár L (2000) Substrate and pH‐dependent contribution of oxyanion binding site to the catalysis of prolyl oligopeptidase, a paradigm of the serine oligopeptidase family. Protein Sci 9:353–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ericsson DJ, Kasrayan A, Johansson P, Bergfors T, Sandström AG, Bäckvall JE, Mowbray SL (2008) X‐ray structure of Candida Antarctica lipase A shows a novel lid structure and a likely mode of inter‐facial activation. J Mol Biol 376:109–119. [DOI] [PubMed] [Google Scholar]
- 11. Larsen NA, Lin H, Wei R, Fischbach MA, Walsh CT (2006) Structural characterization of enterobactin hydrolase IroE. Biochemistry 45:10184–10190. [DOI] [PubMed] [Google Scholar]
- 12. Ordentlich A, Barak D, Kronman C, Ariel N, Segall Y, Velan B, Shafferman A (1998) Functional characteristics of the oxyanion hole in human acetylcholinesterase. J Biol Chem 273:19509–19517. [DOI] [PubMed] [Google Scholar]
- 13. Zhang Y, Kua J, McCammon JA (2002) Role of the catalytic triad and oxyanion hole in acetylcholinesterase catalysis: an ab initio QM/MM study. J Am Chem Soc 124:10572–10577. [DOI] [PubMed] [Google Scholar]
- 14. Mandrich L, Menchise V, Alterio V, De Simone G, Pedone C, Rossi M, Manco G (2008) Functional and structural features of the oxyanion hole in a thermophilic esterase from Alicyclobacillus acidocaldarius . Proteins 71:1721–1731. [DOI] [PubMed] [Google Scholar]
- 15. Lai L, Xu Z, Zhou J, Lee KD, Amidon GL (2008) Molecular basis of prodrug activation by human valacyclovirase, an alpha‐amino acid ester hydrolase. J Biol Chem 283:9318–9327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Charavgi MD, Dimarogona M, Topakas E, Christakopoulos P, Chrysina ED (2013) The structure of a novel glucuronoyl esterase from Myceliophthora thermophila gives new insights into its role as a potential biocatalyst. Acta Crystallogr D Biol Crystallogr 69:63–73. [DOI] [PubMed] [Google Scholar]
- 17. Huang J, Huo YY, Ji R, Kuang S, Ji C, Xu XW, Li J (2016) Structural insights of a hormone sensitive lipase homologue Est22. Sci Rep 6:28550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Goettig P, Brandstetter H, Groll M, Göhring W, Konarev PV, Svergun DI, Huber R, Kim JS (2005) X‐ray snapshots of peptide processing in mutants of tricorninteracting factor F1 from Thermoplasma acidophilum . J Biol Chem 280:33387–33396. [DOI] [PubMed] [Google Scholar]
- 19. Kiss AL, Palló A, NáraySzabó G, Harmat V, Polgár L (2008) Structural and kinetic contributions of the oxyanion binding site to the catalytic activity of acylaminoacyl peptidase. J Struct Biol 162:312–323. [DOI] [PubMed] [Google Scholar]
- 20. Dimitriou PS, Denesyuk A, Takahashi S, Yamashita S, Johnson MS, Nakayama T, Denessiouk K (2017), Alpha/beta‐hydrolases: a unique structural motif coordinates catalytic acid residue in 40 protein fold families. Proteins 85:1845–1855. [DOI] [PubMed] [Google Scholar]
- 21. Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE (2000) The protein data bank. Nucleic Acids Res 28:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Murzin AG, Brenner SE, Hubbard T, Chothia C (1995) SCOP: a structural classification of proteins database for the investigation of sequences and structures. J Mol Biol 247:536–540. [DOI] [PubMed] [Google Scholar]
- 23. Ohara K, Unno H, Hosoya M, Fujino N, Hirooka K, Takahashi S, Yamasita S, Kusunoki M, Nakayama T (2014) Structural insights into the low pH adaptation of a unique carboxylesterase from Ferroplasma: altering the pH optima of two carboxylesterases. J Biol Chem 289:24499–24510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Narasimhan D, Collins GT, Nance MR, Nichols J, Edwald E, Chan J, Ko MC, Woods JH, Tesmer JJ, Sunahara RK (2011) Subunit stabilization and polyethylene glycolation of cocaine esterase improves in vivo residence time. Mol Pharmacol 80:1056–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Szeltner Z, Rea D, Renner V, Fulop V, Polgar L (2002) Electrostatic effects and binding determinants in the catalysis of prolyl oligopeptidase. Site specific mutagenesis at the oxyanion binding site. J Biol Chem 277:42613–42622. [DOI] [PubMed] [Google Scholar]
- 26. Engel M, Hoffmann T, Wagner L, Wermann M, Heiser U, Kiefersauer R, Huber R, Bode W, Demuth HU, Brandstetter H (2003) The crystal structure of dipeptidyl peptidase IV (CD26) reveals its functional regulation and enzymatic mechanism. Proc Natl Acad Sci U S A 100:5063–5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kim HK, Liu JW, Carr PD, Ollis DL (2005) Following directed evolution with crystallography: structural changes observed in changing the substrate specificity of dienelactone hydrolase. Acta Crystallogr D Biol Crystallogr 61:920–931. [DOI] [PubMed] [Google Scholar]
- 28. Jansson A, Niemi J, Mantsala P, Schneider G (2003) Crystal structure of aclacinomycin methylesterase with bound product analogues: implications for anthracycline recognition and mechanism. J Biol Chem 278:39006–39013. [DOI] [PubMed] [Google Scholar]
- 29. Ghosh D, Erman M, Sawicki M, Lala P, Weeks DR, Li N, Pangborn W, Thiel DJ, Jornvall H, Gutierrez R, Eyzaguirre J (1999) Determination of a protein structure by iodination: the structure of iodinated acetylxylan esterase. Acta Crystallogr D Biol Crystallogr 55:779–784. [DOI] [PubMed] [Google Scholar]
- 30. Wei Y, Swenson L, Castro C, Derewenda U, Minor W, Arai H, Aoki J, Inoue K, ServinGonzalez L, Derewenda ZS (1998) Structure of a microbial homologue of mammalian platelet‐activating factor acetylhydrolases: Streptomyces exfoliatus lipase at 1.9 Å resolution. Structure 6:511–519. [DOI] [PubMed] [Google Scholar]
- 31. Uppenberg J, Hansen MT, Patkar S, Jones TA (1994) The sequence, crystal structure determination and refinement of two crystal forms of lipase B from Candida Antarctica . Structure 2:293–308. [DOI] [PubMed] [Google Scholar]
- 32. Bruner SD, Weber T, Kohli RM, Schwarzer D, Marahiel MA, Walsh CT, Stubbs MT (2002) Structural basis for the cyclization of the lipopeptide antibiotic surfactin by the thioesterase domain SrfTE. Structure 10:301–310. [DOI] [PubMed] [Google Scholar]
- 33. Pletnev V, Addlagatta A, Wawrzak Z, Duax W (2003) Three‐dimensional structure of homodimeric cholesterol esterase‐ligand complex at 1.4 Å resolution. Acta Crystallogr D Biol Crystallogr 59:50–56. [DOI] [PubMed] [Google Scholar]
- 34. Braaz R, Handrick R, Jendrossek D (2003) Identification and characterisation of the catalytic triad of the alkaliphilic thermo‐tolerant PHA depolymerase PhaZ7 of Paucimonas lemoignei . FEMS Microbiol Lett 224:107–112. [DOI] [PubMed] [Google Scholar]
- 35. Hisano T, Kasuya K, Tezuka Y, Ishii N, Kobayashi T, Shiraki M, Oroudjev E, Hansma H, Iwata T, Doi Y, Saito T, Miki K (2006) The crystal structure of polyhydroxybutyrate depolymerase from Penicillium funiculosum provides insights into the recognition and degradation of biopolyesters. J Mol Biol 356:993–1004. [DOI] [PubMed] [Google Scholar]
- 36. Papageorgiou AC, Hermawan S, Singh CB, Jendrossek D (2008) Structural basis of poly(3‐hydroxybutyrate) hydrolysis by PhaZ7 depolymerase from Paucimonas lemoignei . J Mol Biol 382:1184–1194. [DOI] [PubMed] [Google Scholar]
- 37. Nardini M, Ridder IS, Rozeboom HJ, Kalk KH, Rink R, Janssen DB, Dijkstra BW (1999) The x‐ray structure of epoxide hydrolase from Agrobacterium radiobacter AD1. An enzyme to detoxify harmful epoxides. J Biol Chem 274:14579–14586. [PubMed] [Google Scholar]
- 38. De Simone G, Galdiero S, Manco G, Lang D, Rossi M, Pedone C (2000) A snapshot of a transition state analogue of a novel thermophilic esterase belonging to the subfamily of mammalian hormone‐sensitive lipase. J Mol Biol 303:761–771. [DOI] [PubMed] [Google Scholar]
- 39. Bahl CD, Hvorecny KL, Bridges AA, Ballok AE, Bomberger JM, Cady KC, O'Toole GA, Madden DR (2014) Signature motifs identify an Acinetobacter Cif virulence factor with epoxide hydrolase activity. J Biol Chem 289:7460–7469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Medrano FJ, Alonso J, García JL, Romero A, Bode W, GomisRüth FX (1998) Structure of proline iminopeptidase from Xanthomonas campestris pv. citri: a prototype for the prolyl oligopeptidase family. EMBO J 17:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lai KK, Stogios PJ, Vu C, Xu X, Cui H, Molloy S, Savchenko A, Yakunin A, Gonzalez CF (2011) An inserted α/β sub‐domain shapes the catalytic pocket of Lactobacillus johnsonii cinnamoyl esterase. PLoS One 6:e23269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tyukhtenko S, Karageorgos I, Rajarshi G, Zvonok N, Pavlopoulos S, Janero DR, Makriyannis A (2016) Specific inter‐residue interactions as determinants of human monoacylglycerol lipase catalytic competency: a role for global conformational changes. J Biol Chem 291:2556–2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bell PA, Kasper CB (1993) Expression of rat microsomal epoxide hydrolase in Escherichia coli. Identification of a histidyl residue essential for catalysis. J Biol Chem 268:14011–14017. [PubMed] [Google Scholar]
- 44. Porter JL, Boon PL, Murray TP, Huber T, Collyer CA, Ollis DL (2015) Directed evolution of new and improved enzyme functions using an evolutionary intermediate and multi‐directional search. ACS Chem Biol 10:611–621. [DOI] [PubMed] [Google Scholar]
- 45. Bordes F, Barbe S, Escalier P, Mourey L, André I, Marty A, Tranier S (2010) Exploring the conformational states and rearrangements of Yarrowia lipolytica lipase. Biophys J 99:2225–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Roussel A, Canaan S, Egloff MP, Rivière M, Dupuis L, Verger R, Cambillau C (1999) Crystal structure of human gastric lipase and model of lysosomal acid lipase, two lipolytic enzymes of medical interest. J Biol Chem 274:16995–17002. [DOI] [PubMed] [Google Scholar]
- 47. Ronning DR, Klabunde T, Besra GS, Vissa VD, Belisle JT, Sacchettini JC (2000) Crystal structure of the secreted form of antigen 85C reveals potential targets for mycobacterial drugs and vaccines. Nat Struct Biol 7:141–146. [DOI] [PubMed] [Google Scholar]
- 48. Liao DI, Breddam K, Sweet RM, Bullock T, Remington SJ (1992) Refined atomic model of wheat serine carboxypeptidase II at 2.2Å resolution. Biochemistry 31:9796–9812. [DOI] [PubMed] [Google Scholar]
- 49. Schleberger C, Sachelaru P, Brandsch R, Schulz GE (2007) Structure and action of a C‐C bond cleaving alpha/beta‐hydrolase involved in nicotine degradation. J Mol Biol 367:409–418. [DOI] [PubMed] [Google Scholar]
- 50. Rudenko G, Bonten E, D'Azzo A, Hol WGJ (1995) Three‐dimensional structure of the human protective protein: structure of the precursor form suggests a complex activation mechanism. Structure 3:1249–1259. [DOI] [PubMed] [Google Scholar]
- 51. Brim RL, Nance MR, Youngstrom DW, Narasimhan D, Zhan CG, Tesmer JJ, Sunahara RK, Woods JH (2010) A thermally stable form of bacterial cocaine esterase: a potential therapeutic agent for treatment of cocaine abuse. Mol Pharmacol 77:593–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Schiefner A, Gerber K, Brosig A, Boos W (2014) Structural and mutational analyses of Aes, an inhibitor of MalT in Escherichia coli . Proteins 82:268–277. [DOI] [PubMed] [Google Scholar]
- 53. Faulds CB, Molina R, Gonzalez R, Husband F, Juge N, SanzAparicio J, Hermoso JA (2005) Probing the determinants of substrate specificity of a feruloyl esterase, AnFaeA, from Aspergillus niger . FEBS J 272:4362–4371. [DOI] [PubMed] [Google Scholar]
- 54. Chourasia M, Sastry GM, Sastry GN (2011) Aromatic‐Aromatic Interactions Database, A(2)ID: an analysis of aromatic π‐networks in proteins. Int J Biol Macromol 48:540–552. [DOI] [PubMed] [Google Scholar]
- 55. Madhusudan Makwana K, Mahalakshmi R (2015) Implications of aromatic‐aromatic interactions: From protein structures to peptide models. Protein Sci 24:1920–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Truongvan N, Chung HS, Jang SH, Lee C (2016) Conserved tyrosine 182 residue in hyper‐thermophilic esterase EstE1 plays a critical role in stabilizing the active site. Extremophiles 20:187–193. [DOI] [PubMed] [Google Scholar]
- 57. Burley SK, Petsko GA (1985) Aromatic‐aromatic interaction: a mechanism of protein structure stabilization. Science 229:23–28. [DOI] [PubMed] [Google Scholar]
- 58. Kannan N, Vishveshwara S (2000) Aromatic clusters: a determinant of thermal stability of thermophilic proteins. Protein Eng 13:753–761. [DOI] [PubMed] [Google Scholar]
- 59. Lanzarotti E, Biekofsky RR, Estrin DA, Marti MA, Turjanski AG (2011) Aromatic‐aromatic interactions in proteins: beyond the dimer. J Chem Inf Model 51:1623–1633. [DOI] [PubMed] [Google Scholar]
- 60. Haruki M, Oohashi Y, Mizuguchi S, Matsuo Y, Morikawa M, Kanaya S (1999) Identification of catalytically essential residues in Escherichia coli esterase by site‐directed mutagenesis. FEBS Lett 454:262–266. [DOI] [PubMed] [Google Scholar]
- 61. Li C, Li JJ, Montgomery MJ, Wood SP, Bugg TDH (2006) Catalytic role for arginine 188 in the C‐C hydrolase catalytic mechanism for Escherichia coli MhpC and Burkholderia xenovorans LB400 BphD. Biochemistry 45:12470–12479. [DOI] [PubMed] [Google Scholar]
- 62. Horsman GP, Bhowmik S, Seah SY, Kumar P, Bolin JT, Eltis LD (2007) The tautomeric half‐reaction of BphD, a C‐C bond hydrolase. Kinetic and structural evidence supporting a key role for histidine 265 of the catalytic triad. J Biol Chem 282:19894–19904. [DOI] [PubMed] [Google Scholar]
- 63. Janin J, Chothia C (1980) Packing of alpha‐helices onto beta‐pleated sheets and the anatomy of alpha/beta proteins. J Mol Biol 143:95–128. [DOI] [PubMed] [Google Scholar]
- 64. Burnett RM, Darling GD, Kendall DS, LeQuesne ME, Mayhew SG, Smith WW, Ludwig ML (1974) The structure of the oxidized form of clostridial flavodoxin at 19A resolution. J Biol Chem 249:4383–4392. [PubMed] [Google Scholar]
- 65. McKary MG, Abendroth J, Edwards TE, Johnson RJ (2016) Structural basis for the strict substrate selectivity of the mycobacterial hydrolase LipW. Biochemistry 55:7099–7111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zhang XF, Yang GY, Zhang Y, Xie Y, Withers SG, Feng Y (2016) A general and efficient strategy for generating the stable enzymes. Sci Rep 6:33797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Auldridge ME, Guo Y, Austin MB, Ramsey J, Fridman E, Pichersky E, Noel JP (2012) Emergent decarboxylase activity and attenuation of α/β‐hydrolase activity during the evolution of methylketone biosynthesis in tomato. Plant Cell 24:1596–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Knapik AA, Petkowski JJ, Otwinowski Z, Cymborowski MT, Cooper DR, Majorek KA, Chruszcz M, Krajewska WM, Minor W (2012) A multi‐faceted analysis of RutD reveals a novel family of α/β hydrolases. Proteins 80:2359–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bezerra GA, Dobrovetsky E, Seitova A, Fedosyuk S, Dhe‐Paganon S, Gruber K (2015) Structure of human dipeptidyl peptidase 10 (DPPY): a modulator of neuronal Kv4 channels. Sci Rep 5:8769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Xie Y, Takemoto C, Kishishita S, UchikuboKamo T, Murayama K, Chen L, Liu ZJ, Wang BC, Manzoku M, Ebihara A, Kuramitsu S, Shirouzu M, Yokoyama S (2007) Structure of the minimized alpha/beta‐hydrolase fold protein from Thermus thermophilus HB8. Acta Crystallogr F 63:993–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Discovery Studio Modeling Environment, Release 2017. (2016). San Diego: Dassault Systèmes.
- 72. Derewenda ZS, Lee L, Derewenda U (1995) The occurrence of CH…O hydrogen bonds in proteins. J Mol Biol 252:248–262. [DOI] [PubMed] [Google Scholar]
- 73. Lehtonen JV, Still DJ, Rantanen VV, Ekholm J, Björklund D, Iftikhar Z, Huhtala M, Repo S, Jussila A, Jaakkola J, Pentikäinen O, Nyrönen T, Salminen T, Gyllenberg M, Johnson M (2004) BODIL: a molecular modeling environment for structure‐function analysis and drug design. J Comput Aided Mol Des 18:401–419. [DOI] [PubMed] [Google Scholar]
- 74. Kraulis PJ (1991) MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J Appl Cryst 24:946–950. [Google Scholar]
- 75. Spiller B, Gershenson A, Arnold FH, Stevens RC (1999) A structural view of evolutionary divergence. Proc Natl Acad Sci U S A 96:12305–12310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Zhu X, Larsen NA, Basran A, Bruce NC, Wilson IA (2003) Observation of an arsenic adduct in an acetyl esterase crystal structure. J Biol Chem 278:2008–2014. [DOI] [PubMed] [Google Scholar]
- 77. Murayama K, Shirouzu M, Terada T, Kuramitsu S, Yokoyama S (2005) Crystal structure of TT1662 from Thermus thermophilus HB8: a member of the alpha/beta hydrolase fold enzymes. Proteins 58:982–984. [DOI] [PubMed] [Google Scholar]
- 78. Goettig P, Groll M, Kim JS, Huber R, Brandstetter H (2002) Structures of the tricorninteracting aminopeptidase F1 with different ligands explain its catalytic mechanism. EMBO J 21:5343–5352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Zhang R, Koroleva O, Collert F, Joachimiak A, Midwest Center for Structural Genomics (in press). 1.5 Å crystal structure of the cephalosporin C deacetylase.
- 80. Oakley AJ, Klvana M, Otyepka M, Nagata Y, Wilce MC, Damborsky J (2004) Crystal structure of haloalkane dehalogenase LinB from Sphingomonas paucimobilis UT26 at 0.95 A resolution: dynamics of catalytic residues. Biochemistry 43:870–878. [DOI] [PubMed] [Google Scholar]
- 81. Horsman GP, Ke J, Dai S, Seah SY, Bolin JT, Eltis LD (2006) Kinetic and structural insight into the mechanism of BphD, a C‐C bond hydrolase from the biphenyl degradation pathway. Biochemistry 45:11071–11086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Agarwal V, Lin S, Lukk T, Nair SK, Cronan JE (2012) Structure of the enzyme‐acyl carrier protein (ACP) substrate gatekeeper complex required for biotin synthesis. Proc Natl Acad Sci U S A 109:17406–17411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Seetharaman J, Lew S, Wang D, Kohan E, Patel D, Whitehead T, Fleishman S, Ciccosanti C, Xiao R, Everett JK, Acton TB, Montelione GT, Tong L, Hunt JF, Northeast Structural Genomics Consortium. Crystal Structure of Engineered Protein. Northeast Structural Genomics Consortium Target OR94.
- 84. Zou J, Hallberg BM, Bergfors T, Oesch F, Arand M, Mowbray SL, Jones TA (2000) Structure of Aspergillus niger epoxide hydrolase at 1.8 Å resolution: implications for the structure and function of the mammalian microsomal class of epoxide hydrolases. Structure 8:111–122. [DOI] [PubMed] [Google Scholar]
- 85. Hofmann B, Tolzer S, Pelletier I, Altenbuchner J, van Pee KH, Hecht HJ (1998) Structural investigation of the cofactorfree chloroperoxidases. J Mol Biol 279:889–900. [DOI] [PubMed] [Google Scholar]
- 86. Bellizzi JJ III, Widom J, Kemp C, Lu JY, Das AK, Hofmann SL, Clardy J (2000) The crystal structure of palmitoyl protein thioesterase 1 and the molecular basis of infantile neuronal ceroid lipofuscinosis. Proc Natl Acad Sci U S A 97:4573–4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Devedjiev Y, Dauter Z, Kuznetsov SR, Jones TL, Derewenda ZS (2000) Crystal structure of the human acyl protein thioesterase I from a single X‐ray data set to 1.5 Å. Structure 8:1137–1146. [DOI] [PubMed] [Google Scholar]
- 88. Padmanabhan B, Kuzuhara T, Adachi N, Horikoshi M (2004) The crystal structure of CCG1/TAF(II)250‐interacting factor B (CIB). J Biol Chem 279:9615–9624. [DOI] [PubMed] [Google Scholar]
- 89. Bourne PC, Isupov MN, Littlechild JA (2000) The atomic‐resolution structure of a novel bacterial esterase. Structure 8:143–151. [DOI] [PubMed] [Google Scholar]
- 90. Kawasaki K, Kondo H, Suzuki M, Ohgiya S, Tsuda S (2002) Alternate conformations observed in catalytic serine of Bacillus subtilis lipase determined at 1.3 Å resolution. Acta Crystallogr D Biol Crystallogr 58:1168–1174. [DOI] [PubMed] [Google Scholar]
- 91. Roussel A, Yang Y, Ferrato F, Verger R, Cambillau C, Lowe M (1998) Structure and activity of rat pancreatic lipase‐related protein 2. J Biol Chem 273:32121–32128. [DOI] [PubMed] [Google Scholar]
- 92. Schmidt A, Gruber K, Kratky C, Lamzin VS (2008) Atomic resolution crystal structures and quantum chemistry meet to reveal subtleties of hydroxynitrile lyase catalysis. J Biol Chem 283:21827–21836. [DOI] [PubMed] [Google Scholar]
- 93. Janda IK, Devedjiev Y, Cooper DR, Chruszcz M, Derewenda U, Gabrys A, Minor W, Joachimiak A, Derewenda ZS (2004) Harvesting the high‐hanging fruit: the structure of the YdeN gene product from Bacillus subtilis at 1.8 angstroms resolution. Acta Crystallogr D Biol Crystallogr 60:1101–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Arndt JW, Schwarzenbacher R, Page R, Abdubek P, Ambing E, Biorac T, Canaves JM, Chiu HJ, Dai X, Deacon AM, DiDonato M, Elsliger MA, Godzik A, Grittini C, Grzechnik SK, Hale J, Hampton E, Han GW, Haugen J, Hornsby M, Klock HE, Koesema E, Kreusch A, Kuhn P, Jaroszewski L, Lesley SA, Levin I, McMullan D, McPhillips TM, Miller MD, Morse A, Moy K, Nigoghossian E, Ouyang J, Peti WS, Quijano K, Reyes R, Sims E, Spraggon G, Stevens RC, van den Bedem H, Velasquez J, Vincent J, von Delft F, Wang X, West B, White A, Wolf G, Xu Q, Zagnitko O, Hodgson KO, Wooley J, Wilson IA (2005) Crystal structure of an alpha/beta serine hydrolase (YDR428C) from Saccharomyces cerevisiae at 1.85 Å resolution. Proteins 58:755–758. [DOI] [PubMed] [Google Scholar]
- 95. Bartlam M, Wang G, Yang H, Gao R, Zhao X, Xie G, Cao S, Feng Y, Rao Z (2004) Crystal structure of an acylpeptide hydrolase/esterase from Aeropyrum pernix K1. Structure 12:1481–1488. [DOI] [PubMed] [Google Scholar]
- 96. Legler PM, Kumaran D, Swaminathan S, Studier FW, Millard CB (2008) Structural characterization and reversal of the natural organophosphate resistance of a D‐type esterase, Saccharomyces cerevisiae S‐formyl‐glutathione hydrolase. Biochemistry 47:9592–9601. [DOI] [PubMed] [Google Scholar]
- 97. Gorman J, Shapiro L, Burley SK, Midwest Center for Structural Genomics (in press). Structural Genomics target NYSGRCT920 related to A/B‐hydrolase fold.
- 98. Osipiuk J, Xu X, Zheng H, Savchenko A, Edwards A, Joachimiak A, Midwest Center for Structural Genomics (in press). Crystal structure of hypothetical protein Atu1826, a putative alpha/beta hydrolase from Agrobacterium tumefaciens.
- 99. Mirza IA, Nazi I, Korczynska M, Wright GD, Berghuis AM (2005) Crystal structure of homoserine transacetylase from Haemophilus influenzae reveals a new family of alpha/beta‐hydrolases. Biochemistry 44:15768–15773. [DOI] [PubMed] [Google Scholar]
- 100. Lin S, Hanson RE, Cronan JE (2010) Biotin synthesis begins by hijacking the fatty acid synthetic pathway. Nat Chem Biol 6:682–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Tomczyk NH, Nettleship JE, Baxter RL, Crichton HJ, Webster SP, Campopiano DJ (2002) Purification and characterisation of the BIOH protein from the biotin biosynthetic pathway. FEBS Lett 513:299–304. [DOI] [PubMed] [Google Scholar]
- 102. Hwang J, Kim Y, Kang HB, Jaroszewski L, Deacon AM, Lee H, Choi WC, Kim KJ, Kim CH, Kang BS, Lee JO, Oh TK, Kim JW, Wilson IA, Kim MH (2011) Crystal structure of the human N‐Myc downstream‐regulated gene 2 protein provides insight into its role as a tumor suppressor. J Biol Chem 286:12450–12460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Fabrichny IP, Leone P, Sulzenbacher G, Comoletti D, Miller MT, Taylor P, Bourne Y, Marchot P (2007) Structural analysis of the synaptic protein neuroligin and its beta‐neurexin complex: determinants for folding and cell adhesion. Neuron 56:979–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Kellett WF, Brunk E, Desai BJ, Fedorov AA, Almo SC, Gerlt JA, Rothlisberger U, Richards NG (2013) Computational, structural, and kinetic evidence that Vibrio vulnificus FrsA is not a cofactor‐independent pyruvate decarboxylase. Biochemistry 52:1842–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Wilson RA, Maughan WN, Kremer L, Besra GS, Fütterer K (2004) The structure of mycobacterium tuberculosis MPT51 (FbpC1) defines a new family of non‐catalytic alpha/beta hydrolases. J Mol Biol 335:519–530. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting information