

Abstract
Oligomerization of γ‐Synuclein is known to have implications for both neurodegeneration and cancer. Although it is known to co‐exist with the fibrillar deposits of α‐Synuclein (Lewy bodies), a hallmark in Parkinson's disease (PD), the effect of potential therapeutic modulators on the fibrillation pathway of γ‐Syn remains unexplored. By a combined use of various biophysical tools and cytotoxicity assays we demonstrate that the flavonoid epigallocatechin‐3‐gallate (EGCG) significantly suppresses γ‐Syn fibrillation by affecting its nucleation and binds with the unstructured, nucleus forming oligomers of γ‐Syn to modulate the pathway to form α‐helical containing higher‐order oligomers (~158 kDa and ~ 670 kDa) that are SDS‐resistant and conformationally restrained in nature. Seeding studies reveal that these oligomers although “on‐pathway” in nature, are kinetically retarded and rate‐limiting species that slows down fibril elongation. We observe that EGCG also disaggregates the protofibrils and mature γ‐Syn fibrils into similar SDS‐resistant oligomers. Steady‐state and time‐resolved fluorescence spectroscopy and isothermal titration calorimetry (ITC) reveal a weak non‐covalent interaction between EGCG and γ‐Syn with the dissociation constant in the mM range (K d ~ 2–10 mM). Interestingly, while EGCG‐generated oligomers completely rescue the breast cancer (MCF‐7) cells from γ‐Syn toxicity, it reduces the viability of neuroblastoma (SH‐SY5Y) cells. However, the disaggregated oligomers of γ‐Syn are more toxic than the disaggregated fibrils for MCF‐7cells. These findings throw light on EGCG‐mediated modulation of γ‐Syn fibrillation and suggest that investigation on the effects of such modulators on γ‐Syn fibrillation is critical in identifying effective therapeutic strategies using small molecule modulators of synucleopathies.
Keywords: γ‐Synuclein, EGCG, aggregation, oligomerization, fibril, inhibition mechanism, toxicity, disaggregation, synucleopathies
Abbreviations and symbols
- EGCG
epigallocatechin‐3‐gallate
- TEM
transmission electron microscopy
- SEC
size‐exclusion chromatography
- HPLC
high‐performance liquid chromatography
- CD
circular dichroism
- γ‐Syn
γ‐synuclein
- α‐Syn
α‐synuclein
- TCSPC
time‐correlated single photon counting
- DLS
dynamic light scattering
- SDS‐PAGE
sodium dodecyl sulphate polyacrylamide gel electrophoresis
- ITC
isothermal titration calorimetry
- MTT
3‐(4,5‐dimethylthiazol‐2‐Yl)‐2,5‐Diphenyltetrazolium Bromide
- ANS
(1‐anilino‐8‐sulfonic acid)
- Rh
hydrodynamic radius
- Rs
Stoke's radius
- ThT
thioflavin T
- DMEM
Dulbecco's modified Eagle's medium
- ESI‐MS
electrospray‐ionisation mass spectrometry
- AFM
atomic force microscopy
- LDH
lactate‐dehydrogenase
- τ
time decay constant
- α
amplitude
- τm
average lifetime
- kq
bimolecular quenching constant
Introduction
Rising evidence of toxicity associated with the soluble oligomeric species formed during amyloidogenesis has shifted the focus from inhibiting the amyloid plaque formation to developing strategies that could modulate the species to become less toxic.1 These oligomers share strikingly similar structural motifs irrespective of their protein sequences, thereby involving similar mechanisms of toxicity.1, 2 Although the high dynamicity of these species pose a challenge in deciphering the exact cascade of pathogenicity, cell‐to‐cell transmission,3 membrane permeabilization, and aberrant signal transduction1, 4 are some of the key features known.
Belonging to the family of Synucleins, which are an important class of intrinsically disordered neural proteins, our study emphasizes the critical role of a highly oligomer forming, yet moderately fibrillogenic member of the family “γ‐Synuclein,”5 which despite having a plethora of pathogenic implications in both neurodegeneration6 and cancer,7 remains poorly studied. The involvement of γ‐Syn in the etiology of the diseases is speculated to result from its inherent tendency to self‐associate and form oligomers that are capable of cellular invasion as well as in facilitating the aggregation of other amyloidogenic proteins like α‐Syn, a hallmark in Parkinson's disease (PD).8 Nevertheless, γ‐Syn deposits have also been traced in Lewy bodies in conjunction with the fibrillar deposits of α‐Syn.9 Several factors like high concentration,5 oxidation8 and dissociation of γ‐Syn from its interaction partners10 are reported to facilitate oligomerization. Also, the over expressed γ‐Syn is considered a prognostic marker in multiple invasive cancers as well as neurodegenerative conditions where it enhances the debilitating effects by causing death of motor neurons6 and promoting metastasis.11, 12, 13
In the light of the current scenario, it is important to employ a strategy for regulating the toxicity of these resultant oligomeric species and investigate the effect of various therapeutic modulators on the fibrillation process of γ‐Syn. Several strategies have so far been employed to understand the series of amyloid forming events and modulation of the fibrillation pathway of a number of proteins. The use of neurotransmitters like dopamine and calcium,14 peptide inhibitors15 and naturally occurring small molecules like polyphenols16 have been investigated but studies on the role of such modulators in the fibril‐forming pathway of γ‐Syn are non‐existent. The major green tea polyphenol epigallocatechin‐3‐gallate has a well‐established neuroprotective17 as well as anticancer properties.18 EGCG inhibits the fibrillation of a wide range of amyloidogenic proteins like α‐Syn, Aβ peptide, Huntingtin, etc.19, 20 The most prevalent characteristic of EGCG inhibited fibrillation pathway is the formation of non‐toxic, stable oligomeric species or benign protein aggregates which do not further participate in the polymerization process thus rendering them off‐pathway.21, 22 Cross‐linking of amyloid fibrils by oxidized EGCG,22 covalent modification with the sulfhydryl groups on the protein23 and formation of a molecular zipper by EGCG to facilitate the assembly of EGCG‐containing oligomers20 are the proposed mechanisms of EGCG mediated inhibition.
In order to control the debilitating effects of γ‐Syn oligomers and prevent the onset of underlying diseases, it is important to gain an in‐depth understanding of the effects of such modulators on the oligomerization and fibrillation pathway of γ‐Syn. It is proposed that co‐localization of γ‐Syn with its most fibrillogenic counterpart α‐Syn could make it an important target of inhibition and thus necessitates investigation of such inhibitors reported to act on α‐Syn fibrillation pathway for γ‐Syn fibrillation as well. In this study, we investigate the modulating effects of a widely reported anti‐amyloidogenic polyphenol EGCG on the oligomerization and fibrillation propensity of γ‐Syn to provide an insight into the underlying mechanism by addressing the following questions. Does EGCG modulate the fibrillation pathway of γ‐Syn? How does it modulate the pathway and what is the mode of interaction with γ‐Syn? Lastly, what are the cytotoxic effects of the EGCG generated γ‐Syn species?
Our study provides the first evidence of significant suppression and modulation of γ‐Syn fibrillation pathway by EGCG. We report that EGCG retards nucleus formation thus decelerating fibril polymerization and mediates its inhibitory effect by modulating the on‐going fibrillation pathway to form two separate populations of SDS‐resistant higher‐order γ‐Syn oligomers (~ 4 and 10 mer), that are conformationally restrained and gain an α‐helical propensity during fibrillation. Seeding studies further reveal that these oligomers are on‐pathway in nature and are kinetically retarded species (increased lag time) that fail to build‐up into mature fibrils in the pathway. We observe that the pronounced effect of EGCG is mediated by weak, non‐covalent interactions (K d ~ mM), that points to the importance of weak binding interactions in modulating the γ‐Syn fibrillation pathway. We also demonstrate that EGCG attenuates the protofibrillar stages of the pathway and disaggregates protofibrils as well as the mature fibrils into similar kind of SDS‐resistant oligomers. Interestingly, we observe that EGCG‐generated oligomers are differentially toxic to the breast cancer (MCF‐7) and neuroblastoma (SH‐SY5Y) cells, where they completely rescue the MCF‐7 cells from γ‐Syn toxicity but are more toxic than the untreated γ‐Syn oligomers for the SH‐SY5Y cells. This indicates the critical role of γ‐Syn and complexities it may impart in the treatment of amyloidogenic diseases. However, the disaggregated γ‐Syn oligomers are highly toxic to MCF‐7 cells relative to the disaggregated γ‐Syn fibrils that highlights an early gain of toxicity of γ‐Syn which is governed by the morphology of the species formed.
The results presented delineate the EGCG‐mediated mechanism of γ‐Syn fibrillation and point to the differences observed for its effect toward α‐ and γ‐Syn in the form of modulation of fibrillation pathway and toxicity. Due to the co‐existence of α‐ and γ‐Syn in the cellular milieu, it further necessitates an in‐depth investigation of small molecule modulators like EGCG on the fibrillation properties of the synucleins involved in synucleopathies.
Results and Discussion
EGCG inhibits γ‐Syn fibrillation in a concentration‐dependent manner
In previous studies, EGCG has been reported to inhibit fibrillation of many other amyloidogenic proteins such as α‐Synuclein and Aβ peptide at a ratio of 5–10‐fold molar excess over protein concentration,20, 22 whereas the effects of EGCG on γ‐Syn is still unknown. The effect of EGCG on the fibrillation kinetics of γ‐Syn was analyzed by Thioflavin T binding assay. ThT is a benzothiazole dye that fluoresces strongly upon binding with the cross β‐sheet structures which are a signature for amyloid fibrils.24 The ThT fluorescence was found to increase by almost 8‐fold in magnitude upon binding with γ‐Syn fibrils formed in the absence of EGCG with an apparent fibrillation rate (k app) of 0.36 ± 0.02 h−1. Incubation of γ‐Syn in the presence of increasing concentration of EGCG (5–50 μM) resulted in a concentration‐dependent decrease in ThT fluorescence [Fig. 1(A) and (B)] with negligible rise in ThT fluorescence recorded in the presence of EGCG at higher concentrations (>30 μM). The decrease in the apparent fibrillation rate (k app) of γ‐Syn with respect to EGCG is shown in Figure 1(C). To further ascertain that the decrease in ThT fluorescence in the presence of EGCG is due to fibril inhibition and not due to interference between ThT and EGCG, the control containing EGCG and ThT in buffer alone was also monitored, which showed negligible fluorescence at all time‐points during fibrillation [Fig. 1(B), inset]. Additionally, the fibrillation kinetics of γ‐Syn were monitored by extrinsic ThT assay after removing the unbound EGCG by centrifugation wash protocol as previously mentioned22 with slight modifications (see Supplementary Material S2). We observed a reduced ThT fluorescence in the presence of EGCG as compared to untreated γ‐Syn, clearly indicating a significant suppression of γ‐Syn fibrillation by EGCG (Supporting Information Fig. S2).
Figure 1.
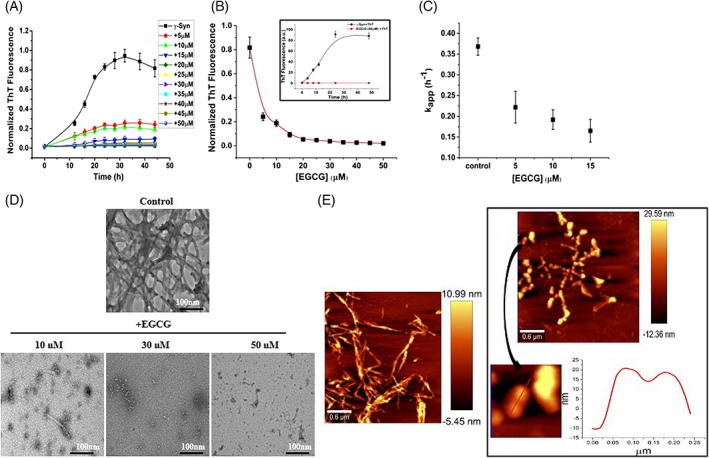
EGCG suppresses γ‐Syn fibrillation in a concentration‐dependent manner. Fibrillation kinetics of γ‐Syn (75 μM) studied using thioflavin T (ThT) assay in the presence of increasing concentration of EGCG (5–50 μM). (A) Decrease in ThT fluorescence with respect to time of fibrillation and (B) with respect to EGCG concentration, (inset) control of ThT and EGCG showing absence of interference between the two. The error bars represent the standard deviation from three independent aggregation reactions, ±SD (n = 3). (C) The apparent rate of fibrillation (kapp) is shown to decrease with an increasing concentration of EGCG. D) Transmission electron microscopy images of γ‐Syn fibrils formed at the end of fibrillation in the presence of increasing concentration of EGCG (Scale bars, 100 nm). E) Atomic force microscopy images showing significant disappearance of mature γ‐Syn fibrils in the presence of EGCG (50 μM) with an appearance of self‐associated large spherical oligomers clearly depicted in the cross section of the oligomers (right panel; below).
Interestingly, EGCG suppresses γ‐Syn fibrillation by 50% even at a 15‐fold less stoichiometry and a ratio of less than 1:1 is needed to bring complete inhibition [Fig. 1(A)]. Effective inhibition of γ‐Syn fibrillation by a significantly lower concentration of EGCG suggests that it acts on the nucleation phase of the pathway where it interacts with the nucleus or seeds present in low concentration as reported for small molecule mediated inhibition of Aβ fibrillation.25
The effect of EGCG on the morphology of γ‐Syn fibrils was studied by negatively stained transmission electron microscopy and atomic force microscopy. Incubating the monomeric γ‐Syn with EGCG (5–50 μM) during fibrillation showed a concentration dependent disappearance of the γ‐Syn fibrils [Fig. 1(D)] corresponding well with the observed decrease in ThT fluorescence [Fig. 1(A)]. AFM images of γ‐Syn fibrils formed both in the absence and presence of EGCG (50 μM), demonstrated a significant suppression of fibril formation in the presence of EGCG. γ‐Syn in the absence of EGCG formed mature intertwined fibrils with an average height of ~11 nm. However, in the presence of EGCG, no mature fibrils were observed and highly populated large spherical oligomers with an average height of 26.2 nm and sparsely populated smaller oligomers with an average height of 12.3 nm were observed. These oligomers were found to self‐associate with each other giving rise to a beaded appearance as shown in Figure 1(E).
The significant suppression of γ‐Syn fibrillation by EGCG at substoichiometric ratios and the known use of EGCG in Phase IV clinical trials26 highlight the physiological relevance of these findings. Several studies on the bioavailability of EGCG have demonstrated its ability to cross the blood brain barrier26 and get absorbed by the digestive tract of the human body upon oral administration27 at a dosage of around 300 mg/day.28 Thus, it is noteworthy that the concentrations used in the study are within the safety limits and suggest that consumption of 1–6 cups of green tea/day would sufficiently provide the effective concentration of EGCG required for inhibition of fibrillation as used in this study.
EGCG disaggregates mature γ‐Syn fibrils and attenuates fibril elongation
To probe the effect of EGCG on different stages of γ‐Syn fibrillation, EGCG (50 μM) was added at different time intervals (8 h, 24 h, and 48 h) which respectively correspond to late lag, log and saturation stage of γ‐Syn fibrillation under conditions used. The ThT fluorescence was found to decrease substantially at all time‐points when EGCG was added [Fig. 2(A)], indicating attenuation of fibrillation in the presence of EGCG. The decrease in ThT fluorescence upon addition of EGCG on preformed fibrils also suggests disaggregation of mature γ‐Syn fibrils [Fig. (2B]). The TEM and AFM images of the fibrils, formed upon addition of EGCG at late lag, log and saturation phase (8 h, 24 h, and 48 h, respectively) of γ‐Syn fibrillation, reveal the presence of shorter and disintegrated fragments indicating the disaggregation of protofibrils and pre‐formed γ‐Syn fibrils [Fig. 2(C) and (D), respectively]. In AFM images, along with the disintegrated broken fragments, spherical oligomers were also observed [yellow encircled, Fig. 2(D)].
Figure 2.
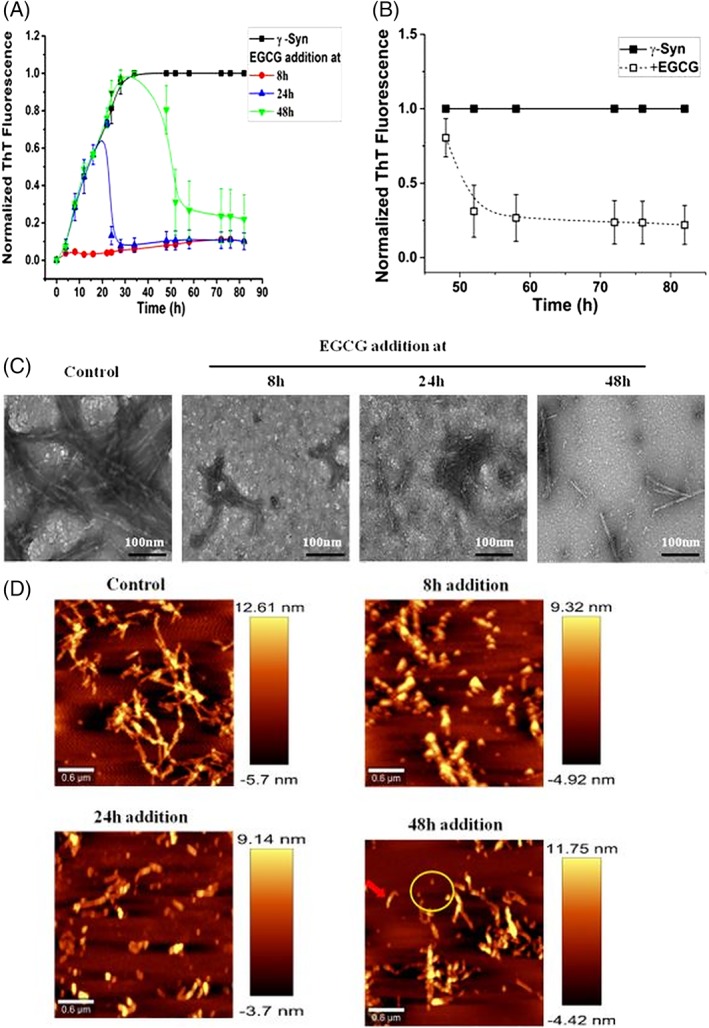
EGCG attenuates γ‐Syn fibrillation at all the stages of the pathway and disaggregates the pre‐existing γ‐Syn fibrils. (A) Thioflavin T assay showing decrease in ThT fluorescence and inhibition of γ‐Syn fibrillation at all three stages upon addition of EGCG at 8 h, 24 h, and 48 h. (B) EGCG‐mediated disaggregation of preformed γ‐Syn fibrils depicted by a continuous decrease in the ThT fluorescence upon addition of EGCG. The error bars represent ±SD (n = 3). (C) Transmission electron microscopy images of γ‐Syn fibrils formed at different stages of EGCG‐mediated inhibition. (Left to right): Control, EGCG added at 8 h, 24 h, and 48 h, respectively (scale bars, 100 nm). (D) AFM images showing disaggregation of γ‐Syn fibrils upon the addition of EGCG at different stages of fibrillation. (Left to right): Control, EGCG added at 8 h, 24 h, and 48 h, respectively. Addition of EGCG at 48 h shows broken fibrils (red arrow) and disaggregated spherical oligomers (yellow encircled).
EGCG modulates the γ‐Syn fibrillation pathway to form SDS‐resistant large spherical oligomers
To investigate the effect of EGCG on the population of various γ‐Syn species formed during aggregation and its effect on the modulation of γ‐Syn fibrillation pathway size‐exclusion chromatography (SEC) was employed. The incubated samples of γ‐Syn formed both in the absence and presence of EGCG (50 μM) during fibrillation were withdrawn at regular time intervals (0, 24, and 48 h) and the soluble fractions obtained after centrifugation were subjected to SEC at an absorbance of 275 nm [Fig. 3(A)]. Freshly prepared γ‐Syn solution eluted with a major monomeric peak at ~15.6 min and a small oligomeric peak at ~10.5 min, corresponding to an apparent molecular weight of ~44 kDa and ~670 kDa (~15 mer), respectively as calculated from the standard calibration curve (Supporting Information Fig. S3). The overestimated apparent molecular weight of monomeric γ‐Syn instead of its actual molecular weight of 13.4 kDa results from the natively unfolded nature of the intrinsically disordered proteins.10 The SEC profiles of γ‐Syn during aggregation showed an increase in the oligomeric peak area during the exponential phase (24 h) of fibrillation, whereas during the end of fibrillation (48 h) the soluble protein was significantly reduced eluting as monomers with only a negligible trace of oligomers present [Fig. 3(A)].
Figure 3.
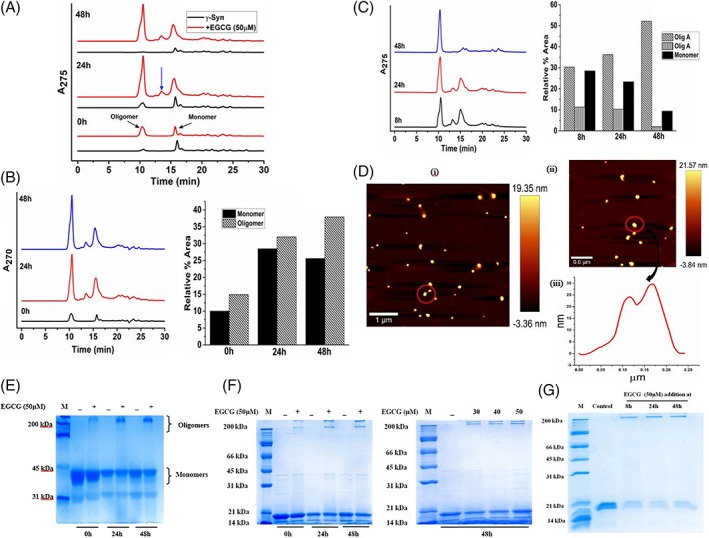
EGCG modulates the fibrillation pathway of γ‐Syn to form SDS‐resistant large spherical oligomers. (A) SEC‐HPLC of γ‐Syn during fibril formation monitored at regular time intervals 0 h, 24 h, and 48 h (bottom to top), respectively, showing formation of two types of oligomers in the presence of EGCG which are absent in the untreated population. The formation of second type of oligomer at 24 h of fibrillation is shown with a blue arrow. (B) The SEC profile at wavelength of 270 nm of EGCG (left panel) and the relative percent area distribution of the different γ‐Syn species (right panel) indicating preferential binding of EGCG with γ‐Syn oligomers. (C) SEC‐profile (left panel) and percent area distribution (right panel) of γ‐Syn species monitored during fibrillation with EGCG added at 8 h, 24 h, and 48 h depicting disaggregation of pre‐existing fibrils of γ‐Syn into oligomeric forms, designated in the plot as olig A and olig B corresponding to apparent molecular weights of ~670 kDa and ~158 kDa, respectively. (D) AFM measurements of EGCG‐generated purified oligomers (i and ii) demonstrate comet‐like formations (red encircled) indicating their self‐associating tendency with an average height of 21.6 nm. (iii) The cross section of the oligomers shows two peaks indicating the self‐association of oligomers. (E) Polyacrylamide gel electrophoresis under native condition and (F) under denaturing conditions at different times of fibrillation (left panel) in the presence of EGCG (50 μM) and in the presence of increasing concentration of EGCG (right panel), demonstrating the formation of SDS‐stable oligomeric species in the presence of EGCG. (G) SDS‐PAGE of disaggregated oligomers showing EGCG‐mediated formation of SDS‐resistant oligomers. The molecular weight marker lane in all the gels is depicted as M.
As shown in Figure 3(A), on addition of EGCG (50 μM), an immediate rise in the oligomer peak eluting at ~10.3 min was observed. During the exponential phase of fibrillation (24 h), the EGCG treated γ‐Syn eluted with the appearance of another significant peak at ~13.4 min corresponding to an apparent molecular weight of ~158 kDa, suggesting the formation of second type of oligomer (~ 4mer) in the presence of EGCG which otherwise was absent in the SEC profiles of untreated γ‐Syn species. These results indicate EGCG mediated formation of γ‐Syn oligomers during fibrillation.
There were appearances of other small and multiple peaks eluting immediately after the major monomeric peak (~10.5 min) which were visibly reduced in the presence of EGCG and showed decreased absorbance with an increasing time of fibrillation, suggesting the presence of small fractions of various conformations which are getting retarded for a longer period of time in the column. The possibility that these peaks were a resultant of different γ‐Syn conformations was further validated by estimating the hydrodynamic radii of γ‐Syn using the empirical equations established for calculating the hydrodynamic properties of a protein from the apparent molecular weights obtained from SEC.5 Stoke's radius (R s) of the monomeric γ‐Syn as calculated using the apparent molecular mass (~44 kDa) was found to be ~28.7 Å, which is smaller than that expected for a completely random coil protein and is close to the previously reported value of ~30 Å for monomeric γ‐Syn.5 The reduced hydrodynamic radius (R h) of the monomeric γ‐Syn indicates compactness in the overall structure which in turn suggests a high self‐associating tendency of γ‐Syn, also reported in previous studies.5 However, we observe slight difference between R s observed by us and previous reports which could arise due to the differences in the protein concentrations used, emphasizing the role of concentration‐dependent self‐association of γ‐Syn.
The EGCG‐mediated formation of higher‐order γ‐Syn oligomers was further confirmed by Rayleigh scattering (Supporting Information Fig. S4). Monomeric γ‐Syn was incubated with an increasing concentration of EGCG (10, 30, and 50 μM) and the scattering intensities were recorded at regular time intervals during fibrillation. During the initial stages of fibrillation, an early rise in the scattering intensity with the increasing concentration of EGCG indicated the formation of higher‐order oligomers by EGCG, while the overall reduced scattering in the presence of EGCG validated suppression of γ‐Syn fibrillation (Supporting Information Fig. S4). The formation of higher‐order oligomers by EGCG were further confirmed by monitoring the scattering intensity of the soluble γ‐Syn species (obtained after centrifugation) and a concentration dependent increase in the scattering intensity in the presence of EGCG clearly demonstrated the formation of soluble and higher order oligomers by EGCG (Supporting Information Fig. S4).
To further investigate the binding of EGCG to different species of γ‐Syn, the absorbance was also recorded at 270 nm where EGCG absorbs [Fig. 3(B)]. However, the contribution by the tyrosine present in γ‐Syn in this region cannot be ruled out. The quantitation of the relative area under the respective peaks at all the time intervals (0, 24, and 48 h), showed an increase in the oligomeric peak area as compared to the area covered by the monomeric peak, thereby indicating an increased affinity of EGCG for γ‐Syn oligomers.
Additionally, to investigate the disaggregated products of the protofibrillar and fibrillar species of γ‐Syn, the SEC profiles of γ‐Syn formed upon addition of EGCG (50 μM) at the late lag (8 h), log (24 h), and exponential phase (48 h) of the pathway were also monitored at 275 nm [Fig. 3(C)]. The incubation samples with EGCG added at different time intervals were withdrawn at the end of fibrillation and the samples were similarly centrifuged to analyze the soluble aggregates of the pathway. The SEC profiles clearly demonstrated the formation of γ‐Syn oligomers at all the three stages of EGCG addition. The increased oligomeric peak area [Fig. 3(C)] even at 48 h of EGCG addition further validated the EGCG‐mediated disaggregation of γ‐Syn fibrils into higher‐ordered oligomers (~ 670 kDa). The AFM studies of purified γ‐Syn oligomers corresponding to an apparent molecular weight of ~ 670 kDa as observed in SEC [Fig. 3(A)] formed in the presence of EGCG (50 μM) revealed that the oligomers were large and spherical in nature with an average height of ~ 21 nm [Fig. 3(D)]. Interestingly, we observed comet‐like formations or a tailing effect [Fig. 3(D)] which indicated the self‐associating tendency of the γ‐Syn oligomers.
The native‐PAGE analysis further validated the SEC observations [Fig. 3(E)], where bands migrating just below the stacking gel were visualized only in the EGCG‐treated samples and showed large intensities with increasing time of fibrillation. To decipher the nature of the oligomers formed, electrophoresis performed under denaturing conditions as shown in Figure 3(F) showed the formation of two discrete bands in EGCG‐treated γ‐Syn samples at regular intervals of fibrillation (left panel) as well as in the presence on increasing concentration of EGCG upon saturation (48 h) (right panel), depicting the formation of SDS‐stable γ‐Syn oligomers in the presence of EGCG with an apparent molecular weight of ~200 kDa and above, not seen in the untreated γ‐Syn samples. A similar observation was made with the disaggregated species of γ‐Syn [Fig. 3(G)], where SDS‐stable oligomers were formed in the presence of EGCG.
The data demonstrates that EGCG diverts the on‐going fibrillation pathway and disaggregates the existing fibrils into two similar kinds of SDS‐resistant higher ordered oligomers with apparent molecular weights of ~158 kDa and ~670 kDa [Fig. 3(A), (C), (F), and (G)], with the formation of larger oligomers (~ 670 kDa) favored over the smaller ones. EGCG‐mediated self‐association of the γ‐Syn oligomers into large spherical structures (average height of ~21 nm) as seen in the AFM images, possibly imparts structural stability, thus making the oligomers resistant against denaturing by SDS. A similar effect of EGCG‐mediated disaggregation of the pre‐existing fibrils and protofibrils of γ‐Syn into SDS‐ resistant oligomers [Fig. 3(C) and (G), respectively] without forming amorphous aggregates, has also been reported for polyphenol Baicalein on mature fibrils of α‐Syn.29 However, the formation of amorphous aggregates upon fibrillar disaggregation has been reported to be one of the effects of EGCG on mature fibrils of other amyloidogenic proteins like α‐Syn and Aβ‐peptide.20, 30 The gradual build up of γ‐Syn oligomers during the exponential phase of the pathway and their subsequent decline by the end of fibrillation in the absence of EGCG [Fig. 3(A)] further sheds light on the transient nature of these highly reactive species which could remain in a dynamic equilibrium with the monomer and disassemble into unstructured monomers under denaturing conditions unlike the EGCG generated oligomers [Fig. 3(F)]. The transient nature of these fibril forming oligomers have also recently been reported for tau protein.31 Although the exact mechanism of EGCG induced stability of γ‐Syn is still unclear and a residue level information is currently unavailable, the effect of EGCG could be understood in light of a previous study where it is reported that the hydroxyl‐rich aromatic structure of EGCG could form stable hydrogen bonds with the unfolded polypeptide such as α‐Synuclein and Aβ‐peptide, thus establishing that intermolecular interactions facilitate the self‐assembly of the EGCG stabilised oligomers.20
EGCG‐generated oligomers act as partial templates for γ‐Syn polymerization and inhibit γ‐Syn elongation
In order to further investigate whether the EGCG‐generated oligomers act as template for γ‐Syn polymerization and inhibit γ‐Syn elongation, a seeding assay was carried out. The fibrillation kinetics in the presence of EGCG (50 μM) treated and an untreated seeds (20% v/v) were monitored at regular intervals under shaking conditions using ThT fluorescence assay. As shown in Figure 4(A), γ‐Syn seed fibrils formed after 48 h of incubation, reduced the lag time by ~ 10 h, compared to the non‐seeded γ‐Syn fibrillation, indicating nucleation‐dependent polymerization of γ‐Syn as also seen for its fibrillogenic counterpart α‐Syn.5 The EGCG‐treated seeds interestingly showed an intermediate kinetic characteristic with a reduced lag time of ~ 6 h with respect to non‐seeded γ‐Syn kinetics and increase in the lag time by ~ 4 h with respect to the γ‐Syn seeded fibrillation. Further, fibrillation kinetics in the presence of untreated γ‐Syn seeds when supplemented with EGCG (50 μM), led to a significantly reduced ThT fluorescence, indicating inhibition of fibril elongation by EGCG. To further rule out the seeding effects of untreated residual fibrils of γ‐Syn and obtain EGCG enriched oligomers, the 24 h incubation samples containing the EGCG‐generated oligomeric species in the soluble fraction of the fibrillation reaction (obtained after centrifugation) were used as seeds (see Supplementary material 4, Supporting Information Fig. S5). Since, the fibrillation kinetics of γ‐Syn around 24 h of incubation, under the given experimental conditions correspond to the oligomerization stage of γ‐Syn, a change in the lag time of γ‐Syn fibrillation in the presence of preformed EGCG‐generated oligomers, would reflect the on‐/off‐pathway nature of these species. The increased lag time in the presence of EGCG‐generated seeds with respect to the untreated γ‐Syn seeds (Supporting Information Fig. S5) suggests that the EGCG‐generated oligomers are partial templates for monomer addition for fibril polymerization. The intermediate lag time suggests that the EGCG‐generated oligomers increase the kinetic barrier for nucleus formation and, thus, delay fibril elongation. It is interesting to note that EGCG has been previously reported to modulate α‐Syn fibrillation pathway into SDS‐resistant, higher order oligomers,20 as also observed for γ‐Syn but the EGCG‐generated α‐Syn oligomers were reported to be off‐pathway20 in nature unlike the oligomers of γ‐Syn which are kinetically retarded and possess characteristics of “on‐pathway” species. Such contrasting effects of a polyphenol on two closely related members of the same family indicate the involvement of complex modulating mechanisms that needs to be further investigated.
Figure 4.
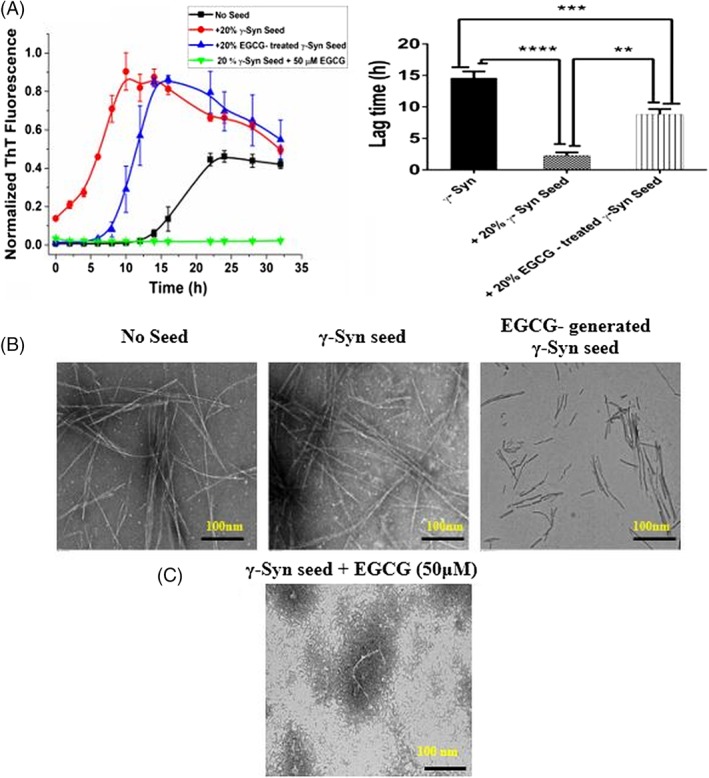
EGCG inhibits γ‐Syn elongation and forms kinetically retarded on‐pathway oligomers: (A) (Left panel) Fibrillation kinetics of γ‐Syn in the presence of EGCG (50 μM) treated and untreated seeds (20% v/v) shows an increased lag time of fibrillation in the presence of EGCG generated seeds as compared to the untreated γ‐Syn seeds. Negligible ThT fluorescence in the presence of EGCG (50 μM) added to the seeded γ‐Syn fibrillation (green line) shows elongation inhibition. The data have been normalized with respect to the maximum fluorescence intensity recorded in the seeded γ‐Syn and the error bars represent ±SD (n = 3). (Right panel) Bar graph showing the significant difference in the lag time in the EGCG‐generated seeds as compared to the seeds formed without EGCG (**P < 0.001) and between non‐seeded γ‐Syn and EGCG‐generated γ‐Syn seeds (***P < 0.0005). The lag time of fibrillation in the presence of untreated γ‐Syn seeds was significantly shorter than the non‐seeded kinetics (****P < 0.0001). (B) The TEM images showing the morphology of the fibrils formed (left to right: no seed, γ‐Syn seed, EGCG‐treated γ‐Syn seeds) (Scale bars, 100 nm). (C) TEM image showing formation of short, broken fragments upon addition of EGCG (50 μM) to seeded γ‐Syn pathway indicating inhibition of fibril elongation by EGCG.
The TEM images of the fibrils formed in the absence and presence of EGCG treated seeds [Fig. 4(B)] showed self‐seeding capacity of γ‐Syn by giving rise to thick intertwined mature fibrils whereas only short and partially polymerized fragments were observed in the presence of EGCG‐treated seeds. This further suggests that the EGCG‐generated oligomers are kinetically compromised species for fibril maturation. Additionally, the formation of numerous short and broken fragments upon addition of EGCG (50 μM) to the γ‐Syn seeded fibrillation reaction [Fig. 4(C)] establish the inhibition of γ‐Syn fibril elongation by EGCG, corresponding with the negligible ThT fluorescence as observed in Figure 4(A).
EGCG delays β‐sheet formation and generates α‐helical containing oligomers during fibrillation
The shift in the equilibrium toward formation of oligomers in the presence of EGCG (results above) also suggested that EGCG favors conformations that have less likelihood of being the precursors for γ‐Syn fibrillation. To gain insight into the effect of EGCG on γ‐Syn structure and fibrillation, far‐UV circular dichroism (CD) spectroscopy of the γ‐Syn species incubated with an increasing concentration of EGCG (10, 30 and 50 μM) under native and fibrillating conditions was carried out. Untreated and freshly prepared solutions of γ‐Syn showed far‐UV CD spectra of a typical natively unfolded conformation with a large negative ellipticity at ~200 nm and a small value at ~222 nm, which was also maintained at all the concentrations of EGCG used [Fig. 5(A)]. The far‐UV CD spectra of γ‐Syn, monitored after 24 h and 48 h of fibrillation [Fig. 5(B)], showed a shift in the negative ellipticity to 218 nm, indicating a structural transition from the natively unfolded conformation to a characteristic β‐sheet structure. This structural transition was found to be significantly delayed in the presence of EGCG in a concentration‐dependent manner, with an increase in negative ellipticity at ~208 nm and ~221 nm at higher concentration of EGCG (50 μM), indicating the formation of an α‐helical like structure by EGCG [Fig. 5(B)].
Figure 5.
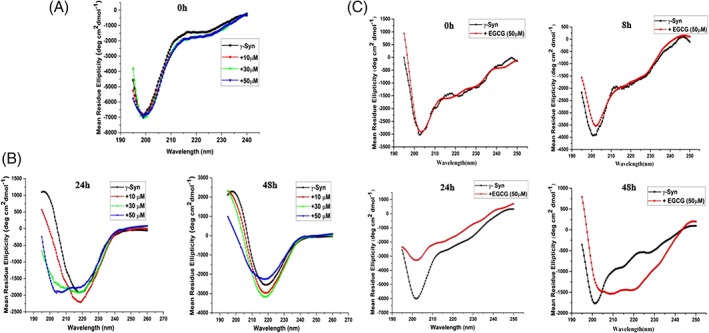
EGCG stabilizes the natively unfolded structure of γ‐Syn and imparts an α‐helical structure during fibrillation. (A) and (B) The far‐UV CD of γ‐Syn in the absence and presence of EGCG both under native and fibrillating conditions, respectively, were monitored at 25°C using a quartz cuvette of 1 mm path length. (A) Far‐UV CD spectra of γ‐Syn (0.5 mg/mL) dissolved in 20 mM phosphate buffer, pH ‐7.4 in the presence of increasing concentration of EGCG (10, 30, and 50 μM), monitored under native conditions. (B) Far‐UV CD spectra of γ‐Syn (0.3 mg/mL) in the presence of increasing concentrations of EGCG (10, 30, and 50 μM) monitored at time‐intervals of 24 h (left panel) and 48 h (right panel) during fibrillation showing a shift in the negative ellipticity from 200 nm to ~ 218 nm in the untreated γ‐Syn, depicting the formation of characteristic β‐sheet conformation. (C) Far‐UV CD spectra of γ‐Syn oligomers in the presence of EGCG (50 μM) during fibrillation (left to right: 0 h, 8 h, 24 h, and 48 h) were monitored at 25°C using a cuvette of 0.1 mm path length. Far‐UV CD spectra of EGCG‐treated oligomers at 48 h showing double minima at ~208 nm and ~220 nm indicating an α‐helical structure.
In order to further characterize the structure of γ‐Syn oligomers formed both in the absence and presence of EGCG, far‐UV CD measurements of the soluble oligomers obtained after centrifugation of the fibrillation samples were carried out as a function of time [Fig. 5(C)]. The CD spectra of soluble γ‐Syn oligomers measured at different time intervals, i.e., 0 h, 8 h, 24 h, and 48 h, showed spectra characteristic of a natively unfolded protein, with a negative peak at ~200 nm. The differences in the intensities of the negative ellipticity indicated slight perturbations in the structure. In contrast, the EGCG generated oligomers during aggregation were found to gain secondary structure showing a decrease in ellipticity at ~200 nm and appearance of negative ellipticity with two minima at 208 nm and 220 nm, indicating a propensity for α‐helical conformation. It is to be noted that the far‐UV CD spectra of the EGCG generated oligomers show slight deviations from a characteristic far‐UV CD spectra of a perfectly α‐helical peptide which could arise due to the combination of peaks contributed by other soluble aggregated forms of the species populated during fibrillation. The formation of an α‐helical structure by small molecule ligands has also been reported in Aβ‐peptide, that also reduces Aβ‐ oligomer toxicity.32 The increased tendency of γ‐Syn to form α‐helical structure in its amyloid forming region has been suggested to be an intrinsic property that can be induced under various conditions making it less prone to fibrillation compared to α‐Syn having high β‐sheet forming propensity in that region.33 Our results clearly demonstrate that the EGCG mediated γ‐Syn oligomerization occurs spontaneously and is relatively faster than γ‐Syn fibrillation alone which agrees with the theory of competitive binding.20, 34 In the light of this theory, we propose that EGCG competes out the self‐associating unfolded γ‐Syn monomers by binding preferentially with the unstructured oligomers of γ‐Syn, and thereby reducing the formation of β‐sheet containing species, and remodeling the pathway to form kinetically retarded α‐helix containing oligomeric species majorly divided into two higher‐order forms.
EGCG‐generated species maintain an overall low‐surface hydrophobicity
It is well established that formation of β‐sheet is a signature for amyloid fibrils35, 36, 37 and an increase in the hydrophobicity is one of the important physicochemical properties that is known to be involved in driving the formation of β‐sheet structures leading to the build‐up of toxic oligomeric species.37, 38, 39, 40 The extent of exposed hydrophobic surface area in γ‐Syn was assessed by ANS binding assay. ANS (1‐anilino‐8‐naphthalenesulfonic acid), a widely used solvent‐sensitive dye is known to fluoresce with a blue‐shifted emission upon binding with hydrophobic surroundings.38 The ANS fluorescence spectra of monomeric γ‐Syn in the presence of increasing concentration of EGCG overlapped with that of the fluorophore in buffer alone condition, with an emission around 525 nm [Fig. 6(A)]. This indicates the stabilization of the natively unfolded conformation of γ‐Syn by EGCG, as also observed in native far‐UV CD [Fig. 5(A)], corresponding to the low mean hydrophobicity of these intrinsically disordered proteins.41 The ANS fluorescence spectra measured at different time intervals (0 h, 24 h, and 48 h) during the course of fibrillation [Fig. 6(B)], showed a concomitant increase in the fluorescence intensity with a characteristic blue shifted emission (λ em at 480 nm) in the untreated γ‐Syn fibrils. The ANS emission spectra obtained in the EGCG‐treated γ‐Syn fibrils were also blue shifted but the increments in the intensity were much reduced at all the time‐points [Fig. 6(B)]. The increase in the ANS fluorescence intensity under fibrillating conditions indicates the formation of a molten‐globule like state, a prerequisite for intrinsically disordered proteins for fibril formation,42 that does not form in the presence of EGCG. These results thus demonstrate that the EGCG modulated species maintain an overall low surface hydrophobicity.
Figure 6.
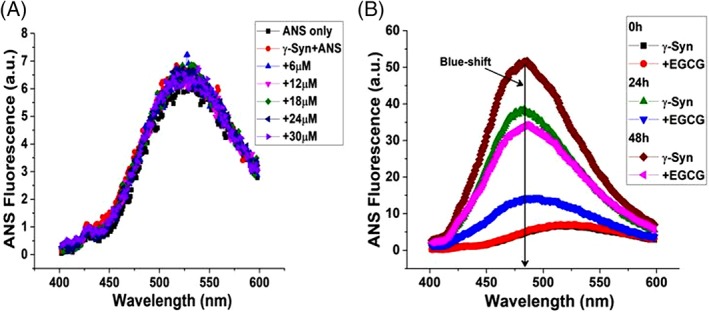
ANS binding to γ‐Syn in the presence of EGCG shows an overall low surface hydrophobicity. (A) Emission spectra of ANS binding to γ‐Syn with increasing concentrations of EGCG. (B) ANS binding to aggregating species of γ‐Syn in the absence and presence of EGCG with a characteristic blue‐shift shown at ~480 nm.
EGCG forms a weakly associated complex with γ‐Syn
To investigate the accessibility of EGCG to the lone tyrosine present in γ‐Syn, tyrosine fluorescence of γ‐Syn in the presence of increasing concentration of EGCG (2–30 μM) was monitored upon excitation at 275 nm with emission intensity recorded from 280 to 400 nm. The single tyrosine in γ‐Syn has an emission maximum at 342 nm due to tyrosinate formation upon excitation as also reported for other proteins containing single tyrosine in the absence of tryptophan.43, 44 A concentration‐dependent decrease in tyrosine fluorescence indicated an interaction between γ‐Syn and EGCG [Fig. 7(A)]. Using the Stern–Volmer quenching equation, the bimolecular quenching constant (k q) was found to be more than 1010 M−1 s−1 (k q = 9.6 × 1012 M−1 s−1) (Supporting Information Fig. S6) indicating a static quenching mechanism and a binding interaction between EGCG and γ‐Syn. Since the single tyrosine of γ‐Syn is located in the C‐terminal domain,5 the higher value of k q suggests a solvent exposed C‐terminal end of γ‐Syn in the presence of EGCG.
Figure 7.
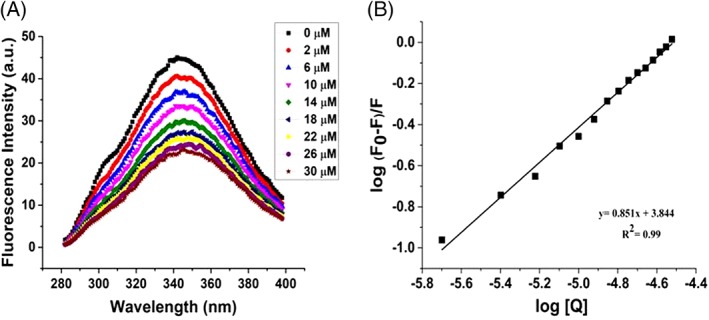
Steady‐state fluorescence shows the formation of weak binding complex between EGCG and γ‐Syn. (A) Steady‐state fluorescence showing quenching of intrinsic tyrosine fluorescence of γ‐Syn (0.3 mg/mL) continuously titrated with an increasing concentration of EGCG (2–30 μM) at 25°C. (B) Modified Stern–Volmer plot of log (F0–F)/F versus log [Q] showing weak binding interaction between γ‐Syn and EGCG (K a = 6.9 × 103M−1).
The binding parameters were evaluated by employing the modified Stern–Volmer equation which resulted in a linear dependence between fluorescence quenching intensity and EGCG concentration from a plot of log (Fo–F/F) versus log (Q)45 [Fig. 7(B)]. γ‐Syn was found to bind with EGCG at an approximately equimolar ratio of 1:1. An interaction with a binding constant (K a) of 6.9×103 M−1 was obtained at 25°C suggesting the role of weak non‐covalent interactions in EGCG‐mediated inhibition of γ‐Syn fibrillation. In addition, the binding of EGCG to the different species of γ‐Syn formed during fibrillation was carried out and the samples of γ‐Syn withdrawn at regular time intervals of 0 h, 24 h, and 48 h were similarly titrated with increasing concentrations of EGCG at 25°C. The modified Stern–Volmer plots showing the binding affinity (K a) of EGCG to different species of γ‐Syn is shown in Figure S7 and Table 1, respectively. EGCG was found to bind with highest affinity with the species formed during exponential phase of fibrillation (24 h) validating the increased affinity of EGCG to γ‐Syn oligomers as revealed by SEC.
Table 1.
Binding of γ‐Syn with Different Aggregating Species of γ‐Syn
| Time (h) | K a (Binding constant) |
|---|---|
| 0 | 1.4 × 104 M−1 |
| 24 | 5.2 ×105 M−1 |
| 48 | 5.1 × 103 M−1 |
EGCG binds to γ‐Syn by a static quenching mechanism and forms conformationally restrained oligomers
To further investigate the binding mechanism between EGCG and γ‐Syn, time resolved fluorescence measurements of the single tyrosine present in γ‐Syn were carried out in the presence of increasing concentration of EGCG (5–25 μM). The fluorescence intensity decay curves and the autocorrelation of the weighted residual mean used for deciding the goodness of the fit are shown in Figure 8(A) and (B), respectively. The decay curves both in the absence and in the presence of EGCG were appropriately described by three discrete decay components distributed into shorter lifetime (τ 1 < 0.5 ns), intermediate lifetime (τ 2 ~2 ns) and longer lifetime (τ 3~ 6 ns). The decay lifetimes observed in the presence of EGCG were found to be only marginally less than the average lifetime (τ m) of native γ‐Syn and the decay curves were indistinguishable from each other yielding the ratio of τ 0/τ ~1(Table 2). This indicates that the changes in the fluorescence intensity were dominantly due to the changes in the decay amplitudes suggesting the existence of static quenching mechanism and a complex formation between EGCG and γ‐Syn, an observation in accordance with the steady‐state fluorescence measurements.
Figure 8.
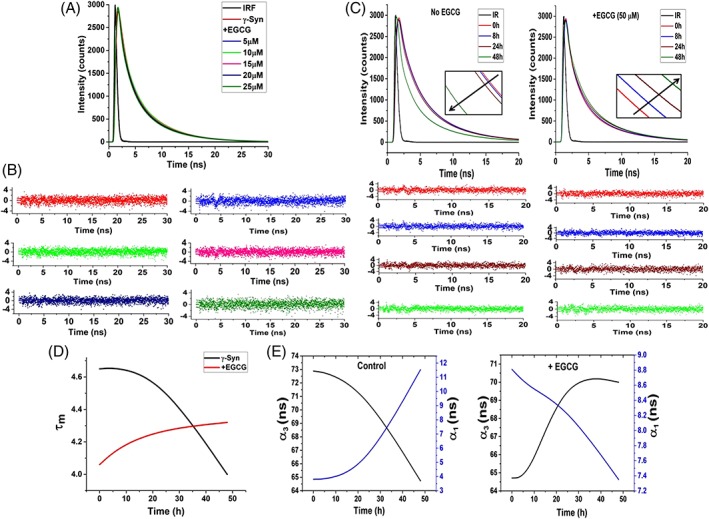
Time‐resolved fluorescence shows static‐quenching mechanism and indicates the formation of conformationally restrained oligomers by EGCG. (A) Time‐resolved fluorescence intensity decay of γ‐Syn under native conditions in the presence of increasing concentrations of EGCG and (B) plots of the autocorrelation function of the weighted residuals used to judge the goodness of the fit (left to right: no EGCG, 5, 10, 15, 20, and to 25 μM EGCG, respectively) . The overlapping decay curves obtained both in the absence and presence of increasing concentration of EGCG depicts a static quenching mechanism. (C) Lifetime decay curves of γ‐Syn oligomers formed at 0, 8, 24, and 48 h of fibrillation both in the absence (left panel) and presence of EGCG (right panel) along with their residuals below their respective decay curves are shown. The time‐dependent decrease in the fluorescence lifetime in the untreated γ‐Syn oligomers and the increase in the fluorescence lifetime in the EGCG‐generated oligomers depicts solvent exposed untreated γ‐Syn oligomers and conformationally restrained EGCG generated γ‐Syn oligomers, respectively. (Inset: shows the change in the fluorescence lifetimes at regular intervals during fibrillation). (D) The plot of average lifetime (τ m) versus the time of fibrillation clearly demonstrates the difference in the decay kinetics in the EGCG‐generated and untreated oligomers. (E) The relationship between the change in the amplitudes of the fastest (τ 1) and slowest (τ 3) time constants (α 1 and α 3, respectively) with respect to the increasing time of fibrillation in the control and EGCG‐treated samples is shown.
Table 2.
Lifetime Decay Parameters of γ‐Syn in the Presence of EGCG at 25°C
| γ‐Syn | τ 1 [ns](α1) | τ 2[ns](α2) | τ 3[ns](α3) | τ m[ns] | χ 2 |
|---|---|---|---|---|---|
| γ‐Syn (control) | 0.41 (4.29) | 2.00 (22.32) | 5.83 (73.38) | 4.74 | 1.04 |
| +5 μM EGCG | 0.35 (4.22) | 1.92 (23.08) | 5.82 (72.70) | 4.68 | 1.07 |
| +10 μM EGCG | 0.38 (4.59) | 1.93 (21.79) | 5.82 (73.61) | 4.72 | 1.09 |
| +15 μM EGCG | 0.36(4.84) | 1.91 (22.97) | 5.82 (72.19) | 4.66 | 1.03 |
| +20 μM EGCG | 0.30 (4.91) | 1.80 (23.06) | 5.77 (72.03) | 4.58 | 1.07 |
| +25 μM EGCG | 0.31 (5.62) | 1.83 (24.01) | 5.78 (70.37) | 4.52 | 1.08 |
The effect of aggregation on the fluorescence lifetime of the lone tyrosine present in γ‐Syn was also probed in order to gain insight into the conformational dynamics of γ‐Syn oligomers formed both in the absence and in the presence of EGCG. The samples of soluble oligomeric species of γ‐Syn characterized by far‐UV CD were also characterized by time‐resolved fluorescence spectroscopy and the differences in the lifetimes between the EGCG generated and native γ‐Syn oligomers were investigated. The fluorescence intensity decay curves at regular time intervals (0 h, 8 h, 24 h, and 48 h) and the autocorrelation of the weighted residual mean used for deciding the goodness of the fit are shown in Figure 8(C). Time‐resolved fluorescence measurements of γ‐Syn oligomers both in the absence and in the presence of EGCG at all the time intervals are appropriately described by three discrete decay components as also observed under native state. The results showing the amplitudes of respective lifetimes along with their mean lifetimes are summarized in Table 3. The fluorescence decay curves showing the time‐dependent changes in the fluorescence lifetimes in the untreated γ‐Syn oligomers and EGCG‐generated oligomers [Fig. 7(C)] demonstrate a time‐dependent increase in the fluorescence lifetime in the EGCG‐generated oligomers while in the untreated γ‐Syn oligomers a decreasing fluorescence lifetime with an increase in the time of fibrillation, evident during the exponential (24 h) and saturation phase (48 h) was observed. These results indicate an increase in the compaction of the overall structure in the EGCG‐generated oligomers during fibrillation. The faster decay of fluorescence lifetime in the untreated γ‐Syn oligomers indicate the increased dynamicity of the untreated γ‐Syn oligomers that become solvent exposed during fibrillation. The changes in the average lifetime with the increasing time of fibrillation in the EGCG untreated and treated γ‐Syn oligomers are clearly depicted in Figure 8(D). The increase in the amplitude (α 1) of the shortest lifetime (τ 1) and a concomitant decrease in the amplitude (α 3) of the longest lifetime (τ 3) in the untreated γ‐Syn oligomers and vice versa in the EGCG‐generated oligomers during fibrillation [Fig. 8(E)] clearly demonstrate that the unfolded state of the untreated γ‐Syn oligomers, which when formed in the presence of EGCG, becomes conformationally more restrained and structured upon prolonged incubation during fibrillation. These observations, thus, further support the results obtained from far‐UV CD of γ‐Syn oligomers [Fig. 5(C)]. The faster fluorescence decay in the EGCG‐generated oligomers during the initial stages of fibrillation suggest that EGCG perturbs the local conformations in the natively unfolded γ‐Syn to become more solvent exposed during early stages of fibrillation to facilitate the formation of conformationally restrained α‐helical structure.
Table 3.
Lifetime Decay Parameters of γ‐Syn Oligomers Formed in the Presence and Absence of EGCG at 25°C
| Time (h) | Sample | τ 1 [ns] (α1) | τ 2 [ns] (α2) | τ 3 [ns] (α3) | τ m [ns] | χ 2 |
|---|---|---|---|---|---|---|
| 0 | γ‐Syn | 0.30 (3.81) | 2.04 (23.31) | 5.72 (72.88) | 4.65 | 1.05 |
| +EGCG | 0.20 (8.81) | 1.63 (26.47) | 5.58 (64.71) | 4.06 | 1.06 | |
| 8 | γ‐Syn | 0.35 (3.82) | 2.08 (23.46) | 5.73 (72.72) | 4.66 | 1.04 |
| +EGCG | 0.23 (8.56) | 1.77 (26.69) | 5.71 (64.74) | 4.18 | 1.06 | |
| 24 | γ‐Syn | 0.25 (4.81) | 2.07 (24.03) | 5.71 (71.15) | 4.57 | 1.10 |
| +EGCG | 0.10 (8.33) | 1.59 (21.09) | 5.58 (70.58) | 4.28 | 1.10 | |
| 48 | γ‐Syn | 0.098 (11.54) | 1.58 (23.74) | 5.606 (64.73) | 4.00 | 1.07 |
| +EGCG | 0.09 (7.35) | 1.78 (23.44) | 5.64 (69.20) | 4.32 | 1.09 |
ITC reveals weak binding affinity between EGCG and γ‐Syn
The binding of EGCG to γ‐Syn was also measured using ITC (Fig. 9). The calorimetric study of the interaction of EGCG with γ‐Syn was carried out at 25°C with a 30‐fold molar excess ratio of EGCG over γ‐Syn. The representative ITC trace is displayed in Figure 9. The dissociation constant (Kd) obtained from the binding isotherm was of weak magnitude and in the mM range (K d = 2.19 mM), thereby substantiating the role of weak non‐covalent interactions in facilitating the binding of EGCG to γ‐Syn. Due to the weak nature of binding interactions, enthalpy and entropy values could not be accurately deduced. The association constant (K a) deduced from the modified Stern–Volmer plot [Fig. 7(B)] obtained from steady‐state fluorescence studies also revealed a weak binding affinity between EGCG and γ‐Syn, although estimating a ~10‐fold higher value than that observed by ITC. This is likely due to the nature of two techniques, whereas fluorescence relies on the single tyrosine probe in γ‐Syn and ITC is based on global heat change measurements. The IDPs have been reported to accomplish interaction with their targets by high specificity and low binding affinity, facilitating specific ligand binding interaction and playing important role in signal transductions.46, 47 The single‐site weak binding interaction of EGCG with γ‐Syn along with a strong suppression of fibrillation in the presence of 15‐fold less concentration of EGCG, highlights the role of weak binding interaction in EGCG‐mediated modulation of γ‐Syn fibrillation.
Figure 9.
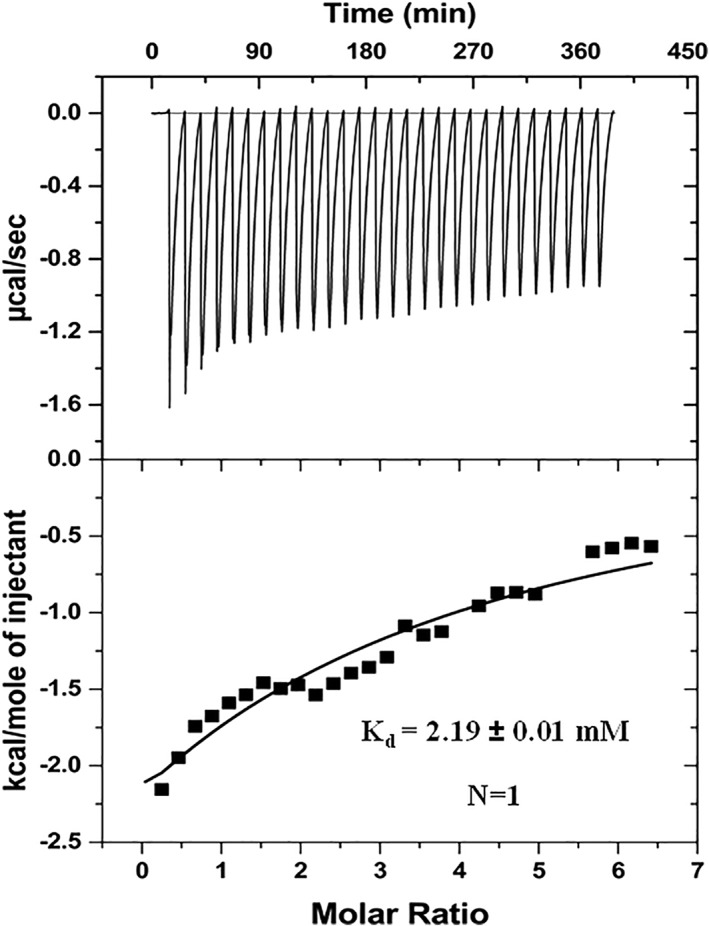
Isothermal titration calorimetry of EGCG interaction with γ‐Syn shows a weak binding interaction. The interaction was carried out at 25°C with a γ‐Syn and EGCG ratio of 1:30. Upper panel: a raw data plot of heat flow against time for titration of EGCG into γ‐Syn and lower panel: plot of total normalized heat released as a function of ligand concentration for the titration. The solid line shows the one‐site fit for the obtained data.
EGCG‐modulated species are differentially toxic to MCF‐7 and SH‐SY5Y cells
One of the important aspects of small molecule mediated modulation of fibrillation pathways is to investigate cytotoxic effects of the species formed as the oligomers and the protofibrillar intermediates formed during amyloidogenesis have previously been reported to be more toxic than the mature fibrils.2, 48, 49 Given the fact that γ‐Syn is involved in both cancer and neurodegeneration, our study compares the cytotoxic effects of EGCG‐generated γ‐Syn oligomers on breast cancer (MCF‐7) and neuroblastoma (SH‐SY5Y) cells, taken as representatives of the diverse diseases, respectively. By measuring the metabolic activity using MTT assay and the non‐viability of these cells by LDH‐cytotoxicity assay, we show that upon treatment of MCF‐7 cells with the protofibrillar and oligomeric species of γ‐Syn generated after 24 h of fibrillation, the MTT reduction was decreased by approximately 20% which also corresponded with reduced cell viability or increased cell death in MCF‐7 cells as revealed by an increased LDH activity [Fig. 10(A)]. On the other hand, the treatment of the cells with the EGCG generated oligomers was found to significantly increase the cell viability, almost completely rescuing the MCF‐7 cells from γ‐Syn toxicity [Fig. 10(A)].
Figure 10.
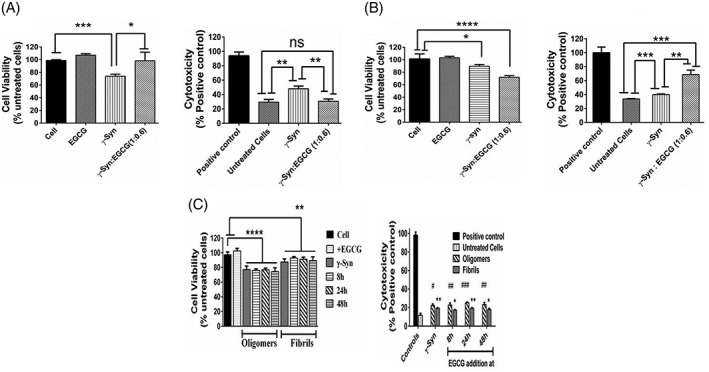
Cytotoxic effects of γ‐Syn. (A) and (B) Effect of EGCG‐generated γ‐Syn oligomers on MCF‐7 and SH‐SY5Y cells, respectively as studied by MTT assay (left panel) and LDH cytotoxicity assay (right panel). (A) MTT assay reveals an increased viability of MCF‐7 cells in the presence of EGCG‐generated oligomers (*P < 0.01 and *** P < 0.0005, related to the cells treated with γ‐Syn oligomers and with respect to the untreated cells, respectively). LDH assay shows an increased cytotoxicity in the presence of γ‐Syn oligomers (**P < 0.005 related to the untreated cells) which is completely rescued in the presence of EGCG‐generated oligomers (**P < 0.005 with respect to the γ‐Syn‐treated cells). On the other hand, (B) MTT assay with SH‐SY5Y cells show a significant reduction in cell viability in the presence of EGCG‐generated oligomers (****P < 0.0001, related to the untreated cells), which is only marginally reduced in the presence of untreated γ‐Syn oligomers (*P < 0.01 with respect to untreated cells). Similarly, LDH assay shows an increased cell death of SH‐SY5Y cells in presence of EGCG‐generated oligomers (***P < 0.0005 and ** P < 0.005 related to untreated cells and γ‐Syn‐treated cells, respectively). The statistical analysis was done using unpaired t‐test. (C) MTT assay (left panel) shows the reduction (****P < 0.0001) in the MTT absorbance with respect to the controls taken as untreated cells and indicate that the disaggregated oligomers are significantly toxic to cells, whereas the whole population of disaggregated fibrils formed upon fragmentation are less toxic (**P < 0.001) to the untreated cells. The error bars represent ±SD (n = 3). The statistical analysis was done using one‐way ANOVA. LDH assay (right panel) also reveals an increased cell death in the presence of EGCG‐mediated disaggregated oligomers as compared to disaggregated fibrils (# and * denote the significant difference between the disaggregated oligomers and disaggregated fibrils with respect to untreated cells, respectively). The statistical analysis was done using unpaired t‐test and the error bars represent ±SD (n = 3).
Figure 11.
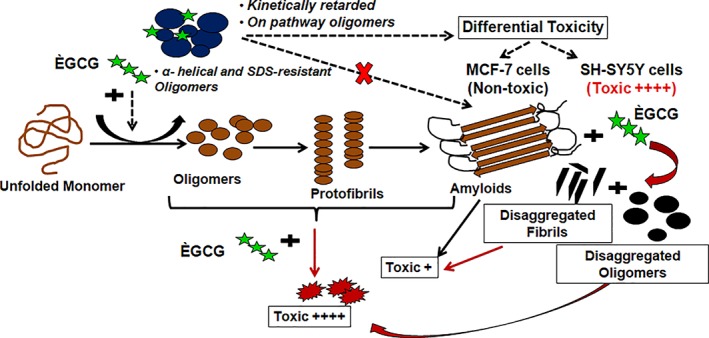
A schematic representation of the mechanism of EGCG‐mediated modulation of γ‐Syn fibrillation pathway. The figure illustrates the mechanism of EGCG‐mediated modulation of γ‐Syn fibrillation pathway. The black arrows (→) represent γ‐Syn fibrillation pathway in the absence of EGCG, the black dashed arrows (→) represent EGCG modulated γ‐Syn fibrillation pathway and the red arrows ( ) depict disaggregation pathway of γ‐Syn in the presence of EGCG. The cytotoxic effects of both EGCG untreated and treated γ‐Syn species are denoted by ++++ and +, for highly toxic and less toxic effects, respectively.
) depict disaggregation pathway of γ‐Syn in the presence of EGCG. The cytotoxic effects of both EGCG untreated and treated γ‐Syn species are denoted by ++++ and +, for highly toxic and less toxic effects, respectively.
A contrasting effect of γ‐Syn oligomers on the viability of neuronal cells (SH‐SY5Y) was observed. Unlike for of MCF‐7 cells, the γ‐Syn oligomers were found to be only moderately toxic to SH‐SY5Y cells [Fig. 10(B)], whereas the EGCG generated oligomers were found to increase the number of dead cells by almost 50% as compared to the untreated and γ‐Syn treated cells. The contrasting effects of the EGCG generated γ‐Syn oligomers on the MCF‐7 and SH‐SY5Y cells have also been additionally confirmed by trypan blue assay (Supporting Information Fig. S8). The differential toxicity points toward the cell‐specific behavior of γ‐Syn that could possibly play a critical role in the propagation of such diseases. We also speculate that the toxic effect of EGCG‐generated γ‐Syn oligomers on SH‐SY5Y cells could arise due to post‐translational modifications in γ‐Syn which has previously been reported to impart pathogenecity upon modification like oxidation by neurotransmitter dopamine (DA).8 This highlights the complexities in the use of EGCG and other polyphenols in the prevention of synucleopathies and needs further exploration.
Furthermore, the mature fibrils of several amyloidogenic proteins have also been known to disaggregate under several conditions such as prolonged incubation during fibrillation,50 changes in pH51 and chemical denaturants52 as also observed in the presence of EGCG (Figs. 2 and 3). These disaggregated oligomers have also been reported to possess toxic characteristics and promote underlying pathogenic conditions.50 In order to investigate the cytotoxic effect of EGCG‐generated disaggregated γ‐Syn species, the toxicity of the disaggregated oligomers and the whole population of disaggregated γ‐Syn fibrils containing various forms of γ‐Syn disaggregates were assessed separately on MCF‐7 cells. As the EGCG‐diverted γ‐Syn oligomers were found to be protective on MCF‐7 cells [Fig. 10(A)], it was interesting to compare the cytotoxic effects of the disaggregated oligomers on the same. To investigate this, the disaggregated γ‐Syn species formed after addition of EGCG (50 μM) at 8 h, 24 h, and 48 h (corresponding to lag, log, and saturation phase, respectively) of fibrillation, were withdrawn at the end of fibrillation (~72 h) and were separated into two groups wherein one group was centrifuged to obtain only the disaggregated oligomers and the other was used for the assay as such. The disaggregated oligomers at all the time intervals (8 h, 24 h, and 48 h) were found to be equally toxic to MCF‐7 cells as the untreated γ‐Syn oligomers, as revealed by both MTT and LDH assay [Fig. 10(C)]. On the other hand, the disaggregated fibrils were found to be comparatively less toxic than the γ‐Syn oligomers. This indicates an early gain of toxic characteristics by γ‐Syn which cannot be reversed upon fibril disaggregation by EGCG. Interestingly, the significant toxicity of the disaggregated oligomers observed against the partial toxicity of the fragmented fibrils [Fig. 10(C)] suggests potential permeation of the disaggregated oligomers into the cell membrane causing cell death as compared to the disaggregated fibrils, therefore, highlighting the critical role of the length and physical structure of the amyloid species that could possibly affect their cellular permeability and in turn govern their cytotoxicity. A relationship between the fibril length and the amyloid toxicity has also been reported previously for amyloidogenic proteins like β2m, lysozyme and α‐Syn where a reduction in the fibril length by fragmentation was associated with increased cytotoxicity.53 These results suggest that several factors such as stage of inhibition, morphology of aggregating species as well as the cell type together play a role in governing the toxicity of these species. This would, thus also have a strong bearing on transmission of toxic species from one cell to another.
Conclusion
This study gives us an insight into the modulating effects of EGCG on the fibrillation pathway of highly oligomer forming human γ‐Syn and demonstrates that EGCG significantly suppresses γ‐Syn fibrillation and also leads to fibril disaggregation by modulating the pathway to form two major populations of large‐sized, SDS‐ resistant oligomeric species. The formation of such oligomers, which are kinetically retarded on pathway species, possess an increased α‐helical propensity is responsible for inhibition of γ‐Syn fibrillation by EGCG. A detailed investigation on the differential toxicity of these oligomers on breast cancer (MCF‐7) and neuroblastoma (SH‐SY5Y) cells observed in this study may lead to better understanding of the role of γ‐Syn in disease etiology. Lastly, resultant toxic disaggregates including the disaggregated oligomers and fragmented fibrils indicate an early gain of toxicity by γ‐Syn which is not reversed by EGCG suggesting that modulators that could inhibit the early stages of γ‐Syn fibrillation without leading to disaggregation may prove to be potential candidates for preventing synucleopathies. The differences observed in the mechanism of EGCG‐mediated modulation of γ‐Syn fibrillation in this study from that previously reported for α‐Syn indicate a critical role of γ‐Syn in underlying pathogenesis and suggest that an in‐depth investigation on the effects of such modulators on γ‐Syn fibrillation will help to better understand the events leading to the onset of synucleopathies and help in the development of effective intervention strategies. A schematic representation of the mechanism of EGCG –mediated modulation of γ‐Syn fibrillation is summarized in Figure 1.
Materials and Methods
Materials
EGCG, thioflavin T and MTT were obtained from Sigma. All other chemicals were of analytical grade and were purchased from Sisco Research laboratory Pvt. Ltd. and Merck. A plasmid containing the full length human γ‐syn was obtained as a gift from Dr Peter T. Lansbury (Harvard Medical School, Cambridge, MA).
Purification of human γ‐Synuclein
Human wild‐type γ‐Synuclein was expressed in BL‐21 (DE3) cell line and was purified as already described.54 The purified protein instead of being lyophilized was resuspended in 10 mM phosphate buffer, pH 7.4 and stored at −20°C into small aliquots. Protein purity was determined both by SDS‐PAGE analysis and ESI‐MS analysis using a Synapt G2 HDMS instrument (Waters Corp., Manchester, UK) giving a molecular mass of 13,321 Da10 and the parameters used for the analysis are mentioned in methods (see Supplementary material 1). The ESI‐MS spectrum of purified γ‐syn is shown in Figure S1. The protein concentration was estimated using the molar extinction coefficient of 1400 M−1 cm−1 at 275 nm.
Fibril formation and ThT assays
γ‐Syn, Thioflavin T, and EGCG were dissolved in fibrillation buffer containing 20 mM phosphate, 100 mM NaCl, 0.02% NaN3 at pH ‐7.4. γ‐Syn (1 mg/ml; 75 μM) was mixed with EGCG making the final concentrations (5 ‐ 50μM) in fibrillation buffer containing 20 μM ThT and were incubated at 37°C with shaking at 200 rpm. ThT fluorescence at regular intervals was recorded at 480 nm with an excitation of 445 nm keeping both excitation and emission slit widths of 5 nm in a Cary Eclipse Fluorescence Spectrophotometer. A control sample without EGCG was also prepared. Concentration of ThT and EGCG were measured using molar extinction coefficient value of 35,000 M−1 cm−1 at 412 nm and 11,920 M−1 cm−1 at 270 nm, respectively. Each sample was run in triplicates and the data from the triplicates were averaged before plotting ThT fluorescence with respect to time. The apparent fibrillation rate and the lag time of fibrillation were calculated by fitting the fibrillation kinetics of γ‐Syn into a sigmoidal curve using the empirical formula reported previously.55
Time‐dependent addition of EGCG and disaggregation assay
EGCG (50 μM) was added at 8 h, 24 h, and 48 h during the course of γ‐Syn fibrillation and the ThT was monitored at regular intervals as mentioned above. To negate the dilution effects on fibrillation, buffer of same volume as that of EGCG was added into the control samples.
Seeding experiment
The seeds were prepared by incubating monomeric γ‐Syn (75 μM) for 2 days at 37°C, 200 rpm both in the absence and presence of EGCG (30 μM). The unbound EGCG and soluble aggregates were removed by centrifugation at 14,000 g for 30 min and washed twice with the buffer. The seeds were resuspended in the 20 mM phosphate buffer with 100 mM NaCl, pH 7.4, and the various seed concentrations (% v/v) were added into the monomeric γ‐Syn solution containing 20 μM ThT. The fibrillation kinetics was monitored by recording ThT fluorescence at 480 nm with an excitation at 445 nm in a 96‐well plate using a Varioskan microplate reader (Thermofischer) at a regular interval of 30 min at 280 rpm and diameter of 8 mm.
Electron microscopy measurements
TEM images were collected using a JEOL TEM 2100 microscope operating with an accelerating voltage of 200 kV. Samples were deposited on carbon coated 200 mesh copper grid and negatively stained with 1% uranyl acetate. Human γ‐syn after fibrillation were placed on the carbon coated grid and allowed to dry in the air for overnight. The grid was then negatively stained by adding a drop of 1% uranyl acetate and blotted with a filter paper after 10 s and allowed to dry in air for next 30 min and the samples examined using TEM with images captured using a CCD camera.
Atomic force microscopy
Freshly cleaved mica sheet was glued on a microscopic slide and the mica surface was exfoliated twice before the addition of the sample onto the mica surface. About 10 μl of sample was adsorbed on the mica surface and was allowed to dry for 20 min. The unbound sample and remaining salts were further removed by gently washing the mica surface with de‐ionized water and again allowed to dry overnight. The AFM measurements were made on a WiTech Alpha 300 RA instrument (WITECH Instrument Co., Ltd., Seoul, Korea) in a non‐contact mode. Cantilevers with resonance frequency of 75 kHz and force constant of 40 N/m were used.
Circular dichroism spectroscopy
Far‐UV CD spectra were collected using a JASCO J‐815 CD spectrophotometer equipped with a peltier device. The spectra were collected using a 1 mm path length cuvette from a wavelength of 195–260 nm at a step size of 0.1 nm, bandwidth of 1 nm, and scanning speed 20 nm/min at 25°C. Total of three accumulations were collected. The far‐UV CD spectra of γ‐Syn (0.5 mg/ml) under native conditions in the presence of increasing concentration of EGCG (10, 30, and 50 μM) was carried out in 20 mM phosphate buffer (without salt), pH ‐7.4. The fibrillation reactions were set up in triplicates where γ‐Syn (1 mg/ml) was incubated with varying concentrations of EGCG (10, 30, and 50 μM) in 20 mM phosphate buffer containing 100 mM NaCl, pH ‐7.4. For analysis, the incubation samples were dissolved in a 20 mM phosphate buffer with no salt, bringing the final concentration of γ‐Syn to 0.3 mg/mL and reducing NaCl concentration to 33.3 mM. The background spectra of each EGCG concentration and the buffer were simultaneously subtracted. The data were plotted in mean residue ellipticity units expressed as degree cm2 dmol−1. To monitor the far‐UV CD spectra of the γ‐Syn oligomers, the fibrillation samples withdrawn after 48 h of fibrillation were centrifuged at 14,000 g for 30 min to remove the insoluble aggregates. The far‐UV scans were measured using a cuvette of 0.1 mm path length to reduce interference by 100 mM NaCl present in the fibrillation buffer and the data was normalized with reference to the initial concentration.
ANS binding assay
The incubation samples were withdrawn at different time intervals and ANS spectra was recorded from 400 to 600 nm with an excitation at 375 nm at 25°C and slit widths of 5 nm were used for both excitation and emission. The samples withdrawn were diluted in fibrillation buffer making a final concentration of 0.3 mg/ml keeping final ratio of ANS to protein concentration equal to 5.
Size‐exclusion chromatography
Aliquot of γ‐Syn in the presence of EGCG (50 μM) was withdrawn at different time intervals and the larger aggregates or the insoluble material was removed by centrifugation for 30 min at 14,000 g. For disaggregation studies, the monomeric solution of γ‐Syn (75 μM) was incubated with EGCG (50 μM) added at three different time intervals 8 h, 24 h, and 48 h marking the three stages of fibrillation respectively and the soluble aggregates were collected by centrifugation as mentioned above. The supernatant was further filtered through 0.22 μm filter and 20 μL of supernatant was eluted through BioSep SEC‐S 2000 column (300 × 7.8 mm), from Phenomenex in 20 mM phosphate buffer containing 0.1 M NaCl, pH ‐7.4 using a Waters 2489 UV/vis separations module attached with Waters 515 HPLC pumps. The HPLC system was operated and the data was collected and analyzed by Empower software. The column was eluted at a flow rate of 0.5 mL/min at 25°C and the absorbance of the mobile phase was measured at 275 and 270 nm corresponding to absorbance of γ‐Syn and EGCG, respectively. The column was calibrated using the following standards obtained from Bio‐Rad: thyroglobulin (670 kDa), γ‐globulin (158 kDa), ovalbumin (44 kDa), myoglobin (17 kDa), and vitamin B12 (1.3 kDa). The hydrodynamic properties of the species were calculated using the empirical formula as previously reported.5 The monomer and oligomers formed during the fibrillation was quantified by calculating area under peak using Origin software.
SDS and NATIVE polyacrylamide gel electrophoresis
γ‐Syn samples treated with various concentrations of EGCG (30, 40, and 50 μM) at time points 0 h, 24 h, and 48 h as well as the disaggregation samples were withdrawn and prepared similarly as for size‐exclusion studies. About 10 μL of 5× sample buffer (250 mM Tris, 5% β‐mercaptoethanol, 10% SDS, 50% glycerol, and 0.5% bromophenol blue) was added to supernatant and were boiled at 95°C for 10 min. About 14 μL aliquot was loaded onto a 15% SDS‐polyacrylamide gel. The samples for native‐PAGE analysis were prepared as mentioned above except the sample buffer used was devoid of SDS and β‐mercaptoethanol and also the sample were not subjected to boiling. A 10% native‐gel was prepared for the analysis. Electrophoresis was carried out in 0.75 mm‐thick plates at 80 V and stained with Coomassie Brilliant Blue G. The molecular weight of the protein was determined by using a broad range (6.5–210 kD) protein standard (Bio‐Rad, CA, USA).
Steady‐state fluorescence
The binding of EGCG to γ‐Syn (22.5 μM) was studied by continuously titrating γ‐Syn with an increasing concentration of EGCG (2–30 μM) at 37°C and the binding was ascertained by the quenching of tyrosine fluorescence upon titration with EGCG. The association constant K a was calculated using the modified Stern–Volmer quenching equation:
| (1) |
| (2) |
where K sv is Stern–Volmer constant for static quenching, F o and F is the fluorescence intensity in the absence and in the presence of quencher, respectively, K a is the association constant, n is the number of binding sites, and Q denotes the quencher concentration.56
Time‐resolved fluorescence and data analysis
Time‐resolved fluorescence intensity was measured by the time‐correlated single photon counting (TCSPC) method using a model F900 fluorescence lifetime spectrometer (Edinburgh Instruments, UK). The lifetime of the single tyrosine present in γ‐Syn was recorded with 75 μM γ‐Syn in the presence of increasing concentrations of EGCG (5–25 μM), excited at 284 nm wavelength using a Nano LED pulsed laser. The emission was recorded at emission maxima (λ max) of 342 nm as determined from the steady‐state fluorescence spectra. The instrument response factor (IRF) was typically 1.5 ns (fwhm) as recorded by scattering a dilute suspension of colloidal silica (Ludox). To determine the decay constants a peak count of 3000 was collected for total decay duration of 50 ns. The data were recorded into 4096 channels at a timing resolution of 0.012 ns/channel.
Data analysis and curve fitting
The resulting data were analyzed by using the FAST software provided with the instrument. Non‐linear least squares method was employed which assumes the fluorescence decays to be the sum of discrete exponential components expressed by the equation as reported57, 58, 59
| (3) |
where n is the number of discrete exponentials, i is the index, and α i and τ i are the amplitudes and lifetimes, respectively. The average of fluorescence lifetime was calculated as
| (4) |
The goodness of the fit was decided by the reduced χ 2 and from the randomness of the weighted residuals distributions.
ITC
The ITC experiments for defining the thermodynamic parameters of EGCG binding to γ‐Syn were carried out on VP‐ITC model from Microcal at 25°C, pH ‐7.4. The γ‐Syn sample was centrifuged at 14,000 g for 20 min to remove any high order suspended particles and the supernatant was filtered through 0.22 μm filter. All the samples before loading into the calorimeter cell were degassed for 10 min using the Thermo Vac degassing unit supplied by Microcal to ensure that no bubbles were formed. Sample cell containing γ‐Syn (0.22 mM) was continuously titrated with 9 μL of EGCG (6.6 mM) with a uniform interval of 800 s with a rotating syringe at 329 rpm. Corresponding control experiments for determining the heat of dilution of EGCG was performed by titrating EGCG into the buffer under identical conditions. The heat associated with the EGCG‐buffer reaction was subtracted from EGCG‐ γ‐Syn reaction to obtain the actual heat of interaction. The data were analyzed using Origin7 software provided by Microcal.
MTT cytotoxicity assay
The γ‐Syn fibrils were formed by incubating γ‐Syn (75 μM) in the absence and the presence of EGCG (50 μM) in fibrillation buffer at 37°C with shaking at 200 rpm. For estimating the toxic effects of disaggregated oligomers, the fibrillation reaction was set as described in materials and methods. At the end of the fibrillation reaction, the samples were divided into two groups where one group was centrifuged at 14,000 g for 30 min to remove the insoluble aggregates, leaving only the soluble disaggregated oligomers and another was used as such without centrifugation containing the whole of soluble and insoluble disaggregated species. The buffer without protein and with or without EGCG was maintained as controls. The incubation samples were withdrawn at 24 h of incubation for assessing the toxicity of EGCG‐generated oligomers and at the end of fibrillation for assessing toxicity of disaggregated oligomers as well as fibrils formed at different stages upon EGG addition. A 7‐fold dilution of the incubation samples were applied to human breast cancer cell line (MCF‐7) and neuroblastoma cell line (SH‐SY5Y). The cells were cultured in DMEM but for growing SH‐SY5Y, the media was supplemented with 10% l‐glutamine. Cells were plated at a density of 20,000 cells/well on 96‐well plates in 200 μL of media. The cells were grown for 24 h at 37°C to allow the cells to reach the exponential phase. After 24 h the protein aggregates were added and the cells were kept for incubation for 2 days at 37°C. Cytotoxicity was measured by using 3‐[4, 5‐dimethylthiazol‐2‐yl]‐2, 5‐diphenyltetra‐zolium bromide (MTT) toxicity assay. About 20 μL of MTT (5 mg/ml) was added to each well containing 200 μL of media. MTT addition was done under dark and the plates after MTT addition were incubated for 4 h in the incubator. After completion of 4 h, the MTT crystals formed were dissolved using 200 μL of 100% DMSO and the readings were immediately recorded at 570 nm and background was corrected for 650 nm. The assays were performed in three independent reactions and statistical analysis was done using unpaired t‐test for the assessing the toxicity of EGCG‐generated oligomers and for assessing the toxicity of disaggregated species, one‐way ANOVA was employed.
LDH cytotoxicity assay
The LDH cytotoxicity assay measures the non‐viable or dead cells by monitoring the extent of release of cytoplasmic LDH into the culture medium upon cell death due to damage of the plasma membrane. An increase in absorbance of a red formazan product at 490 nm directly indicates an increased LDH activity which in turn corresponds to the number of lysed cells. For LDH assay the fibrils were formed by incubating γ‐Syn (150 μM) in the absence and presence of EGCG (100 μM), keeping the ratio of γ‐Syn: EGCG (1:0.6) as used in MTT‐assay and the incubation samples for the assay were then similarly prepared as for the MTT assay. MCF‐7 and SH‐SY5Y cells were plated out at a density of 50,000 cells per well in a 200 μL of media in a 96‐well plate. The cytotoxicity of γ‐Syn species formed in the absence and presence of EGCG was quantified using Pierce™ LDH Cytotoxicity Assay Kit (Thermo Fischer Scientific) and the % cytotoxicity was calculated using the manufacturer's protocol.
Supplementary materials
Supplementary methods: ESI‐MS analysis of purified γ‐Syn, Extrinsic ThT assay with centrifuged γ‐Syn fibrils, Rayleigh scattering, Seeding experiment, Trypan Blue assay for cell viability.
Supplementary figures: Mass spectrometry analysis of purified γ‐Syn by ESI‐MS, extrinsic ThT assay of γ‐Syn in the presence and absence of EGCG (50 μM), calibration curve of protein standards of known molecular weights obtained by size‐exclusion chromatography, Rayleigh scattering of γ‐Syn species formed in the absence and presence of EGCG during fibrillation, effect of EGCG‐generated seeds on γ‐Syn fibrillation pathway, Stern–Volmer quenching plot for the evaluation of bimolecular quenching constant (k q) of EGCG binding to γ‐Syn at 25°C, modified Stern–Volmer plots showing binding affinities of EGCG to different aggregating species of γ‐Syn, cytotoxic effects of γ‐Syn on MCF‐7 and SH‐SY5Y cells studied by trypan blue assay.
Supporting information
Appendix S1: Supporting Information
Acknowledgments
The authors would like to thank Dr Peter T. Lansbury (Harvard Medical School, Cambridge, MA) for the clone. The authors would like to acknowledge Dr Gajender Saini, Saroj K. Jha, and Dr Neetu Singh of AIRF, JNU, for technical assistance of TEM, AFM, and TCSPC experiments, respectively. We sincerely extend our gratitude to Prof. Sobhan Sen of School of Physical Sciences, JNU for helping with the analysis of the TCSPC data. We also thank DBT‐BUILDER project (BT/PR/5006/INF/2012) for the instrumentation facility (HPLC, DLS, and Varioskan plate reader). We further thank UPE‐II and DST‐PURSE grants (DST/SR/PURSE II/11) for the consumables. SR would like to thank Prof. Vladimir Uversky for fruitful discussion on the work. SR further thanks CSIR, Govt. of India for the research fellowship.
Short statement: Oligomerization of neural protein γ‐Synuclein that is involved in both neurodegeneration and cancer and coexists with the fibrillar plaques of α‐Synuclein, is a hallmark in Parkinson's disease. We show that the polyphenol EGCG significantly suppresses γ‐Syn fibrillation and modulates the pathway to form α‐helical containing higher‐order oligomers that are differentially toxic for neuronal and breast cancer cells. Investigating the effect of such small molecule modulators on the fibrillation pathway of γ‐Syn is, therefore, critical for understanding synucleopathies.
References
- 1. Glabe CG (2006) Common mechanisms of amyloid oligomer pathogenesis in degenerative disease. Neurobiol Aging 27:570–575. [DOI] [PubMed] [Google Scholar]
- 2. Ferreira ST, Vieira MN, De Felice FG (2007) Soluble protein oligomers as emerging toxins in Alzheimer's and other amyloid diseases. IUBMB Life 59:332–345. [DOI] [PubMed] [Google Scholar]
- 3. Danzer KM, Kranich LR, Ruf WP, Cagsal‐Getkin O, Winslow AR, Zhu L, Vanderburg CR, McLean PJ (2012) Exosomal cell‐to‐cell transmission of alpha synuclein oligomers. Mol Neurodegener 7:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Volles MJ, Lee S‐J, Rochet J‐C, Shtilerman MD, Ding TT, Kessler JC, Lansbury PT (2001) Vesicle permeabilization by protofibrillar α‐synuclein: implications for the pathogenesis and treatment of Parkinson's disease. Biochemistry 40:7812–7819. [DOI] [PubMed] [Google Scholar]
- 5. Uversky VN, Li J, Souillac P, Millett IS, Doniach S, Jakes R, Goedert M, Fink AL (2002) Biophysical properties of the synucleins and their propensities to fibrillate: inhibition of α‐synuclein assembly by β‐ and γ‐synucleins. J Biol Chem 277:11970–11978. [DOI] [PubMed] [Google Scholar]
- 6. Ninkina N, Peters O, Millership S, Salem H, Van der Putten H, Buchman VL (2009) γ‐Synucleinopathy: neurodegeneration associated with overexpression of the mouse protein. Hum Mol Genet 18:1779–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ahmad M, Attoub S, Singh MN, Martin FL, El‐Agnaf OM (2007) γ‐Synuclein and the progression of cancer. FASEB J 21:3419–3430. [DOI] [PubMed] [Google Scholar]
- 8. Surgucheva I, Sharov VS, Surguchov A (2012) γ‐Synuclein: seeding of α‐synuclein aggregation and transmission between cells. Biochemistry 51:4743–4754. [DOI] [PubMed] [Google Scholar]
- 9. Galvin JE, Uryu K, Lee VM‐Y, Trojanowski JQ (1999) Axon pathology in Parkinson's disease and Lewy body dementia hippocampus contains α‐, β‐, and γ‐synuclein. Proc Natl Acad Sci USA 96:13450–13455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Golebiewska U, Zurawsky C, Scarlata S (2014) Defining the oligomerization state of γ‐synuclein in solution and in cells. Biochemistry 53:293–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hibi T, Mori T, Fukuma M, Yamazaki K, Hashiguchi A, Yamada T, Tanabe M, Aiura K, Kawakami T, Ogiwara A (2009) Synuclein‐γ is closely involved in perineural invasion and distant metastasis in mouse models and is a novel prognostic factor in pancreatic cancer. Clin Cancer Res 15:2864–2871. [DOI] [PubMed] [Google Scholar]
- 12. Jiang Y, Liu YE, Goldberg ID, Shi YE (2004) γ Synuclein, a novel heat‐shock protein‐associated chaperone, stimulates ligand‐dependent estrogen receptor α signaling and mammary tumorigenesis. Cancer Res 64:4539–4546. [DOI] [PubMed] [Google Scholar]
- 13. Zhang H, Kouadio A, Cartledge D, Godwin AK (2011) Role of gamma‐synuclein in microtubule regulation. Exp Cell Res 317:1330–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jain MK, Bhat R (2014) Modulation of human α‐synuclein aggregation by a combined effect of calcium and dopamine. Neurobiol Dis 63:115–128. [DOI] [PubMed] [Google Scholar]
- 15. Findeis MA, Musso GM, Arico‐Muendel CC, Benjamin HW, Hundal AM, Lee J‐J, Chin J, Kelley M, Wakefield J, Hayward NJ (1999) Modified‐peptide inhibitors of amyloid β‐peptide polymerization. Biochemistry 38:6791–6800. [DOI] [PubMed] [Google Scholar]
- 16. Ngoungoure VLN, Schluesener J, Moundipa PF, Schluesener H (2015) Natural polyphenols binding to amyloid: a broad class of compounds to treat different human amyloid diseases. Mol Nutr Food Res 59:8–20. [DOI] [PubMed] [Google Scholar]
- 17. Roberts BE, Shorter J (2008) Escaping amyloid fate. Nat Struct Mol Biol 15:544–546. [DOI] [PubMed] [Google Scholar]
- 18. Azam S, Hadi N, Khan NU, Hadi SM (2004) Prooxidant property of green tea polyphenols epicatechin and epigallocatechin‐3‐gallate: implications for anticancer properties. Toxicol In Vitro 18:555–561. [DOI] [PubMed] [Google Scholar]
- 19. Ehrnhoefer DE, Duennwald M, Markovic P, Wacker JL, Engemann S, Roark M, Legleiter J, Marsh JL, Thompson LM, Lindquist S (2006) Green tea (−)‐epigallocatechin‐gallate modulates early events in huntingtin misfolding and reduces toxicity in Huntington's disease models. Hum Mol Genet 15:2743–2751. [DOI] [PubMed] [Google Scholar]
- 20. Ehrnhoefer DE, Bieschke J, Boeddrich A, Herbst M, Masino L, Lurz R, Engemann S, Pastore A, Wanker EE (2008) EGCG redirects amyloidogenic polypeptides into unstructured, off‐pathway oligomers. Nat Struct Mol Biol 15:558–566. [DOI] [PubMed] [Google Scholar]
- 21. Bieschke J, Russ J, Friedrich RP, Ehrnhoefer DE, Wobst H, Neugebauer K, Wanker EE (2010) EGCG remodels mature α‐synuclein and amyloid‐β fibrils and reduces cellular toxicity. Proc Natl Acad Sci USA 107:7710–7715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Palhano FL, Lee J, Grimster NP, Kelly JW (2013) Toward the molecular mechanism (s) by which EGCG treatment remodels mature amyloid fibrils. J Am Chem Soc 135:7503–7510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ishii T, Mori T, Tanaka T, Mizuno D, Yamaji R, Kumazawa S, Nakayama T, Akagawa M (2008) Covalent modification of proteins by green tea polyphenol (−)‐epigallocatechin‐3‐gallate through autoxidation. Free Radicals Biol Med 45:1384–1394. [DOI] [PubMed] [Google Scholar]
- 24. Ban T, Hamada D, Hasegawa K, Naiki H, Goto Y (2003) Direct observation of amyloid fibril growth monitored by thioflavin T fluorescence. J Biol Chem 278:16462–16465. [DOI] [PubMed] [Google Scholar]
- 25. Necula M, Kayed R, Milton S, Glabe CG (2007) Small molecule inhibitors of aggregation indicate that amyloid β oligomerization and fibrillization pathways are independent and distinct. J Biol Chem 282:10311–10324. [DOI] [PubMed] [Google Scholar]
- 26. Pervin M, Unno K, Nakagawa A, Takahashi Y, Iguchi K, Yamamoto H, Hoshino M, Hara A, Takagaki A, Nanjo F (2017) Blood brain barrier permeability of (−)‐epigallocatechin gallate, its proliferation‐enhancing activity of human neuroblastoma SH‐SY5Y cells, and its preventive effect on age‐related cognitive dysfunction in mice. Biochem Biophys Rep 9:180–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nakagawa K, Miyazawa T (1997) Chemiluminescence–high‐performance liquid chromatographic determination of tea catechin, (−)‐epigallocatechin 3‐gallate, at picomole levels in rat and human plasma. Anal Biochem 248:41–49. [DOI] [PubMed] [Google Scholar]
- 28. Yates AA, Erdman JW Jr, Shao A, Dolan LC, Griffiths JC (2017) Bioactive nutrients‐time for tolerable upper intake levels to address safety. Regul Toxicol Pharmacol 84:94–101. [DOI] [PubMed] [Google Scholar]
- 29. Zhu M, Rajamani S, Kaylor J, Han S, Zhou F, Fink AL (2004) The flavonoid baicalein inhibits fibrillation of α‐synuclein and disaggregates existing fibrils. J Biol Chem 279:26846–26857. [DOI] [PubMed] [Google Scholar]
- 30. Stenvang M, Dueholm MS, Vad BS, Seviour T, Zeng G, Geifman‐Shochat S, Søndergaard MT, Christiansen G, Meyer RL, Kjelleberg S (2016) Epigallocatechin gallate remodels overexpressed functional amyloids in Pseudomonas aeruginosa and increases biofilm susceptibility to antibiotic treatment. J Biol Chem 291:26540–26553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kundel F, De S, Flagmeier P, Horrocks MH, Kjaergaard M, Shammas SL, Jackson SE, Dobson CM, Klenerman D (2018) Hsp70 inhibits the nucleation and elongation of tau and sequesters tau aggregates with high affinity. ACS Chem Biol 13:636–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nerelius C, Sandegren A, Sargsyan H, Raunak R, Leijonmarck H, Chatterjee U, Fisahn A, Imarisio S, Lomas D, Crowther D (2009) α‐Helix targeting reduces amyloid‐β peptide toxicity. Proc Natl Acad Sci USA 106:9191–9196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Marsh JA, Singh VK, Jia Z, Forman‐Kay JD (2006) Sensitivity of secondary structure propensities to sequence differences between α‐and γ‐synuclein: Implications for fibrillation. Protein Sci 15:2795–2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gosal WS, Morten IJ, Hewitt EW, Smith DA, Thomson NH, Radford SE (2005) Competing pathways determine fibril morphology in the self‐assembly of β 2‐microglobulin into amyloid. J Mol Biol 351:850–864. [DOI] [PubMed] [Google Scholar]
- 35. Klunk WE, Pettegrew J, Abraham DJ (1989) Quantitative evaluation of Congo red binding to amyloid‐like proteins with a beta‐pleated sheet conformation. J Histochem Cytochem 37:1273–1281. [DOI] [PubMed] [Google Scholar]
- 36. Simmons LK, May PC, Tomaselli KJ, Rydel RE, Fuson KS, Brigham EF, Wright S, Lieberburg I, Becker GW, Brems DN (1994) Secondary structure of amyloid beta peptide correlates with neurotoxic activity in vitro. Mol Pharmacol 45:373–379. [PubMed] [Google Scholar]
- 37. Chiti F, Taddei N, Baroni F, Capanni C, Stefani M, Ramponi G, Dobson CM (2002) Kinetic partitioning of protein folding and aggregation. Nat Struct Mol Biol 9:137–143. [DOI] [PubMed] [Google Scholar]
- 38. Bolognesi B, Kumita JR, Barros TP, Esbjorner EK, Luheshi LM, Crowther DC, Wilson MR, Dobson CM, Favrin G, Yerbury JJ (2010) ANS binding reveals common features of cytotoxic amyloid species. ACS Chem Biol 5:735–740. [DOI] [PubMed] [Google Scholar]
- 39. Pawar AP, DuBay KF, Zurdo J, Chiti F, Vendruscolo M, Dobson CM (2005) Prediction of “aggregation‐prone” and “aggregation‐susceptible” regions in proteins associated with neurodegenerative diseases. J Mol Biol 350:379–392. [DOI] [PubMed] [Google Scholar]
- 40. Campioni S, Mannini B, Zampagni M, Pensalfini A, Parrini C, Evangelisti E, Relini A, Stefani M, Dobson CM, Cecchi C (2010) A causative" link between the structure of aberrant protein oligomers and their toxicity. Nat Chem Biol 6:140–147. [DOI] [PubMed] [Google Scholar]
- 41. Uversky VN (2011) Intrinsically disordered proteins from A to Z. Int J Biochem Cell Biol 43:1090–1103. [DOI] [PubMed] [Google Scholar]
- 42. Uversky VN (2008) Amyloidogenesis of natively unfolded proteins. Curr Alzheimer Res 5:260–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Libertini LJ, Small EW (1985) The intrinsic tyrosine fluorescence of histone H1. Steady state and fluorescence decay studies reveal heterogeneous emission. Biophys J 47:765–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Szabo A, Lynn K, Krajcarski D, Rayner D (1978) Tyrosinate fluorescence maxima at 345 nm in proteins lacking tryptophan at pH 7. FEBS Lett 94:249–252. [DOI] [PubMed] [Google Scholar]
- 45. Ruan K, Li J, Liang R, Xu C, Yu Y, Lange R, Balny C (2002) A rare protein fluorescence behavior where the emission is dominated by tyrosine: case of the 33‐kDa protein from spinach photosystem II. Biochem Biophys Res Commun 293:593–597. [DOI] [PubMed] [Google Scholar]
- 46. Babu MM (2016) The contribution of intrinsically disordered regions to protein function, cellular complexity, and human disease. Biochem Soc Trans 44:1185–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mollica L, Bessa LM, Hanoulle X, Jensen MR, Blackledge M, Schneider R (2016) Binding mechanisms of intrinsically disordered proteins: theory, simulation, and experiment. Front Mol Biosci 3:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Caughey B, Lansbury PT Jr (2003) Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu Rev Neurosci 26:267–298. [DOI] [PubMed] [Google Scholar]
- 49. Bucciantini M, Giannoni E, Chiti F, Baroni F, Formigli L, Zurdo J, Taddei N, Ramponi G, Dobson CM, Stefani M (2002) Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 416:507–511. [DOI] [PubMed] [Google Scholar]
- 50. Cremades N, Cohen SI, Deas E, Abramov AY, Chen AY, Orte A, Sandal M, Clarke RW, Dunne P, Aprile FA (2012) Direct observation of the interconversion of normal and toxic forms of α‐synuclein. Cell 149:1048–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Picotti P, De Franceschi G, Frare E, Spolaore B, Zambonin M, Chiti F, de Laureto PP, Fontana A (2007) Amyloid fibril formation and disaggregation of fragment 1‐29 of apomyoglobin: insights into the effect of pH on protein fibrillogenesis. J Mol Biol 367:1237–1245. [DOI] [PubMed] [Google Scholar]
- 52. Calamai M, Canale C, Relini A, Stefani M, Chiti F, Dobson CM (2005) Reversal of protein aggregation provides evidence for multiple aggregated states. J Mol Biol 346:603–616. [DOI] [PubMed] [Google Scholar]
- 53. Xue W‐F, Hellewell AL, Gosal WS, Homans SW, Hewitt EW, Radford SE (2009) Fibril fragmentation enhances amyloid cytotoxicity. J Biol Chem 284:34272–34282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Volles MJ, Lansbury PT (2007) Relationships between the sequence of α‐synuclein and its membrane affinity, fibrillization propensity, and yeast toxicity. J Mol Biol 366:1510–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Uversky VN, Li J, Fink AL (2001) Evidence for a partially folded intermediate in α‐synuclein fibril formation. J Biol Chem 276:10737–10744. [DOI] [PubMed] [Google Scholar]
- 56. Lackowicz JR. Principles of fluorescence spectroscopy. (New York), Chapter 5: Plenum Press, 1983;p. 111–150. [Google Scholar]
- 57. Nath S, Meuvis J, Hendrix J, Carl SA, Engelborghs Y (2010) Early aggregation steps in α‐synuclein as measured by FCS and FRET: evidence for a contagious conformational change. Biophys J 98:1302–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Neyroz P, Zambelli B, Ciurli S (2006) Intrinsically disordered structure of Bacillus pasteurii UreG as revealed by steady‐state and time‐resolved fluorescence spectroscopy. Biochemistry 45:8918–8930. [DOI] [PubMed] [Google Scholar]
- 59. Sahay S, Anoop A, Krishnamoorthy G, Maji SK (2014) Site‐specific fluorescence dynamics of α‐synuclein fibrils using time‐resolved fluorescence studies: Effect of familial Parkinson's disease‐associated mutations. Biochemistry 53:807–809. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting Information