

Abstract
Apolipoprotein‐D is a glycosylated tetrameric lipocalin that binds and transports small hydrophobic molecules such as progesterone and arachidonic acid. Like other lipocalins, apolipoprotein‐D adopts an eight‐stranded β‐barrel fold stabilized by two intramolecular disulphide bonds, with an adjacent α‐helix. Crystallography studies of recombinant apolipoprotein‐D demonstrated no major conformational changes upon progesterone binding.
Amide hydrogen‐deuterium exchange mass spectrometry (HDX‐MS) reports structural changes of proteins in solution by monitoring exchange of amide hydrogens in the protein backbone with deuterium. HDX‐MS detects changes in conformation and structural dynamics in response to protein function such as ligand binding that may go undetected in X‐ray crystallography, making HDX‐MS an invaluable orthogonal technique. Here, we report an HDX‐MS protocol for apolipoprotein‐D that solved challenges of high protein rigidity and low pepsin cleavage using rigorous quenching conditions and longer deuteration times, yielding 85% sequence coverage and 50% deuterium exchange. The relative fractional deuterium exchange of ligand‐free apolipoprotein‐D revealed apolipoprotein‐D to be a highly structured protein. Progesterone binding was detected by significant reduction in deuterium exchange in eight peptides. Stabilization of apolipoprotein‐D dynamics can be interpreted as a combined orthosteric effect in the ligand binding pocket and allosteric effect at the N‐terminus and C‐terminus. Together, our experiments provide insight into apolipoprotein‐D structural dynamics and map the effects of progesterone binding that are relayed to distal parts of the protein. The observed stabilization of apolipoprotein‐D dynamics upon progesterone binding demonstrates a common behaviour in the lipocalin family and may have implications for interactions of apolipoprotein‐D with receptors or lipoprotein particles.
Statement: We reveal for the first time how apolipoprotein‐D, which is protective in Alzheimer's disease, becomes more ordered when bound to a molecule of steroid hormone. These results significantly extend the understanding of apolipoprotein‐D structure from X‐ray crystallography studies by incorporating information on how protein motion changes over time. To achieve these results an improved protocol was developed, suitable for proteins similar to apolipoprotein‐D, to elucidate how proteins change flexibility when binding to small molecules.
Keywords: apolipoprotein‐D, HDX‐MS, lipocalin, lipocalin structure, apolipoprotein structure, ligand, mass spectrometry, lipid
Abbreviations
- AGP
α1‐acid glycoprotein
- apoD
apolipoprotein‐D
- BCF
breast cyst fluid
- GuHCl
Guanidine hydrochloride
- HDX‐MS
amide hydrogen‐deuterium exchange mass spectrometry
- LC–MS
Liquid chromatography‐mass spectrometry
- SDS
sodium dodecyl sulphate
- TCEP
Tris(2‐carboxyethyl)phosphine hydrochloride
Introduction
Apolipoprotein‐D (apoD) is a glycosylated member of the lipocalin family and functions as lipid transporter and antioxidant. As a lipocalin, apoD folds into an eight‐stranded β‐barrel with an adjacent α‐helix as shown by X‐ray crystallography (PDB ID 2hzr)1 and forms a tetramer when not associated with lipoprotein particles.2 Two intramolecular disulphide bonds stabilize the apoD structure and post‐translational modifications include two N‐linked glycosylation (Asn‐45 and Asn‐78, colored orange in Fig. 1) and a pyroglutamate (Q‐1).3, 4 Through a conserved methionine residue (Met‐93, colored blue in Fig. 1), apoD can reduce peroxidised lipids.5 This antioxidant function of apoD has been demonstrated in postmortem human Alzheimer's disease (AD) brain.6 ApoD contains three large hydrophobic loops at the entrance of the ligand binding pocket that are thought to facilitate binding to ligands, membranes and lipoprotein particles.1, 7 In the preformed β‐barrel ligand binding pocket, apoD specifically binds the small hydrophobic molecules progesterone and arachidonic acid with high affinity.8 Retinoic acid and certain fatty acids bind to apoD with lower affinity.9 X‐ray crystallography structures at a resolution of 1.8 Å (PDB ID 2hzq)1 have shown that progesterone is stacked in the ligand binding pocket by two aromatic residues, Phe‐89 and Trp‐127. Three tyrosine residues, Tyr‐22, Tyr‐46, and Tyr‐98, build a hydrogen bond network with progesterone. Interestingly, comparison of the crystal structures of ligand‐free and progesterone‐bound apoD showed that the binding pocket does not undergo major conformational changes upon progesterone binding.1 The root mean square deviation of the protein backbone in the ligand‐free and progesterone‐bound state was 0.38 Å and only three amino acid side chains (Asn‐45, Asn‐58, and Tyr‐46) showed conformational changes.
Figure 1.
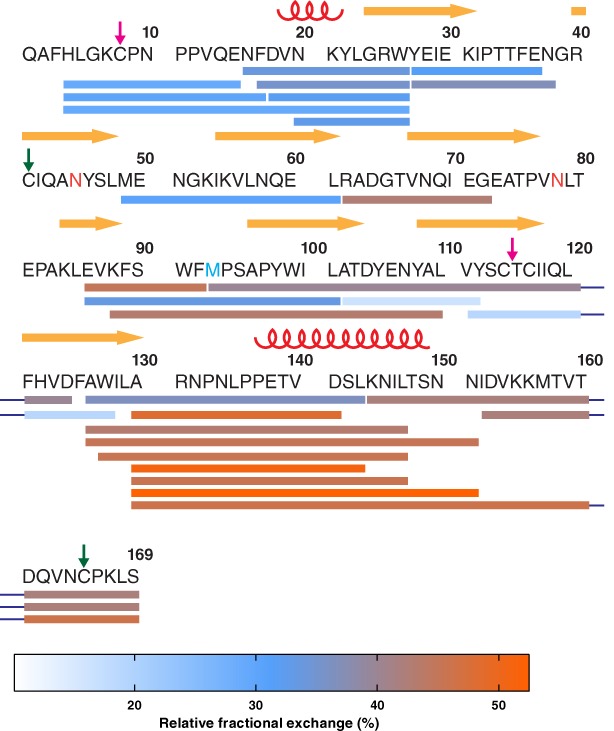
Peptide coverage and overview of protection over the mature apoD sequence. Pepsin‐digested peptides, indicated by bars under the sequence, were detected in each deuteration experiment. The identified peptides cover 85% of the mature apoD sequence. Secondary structure elements and disulphide bonds are marked by red coils (α‐helices), yellow arrows (β‐strands), and coloured arrows (disulphide bonds). Asn‐45 and Asn‐78, which carry the glycosylations, are coloured in orange and the redox‐active Met‐93 in light blue. Each peptide was colour‐coded according to the heat map of relative fractional deuterium exchange after 120 min (colour‐bar below). Overall, the N‐terminus was found to be more protected than the C‐terminus.
X‐ray crystallography provides high resolution snapshots of conformational end states of proteins. However, these snapshots represent states under non‐native crystallization conditions and consequently do not offer complete insights into protein function in solution. Amide hydrogen‐deuterium exchange mass spectrometry (HDX‐MS), in contrast, allows the monitoring of conformational fluidity and structural dynamics of proteins in solution, using the exchange of backbone amide hydrogens to deuterons as conformational probes.10 Following deuterium exchange and proteolysis by pepsin protease, liquid chromatography coupled to mass spectrometry (LC–MS) localizes and quantifies the deuterium exchange in pepsin‐cleaved peptides.10, 11, 12 The rate of deuterium exchange at a certain pH and temperature largely depends on hydrogen bond propensities and to a lesser degree solvent accessibility across the protein.13, 14 Protection from deuterium exchange is due to amide hydrogens engaging in stable intramolecular hydrogen bond networks, which slows their exchange to deuterium. Therefore, a folded protein shows slower deuterium exchange than chemically possible, even if all amide hydrogens are solvent exposed. Thus, deuterium exchange can reflect changes in conformation and in structural dynamics of proteins as a result of interaction with binding partners. In conjunction with X‐ray crystallography structures, HDX‐MS provides insights into the dynamics of the protein in solution. This can be easily extended to map binding sites of ligands and identify orthosteric and allosteric sites.15 Communication between orthosteric and allosteric sites via hydrogen bond networks can then be inferred.16 These aspects make HDX‐MS an invaluable tool in a biochemist's toolbox.
Here, we analysed the deuterium exchange of ligand‐free, tetrameric apoD, purified from human breast cyst fluid (BCF). This enabled the characterization of structural features of native apoD using nonrecombinant protein in solution. We next compared the deuterium exchange of ligand‐free apoD with apoD bound to progesterone, an established ligand of apoD. In order to analyse apoD by HDX‐MS, we established an HDX‐MS protocol for apoD which increased the extent of pepsin cleavage, improved sequence coverage and enhanced deuterium exchange. This protocol is likely to be of use for other lipocalin family members and generally for heavily glycosylated proteins with rigid structures. Our results add to our understanding of native apoD structure and dynamics beyond what static crystal structures reveal. Furthermore, apoD structural dynamics may also be exemplary for common structural processes upon ligand binding within the lipocalin family.
Results and Discussion
Initial deuteration results using native tetrameric glycosylated apoD, purified from human BCF, showed that apoD was resistant to pepsin proteolysis and thus exhibited only limited proteolytic cleavage by pepsin following deuterium exchange, resulting in low sequence coverage of initially ~60% (data not shown). Optimising conditions for quenching deuterium exchange through addition of the chaotrope guanidine hydrochloride (GuHCl) and the reducing agent Tris(2‐carboxyethyl)phosphine hydrochloride (TCEP) as well as increasing the duration of pepsin proteolysis to 7 min improved sequence coverage to 85% of the mature apoD sequence (Fig. 1). Due to heterogeneous glycosylation, deuterium exchange of glycosylated apoD peptides (i.e. peptides including Asn‐45 and Asn‐78) was not quantified. Furthermore, we initially observed low deuterium exchange of apoD during a 10 min time‐course at room temperature (data not shown), suggesting a highly rigid structure. We therefore increased the deuteration time‐course to 120 min and increased the temperature to 37°C to identify additional reporter amides. This allowed us to monitor differences in deuterium exchange between ligand‐free and ligand‐bound apoD.
Two other lipocalin family members have previously been studied with HDX‐MS, β‐lactoglobulin17 and α1‐acid glycoprotein (AGP).18 β‐lactoglobulin, a homodimer,19 showed little cleavage by pepsin, leading to low sequence coverage.17 In the case of AGP, a monomer,20 quenching conditions comparable to this study were used.18 Both proteins contain disulphide bonds,21 as do many other lipocalins20 including apoD.22 Therefore, our changes to the HDX‐MS protocol are likely to be applicable across the lipocalin family and, more broadly, for proteins with inherently rigid structures and intra‐subunit disulphide bonds.
ApoD is a highly structured protein with high protection from deuterium exchange
To gain insights into the dynamics of ligand‐free apoD, the relative fractional exchange per peptide was calculated by dividing the absolute deuterium uptake by the number of exchangeable backbone amides for each peptide and time point [Fig. 2(A)]. Relative fractional uptake allows comparison of deuterium exchange of peptides of differing length across a protein sequence as the uptake is normalized to the number of exchangeable hydrogens. Overall, apoD showed a maximum relative fractional exchange of 52%, out of a maximal 90% deuterium exchange and without back‐exchange correction. The low relative exchange points to a stable lipocalin fold of apoD, consisting of an eight‐stranded β‐barrel and an adjacent α‐helix without any disordered segments (Fig. 1, indicated by yellow arrows and red coils above the primary sequence). Additionally, protein glycosylation has been shown to confer a stabilizing effect on protein structure, reflected in reduced dynamics at and around the glycosylation site.23 Therefore, it is likely that the extensive glycosylation of apoD reduces the structural dynamics of apoD leading to low deuterium exchange.23 Interestingly, peptides 4–26, 93–124, 130–143, 130–147, 130–151, 130–169, 144–169, and 152–169 [marked with asterisks, Fig. 2(A)] showed characteristic bimodal exchange at the three time points examined. Bimodal exchange reflects ensemble behaviour and the existence of at least two slowly interchanging conformations in solution.24 This behaviour is exemplarily showed for peptide 130–169 in Supporting Information Figure S1.
Figure 2.
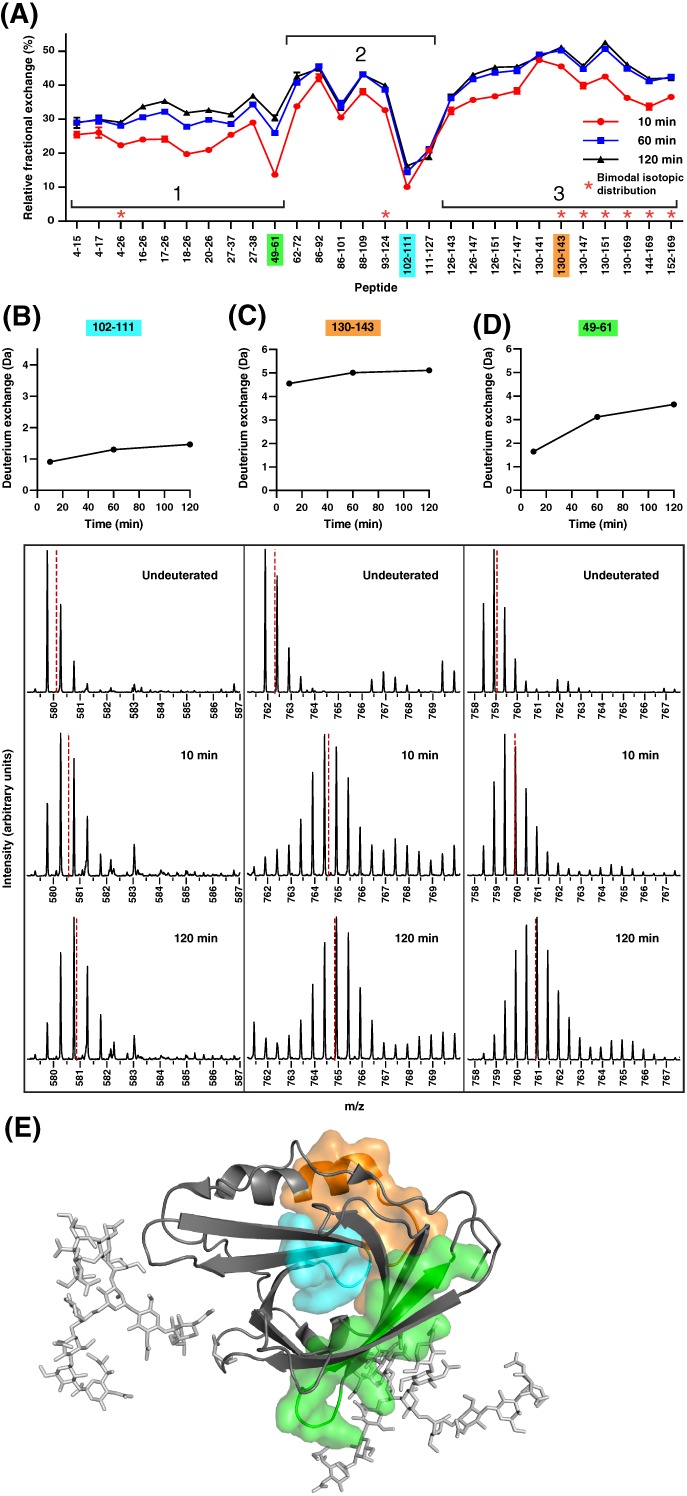
Relative fractional deuterium exchange of the apo‐form of apoD reveals differences in exchange and dynamics. (A) Relative fractional exchange of ligand‐free apoD was calculated by normalizing the absolute deuterium exchange after 10, 60, and 120 min to exchangeable amides per peptide. Colour‐coded peptides show different behaviours in deuterium exchange extent or dynamics. The graph depicts means of triplicates; error bars show standard deviation and are not depicted when smaller than the symbols. Asterisks denote peptides showing bimodal exchange profiles. (B–D) Absolute deuterium exchange over time of marked peptides showed low deuterium exchange (102–111), high deuterium exchange (130–143), low dynamics (102–111) and high dynamics (49–61). Graphs show means of triplicates; error bars show standard deviation and are not depicted when smaller than the symbols. Y‐axes are normalized to half of the number of exchangeable hydrogens. The spectra of undeuterated peptides and after 10 and 120 min of deuteration are shown below indicating the centroid shift. (E) Mapped peptides 102–111 (blue, low exchange and low dynamics), 130–143 (orange, high exchange) and 49–61 (green, high dynamics) are mapped onto the crystal structure of apoD.
In some areas with overlapping peptides, the relative fractional uptake of overlapping peptides seemingly do not match (for instance peptide 86–92 with higher relative fractional uptake and 86–101 with lower relative fractional uptake). This can be explained by differences in the number of exchangeable amide hydrogens. The absolute uptake for peptide 86–92 (3.4 Da after 120 min) is lower than for peptide 86–101 (4.3 Da after 120 min) but due to the different number of exchangeable hydrogens (6 vs 13), the relative fractional uptake is higher for peptide 86–92 than for peptide 86–101.
We observed low relative deuterium exchange in peptide 102–111 (blue) and high deuterium exchange in peptide 130–143 (orange) [Fig. 2(A)]. For both peptides, the absolute deuterium exchange over time and the mass spectra for undeuterated, 10 min deuteration and 120 min deuteration are shown in Figure 2(B,C). Mapping the peptides onto the apoD crystal structure [Fig. 2(E)] showed that the peptide 102–111 (blue, exhibiting low exchange) is part of the β‐barrel and encompasses a short hairpin loop. It is likely that this part of apoD is in a rigid conformation with stable backbone hydrogen bonds maintaining the structure in a predominantly closed state and therefore remains unavailable for deuterium exchange. In contrast, peptide 130–143 (orange, exhibiting high exchange) is situated in a larger loop and includes part of the adjacent α‐helix. The high deuterium exchange in this peptide is indicative of a less structured part of apoD. The difference in deuterium exchange behaviour between the peptides 102–111 and 130–143 corresponds to the difference in b‐factors observed in the crystal structure for these regions.1 Peptide 102–111 has an average b‐factor of 16.9 Å2, compared to 22.0 Å2 for peptide 130–143.
We additionally observed differences in dynamics over the time course between the peptides 102–111 and 49–61 [blue and green, Fig. 2(B,D)]. Peptide 102–111 [blue, Fig. 2(B)] showed almost no changes in deuteration over the deuteration time course. As described above, peptide 102–111 encompasses β‐strands and a short hairpin loop [Fig. 2(E)], a highly structured region with low structural dynamics. In contrast, the deuterium exchange of peptide 49–61 [green, Fig. 2(D)] increased over the time frame of the experiment, indicating high dynamics. The peptide 49–61 [green, Fig. 2(E)] is part of the β‐barrel and encompasses a turn at the closed end of the barrel and a complete β‐strand running from the back to the front of the ligand pocket. Increasing exchange over the time‐course in this protein area indicates higher dynamics with a transition of the protein from a closed to a transient exchangeable state within the timescale of our experiment.25
To gain differential insights into the overall deuteration behaviour of apoD, we generated a heat map to visually assess the deuterium exchange across the apoD sequence (Fig. 1). The relative fractional deuterium exchange after 120 min of each peptide was colour‐coded from 0% to a maximum exchange of 52%. In addition, nine non‐overlapping peptides covering 83% of apoD were selected and the relative fractional deuterium exchange after 120 min was mapped onto the crystal structure of apoD. These results are displayed by colour‐coding and cartoon ‘putty’ representation [Fig. 3(A)]. In the putty style, the diameter of the tubular spine representing the protein backbone is proportional to the relative fractional deuterium exchange. Regions with no peptides detected were set to blue and appear thinnest in the putty representation. This analysis showed that the overall relative fractional exchange of apoD was divided. The N‐terminus with the short 310‐helix and the first three β‐strands showed relatively low deuterium exchange indicating structural protection from deuterium exchange. The glycosylations can contribute to low exchange due to reducing structural dynamics in these areas.23 Additionally, the observed low deuterium exchange in the first three β‐strands indicates that the modelled tetramer inter‐subunit interface in this area is stable and consequently, resistant to deuterium exchange.2
Figure 3.
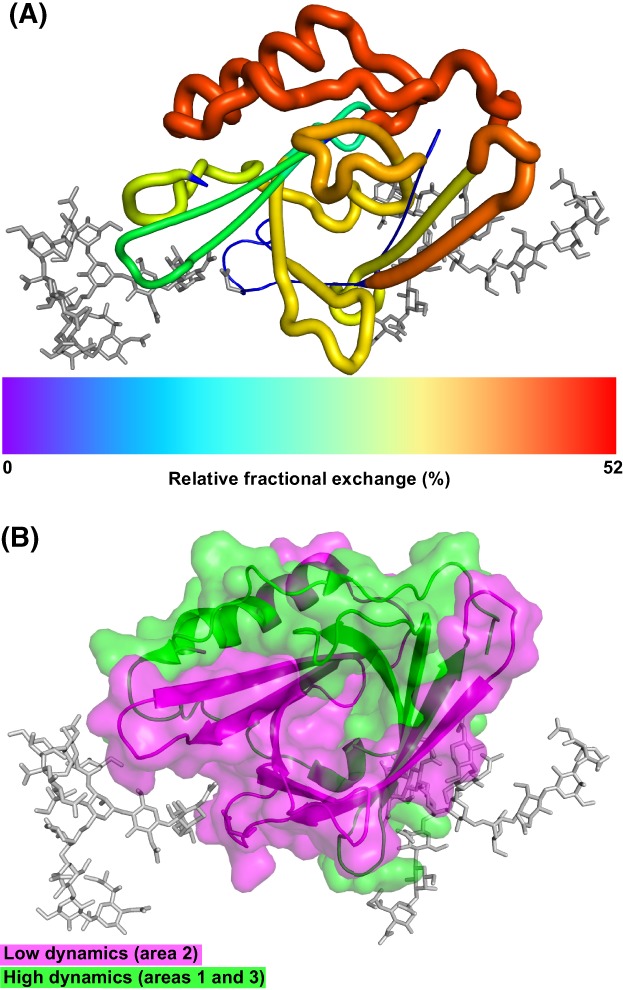
Overview of exchange and dynamics over the apoD structure. (A) Relative fraction deuterium exchange after 120 min of non‐overlapping peptides in putty representation and colour‐coded. (B) Areas with low (area 2, magenta) and high dynamics (areas 1 and 3, green) as identified in Figure 2(A).
In contrast, the C‐terminal α‐helix and disordered region showed relatively high deuterium exchange which indicates lower structural protection. The high exchange in this area can also point toward a more fluid inter‐subunit interface within the apoD tetramer.2
We next identified three overall regions with high (areas 1 and 3) and low dynamics (area 2), marked in Figure 2(A). These areas were then mapped onto the crystal structure in green (area 1: 4–61, area 3: 128–169) and magenta (area 2: 62–127), respectively [Fig. 3(B)]. It appeared that the N‐terminus and C‐terminus (green) both display high structural dynamics. The area with low structural dynamics (magenta) tended to cover mostly the β‐barrel ligand binding pocket which indicates the more stably folded core of apoD.
Progesterone binding induces stabilization of the apoD ligand binding pocket
To characterize effects of progesterone binding on apoD, we pre‐incubated apoD with progesterone, performed HDX‐MS and compared the deuterium exchange of the progesterone‐bound with the ligand‐free form of apoD. A difference plot [Fig. 4(A)] shows the differences in deuterium exchange in all detected peptides. Considering a difference in absolute deuterium uptake of > ±0.5 Da for at least one time point as significant, differences were observed in nine peptides. Out of these nine peptides, eight peptides showed reduced deuterium exchange in the progesterone‐bound form of apoD over the entire time‐course of the experiment (10–120 min). Peptide 49–61, in contrast, showed increased deuterium exchange upon progesterone binding. The absolute deuterium exchange graphs for all nine peptides are shown in Figure 4(B).
Figure 4.
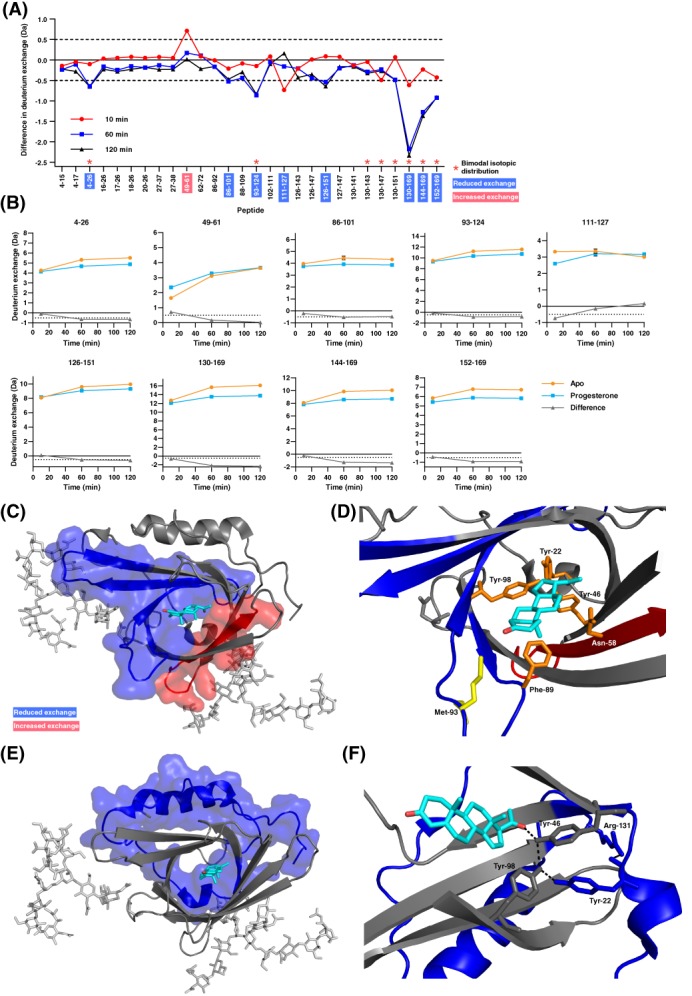
Binding of progesterone induces significant orthosteric and allosteric changes in deuterium exchange of apoD. (A) Difference plot generated by subtracting the absolute deuterium exchange of ligand‐free apoD from progesterone‐bound apoD. Differences > ±0.5 Da are considered significant and were found in nine peptides (marked red: Increased exchange, blue: decreased exchange upon progesterone binding). Asterisks denote peptides showing bimodal exchange profiles in both the apo‐form and in the presence of progesterone. (B) Absolute deuterium exchange over time of nine peptides showing significant changes upon progesterone binding. The graphs show means of triplicates. Y‐axes are normalised to half of the number of exchangeable hydrogens. (C) Significant orthosteric changes in apoD upon progesterone binding. (D) Zoom of the ligand binding pocket. Asn‐58 is located in peptide 49–61 which shows increased deuterium exchange upon progesterone binding. Met‐93 and Phe‐98 are located in peptides which show decreased deuterium exchange upon progesterone binding. (E) Significant allosteric changes in apoD upon progesterone binding. (F) Hydrogen bond network between progesterone, Tyr‐46, Tyr‐22, Tyr‐98, and Arg‐131.
According to Konerman et al.26 ligand binding can cause three different HDX scenarios: In a type 0 scenario, ligand binding causes no effect on deuterium exchange. In a Type 1 scenario, ligand binding causes a strengthening of intramolecular hydrogen bonding, leading to decreased deuterium exchange. In a Type 2 scenario, ligand binding causes a weakening of intramolecular hydrogen bonding, leading to increased deuterium exchange. In the case of apoD, deuterium exchange upon progesterone binding shows both Type 1 and 2 scenarios, illustrating a mixed deuterium exchange response and differential free energy shifts, in different parts of the protein.
Mapping the differences in deuterium exchange onto the crystal structure revealed that changes take place in the ligand binding pocket upon progesterone binding [Fig. 4(C)]. The peptides 86–101 and 93–124, which show decreased deuterium exchange, encompass four β‐strands with two large hydrophobic turns indicating a stabilization upon progesterone binding (blue surface representation). In contrast, peptide 49–61, which comprises one β‐strand within the binding pocket, showed increased deuterium exchange suggesting a destabilization upon progesterone binding (red surface representation). When comparing the average B‐factors of the apoD crystal structure in the region of peptide 49–61, the average B‐factor in the progesterone‐bound structure (25.7 Å2) is higher than in the apo‐form structure (24.6 Å2)1 which supports our observation of a destabilization. Interestingly, in peptide 49–61, the difference in hydrogen exchange between the apo‐form and progesterone‐bound apoD diminishes over the course of the experiment. ApoD in the apo‐form initially has a lower exchange at 10 min but the slope of the exchange curve over time is steeper than in the progesterone‐bound form. Therefore, the deuterium exchange converges with time over the experiment. This behaviour can be interpreted as a weakening of hydrogen bonds upon progesterone binding.
Closer examination of the changes within the ligand binding pocket indicates two crucial first‐shell amino acids of apoD that make contact with progesterone are located in the stabilized regions: Phe‐89 and Tyr‐98 [Fig. 4(D)]. Phe‐89, together with Trp‐127, sandwiches progesterone in the ligand binding pocket.1 The Tyr‐98 sidechain is involved in a hydrogen bond network with progesterone, Tyr‐22, and Tyr‐46.1 Our data indicate that Phe‐89 and Tyr‐98 are responsible for conveying the binding of progesterone to stabilization of the protein backbone. No coverage was obtained for Tyr‐46. Peptide 49–61, which showed increased deuterium exchange upon progesterone binding, encompasses Asn‐58. In ligand‐free apoD, Asn‐58 forms a hydrogen bond with Tyr‐46 via a water molecule. Upon progesterone binding, however, this hydrogen bond breaks due to rotation of the Asn‐58 side chain.1 The breakage of a hydrogen bond and the accompanying conformational change may cause the destabilization of the protein backbone, resulting in an increase in deuterium exchange.
Remarkably, other important features of apoD are located within the stabilized regions [Fig. 4(C,D)]. Two of the large hydrophobic turns in the stabilized area at the entry of the apoD ligand binding pocket are hypothesized to be involved in binding to membranes and lipoprotein particles as well as in facilitating the exchange and release of ligands.1, 7 In addition, the redox‐active Met‐935 is located in this stabilized area. Therefore, stabilization of apoD in this area could modify these aspects of apoD function.
Allosteric effects of apoD upon progesterone binding
Differences in apoD deuterium exchange were not only observed orthosterically in the ligand binding pocket but also allosterically in distal parts of the protein. The N‐terminal peptide 4–26 and a large C‐terminal part of apoD (peptides covering amino acids 126–169) showed reduced deuterium exchange, indicating a stabilizing effect, upon binding of progesterone [Fig. 4(E,F)]. The allosteric changes of structural dynamics in the N‐terminal peptide may be conveyed through Tyr‐22 in the second shell. Tyr‐22 is in a hydrogen bond network with Tyr‐46 which in turn forms the direct hydrogen bond with progesterone and Tyr‐98.1 Furthermore, a disulphide bond connects Cys‐114 in the β‐barrel pocket to the N‐terminal Cys‐8. This covalent connection may allow allosteric communication of protein stabilization. Intriguingly, the N‐terminal peptide includes the 310‐helix at the bottom of the apoD ligand binding pocket which is thought to be involved in binding of apoD to its potential receptor basigin.27 It is therefore possible that the observed allosteric changes in this area have implications for apoD function. Stabilization of this area upon ligand binding could enhance recognition and binding to basigin and therefore facilitate uptake of ligand‐bound apoD into a cell.
The communication of structural changes from the ligand binding pocket to the C‐terminus may be transmitted by hydrophobic contacts between the ligand pocket and the α‐helix in the C‐terminus and via a cluster of hydrogen bonds between Tyr‐22 and Arg‐131 [Fig. 4(F)].
It is noteworthy that the peptides in the C‐terminal region exhibited a large absolute change in deuterium exchange upon binding of progesterone. A large number of peptides in the N‐ and C‐termini of apoD showed characteristic bimodal envelope profiles upon deuterium exchange at the time points examined (Supporting Information Fig. S1). The two envelopes reflect a lower exchanging conformation in equilibrium with a greater exchange conformation in slow timescales (milliseconds and slower) in solution. Progesterone binding increased the relative abundance of the lower exchanging conformation and overall shifted the centroid lower upon progesterone binding. The presence of bimodal exchange is characteristic of EX1 deuterium exchange kinetics28 and explains how ligand (progesterone) binding promotes allostery through conformational selection.
The observed differences in deuterium exchange of overlapping peptides 130–151, 130–169, and 152–169 seemingly do not match, the long peptide 130–169 with a higher difference than two overlapping peptides added together (130–151 plus 152–169). The increase in relative intensity of the lower exchanging population upon progesterone binding is suggestive of allosteric effects of progesterone binding at the N‐terminus and C‐terminus.
While X‐ray crystallography of apoD did not reveal major conformational changes upon progesterone binding,1 our HDX‐MS results show changes in protein dynamics in the ligand binding pocket as well as in N‐terminal and C‐terminal regions. Crystallography is a static snapshot of dynamic regions and changes upon ligand binding may therefore be masked.29 HDX‐MS, in contrast, detects protein structural dynamics and complements crystallography. In fact, HDX‐MS and X‐ray crystallography are often used alongside each other30, 31 and HDX‐MS has been shown to detect allosteric changes in protein dynamics that are not recognized as conformational changes in crystallography.32 Notably, in the present HDX‐MS study, we examined native, fully glycosylated apoD.
Our experiments showed that progesterone binding leads to a stabilization of the apoD pocket as well as of distal regions. Interestingly, HDX‐MS studies of AGP have shown a similar process upon ligand binding. More specifically, binding of progesterone to AGP induced stabilization of the β‐barrel ligand binding pocket but not of the adjacent α‐helix.18 In contrast, binding of propranolol to AGP resulted in a stabilization of both the β‐barrel and α‐helical regions as we observed in apoD. This stabilization effect upon ligand binding could therefore be common to lipocalins.33 In fact, this feature makes lipocalins an interesting target for protein engineering: By altering the ligand binding pocket, lipocalins can be designed to bind small target ligands as so‐called anticalins, that are analogous to antibodies.34 HDX‐MS may be a suitable tool to analyse the anticalin engineering process.
In conclusion, our HDX‐MS protocol with modified exchange and quenching conditions achieved high sequence coverage and is suitable for lipocalins and other highly stable proteins. Characterizing the deuterium exchange behaviour of native, glycosylated apoD highlighted the rigid and well‐ordered structure of apoD. HDX‐MS revealed allosteric and orthosteric changes in apoD upon progesterone binding, extending our knowledge of apoD structure from X‐ray crystallography.1 Allosteric and orthosteric stabilization, as seen in apoD, may be a common effect in lipocalins in response to ligand binding.
Material and Methods
Chemicals and reagents
Chemicals for protein purification were biotechnology grade or similar and chemicals for LC–MS were LC–MS grade or higher. Tris, piperazine, progesterone, and glycerol were sourced from Sigma Aldrich (Castle Hill, NSW, Australia), NaCl from Astral Scientific (Gymea, NSW, Australia), TCEP from Thermo Fisher Scientific (Singapore), D2O from Cambridge Isotope Laboratories (Massachusetts), mono‐sodium and di‐sodium phosphate from Ajax Finechem and Sigma Aldrich (Castle Hill, NSW, Australia), GuHCl from Sigma Aldrich (Singapore) and LC–MS mobile phases from Merck Millipore.
ApoD purification
ApoD was purified, as previously described,2 from breast cyst fluid, which was collected by point‐of‐care ultrasound with aspiration of cyst, mixed with protease inhibitor cocktail (Sigma, 1:100 dilution) and stored at −80°C after removal of particulate by centrifugation at 23 k × g for 20 min. BCF aliquots were thawed on ice and applied to a GE HiTrap Anion exchange (ANX) Sepharose FF 5 ml column equilibrated into IEX buffer (20 mM piperazine, pH 5.0). Proteins were eluted with a 0–50% gradient using high salt buffer (20 mM piperazine, 1 M NaCl, pH 5.0) over 4 CV. Fractions containing apoD were identified by Coomassie SDS‐PAGE, pooled, concentrated using Amicon Ultra concentrators with an Ultracel‐10 membrane and buffer exchanged to 50 mM phosphate buffer, 150 mM NaCl, 3% glycerol, pH 7.4. Subsequently, apoD was purified in a polishing SEC step using a Superdex 200 16/600 column at a flowrate of 0.75 ml/min, pure apoD fractions were pooled and concentrated. ApoD concentration were measured a Pierce BCA assay (ThermoFisher) with bovine serum albumin (BSA) serial dilution as standard curve according to the manufacturer's instructions.
Amide hydrogen‐deuterium exchange
Deuterated buffer was prepared by vacuum‐drying 50 mM phosphate buffer, 150 mM NaCl, pH 7.4 and reconstituting with 99.9% deuterium oxide. Deuterium exchange was performed in triplicates on ligand‐free apoD and progesterone‐bound apoD. Ligand‐free apoD (5 μl, 118.4 μM) or progesterone‐bound apoD (4.5 μl apoD, 118.4 μM, preincubated with 3x molar excess of progesterone, 0.5 μl, 3 mM, in ethanol) was mixed with 45 μl deuterated buffer and incubated for 10, 60, and 120 min at 37°C. Deuterium exchange reactions were quenched by addition of prechilled quench solution containing 0.1% trifluoroacetic acid, 3.87 M GuHCl and 147 mM TCEP to lower the pHread to 2.5 for a final volume of 100 μl, prior to injection into the LC–MS system.
Liquid chromatography‐mass spectrometry
The instrument setup and configuration was as previously described.35 The total volume of quenched deuterium exchange reaction (100 μl) was injected into a nanoACQUITY UltraPerformance liquid chromatography (UPLC) system (Waters, Milford, MA). After proteolysis in an immobilised pepsin column (Poroszyme), kept at 12°C, with 0.1% (v/v) formic acid in water at pH ~2.5 at a flow rate of 100 μl/min, the peptides were trapped in a UPLC VanGuard C18 column for 7 min and then separated on a ACQUITY UPLC BEH C18 reversed‐phase column (Waters), kept at 4°C, and eluted using a water‐acetonitrile gradient of 8–40% (v/v) acetonitrile, 0.1% (v/v) formic acid, pH ~2.5 at a flow‐rate of 40 μl/min. Eluted peptides were detected using a SYNAPT G2‐Si mass spectrometer acquiring in MSE mode with continuous calibration using 200 fmol/μl of Glu‐fibrinopeptide B at a flow‐rate of 1 μl/min. The MSE data for undeuterated samples were searched against a database with the input sequence of human mature apoD (Uniprot P05090, amino acids 21–189) to generate a list of peptides using ProteinLynx Global SERVER software (PLGS v3.0; Waters). Peptides identified by PLGS were further filtered and analysed using the semi‐automated analysis software DynamX (v3.0; Waters) to obtain the deuteration profiles of the experimental samples. Relative fractional uptake was calculated by dividing the absolute uptake by the number of exchangeable amide hydrogens. The indicated measurement uncertainties are 1σ.
Data analysis and visualization
Graphs were created using GraphPad Prism 7. Error bars show standard deviation and are not depicted when smaller than the symbols. A difference in absolute deuterium uptake of > ±0.5 Da of at least one time point was considered significant. Structure visualization was done in PyMOL using the protein structures 2hzr (ligand‐free) and 2hzq (progesterone‐bound).1 These structures were aligned with glycosylated models from molecular dynamics simulation.36
Conflict of interest
The authors confirm that this article content has no conflicts of interest.
Supporting information
Fig S1: Mass spectra for peptide 130–169 show bimodal isotopic distribution. Peptide 130–169 shows a bimodal isotopic distribution indicating at least two conformational isomers. Progesterone binding shifts the distribution towards the lower exchanging conformation.
Acknowledgments
Thanks to Tony Palasovski for collecting the breast cyst fluid and to Brett Garner for manuscript reviewing.
Madhubrata Ghosh's current address is Institute of Bioengineering and Nanotechnology, Singapore.
References
- 1. Eichinger A, Nasreen A, Hyun JK, Skerra A (2007) Structural insight into the dual ligand specificity and mode of high density lipoprotein association of apolipoprotein D. J Biol Chem 282:31068–31075. [DOI] [PubMed] [Google Scholar]
- 2. Kielkopf CS, Low JKK, Mok YF, Bhatia S, Palasovski T, Oakley AJ, Whitten AE, Garner B, Brown SHJ (2018) Identification of a novel tetrameric structure for human apolipoprotein‐D. J Struct Biol 203:205–218. [DOI] [PubMed] [Google Scholar]
- 3. Yang CY, Gu ZW, Blanco‐Vaca F, Gaskell SJ, Yang M, Massey JB, Gotto AM Jr, Pownall HJ (1994) Structure of human apolipoprotein D: locations of the intermolecular and intramolecular disulfide links. Biochemistry 33:12451–12455. [DOI] [PubMed] [Google Scholar]
- 4. Pa S, Ca S, Collet X, Fielding CJ, Burlingame AL (1995) Site‐specific detection and structural characterization of the glycosylation of human plasma proteins lecithin:cholesterol acyltransferase and apolipoprotein D using HPLC/electrospray mass spectrometry and sequential glycosidase digestion. Protein Sci 4:791–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bhatia S, Knoch B, Wong J, Kim WS, Else PL, Oakley AJ, Garner B (2012) Selective reduction of hydroperoxyeicosatetraenoic acids to their hydroxy derivatives by apolipoprotein D: implications for lipid antioxidant activity and Alzheimer's disease. Biochem J 442:713–721. [DOI] [PubMed] [Google Scholar]
- 6. Bhatia S, Jenner AM, Li H, Ruberu K, Spiro AS, Shepherd CE, Kril JJ, Kain N, Don A, Garner B (2013) Increased apolipoprotein D dimer formation in alzheimer's disease hippocampus is associated with lipid conjugated diene levels. J Alzheimers Dis 35:475–486. [DOI] [PubMed] [Google Scholar]
- 7. Grzyb J, Latowski D, Strzalka K (2006) Lipocalins ‐ a family portrait. J Plant Physiol 163:895–915. [DOI] [PubMed] [Google Scholar]
- 8. Vogt M, Skerra A (2001) Bacterially produced apolipoprotein D binds progesterone and arachidonic acid, but not bilirubin or E‐3M2H. J Mol Recognit 14:79–86. [DOI] [PubMed] [Google Scholar]
- 9. Ruiz M, Sanchez D, Correnti C, Strong RK, Ganfornina MD (2013) Lipid‐binding properties of human ApoD and lazarillo‐related lipocalins: functional implications for cell differentiation. FEBS J 280:3928–3943. [DOI] [PubMed] [Google Scholar]
- 10. Rogero JR, Englander JJ, Englander SW (1986) Individual breathing reactions measured by functional labeling and hydrogen exchange methods. Methods Enzymol 131:508–517. [DOI] [PubMed] [Google Scholar]
- 11. Konermann L, Pan J, Liu YH (2011) Hydrogen exchange mass spectrometry for studying protein structure and dynamics. Chem Soc Rev 40:1224–1234. [DOI] [PubMed] [Google Scholar]
- 12. Wales TE, Engen JR (2006) Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom Rev 25:158–170. [DOI] [PubMed] [Google Scholar]
- 13. Englander SW, Kallenbach NR (1983) Hydrogen exchange and structural dynamics of proteins and nucleic acids. Q Rev Biophys 16:521–655. [DOI] [PubMed] [Google Scholar]
- 14. Hoofnagle AN, Resing KA, Ahn NG (2003) Protein analysis by hydrogen exchange mass spectrometry. Annu Rev Biophys Biomol Struct 32:1–25. [DOI] [PubMed] [Google Scholar]
- 15. Chandramohan A, Krishnamurthy S, Larsson A, Nordlund P, Jansson A, Anand GS (2016) Predicting allosteric effects from orthosteric binding in Hsp90‐ligand interactions: implications for fragment‐based drug design. PLoS Comput Biol 12:e1004840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Offenbacher AR, Iavarone AT, Klinman JP (2018) Hydrogen‐deuterium exchange reveals long‐range dynamical allostery in soybean lipoxygenase. J Biol Chem 293:1138–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hu W, Liu J, Luo Q, Han Y, Wu K, Lv S, Xiong S, Wang F (2011) Elucidation of the binding sites of sodium dodecyl sulfate to beta‐lactoglobulin using hydrogen/deuterium exchange mass spectrometry combined with docking simulation. Rapid Commun Mass Spectrom 25:1429–1436. [DOI] [PubMed] [Google Scholar]
- 18. Huang RY, Hudgens JW (2013) Effects of desialylation on human alpha1‐acid glycoprotein‐ligand interactions. Biochemistry 52:7127–7136. [DOI] [PubMed] [Google Scholar]
- 19. Mercadante D, Melton LD, Norris GE, Loo TS, Williams MA, Dobson RC, Jameson GB (2012) Bovine beta‐lactoglobulin is dimeric under imitative physiological conditions: dissociation equilibrium and rate constants over the pH range of 2.5‐7.5. Biophys J 103:303–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Akerstrom B, Flower DR, Salier JP (2000) Lipocalins: unity in diversity. Biochim Biophys Acta 1482:1–8. [DOI] [PubMed] [Google Scholar]
- 21. Schonfeld DL, Ravelli RB, Mueller U, Skerra A (2008) The 1.8‐A crystal structure of alpha1‐acid glycoprotein (Orosomucoid) solved by UV RIP reveals the broad drug‐binding activity of this human plasma lipocalin. J Mol Biol 384:393–405. [DOI] [PubMed] [Google Scholar]
- 22. Nasreen A, Vogt M, Kim HJ, Eichinger A, Skerra A (2006) Solubility engineering and crystallization of human apolipoprotein D. Protein Sci 15:190–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Joao HC, Dwek RA (1993) Effects of glycosylation on protein structure and dynamics in ribonuclease B and some of its individual glycoforms. Eur J Biochem 218:239–244. [DOI] [PubMed] [Google Scholar]
- 24. Kaltashov IA, Eyles SJ (2005) Mass spectrometry in biophysics: conformation and dynamics of biomolecules. 1st ed. Hoboken, N.J: John Wiley. [Google Scholar]
- 25. Morgan CR, Engen JR (2009) Investigating solution‐phase protein structure and dynamics by hydrogen exchange mass spectrometry. Curr Protoc Protein Sci Chapter 17:Unit 17(6):1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Konermann L, Rodriguez AD, Sowole MA (2014) Type 1 and Type 2 scenarios in hydrogen exchange mass spectrometry studies on protein‐ligand complexes. Analyst 139:6078–6087. [DOI] [PubMed] [Google Scholar]
- 27. Najyb O, Brissette L, Rassart E (2015) Apolipoprotein D internalization is a basigin‐dependent mechanism. J Biol Chem 290:16077–16087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang LC, Morgan LK, Godakumbura P, Kenney LJ, Anand GS (2012) The inner membrane histidine kinase EnvZ senses osmolality via helix‐coil transitions in the cytoplasm. EMBO J 31:2648–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nussinov R, Tsai CJ (2015) Allostery without a conformational change? Revisiting the paradigm. Curr Opin Struct Biol 30:17–24. [DOI] [PubMed] [Google Scholar]
- 30. Kim MS, Jeong J, Jeong J, Shin DH, Lee KJ (2013) Structure of Nm23‐H1 under oxidative conditions. Acta Cryst D 69:669–680. [DOI] [PubMed] [Google Scholar]
- 31. Dai H, Case AW, Riera TV, Considine T, Lee JE, Hamuro Y, Zhao H, Jiang Y, Sweitzer SM, Pietrak B, Schwartz B, Blum CA, Disch JS, Caldwell R, Szczepankiewicz B, Oalmann C, Ng PY, White BH, Casaubon R, Narayan R, Koppetsch K, Bourbonais F, Wu B, Wang J, Qian D, Jiang F, Mao C, Wang M, Hu E, Wu JC, Perni RB, Vlasuk GP, Ellis JL (2015) Crystallographic structure of a small molecule SIRT1 activator‐enzyme complex. Nat Commun 6:7645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Trelle MB, Hirschberg D, Jansson A, Ploug M, Roepstorff P, Andreasen PA, Jorgensen TJ (2012) Hydrogen/deuterium exchange mass spectrometry reveals specific changes in the local flexibility of plasminogen activator inhibitor 1 upon binding to the somatomedin B domain of vitronectin. Biochemistry 51:8256–8266. [DOI] [PubMed] [Google Scholar]
- 33. Schiefner A, Skerra A (2015) The menagerie of human lipocalins: a natural protein scaffold for molecular recognition of physiological compounds. Acc Chem Res 48:976–985. [DOI] [PubMed] [Google Scholar]
- 34. Skerra A (2000) Lipocalins as a scaffold. Biochim Biophys Acta 1482:337–350. [DOI] [PubMed] [Google Scholar]
- 35. Chandramohan A, Tulsian NK, Anand GS (2017) Dissecting orthosteric contacts for a reverse‐fragment‐based ligand design. Anal Chem 89:7876–7885. [DOI] [PubMed] [Google Scholar]
- 36. Oakley AJ, Bhatia S, Ecroyd H, Garner B (2012) Molecular dynamics analysis of apolipoprotein‐D‐lipid hydroperoxide interactions: mechanism for selective oxidation of Met‐93. PLoS One 7:e34057. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1: Mass spectra for peptide 130–169 show bimodal isotopic distribution. Peptide 130–169 shows a bimodal isotopic distribution indicating at least two conformational isomers. Progesterone binding shifts the distribution towards the lower exchanging conformation.