

Because non-homeostatic proliferation increases anabolic demand, tumor cells reprogram metabolism in ways that support growth. Although tumor cells retain some metabolic flexibility, the constitutive activation of oncogenes and mutation or loss of tumor suppressors limits their metabolic choices and creates nutrient dependencies not present in their normal counterparts (1–8). Identifying and targeting these differences in metabolic wiring will likely be an effective means to limit tumor growth while sparing normal cells. Clearly, different oncogenic mutations activate distinct downstream gene expression programs that drive metabolic reprogramming in ways that favor certain biosynthetic routes. At the same time, oncogenic events occur in the divergent epigenetic landscapes associated with different tissues of origin. As normal lung and pancreatic cells from the same individual contain identical genomes yet exhibit markedly different gene expression patterns, it stands to reason that tumor cells with shared oncogenic drivers but different tissue origins would do the same. Indeed, a tumor’s metabolic signature is more closely aligned with its tissue of origin than with tumors from other tissues (9,10). The tumor microenvironment also provides context that influences metabolic reprogramming by oncogenes. For example, pancreatic ductal adenocarcinomas (PDAC) induce significant stromal desmoplasia resulting in high interstitial pressure and inadequate perfusion (11). Nutrient scavenging strategies such as autophagy and macropinocytosis are likely to be selected for in this cancer class where access to plasma-derived nutrients is limited (12–14). Similarly, spatial metabolic heterogeneity has been noted within individual lung tumors with poorly perfused tumor sections exhibiting distinct metabolic strategies and gene expression profiles compared to better perfused areas (15). In summary, the simplistic view that “cancer metabolism” is defined by aerobic glycolysis (the Warburg effect) and the use of glutamine to replenish TCA cycle intermediates is without a doubt incorrect, failing to capture the complexity, heterogeneity, and context dependence of actual tumor metabolism (8). A recent study by Mayers and colleagues (16) drives this point home, providing robust evidence that the same oncogenic mutations re-program metabolism in distinct ways when they occur in different organs. At the same time, this study also highlights a new metabolic flux in KRAS-driven lung tumors that could be amenable to therapeutic targeting.
Inspired by prior work suggesting that MYC-driven changes in the metabolome differ depending on which tissue hosts the tumor (17), Mayers et al. took advantage of the fact that both non-small cell lung cancers (NSCLC) and PDAC commonly exhibit activating mutations in KRAS (e.g., KRASG12D) and loss of the tumor suppressor TP53, both proteins known to modulate metabolic fluxes in cancer cells (2,6,18). Upon Cre expression in either the lung (via adenovirus) or the pancreas (transgenic expression), KrasLSLG12D/+; Trp53flox/flox mice develop NSCLC (KP model) or PDAC (KP−/− model) with similar features to the respective human disease. Using a single genetically engineered mouse model with differential expression of Cre permitted an ‘apples to apples’ comparison of the metabolic strategies utilized by tumors arising in these distinct anatomical sites. By studying autochthonous tumors in vivo, it was further ensured that nutrient and oxygen levels were in the physiologic range; metabolic fluxes in KP NSCLC cells differ significantly in vitro and in vivo (19). The Vander Heiden group had also previously reported that circulating branched chain amino acids (BCAAs) increase in KP−/−C mice with PDAC as a consequence of muscle breakdown while plasma BCAAs decrease in the KP NSCLC model (20). The current study (16) sought to explain this difference, uncovering a reciprocal increase in BCAA levels in lung but not pancreatic tumor tissue. This result suggested that NSCLC and PDAC tumor cells with activated KRAS and loss of TP53 exhibit differential uptake and utilization of BCAAs.
The BCAAs, leucine, isoleucine, and valine, are essential amino acids and must be acquired from the diet. Once they enter cells, BCAAs are either incorporated directly into proteins or metabolized (21) (Figure 1). In a reversible reaction catalyzed by the enzyme branched chain amino acid transferase (BCAT), the BCAA amino group is transferred to α-ketoglutarate to produce glutamate and a branched chain α-ketoacid (BCKA). BCKAs can be converted back to BCAAs or oxidized through a series of additional enzymatic steps and eventually enter the TCA cycle if cells express the required complement of enzymes. BCKAs as well as certain other BCAA metabolites can also be secreted and used by other tissues. To understand why mice with NSCLC and PDAC exhibited differences in plasma BCAA levels, Mayers and coworkers tracked the fate of dietary BCAAs in normal tissues and NSCLC or PDAC tumors by feeding mice a diet containing U-13C-labeled leucine and valine. These metabolic tracing studies revealed that, relative to normal lung, NSCLC convert more dietary free BCAA into protein and use BCAT to produce more of the leucine-derived BCKA α-ketoisocaproate (KIC); BCAA carbon entry into the TCA cycle was not altered by KRAS expression or TP53 loss. In contrast, when KP−/−C PDAC cells were compared to normal pancreas, less dietary BCAA carbon ended up in proteins and production of KIC from free BCAAs in plasma did not increase significantly. In addition, less labeled free BCAA carbon entered the TCA cycle in PDAC than in normal tissue. Consistent with the observed changes in labeled KIC in NSCLC, BCAT1 and BCAT2 protein levels were increased relative to normal tissue; BCAT1 protein levels were decreased in PDAC tumors although BCAT2 was up-regulated. U-15N-leucine labeling studies showed that the increase in BCAT1/2 protein in NSCLC correlated with increased use of BCAAs as a nitrogen source for nucleotide synthesis. Aspartate, which in these KP NSCLCs was produced from BCAT-dependent glutamate production, is a critical precursor for nucleotide synthesis in proliferating cells (22,23). Taken together, these results indicate that KRAS activation and TP53 deletion in NSCLC increase the uptake of BCAA from plasma to provide precursors for protein and nucleotide synthesis. PDAC cells, in contrast, did not increase their use of plasma-derived BCAAs for protein synthesis or BCKA production.
Figure 1.
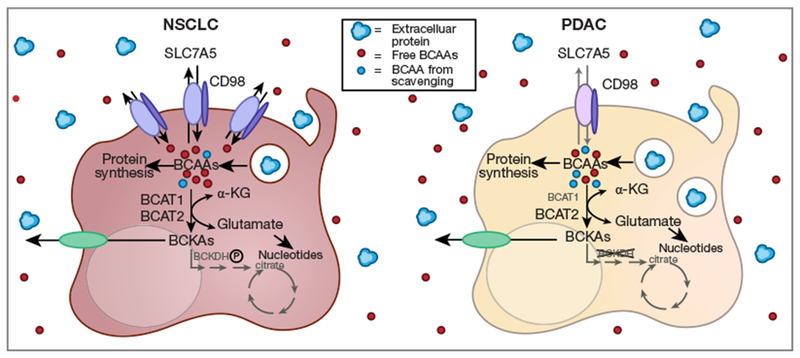
Differential acquisition of BCAAs in KP NSCLC and KP−/−C PDAC as described by Mayers et al. (16). Relative to normal tissue, SLC7A5 and BCAT1 were over-expressed in NSCLC but not PDAC; BCAT2 protein levels increased in both NSCLC and PDAC. BCKDH was inactivated by phosphorylation in NSCLC while in PDAC BCKDH total protein was greatly reduced. Isotopic labeling studies indicated that carbon from circulating BCAAs ended up in protein and BCKAs in NSCLC to a greater extent than in PDAC. Nitrogen from dietary leucine was used for BCAT-dependent nucleotide synthesis in NSCLC. BSA degradation in the lysosome was increased in isolated PDAC cells relative to NSCLC cells suggesting that PDAC cells may rely on macropinocytosis to compensate for reduced import of extracellular BCAAs through SLC7A5. BCAA, branched chain amino acid; NSCLC, non-small cell lung cancer; PDAC, pancreatic ductal adenocarcinoma; BCKDHA, branched chain keto acid dehydrogenase El alpha; BCAT, branched chain amino acid transferase.
It is important to recognize that the failure of PDAC tumor cells to increase utilization of plasma BCAAs does not mean that BCAA transamination is not important in PDAC tumor cells. Indeed, BCAT2 protein levels are significantly increased in KP−/−C PDAC in this study, and BCAT2 over-expression can drive the growth of PDAC cells lacking malic enzyme 2 (16,24). It is interesting to speculate that there may be an advantage to using BCAT and BCAAs rather than other amino acids to produce glutamate. Similar to increases in BCAT1/2 and inhibitory phosphorylation of BCKDH in NSCLC, the up-regulation of BCAT2 and the near complete loss of the branched chain ketoacid dehydrogenase El alpha (BCKDHA) protein in PDAC tumors would raise BCKA levels suggesting that BKCAs may have an important role in both NSCLC and PDAC. Like tumor cells, activated T cells alter their metabolic program to fuel their rapid expansion during an immune response. Activated T cells take up extracellular leucine at an accelerated rate and increase BCAT1 levels 20-fold to produce glutamate, resulting in the release of the leucine-derived BCKA KIC which accumulates because BCKDH activity is again low (25). That BCAAs are also used to produce glutamate in rapidly proliferating lymphocytes suggests that producing BCKAs may be directly or indirectly beneficial for anabolic cells. Intriguingly, a product of valine oxidation that is secreted from muscle cells, 3-hydroxyisobutyrate (3-HIB), promotes fatty acid transport across endothelial cells leading to an increased lipid supply for muscle cells (26). Proliferating cells require fatty acids to produce membranes for daughter cells (5). Although 3-HIB production requires BCKDH activity which is low in T cells and these cancer cells (16) (Figure 1), other BCAA metabolites might have related roles in tissue crosstalk. There are many unknowns and conflicting results regarding the role of BCAAs and their metabolites in controlling feeding behavior and whole body metabolism. Viewing the tumor as an “organ” that may produce and receive signals from other tissues may be important to fully understand the roles of BCAA and their breakdown products in proliferating cells.
The failure of KP−/−C PDAC tumor cells to increase their use of plasma BCAA may stem from the down-regulation of both the BCAA transporter protein, SLC7A5, and BCAT1 following transformation by KRAS activation and TP 53 deletion (16). Over-expressing SLC7A5 in cell lines derived from KP−/−C PDAC tumors increased leucine uptake slightly in vitro but did not confer a growth advantage in culture. However, SLC7A5 may increase PDAC tumor growth in vivo as subcutaneous tumors formed by one clone of SLC7A5 over-expressing PDAC cells narrowly missed being called as significantly larger than controls (P=0.06). A couple of caveats to this experiment further suggest that low SLC7A5 expression should not be discounted as limiting for PDAC tumor growth. For one, SLC7A5 is an obligate exchanger and must couple influx of BCAAs to the efflux of another substrate (27). Expressing SLC7A5 alone without concomitant up-regulation of another amino acid transporter or BCAT1 over-expression to create a gradient for BCAA import may have limited BCAA uptake. Moreover, SLC7A5 expression in the absence of the heavy chain SLC3A2 that stabilizes SLC7A5 at the plasma membrane may have compromised cell surface expression (28,29). It is not clear that the over-expression strategy employed was sufficient to raise leucine uptake in PDAC cells to biologically significant degree that would match uptake in NSCLC cells. SLC7A5 is over-expressed in many cancers and is a negative prognostic indicator suggesting that this transporter often plays a key role in oncogenesis (30–34). Rather than suggesting SLC7A5 is not important in PDAC, the work of Mayers et al. implies that low SLC7A5 expression levels in PDAC may result in enhanced sensitivity to inhibitors. JPH203, a small molecule SLC7A5 inhibitor effective as a single agent in some tumors (35) has advanced to a clinical trial (UMIN000016546) and thus the sensitivity of both PDAC and NSCLC to this agent is worth evaluating.
Although SLC7A5 levels were low in KP−/−C pancreatic tumors, RAS-driven cancers have an alternate means of obtaining amino acids: catabolism of extracellular protein via macropinocytosis (12,13,36). Intriguingly, while bovine serum albumin was taken up and degraded in lysosomes in both tumor types, Mayers et al. observed that KP−/−C PDAC cells catabolize albumin to a greater extent than KP NSCLC in cell culture leading them to hypothesize that the relative amount of macropinocytosis in these tumor classes may relate to their differential ability to acquire leucine from the environment (Figure 1). As alluded to above, this hypothesis would make sense given the significant differences in perfusion in these two tumor classes. However, given that albumin is consumed through macropinocytosis-dependent and -independent pathways (37) and that nutrient-sensitive signal transduction pathways regulate macropinocytosis, quantifying the relative dependence of NSCLC and PDAC on amino acids obtained through macropinocytosis will require additional experiments. The effect of adaptation to cell culture on the relative levels of macropinocytosis must also be considered. In our hands, A549 human lung cancer cells with activating mutations in KRAS exhibit as robust macropinocytosis of high molecular weight dextran as pancreatic cancer cell lines with KRAS mutations (unpublished data). In vivo studies in NSCLC to match those recently performed by this group in autochthonous PDAC tumors (38) will certainly be informative.
Perhaps the most striking result in this report was obtained when the authors directly tested their conclusion from stable isotope labeling studies: BCAT activity is essential for the growth of KP NSCLC but not KP−/−C PDAC tumors. They created genetically matched Bcat1/2 null NSCLC and PDAC tumor-derived cell lines differing only in their epigenomes using CRISPR/Cas9 mediated gene editing. Both NSCLC and PDAC cells proliferated normally in vitro despite Bcat1/2 deletion. Strikingly, loss of Bcat1/2 crippled NSCLC tumors grown either subcutaneously or orthotopically in the lungs of syngeneic hosts. In contrast, the subcutaneous growth of PDAC tumors was unaffected by Bcat1/2 deletion. Some growth inhibition was apparent when Bcat1/2 PDAC tumors were grown orthotopically suggesting that BCAT2 up-regulation may be important in these tumor cells in the context of the pancreatic microenvironment. It is worth noting that BCAT2 activity is required even in standard culture medium in the subset of PDAC where malic enzyme 2 is deleted (24), an event not modeled in KP−/−C mice. An important conclusion from the divergent in vitro and in vivo results with BCAT-deficient KP NSCLC cells in Mayers et al. is that in vitro screens designed to elucidate metabolic liabilities in tumor cells may produce misleading results. Cell culture media formulations were developed to maximize cell proliferation, and reducing nutrients to more physiologic levels often uncovers metabolic dependencies that are not apparent when nutrients are present in large excess. A case in point is the similar disconnect in the in vitro and in vivo dependence of tumor cells on glutaminase (GLS), the enzyme that converts glutamine to glutamate. GLS was essential in KP NSCLC cells in vitro, but GLS inhibitors failed to limit tumor growth in vivo (19). It would be informative to test whether culturing KP NSCLC cells in more physiologic levels of nutrients would elicit a proliferation or survival defect upon Bcat1/2 deletion. If so, conducting metabolic screens in more physiologic nutrient levels might dramatically increase their translational value. Alternatively, other differences between the tumor microenvironment and the cell culture dish that could prove more difficult to identify and mimic in vitro might be responsible for the divergence between cell culture and in vivo results. Either way, these studies with Bcat1/2 deleted cells (16) provide a cautionary tale and emphasize the importance of characterizing metabolic pathways in vivo.
The work of Mayers et al. also suggests new therapeutic approaches for NSCLC. With the caveat that a chemical inhibitor and/or conditional alleles of Bcat1/2 will be required to formally confirm that inhibiting BCAT in an established tumor will stop growth or cause tumor regression, BCAT inhibitors could have value in NSCLC. An important first task will be to determine whether both BCAT1 and BCAT2 should be inhibited or whether targeting only one of these paralogs will be sufficient to slow tumor growth while minimizing toxicity. Bcat1 is expressed primarily in the brain of mature healthy mice, but BCAT1 is dramatically induced in activated T cells and BCAT1 is over-expressed in several cancers consistent with Bcat1’s designation as a MYC target gene (25,39–42) (Table 1). IDHWT, but not IDHmut, glioblastomas also require BCAT1 for growth both in vitro in standard medium and in vivo (48). Interestingly, while the levels of the mitochondrial isoform, BCAT2, are not altered by T cell activation (25), Bcat2 is an SREBP1 target gene whose expression is negatively regulated by AMPK, and in certain genetic contexts BCAT2 over-expression can drive PDAC growth while knocking down Bcat2 limits colony formation (24). BCAT2 protein levels did increase in KP−/−C PDAC tumors (16) relative to normal tissue, and the Human Protein Atlas suggests that BCAT2 is expressed at high levels in other human cancers as well. Taken together, these findings suggest that both isoforms of BCAT may contribute to tumor growth, but inhibiting BCAT1 will likely be necessary and might be sufficient to cause tumor growth inhibition in NSCLC. Encouragingly, Bcat1 whole-body knockout mice exhibit no overt defects suggesting that BCAT2 can compensate for the loss of BCAT1 (Table 1) (25). In fact, impairing bcat-1 expression in C. elegans extended maximum lifespan by 25% and mean lifespan by 19%, further supporting that BCAT1 inhibitors may not be toxic (45). In this study in worms, however, the beneficial effects of BCAT-1 loss depended on mTORC1 activation downstream of leucine accumulation. Bcat1−/− T cells also exhibit higher BCAA levels, mTORC1 activation, and increased glycolysis (25). Given that mTORC1 activation and glycolysis are associated with anabolism in many cancer cells, it will be important to empirically test whether small molecule BCAT1 inhibitors would have undesirable effects on tumors or whole body metabolism. It is also important to recognize that more than one node in a pathway and/or compensatory pathways must often be targeted simultaneously for metabolic inhibitors to exhibit sustained effects and to tip the balance from a cytostatic to cytotoxic effect that might lead to tumor regression. Given the dramatic up-regulation of SLC7A5 observed here in NSCLC (16), small molecule inhibitors of SLC7A5 (35) and/or macropinocytosis inhibitors (13) could be logical choices for combination with inhibitors of BCAT1.
Table 1.
Comparison of the two BCAT isoenzymes
| Variables | BCAT1 | BCAT2 |
|---|---|---|
| Other names | BCATc (cytosolic) | BCATm (mitochondrial) |
| Tissue distribution | Brain, gonads, activated CD4+ T-cells (25) | Most tissues, excluding liver (43,44); high in exocrine pancreas (44) |
| Knockout phenotypes | Activated Bcat1−/− murine T-cells exhibit elevated mTORC1 signaling and glycolytic metabolism (25); bcat-1 null C. elegans have an extended lifespan and healthspan (45) | Bcat2−/− mice have high circulating BCAAs, improved insulin sensitivity and glucose tolerance, and elevated energy expenditure (46); Bcat2−/− mice have lower exercise capacity (47); required in subset of PDAC (24) |
| Expression in cancer cells | Over-expressed in: glioblastoma (48); nasopharyngeal carcinoma (39); cancers with elevated MYC? (39) | Over-expressed in: PDAC (KP−/−C model) (16); cancers with active SREBP1 and low AMPK activity? (24) |
BCAT, branched chain amino acid transferase; PDAC, pancreatic ductal adenocarcinoma.
In summary, this study by Mayers et al. establishes that tissue of origin profoundly influences BCAA flux in response to KRAS activation and loss of TP53, suggests a new therapeutic strategy in NSCLC, and demonstrates that in vitro screening of metabolic inhibitors under standard tissue culture conditions is likely a flawed strategy. At the same time, many important questions are raised that will need to be addressed by future studies. Clearly, the successful therapeutic application of metabolic inhibitors in the clinic will require a more complete understanding of the roles of the tumor and tissue microenvironment, oncogenic mutations, and epigenetic landscape in shaping the metabolic choices and dependencies of different tumor classes.
Acknowledgements
Funding: EM Selwan was supported by T32-CA009054-37. AL Edinger was supported by grants from the NIH (R01 GM089919), CDMRP (W81XWH-15-1-0010), and the UC Cancer Research Coordinating Committee (CRR-17-426826).
Footnotes
Provenance: This is an invited Editorial commissioned by the Section Editor Zhen-Yu Lin (Cancer Center, Union Hospital, Huazhong University of Science and Technology, Wuhan, China).
Comment on: Mayers JR, Torrence ME, Danai LV, et al. Tissue of origin dictates branched-chain amino acid metabolism in mutant Kras-driven cancers. Science 2016;353:1161-5.
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Kim SM, Roy SG, Chen B, et al. Targeting cancer metabolism by simultaneously disrupting parallel nutrient access pathways. J Clin Invest 2016;126:4088–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Denicola GM, Cantley LC. Cancer’s fuel choice: new flavors for a picky eater. Mol Cell 2015;60:514–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab 2016;23:27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Selwan EM, Finicle BT, Kim SM, et al. Attacking the supply wagons to starve cancer cells to death. FEBS Lett 2016;590:885–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Röhrig F, Schulze A. The multifaceted roles of fatty acid synthesis in cancer. Nat Rev Cancer 2016;16:732–49. [DOI] [PubMed] [Google Scholar]
- 6.Humpton TJ, Vousden KH. Regulation of cellular metabolism and hypoxia by p53. Cold Spring Harb Perspect Med 2016;6(7). pii: a026146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guo JY, White E. Autophagy, Metabolism, and Cancer. Cold Spring Harb Symp Quant Biol 2017. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vander Heiden MG, DeBerardinis RJ. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017;168:657–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hu J, Locasale JW, Bielas JH, et al. Heterogeneity of tumor-induced gene expression changes in the human metabolic network. Nat Biotechnol 2013;31:522–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gaude E, Frezza C Tissue-specific and convergent metabolic transformation of cancer correlates with metastatic potential and patient survival. Nat Commun 2016;7:13041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Provenzano PP, Cuevas C, Chang AE, et al. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012;21:418–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kamphorst JJ, Nofal M, Commisso C, et al. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res 2015;75:544–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Commisso C, Davidson SM, Soydaner-Azeloglu RG, et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 2013;497:633–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kamphorst JJ, Cross JR, Fan J, et al. Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc Natl Acad Sci USA 2013;110:8882–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hensley CT, Faubert B, Yuan Q, et al. Metabolic heterogeneity in human lung tumors. Cell 2016;164:681–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mayers JR, Torrence ME, Danai LV et al. Tissue of origin dictates branched-chain amino acid metabolism in mutant Kras-driven cancers. Science 2016;353:1161–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yuneva MO, Fan TW, Allen TD, et al. The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab 2012;15:157–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.White E Exploiting the bad eating habits of Ras-driven cancers. Genes Dev 2013;27:2065–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davidson SM, Papagiannakopoulos T, Olenchock BA, et al. Environment impacts the metabolic dependencies of Ras-Driven Non-Small cell lung cancer. Cell Metab 2016;23:517–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mayers JR, Wu C, Clish CB, et al. Elevation of circulating branched-chain amino acids is an early event in human pancreatic adenocarcinoma development. Nat Med 2014;20:1193–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harper AE, Miller RH, Block KP. Branched-Chain Amino Acid Metabolism. Annu Rev Nutr 1984;4:409–54. [DOI] [PubMed] [Google Scholar]
- 22.Birsoy K, Wang T, Chen WW, et al. An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell 2015;162:540–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sullivan LB, Gui DY, Hosios AM, et al. Supporting aspartate biosynthesis is an essential function of respiration in proliferating cells. Cell 2015;162:552–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dey P, Baddour J, Muller F, et al. Genomic deletion of malic enzyme 2 confers collateral lethality in pancreatic cancer. Nature 2017;542:119–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ananieva EA, Patel CH, Drake CH, et al. Cytosolic branched chain aminotransferase (BCATc) regulates mTORC1 signaling and glycolytic metabolism in CD4+ T cells. J Biol Chem 2014;289:18793–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jang C, Oh SF, Wada S, et al. A branched-chain amino acid metabolite drives vascular fatty acid transport and causes insulin resistance. Nat Med 2016;22:421–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fotiadis D, Kanai Y, Palacín M. The SLC3 and SLC7 families of amino acid transporters. Mol Aspects Med 2013;34:139–58. [DOI] [PubMed] [Google Scholar]
- 28.Shishido T, Uno S, Kamohara M, et al. Transformation of BALB3T3 cells caused by over-expression of rat CD98 heavy chain (HC) requires its association with light chain: mis-sense mutation in a cysteine residue of CD98HC eliminates its transforming activity. Int J Cancer 2000;87:311–6. [DOI] [PubMed] [Google Scholar]
- 29.Campbell WA, Thompson NL. Overexpression of LAT1/CD98 light chain is sufficient to increase system L-amino acid transport activity in mouse hepatocytes but not fibroblasts. J Biol Chem 2001;276:16877–84. [DOI] [PubMed] [Google Scholar]
- 30.Rosilio C, Nebout M, Imbert V , et al. L-type amino-acid transporter 1 (LAT1): a therapeutic target supporting growth and survival of T-cell lymphoblastic lymphoma/T-cell acute lymphoblastic leukemia. Leukemia 2015;29:1253–66. [DOI] [PubMed] [Google Scholar]
- 31.Yazawa T, Shimizu K, Kaira K, et al. Clinical significance of coexpression of L-type amino acid transporter 1 (LAT1) and ASC amino acid transporter 2 (ASCT2) in lung adenocarcinoma. Am J Transl Res 2015;7:1126–39. [PMC free article] [PubMed] [Google Scholar]
- 32.Kaira K, Nakamura K, Hirakawa T, et al. Prognostic significance of L-type amino acid transporter 1 (LAT1) expression in patients with ovarian tumors. Am J Transl Res 2015;7:1161–71. [PMC free article] [PubMed] [Google Scholar]
- 33.Sakata T, Ferdous G, Tsuruta T, et al. L-type amino-acid transporter 1 as a novel biomarker for high-grade malignancy in prostate cancer. Pathol Int 2009;59:7–18. [DOI] [PubMed] [Google Scholar]
- 34.Isoda A, Kaira K, Iwashina M, et al. Expression of L-type amino acid transporter 1 (LAT1) as a prognostic and therapeutic indicator in multiple myeloma. Cancer Sci 2014;105:1496–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao Y, Wang L, Pan J. The role of L-type amino acid transporter 1 in human tumors. Intractable Rare Dis Res 2015;4:165–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Palm W Park Y, Wright K, et al. The utilization of extracellular proteins as nutrients is suppressed by mTORC1. Cell 2015;162:259–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Merlot AM, Kalinowski DS, Richardson DR. Unraveling the mysteries of serum albumin-more than just a serum protein. Front Physiol 2014;5:299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Davidson SM, Jonas O, Keibler MA, et al. Direct evidence for cancer-cell-autonomous extracellular protein catabolism in pancreatic tumors. Nat Med 2017;23:235–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou W, Feng X, Ren C, et al. Over-expression of BCAT1, a c-Myc target gene, induces cell proliferation, migration and invasion in nasopharyngeal carcinoma. Mol Cancer 2013;12:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ben-Yosef T, Yanuka O, Halle D, et al. Involvement of Myc targets in c-myc and N-myc induced human tumors. Oncogene 1998;17:165–71. [DOI] [PubMed] [Google Scholar]
- 41.Schuldiner O, Eden A, Ben-Yosef T, et al. ECA39, a conserved gene regulated by c-Myc in mice, is involved in G1/S cell cycle regulation in yeast. Proc Natl Acad Sci U S A 1996;93:7143–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Benvenisty N, Leder A, Kuo A, et al. An embryonically expressed gene is a target for c-Myc regulation via the c-Myc-binding sequence. Genes Dev 1992;6:2513–23. [DOI] [PubMed] [Google Scholar]
- 43.Torres N, López G, De Santiago S, et al. Dietary protein level regulates expression of the mitochondrial branched-chain aminotransferase in rats. J Nutr 1998;128:1368–75. [DOI] [PubMed] [Google Scholar]
- 44.Sweatt AJ, Wood M, Suryawan A, et al. Branched-chain amino acid catabolism: unique segregation of pathway enzymes in organ systems and peripheral nerves. Am J Physiol Endocrinol Metab 2004;286:E64–76. [DOI] [PubMed] [Google Scholar]
- 45.Mansfeld J, Urban N, Priebe S, et al. Branched-chain amino acid catabolism is a conserved regulator of physiological ageing. Nat Commun 2015;6:10043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.She P, Reid TM, Bronson SK, et al. Disruption of BCATm in mice leads to increased energy expenditure associated with the activation of a futile protein turnover cycle. Cell Metab 2007;6:181–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.She P, Zhou Y, Zhang Z, et al. Disruption of BCAA metabolism in mice impairs exercise metabolism and endurance. J Appl Physiol (1985) 2010;108:941–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tönjes M, Barbus S, Park YJ, et al. BCAT1 promotes cell proliferation through amino acid catabolism in gliomas carrying wild-type IDH1. Nat Med 2013;19:901–8. [DOI] [PMC free article] [PubMed] [Google Scholar]