

Abstract
Cancer is the leading cause of death worldwide. Almost 50% of all cancer patients undergo radiation therapy (RT) during treatment, with varying success. The main goal of RT is to kill tumor cells by damaging their DNA irreversibly while sparing the surrounding normal tissue. The outcome of RT is often determined by how tumors recognize and repair their damaged DNA. A growing body of evidence suggests that tumors often show abnormal expression of DNA double-strand break (DSB) repair genes that are absent from normal cells. Defects in a specific DNA repair pathway make tumor cells overly dependent on alternative or backup pathways to repair their damaged DNA. These tumor cell-specific abnormalities in the DNA damage response (DDR) machinery can potentially be used as biomarkers for treatment outcomes or as targets for sensitization to ionizing radiation (IR). An improved understanding of genetic or epigenetic alterations in the DNA repair pathways specific to cancer cells has paved the way for new treatments that combine pharmacological exploitation of tumor-specific molecular vulnerabilities with IR. Inhibiting DNA repair pathways has the potential to greatly enhance the therapeutic ratio of RT. In this review, we will discuss DNA repair pathways in active cells and how these pathways are deregulated in tumors. We will also describe the impact of targeting cancer-specific aberrations in the DDR as a treatment strategy to improve the efficacy of RT. Finally, we will address the current roadblocks and future prospects of these approaches.
Keywords: Ionizing radiation (IR), cancer, DNA damage, DNA repair, synthetic lethality, charged particle therapy (CPT), radiotherapy
Introduction
Cancer is the leading cause of morbidity and mortality worldwide. According to the most recent estimation by the International Agency for Research on Cancer (IARC), in 2012, 8.2 million people worldwide died of cancer, with 14.1 million new cases being reported (1). The etiology of cancer is multifactorial and complex, and it varies widely from patient to patient. Hanahan and Weinberg identified uncontrolled proliferative potential, replicative immortality, and evasion of apoptosis as some of the major hallmarks of cancer cells (2). In the past decade, scientists have made considerable progress towards the treatment and understanding of these hallmarks, and when combined with advances in early detection and various treatment modalities, many cancers have become curable.
Since the discovery of X-rays in 1895, radiation therapy (RT) has constituted an important modality used in curative cancer treatment, in conjunction with surgery and chemotherapy (3). Its low cost and usefulness as a single-modality treatment for early stage cancer patients are the main reasons why RT is an important branch of medical oncology (4,5). Approximately 50% of all cancer patients receive RT in some form during the course of their illness (6), and it is estimated that RT contributes to about 40% of curative treatments (7). In addition to conventional RT, the last decade has seen high energy proton and carbon ion beam radiotherapy [charged particle therapy (CPT)] gain in popularity. The theoretical advantages of CPT over conventional RT are focused dose distribution, high linear energy transfer (LET), and lower normal tissue toxicity. Also, proton and carbon particles have a higher relative biological effectiveness (RBE) than conventional X-rays. Hence, CPT has the potential to increase the efficacy of RT, particularly for controlling unresectable radioresistant tumors.
Cancer radiotherapy kills cancer cells mostly by inducing DNA damage. However, radiation exposure can also damage normal cells or tissues surrounding the tumor, leading to side effects. For example, radiation received by the heart during the treatment of pediatric, lung and breast cancer substantially increases the risk of developing cardiovascular diseases later in life (8). Similarly, studies indicate childhood cancer patients who received radiation to the brain develop cognitive impairment later in life, possibly because of damage to both hippocampal- and non-hippocampal-dependent domains (9,10). Therefore, increasing the therapeutic ratio, that is, maximizing the effects of radiation on cancer cells while minimizing damage to surrounding healthy tissue is the major goal of modern translational radiation oncology. Finding and exploiting genetic, epigenetic or microenvironmental differences between normal and malignant tissues in each individual patient is central to success.
Many studies show that as tumors become more mutagenic, they often become defective in one aspect of DNA repair but usually try to compensate by increasing the levels of certain repair proteins in the same pathway or in a different one. Since these molecular vulnerabilities are present in tumor cells but not in normal cells, targeting these alternative or backup pathways can render the tumor radiosensitive while leaving the normal tissue relatively resistant. Small-molecule inhibitors of DNA repair factors hold great promise for targeting these tumor cell-specific molecular vulnerabilities. These inhibitors can be honed to target a single step or a single protein in a DNA repair pathway that is vital for the survival of cancer cells but not for normal cells. Every tumor is different, and the goal of truly personalized radiation oncology is to understand the tumor genetics and selectively target the tumor-specific deregulated pathways in each cancer patient. However, the development of such a precise approach is offset by several real-world challenges. First, the mutagenic phenotype seen in tumors rarely results from the under- or over-expression of a single protein, and molecular pathogenesis is rarely linked to an isolated step in oncogenic progression (11). In addition, as tumors become increasingly mutagenic, they become more complex and heterogeneous, which often limits the usefulness of targeting a single protein or pathway. Also, the multifunctionality of many DNA repair proteins means inhibition of these factors sometimes cause unexpected toxicity to normal cells (12). For these reasons, despite the early promise and our extensive knowledge of tumor biology, many small molecule inhibitors of DNA repair factors failed in clinical trial and are not routinely used in clinic by the radiation oncologists (13).
Principles of RT
Depending on the type and stage of cancer, radiotherapy (RT) is used before or in combination with other treatment modalities (neoadjuvant and combination therapy). It is also often used for palliative purposes to relieve patients from symptoms caused by cancer. Radiation is delivered to the location of the tumor either externally (external beam radiation delivered to the location of the tumor from outside the body) or internally (brachytherapy delivered by directly implanting radioactive sources into the tumor site).
DNA damage is the primary effect of RT
Damage to the cellular DNA is the most well-known effect of radiation. Regardless of the mode of delivery, low LET ionizing radiation (IR), such as X-rays, gamma rays, and high LET charged particles used in CPT, deposits energy in the cells of the tissues it passes through. This energy deposition induces DNA lesions in the cells, ultimately forcing them to die, stop proliferating, or become senescent. Radiation-induced DNA lesions are multifaceted and can include base modifications such as 8-oxoG (14), single-strand breaks (SSBs), and double-strand breaks (DSBs) (15). Indeed, 1 Gy of low LET γ-radiation induces around 850 pyrimidine lesions, 450 purine lesions, 1,000 SSBs, and 20–40 DSBs in a mammalian cell (16). DNA lesions induced by IR can lead to cytotoxic, mutagenic, and carcinogenic effects if not repaired properly (17–19).
Repair of damaged DNA is of paramount importance
To ensure genomic integrity, cells have developed sophisticated mechanisms for repairing damaged DNA (Table 1). Base excision repair (BER) and SSB repair (SSBR) are essential for repairing damaged bases and SSBs. However, the most lethal types of lesions caused by IR are DSBs. Not only does IR cause DSBs directly, but unrepaired damaged bases and SSBs convert to DSBs when they encounter a replication fork (20). The two major cellular DSB repair pathways are non-homologous end-joining (NHEJ) and homologous recombination (HR). NHEJ is an error-prone repair pathway that functions throughout all cell cycle phases, whereas HR is an error-free pathway that predominantly functions in the late S and G2 phases. Cells respond to DNA breaks by initiating a series of signaling pathways collectively known as DNA damage response (DDR) signaling. DDR contributes to the timely repair of DNA breaks, transient cell cycle arrest, transcriptional and post-transcriptional activation of a wide array of genes, and, under certain circumstances, programmed cell death. Faithful repair of DNA lesions and activation of DDR signaling are critical for cellular survival and prevention of genomic instability.
Table 1.
An overview of type of DNA lesions commonly encountered by the cell and the relevant repair pathways involved in the processing of these lesions
| Type of DNA lesions | Source | DNA damage repair pathway |
|---|---|---|
| Bulky and helix-distorting DNA adducts | UV rays and chemical mutagens like cisplatin | NER, FA pathway |
| Damaged bases and non-helix-distorting DNA lesions | Ionizing radiation and reactive oxygen species | BER. In case of impaired BER, the SSBs are converted to DSBs, which are repaired by HRR during S and G2 phase. This synthetic lethal relationship is exploited by using PARP inhibitors to kill HRR deficient tumors |
| Single-strand breaks | Ionizing radiation and reactive oxygen species | Single-strand break repair |
| Base mismatches, incorrect insertion and deletion of bases | Replication errors | MMR |
| O6-methylguanine adducts | Alkylating agents like temozolomide, reactive oxygen and nitrogen species | Direct DNA repair. In absence of MGMT, the main enzyme in the DR pathway, MMR and NER can process these lesions |
| Double strand breaks | Ionizing radiation, reactive oxygen species and replication stress | DSBs are repaired either by NHEJ or by HRR in a cell cycle specific manner. Some overlap exists in between HRR and NHEJ pathways. DSBs which are not processed by NHEJ can be processed by HRR during S and G2 phase |
| DNA ICLs | Replication events and chemotherapeutic drugs like mitomycin C | NER and FA pathway |
UV, ultraviolet; NER, nucleotide excision repair; FA, Fanconi anemia; BER, base excision repair; MMR, mismatch repair; MGMT, O6-methylguanine-DNA-methyltransferase; NHEJ, non-homologous end joining; HRR, homologous recombination repair; DSB, double-strand break; ICL, interstrand crosslink.
Dysfunctional DNA repair and cancer incidences
Deficiencies in DNA repair factors are a feature common to many cancers (21). Defects in specific DDR and DNA repair genes, such as ataxia telangiectasia mutated (ATM), Nibrin (NBS1), Werner syndrome (WRN), Fanconi anemia (FA), BRCA1 , and BRCA2, give rise to cancer-prone disease syndromes (22–26). In addition to germ line mutations, many cancers show impaired DDR and defective checkpoints either because of somatic mutations (ATM, TP53, and cyclin-dependent kinase inhibitor 2A (CDKN2A), BRCA1/2 being the most frequently mutated genes) or because of differential expression of DDR factors (6). Furthermore, DNA replication stress as a result of oncogene activation causes constitutive activation of the DDR and tumor progression (27). It has long been postulated that defects in both DNA repair and checkpoint responses in tumor cells affect the response to IR and can be exploited for targeted radiosensitization strategies (28) (Table 2A,B). However, RT also induces DNA damage in normal tissues surrounding the tumor, and using inhibitors of important molecules in DSB repair, such as ATM or DNA-dependent protein kinase (DNA-PK), in conjunction with radiotherapy increases the radiosensitivity of normal cells. Nonetheless, some degree of specificity in treatment can be achieved because of the hyper-proliferative nature of cancer cells relative to normal cells, which allows inhibitors to sensitize cancer cells to radiation more than normal cells. Similarly, replicating cancer cells are more sensitive than normal cells to chemical agents that block replication-associated processes and HR. In addition, replication can further aggravate damage in cancer cells that results from the inhibition of repair. Thus, targeting tumor-specific molecular abnormalities or use of replication stress inducing agents which affects hyper-proliferative tumor cells more than normal cells offers the possibility of selective eradication of cancer cells. In the following section, we will discuss the DNA repair pathways commonly used to repair radiation-induced DNA damage and how the inhibition of tumor cell-specific deregulated DNA repair factors and pathways can potentially be exploited to sensitize cancer cells to radiation (Figure 1).
Table 2A.
An overview of most commonly altered DNA damage response signaling genes found in different types of cancer and relevant strategies for therapeutic intervention using small molecule inhibitors
| Molecule | Function(s) in DNA repair | Type of abnormalities |
|---|---|---|
| ATM | Acts as the central activator of the DDR following DSBs | Loss of function mutation; increased copy number and autophosphorylation |
| MRE11 | DNA damage sensor as part of the MRN complex and initial processing of double-strand DNA breaks prior to NHEJ and HR | Decreased expression |
| NBS1 | DNA damage sensor as part of the MRN complex and initial processing of double-strand DNA breaks prior to NHEJ and HR | Polymorphism in NBS1 gene is most common; increased expression in some cases |
| DNA PKcs | Principle kinase in NHEJ pathway of DSB repair | Overexpression |
| Ligase IV/XRCC complex | Ligation of broken ends of DNA during the NHEJ pathway of DSB repair | Polymorphism in LIG4 gene |
| RAD51 | Initiates DNA strand invasion and DNA strand exchange in HRR | Overexpression |
| RAD54 | Facilitates RAD51 mediated DNA strand exchange and promotes branch migration of Holliday junctions | Missense mutations at functional regions of RAD54 |
| RPA | Protects and stabilizes ssDNA after resection step during the initial phase of HRR | Not known |
| BRCA1 and BRCA2 | Initiates HRR in response to DSBs and stabilizes RAD51 binding to the DNA; transcriptional regulation of many DNA repair genes | Germline and somatic loss of function mutation; BRCA1 overexpression |
| c-Abl | Indirectly regulates HRR by phosphorylating RAD51 and catalyzing strand exchange | Overexpression |
| HSP90 | Indirectly regulates HRR by facilitating correct folding and assembly of many proteins involved in HRR | Overexpression |
| APE1 | Acts as the principle endonuclease in recognition and processing of AP sites in BER; also involved in transcriptional regulation of other genes | Overexpression |
| DNA polymerase β (pel β) | Synthesizes nucleotides and removes blocking residues during BER | Overexpression; missense mutations in pol β gene coding for variant proteins |
| FEN1 | Removes 5’ ssDNA overhangs in long patch BER; also important in microhomology-dependent alternative end joining of DSBs. | Overexpression |
| PARP | PARP 1 and PARP 2 are activated in response to SSB, cleaves NAD+ generating nicotinamide and ADP-ribose scaffolds which facilitate recruitment of other factors to the damage site | Not clear |
| CHK1 | Major checkpoint kinase responsible for the G2/M arrest in response to DNA damage | Decreased expression; increased expression and autophosphorylation |
| CDC25 | Activates CDK and regulate entry into and progression through various phases of the cell cycle | Increased expression |
ATM, ataxia telangiectasia mutated; DDR, DNA damage response; CDK, cyclin dependent kinases.
Table 2B.
An overview of most commonly altered DNA damatge response signaling genes found in different types of cancer and relevant strategies for therapeutic intervention using small molecule inhibitors
| Molecule | Relevance in cancer | Therapeutic intervention strategies |
|---|---|---|
| ATM | Loss of function in lung, colorectal, pancreatic, breast, hematopoietic cancers. Hyperactivated in some prostrate, bladder and breast cancer | KU55933 (29,30) |
| MRE11 | Breast, colorectal, gastric, ovarian, head and neck cancer | Mirin as an indirect inhibitor of HRR (31) |
| NBS1 | Increased risk of breast, colorectal, lung, nasopharyngeal carcinoma, acute lymphoblastic leukemia with NBS1 polymorphism. NBS1 overexpression in esophageal, head and neck, lung cancer and hepatomas | Mirin as an inhibitor of MRN complex (31) |
| DNA PKcs | Glioblastoma, gastric, prostrate, lung, esophageal and oral squamous cell carcinoma | Wortmannin (32); LY294002 (33); NU7026 (34,35); CC-115 (12); CC-122 (36); NVP-BEZ235 (37) |
| Ligase IV/XRCC complex | Breast, ovarian and pediatric brain tumors | SCR7 as an inhibitor of NHEJ pathway (38) |
| RAD51 | Pancreatic cancer, breast cancer, prostate cancer, soft tissue sarcoma, and leukemia | RI-1 and its optimized version RI-2 as inhibitors of recombinase function of RAD51 (39,40). This can be of particular interest for radio-immunotherapy and carbon particle therapy |
| RAD54 | Breast cancer, colon cancer, lymphoma | Streptonigrin as an inhibitor of RAD54 ATPase function, thus inhibiting HRR (41) |
| RPA | RPA has been identified as an auto antigen in breast cancer; increased RPA expression as a prognostic biomarker in some colon cancer | Compound 8 as a disruptor of RPA’s N-terminal domain interacting proteins (42) |
| BRCA1 and BRCA2 | Loss of function in breast and ovarian cancer; BRCA1 overexpression in some lung cancers | PARP inhibitors (e.g., iniparib, olaparib and 2nd and 3rd generation inhibitors) are used to treat BRCA deficient cancers using synthetic lethal approach (43–48) |
| c-Abl | Teratoid and malignant rhabdoid tumors | Tyrosine kinase inhibitors like imatinib, dasatinib and bosutinib as indirect inhibitors of HRR (12,49–51) |
| HSP90 | Many solid tumors including breast, ovarian, uterine, oral and renal carcinoma | Different classes of HSP90 inhibitors e.g., 17-DMAG, tanespimycin, retaspimycin, PU-H71, ganetespib as indirect inhibitors of HRR (12,52,53) |
| APE1 | Lung, breast, head and neck, bladder, and hepatocellular, cervical carcinoma. Also overexpressed in germ cell tumors, osteosarcoma, medulloblastoma and many solid tumors; associated with chemo and radio resistance | Methoxyamine (54); lucanthone (55); AR-03 (56); ML199 (57) as inhibitors of DNA repair function of APE1. E330 as an inhibitor of redox function of APE1 (12,58) |
| DNA polymerase β (pol β) | Pol β variant proteins in gastric, colorectal, prostrate, lung, breast, bladder, esophageal cancer. Overexpression in breast, prostate and colon cancer; also associated with chemo- and radio resistance | NCS-666715 and NSC-124854 (59,60) |
| FEN1 | Lung, pancreatic, breast, prostrate, gastric cancer and neuroblastomas | N-hydroxy urea containing compounds (61) |
| PARP | Expression of PARP-1 protein in cancer and its associations with outcome are yet poorly characterized | PARP inhibitors (e.g., iniparib, olaparib and 2nd and 3rd generation inhibitors) are used to treat BRCA deficient cancers using synthetic lethal approach (43–48) |
| CHK1 | Decreased expression in breast cancers; increased expression and activation in lung, liver, colon and cervical cancer. Cancer cells with impaired G1/S checkpoint are over-dependent on G2/M checkpoint to prevent premature mitotic entry and cell death | UCN-01 (62,63), XL-844 (64) and AZD7762 (65) |
| CDC25 | Breast, ovary, thyroid, colorectal, prostrate liver, esophageal and brain cancer | Still under investigation. Some of the reported CDC25 inhibitors belong to various chemical classes including phosphate bioisosteres, electrophilic entities, and quinonoids (66,67) |
ATM, ataxia telangiectasia mutated; DDR, DNA damage response; CDK, cyclin dependent kinases.
Figure 1.
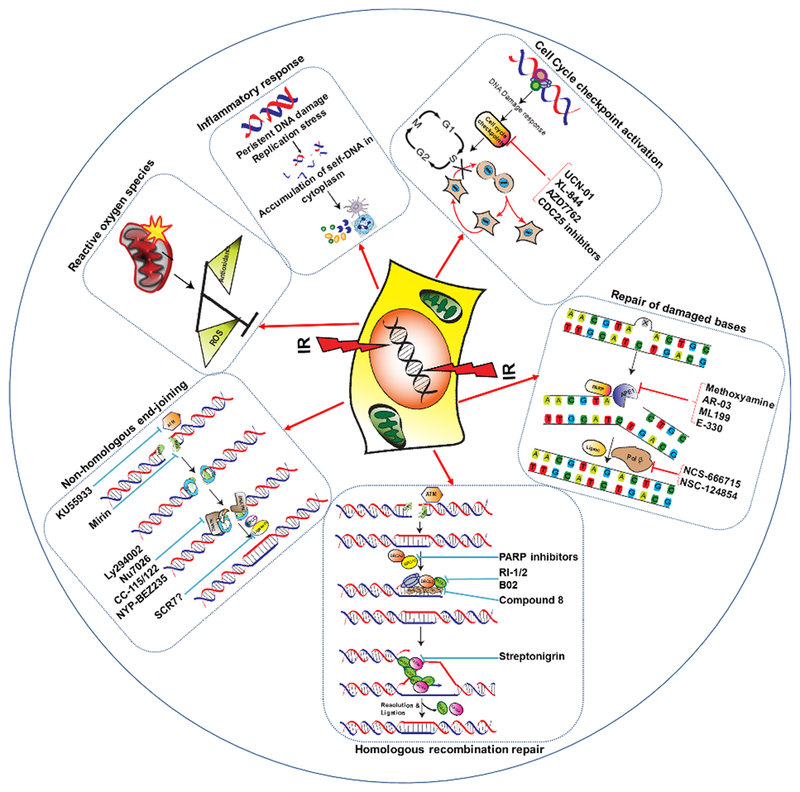
Schematics of direct and indirect effects of ionizing radiation and strategies for the therapeutic intervention. ROS, reactive oxygen species; PARP, poly (ADP-ribose) polymerase; IR, ionizing radiation; ATM, ataxia telangiectasia mutated.
Targeting DNA repair as a cancer therapy
Utilizing synthetic lethality in cancer treatment
In the context of DNA repair, synthetic lethality between two repair pathways arises when the loss of either pathway individually is tolerable, but the simultaneous loss of both pathways is lethal to the cell. It has long been postulated that a synthetic lethal approach could be useful for cancer treatment (68), and utilizing the poly (ADP-ribose) polymerase (PARP)-BRCA interaction is the best example of exploiting the synthetic lethal relationship that has proven clinically useful to control certain types of cancer (43). PARP1 facilitates repair of damaged bases and SSBs by acting as a scaffold for other BER proteins. Inhibiting PARP1 impairs BER, which results in the accumulation of DSBs that have converted from unrepaired SSBs during replication. Normal cells, with intact HR, are not substantially affected (69,70). In contrast, studies have shown that HR-impaired BRCA1/2-deficient tumors are highly sensitive to DNA damage in the presence of PARP inhibitors, both in tumor models in vivo and in the clinic (44–47,71). Since then, different PARP inhibitors, such as olaparib (AZD2461), and iniparib (BSI-201), have been subjects of intense investigation, either as mono- or combination therapies to treat breast cancer patients. However, a phase III trial of iniparib (BSI-201) failed to show major advantages in the treatment of metastatic triple negative breast cancer (TNBC) patients (72), and phase III development of olaparib (AZD-2281) to treat hereditary BRCA1- and BRCA2-associated breast cancer was halted in 2011 (73). These failures can be explained in part by the multifunctionality of the PARP1 protein, insufficient inhibition of PARP 1 DNA repair activity, as well as heavily pretreated patient cohorts and the phenotypical heterogeneity of some cancers (12). Despite these setbacks, 115 different PARP inhibitors are currently in the clinical trial phase (12), and efforts are underway to find more synthetic lethal relationships between DNA repair proteins that can be therapeutically exploited. In addition to BRCA1/2-deficient cancers, PTEN1- and ATM-deficient cancers and colorectal cancers containing microsatellite instabilities are also sensitive to PARP inhibitors (74).
Inhibition of BER in cancer treatment
BER corrects SSBs and non-helix-distorting small base lesions that arise from IR. There are two sub pathways: short-patch BER, where only one nucleotide is replaced, usually during the G1 phase; and long-patch BER, where 2–13 nucleotides are replaced in the S/G2 phase of the cell cycle. The repair mechanism consists of five key enzymatic steps that involve excision of the damaged base by DNA glycosylase, nicking of the damaged DNA strand by AP endonuclease, removal of the sugar fragment by phosphodiesterase, filling of the gap by DNA polymerase, and finally, sealing of the nick by DNA ligase (75,76).
Among the many factors involved in BER, apurinic/apyrimidinic endonuclease 1 (APE1) accounts for over 95% of the total AP endonuclease activity in human cell lines and, besides DNA repair, also performs functions such as redox regulation (mediated through a separate redox domain) and transcriptional regulation of other genes. APE1 dysregulation or upregulation is a common feature in many solid cancers (77,78). Furthermore, overexpression of APE1 is linked with chemo- and radio-resistance of tumors, rapid progression, and overall poor prognosis of the disease (79). Inhibition of APE1, when combined with radiotherapy or DNA damage-inducing chemicals, increases tumor cell killing (80,81). Further, blocking the redox activity of APE1 has been shown to negatively attenuate tumor cell growth and angiogenesis (82,83). Additionally, inhibition of APE1 redox activity and STAT3 activation together causes synthetic lethality in human pancreatic and glioblastoma cell lines (84). Thus, inhibition of APE coupled with radio- or chemo-therapy is suitable for targeting multiple DNA repair and signaling pathways that promote tumor growth and survival. Several inhibitors of APE1 are in various stages of pre-clinical development. A broad spectrum BER inhibitor, methoxyamine (MX, or TRC102), is used in combination treatment for different types of cancer (54). In addition, several non-specific APE inhibitors act as topoisomerase II poisons and can potentially be clinically useful for treating cancers with high topo II expression (54). E3330, a selective inhibitor of APE’s redox function, has shown promise for treating pancreatic cancer and acute lymphoblastic leukemia (12,58). In addition, several direct inhibitors of APE1, like ML199 (57), AR03 (56), and compounds containing 2-methyl-4-amino-6,7-dioxolo-quinoline structure (85), have shown promise in vitro.
DNA polymerase β (pol β) synthesizes nucleotides and removes blocking residues during BER. Like APE1, pol β is often upregulated in many cancers and is linked with resistance to RT and chemotherapeutic agents. Two new inhibitors of pol β, NCS-666715 and NSC-124854, have been shown to sensitize colon cancer cells to DNA damage in vitro and are currently under evaluation in animal models (59,60). Inhibition of another endonuclease, Fenl, which is involved in long-patch BER and overexpressed in many cancers, has shown promise in a cell culture system (12,62). Finally, PARP1 and PARP2 are important sensors for SSBs and facilitate recruitment of additional BER factors, such as XRCC1, to the SSB site (86). As discussed earlier, many inhibitors of PARP1, are already in clinical use, and trials are ongoing for second- and third-generation PARP inhibitors.
Inhibition of DSB repair in cancer treatment
DSBs are the most harmful type of DNA damage induced by IR. One Gy of low LET IR is capable of producing 20–40 DSBs in a cell, as well as around 1,000 SSBs, many of which are converted to DSBs during replication (16). In addition, the reactive oxygen species (ROS) produced as a secondary effect of IR also induces DSBs. In mammalian cells, DSB are repaired by two main pathways: NHEJ and HR.
Inhibition of NHEJ in cancer treatment
NHEJ is the predominant DSB repair pathway in G0/G1 cells but remains active throughout the cell cycle. The main goal of NHEJ-mediated repair is fast rejoining of two broken DNA ends with minimal end processing, regardless of sequence homology. However, NHEJ-mediated repair results in the loss of up to 20 nucleotides from either side of the DSB junction, making it error-prone (87). Classical NHEJ-mediated DNA repair consists of four steps: recognition and binding of broken DNA ends by Ku70/Ku80 heterodimer; recruitment and stabilization of the NHEJ factors, like DNA-PK catalytic subunit (DNA-PKcs) and Artemis, at the DSB for bridging the DNA ends; processing of the DNA ends to make them suitable for ligation, which is mediated by different exo- and endo-nucleases; synthesis of new nucleotides by DNA polymerases μ and λ; and finally, ligation of the two severed DNA ends by ligase TV/XRCC4/XLF complex (88). Many studies have shown that defective NHEJ-mediated DSB repair is associated with increased risk of carcinogenesis, and functional NHEJ is linked with better treatment outcomes (12). On the other hand, overexpression of NHEJ factors or increased NHEJ activity is known to make cancer cells resistant to chemo- or radio-therapy (6,89). Despite our extensive knowledge about the NHEJ pathway, only DNA-PKcs has been exploited as a target for anticancer therapy so far. DNA-PK is overexpressed in many different types of cancer, including gastric cancers, oral squamous cell carcinoma, lung carcinoma, and esophageal cancer (90–92). Recent discoveries also identify DNA-PKcs as a master regulator of cancer metastases (93–95). Two small-molecule pan-PI3K inhibitors, wortmannin and LY294002, have been used to inhibit DNA-PKcs activity in vitro but have toxic off-target effects and are too pharmacologically unstable to be used in human patients (32,33). NU7026 is a more selective and potent DNA-PK inhibitor and has been shown to radiosensitize cancer cells in vitro (34,35). Three other small molecules, CC-115 (dual inhibitor of DNA-PK and mTOR), CC-f 22 (pleiotropic pathway modulator), and NVP-BEZ235 (oral pan-class I PI3K inhibitor), are currently in different phases of clinical trial (12,36,37). In addition to DNA-PK, ligase IV has also been investigated as a target for its anticancer therapeutic value. SCR7, an inhibitor of ligase IV DNA binding activity, has been shown to inhibit NHEJ both in vitro and in vivo. It also potentiates the effect of radiation or chemotherapy by promoting the accumulation of DSBs and inducing apoptosis of cancer cells (38).
Inhibition of HR repair (HRR) in cancer treatment
HR is the predominant DSB repair pathway in the S and G2 phases of the cell cycle, where a homologous sequence of the sister chromatid acts as a template for DNA synthesis and gap filling in an error-free manner. Unrepaired radiation-induced damaged bases and SSBs are converted into DSBs during replication and require HRR to repair (96,97). Furthermore, in vitro studies suggest that HRR is essential for processing clustered DSBs induced by charged particles.
Mechanistically, HRR consists of three steps: first, during the pre-synaptic step, a DSB is recognized and resected to generate single-strand DNA overhangs. These are bound by the protective replication protein A (RPA) to prevent self-annealing. RAD51 and other associated proteins then bind to the DNA ends. Next, during the synaptic step, the resultant nucleoprotein filament invades the sister chromatid or homologous chromosome in search of homologous regions to form heteroduplex DNA. New nucleotides are added to the 3’ end of the invading strand by polymerase η using the sister strand as a template. An extended D-loop is formed which captures the second end of the break resulting in formation of Holliday junction (HJ). Once formed, the HJ undergo branch migration generating varying lengths of heteroduplex DNA. The final post-synaptic step involves resolving the HJ and migration of repaired ends migrate toward each other to restore the duplex DNA (98).
Deregulated HR activity is observed in many cancers, either due to defects in one of the HRR proteins or as a result of compensatory upregulation of HRR proteins following the loss of another DNA repair pathway. Germ line or somatic BRCA1 and BRCA2 mutations, which account for breast and ovarian cancers, have been studied comprehensively and can be sensitized to DNA damage by treatment with PARP inhibitors. Elevation of the HRR pathway has also been correlated with incidences of sporadic breast cancer (99). Deregulated expression of MRN complex proteins, which act as HR damage sensors, is observed in melanoma, ovarian, colorectal, and head and neck cancers (89). Mirin, a selective small-molecule inhibitor of MREll, sensitizes cancer cells to DNA damage, but its mechanism as an inhibitor of HRR is unclear, since MRN complex also acts upstream of NHEJ (31). RPA complex and RAD51 are two essential proteins involved in the HRR process. Targeted inhibition of RPA has been difficult because of its multiple functions beside DNA repair. Recently, efforts have been made to block RPAb N-terminal domain interaction with other proteins, which are essential for DNA repair, without compromising its DNA binding activity (42). RAD51 is upregulated in pancreatic cancer, breast cancer, soft tissue sarcoma, and leukemia (100), and tumor response to radiation correlates directly with RAD51 expression levels (101). Additionally, evidence suggests that the RAD51 -mediated HRR pathway is critical for processing the complex DSBs induced by charged particles, including protons and carbon (12C) ions used in treatment of aggressive and unresectable tumors (102–104). Thus, RAD51 is a valid target for sensitizing cancer cells to radiation. RI-1, a small-molecule inhibitor of RAD51’s recombinase activity, has shown promise in vitro and ex vivo conditions, but further in vivo studies regarding its pharmacologic evaluation and efficacy are needed (39,40,105). Recent studies suggest that the inhibition of RAD52 and RAD54, which are RAD51 paralogs, can be clinically useful for killing BRCA2-deficient tumors (41,106,107). Despite our knowledge of deregulated HRR pathways in many cancers, only inhibitors of c-Abl and HSP90, two proteins that are known to regulate HRR activity only indirectly, are currently in clinic or in advanced clinical trials as anticancer agents (49,52). The non-receptor tyrosine kinase c-Abl is activated by ATM in response to DNA damage and phosphorylates RAD51, and treatment with tyrosine kinase inhibitor imatinib sensitizes the cancer cells to radiation (50). Furthermore, inhibiting RAD51 has proven effective in killing imatinib-resistant CML cells (51). HSP90 is a molecular chaperone that corrects folding of many proteins including RAD51. An HSP90 inhibitor, 17-allylamino-17-demethoxygeldanamycin, radiosensitizes human tumor cell lines by inhibiting RAD51-mediated HRR pathway (53). However, better small molecule inhibitors of HRR factors with higher specificity and improved efficacy are need to be developed to take full advantage of deregulated HRR pathway seen in many tumors.
Inhibition of cell cycle checkpoints in cancer treatment
Besides DNA repair, activation of cell cycle checkpoints is a vital part in the DDR signaling that has been investigated as a potential target for cancer therapy. Unlike normal cells, cancer cells often show impaired G1/S cell cycle checkpoint activation and depend heavily on S and G2 checkpoints to prevent premature mitotic entry with unrepaired DNA damage. Thus, inhibition of the G2 block using checkpoint inhibitors will augment DNA damage in cancer cells and ultimately result in selective killing of tumor cells (108). Among checkpoint inhibitors, UCN-01 (a dual CHK1/CHK2 inhibitor) showed promise in pre-clinical models but was not clinically successful because of poor bioavailability and unacceptable side effects (62,63). Similarly, other CHK1/CHK2 inhibitors, like XL-844 and AZD7762, failed to show substantial improvement in clinical trials (63–65). Recently, inhibitors of CDC25 phosphatase, a downstream substrate of ATM kinase and a regulator of the cell cycle, are being tested as therapeutic targets in oncology (66,67).
ROS as secondary effects of radiation
In addition to directly inducing nuclear DNA damage, IR exposure leads to increased production of ROS that may damage nucleic acids, proteins, and lipids (Radiobiology for the Radiologist, 7th Edition by Eric J. Hall and Amato J. Giaccia), leading to persistent cellular oxidative stress even after initial radiation exposure. Radiation also results in excess production of ROS by mitochondria, which can result in cell death (109). Thus, IR induces damage in nuclear and extranuclear DNA (e.g., mitochondria) not only by direct deposition of energy but also by excess production of ROS. Enhanced ROS levels induce cellular oxidative stress even long after radiation exposure, often limiting cellular proliferation and inducing cell death or apoptosis.
Inhibition of direct DNA repair (DR) in cancer treatment
Clinical alkylating agents like temozolomide (TMZ) or radiation-induced ROS generate adducts that can compromise genomic integrity (110). O6-meG adducts formed in DNA are mutagenic, block DNA polymerase activity, and are considered cytotoxic. DR of O6-meG adducts is mediated by a single enzyme, O6-methylguanine-DNA-methyltransferase (MGMT), using a non-reversible chemical reaction that does not require a nucleotide template, breakage of the phosphodiester backbone, or new DNA synthesis (110). Increased expression of MGMT negatively affects cancer prognosis. Almost 90% of recurrent gliomas show overexpression of MGMT and are resistant to chemotherapy (111). Conversely, targeted inhibition of MGMT increases cancer cells’ sensitivity to alkylating agents. O6-benzylguanine (O6-BG) and its derivative, O6-(4-bromothenyl) guanine, are currently being investigated for the treatment of TMZ-resistant gliomas in pre-clinical and clinical settings in combination with RT(111).
Charged particles in cancer treatment
Advantages of CPT over conventional RT
Charged particle radiation using protons and nuclei of carbon, neon, helium, and silicon has greater potential to control tumors than X-ray radiation. The theoretical advantages of CPT are better spatial selectivity in dose deposition and better efficiency in cell killing. This is because charged particle radiation has a Bragg peak, which maximizes the dose distribution within the target volume (clinical tumor volume or CTV) while minimizing the effect on the surrounding normal tissue (112). Thus, proton and carbon particles have a higher RBE than conventional X-rays. Depending on the biological endpoint used to measure RBE, proton beams have RBE values between 1.0 and 1.2 (113), and carbon ion beams have RBE ranging from 1.5 to 5 (114). Carbon and proton beams are better equipped to kill cancer stem cells (CSCs) associated with colon, pancreatic, and mammary carcinoma (115–118), which are responsible for making tumors radio- and chemoresistant (119). Despite these advantages, only protons and, to some extent, carbon ions have been used clinically for treating cancer patients because of the high capital and operational costs associated with CPT. Currently, only 49 proton (14 in the United States) and 6 carbon ion (none in the United States) treatment centers operate worldwide, mostly in Asia and Europe.
Use of proton beams in cancer treatment
As of 2013, 93,895 patients have received proton therapy during cancer treatment. A significant proportion of these patients are children. The National Cancer Institute reported 10,380 new cases and more than 1,200 deaths from pediatric cancer (age 0–14) in 2016 (https://www.cancer.gov/types/childhood-cancers). Leukemia, brain and central nervous system (CNS) tumors, soft tissue sarcoma, and neuroblastoma are the most common types of childhood cancers. Although pediatric cancer patients are relatively few in number (1 % of total cancer), there is great concern regarding the long-term effects of high dose radiation on surrounding normal tissues that are still developing in children (120). Because of its efficacy in sparing normal tissue, proton therapy has been used to treat pediatric cancers, including Hodgkin’s lymphoma, tumors of the skull base, medulloblastoma, rhabdomyosarcoma, ependymomas, and low-grade astrocytomas. Although the full effect of proton therapy will not be clear for many years, initial studies and dosimetry analyses show that proton therapy is well tolerated, shows better local control of tumors, and has a lower risk of normal tissue toxicity and secondary cancers than conventional RT (121–123). Proton therapy has also been used extensively to treat ocular melanomas with favorable treatment outcomes (95% local control rate and a 90% eye retention rate); it has been particularly useful in the treatment of large ocular melanomas not approachable via brachytherapy (124). Proton therapy has also proven effective in controlling spinal cord tumors, including chordomas and meningiomas, with 80–90% local control and tolerable toxicity (124). Proton therapy has also been used to treat prostate cancer patients with treatment outcomes similar to intensity-modulated RT (IMRT) but with less normal tissue toxicity (124). Initial results also indicate that proton therapy can be clinically useful for treating head and neck cancers (125–127) and breast cancer (128), achieving the same local control of the disease as conventional RT but with lower toxicity to surrounding normal tissues.
Use of carbon beams in cancer treatment
In recent years, carbon ion radiotherapy has gained immense popularity because it provides sharp lateral penumbra, sharp distal dose fall off, less dependence on hypoxic conditions, less dependence on cell cycle phase, and higher RBE, which may be advantageous for treating radioresistant tumors that require superior dose conformity while reducing the collateral effect on surrounding normal tissues (129,130). As of 2014, almost 15,000 cancer patients have been treated with carbon ions (131), and new carbon ion treatment centers are in different stages of development in the United States, Australia, South Korea, Taiwan, and China (132). Two independent studies performed among pediatric cancer patients with chordoma, chondrosarcoma, osteosarcoma, skull base, head and neck, and CNS tumors found carbon ion radiation to be effective in disease control and well tolerated by patients (133,134). Carbon ion radiotherapy has also demonstrated efficacy in treating bone and soft tissue sarcomas, including large, unresectable sarcomas and sacral chordomas, which are normally radioresistant and have a poor prognosis (135–138). Two different studies conducted among advanced, radioresistant head and neck cancer patients also concluded that carbon ion RT substantially improved patient survival (139,140). Another study conducted among 64 patients found carbon particle radiation to be an effective and safe treatment option for hepatocellular carcinoma, which can only be partially treated with surgery and has low tolerance for conventional RT (141). Pancreatic cancer is the fourth most common cause of cancer death in the United States, with a survival rate of less than 10%. A study conducted by National Institute of Radiological Sciences (NIRS), Japan found that preoperative, short-course carbon ion radiotherapy followed by surgery substantially increased 5-year survival rate in pancreatic cancer patients without any increase in toxicity (142). Excellent local control of the disease and a 59% 5-year survival rate were also obtained when locally recurrent, radioresistant renal cancer patients were treated with an escalating dose of carbon ion radiation (143). Lung cancer is the leading cause of cancer-related death worldwide (144). Hypofractionated carbon ion radiation of unresectable non-small cell lung tumors resulted in almost 90% local control of the disease and improved 5-year survival rate (71,145). More clinical trials assessing the efficacy of carbon ion therapy in lung cancer patients are ongoing (132).
Inhibition of DNA repair coupled with charged particle RT
Recent pre-clinical studies demonstrated that inhibition of NHEJ using NU7026, a potent DNA-PK inhibitor, can further sensitize non-small cell lung cancer cells to carbon ion radiation (34,146). However, carbon ions’ high LET induces clustered DNA damage, a class of lesions comprising DSBs, SSBs, and abasic sites within one or two helical turns of DNA, induced by a single radiation track (147). Evidence suggests that HRR, but not the NHEJ pathway, is critical for processing DSBs induced by heavy ions, including carbon ions (102–104). Thus, inhibition of HRR factors can potentially be used to sensitize tumors to carbon particle therapy. Further understanding of the radiobiology of carbon ion particles, including the nature of the DNA lesions induced, the pathway used for repairing DNA lesions, and the fidelity of the DNA lesions repaired, is needed to achieve the greatest benefits of carbon particle therapy.
Combining radiation and immunotherapy approaches for cancer treatment
In 1980, Stone and colleagues observed that the host’s immune competence often determines tumor response to RT. Yet, deliberate efforts to channel RT to hone the immune system against tumor cells have only started in the last decade. Tumor cells are known to thwart the immune system by attracting immunosuppressive cells or factors in the microenvironment. Ipilimumab and pembrolizumab, two monoclonal antibodies that have been used clinically to treat metastatic melanoma, target the immune suppressive factors cytotoxic T-lymphocytes-associated protein 4 (CLTA4) and programmed cell death protein 1 (PD1), respectively (148). Limited availability of dendritic cells (DCs) to present the tumor antigens also contributes to the immune system’s tolerance of tumors (149). Recent pre-clinical studies have found that dying tumor cells exposed to RT accumulate cytosolic DNA, which activates the stimulator of interferon genes (STING) pathway (150,151). STING activation is critical for the expression of inflammatory cytokines and chemokines, specifically type I interferon (IFN), which in turn recruits DCs to the tumor and activates them. Activated DCs are capable of presenting the tumor antigens to T cells, which primes the T cells to selectively kill tumor cells (150,151). Thus, radiation can activate the cell-mediated immune response against tumors by enhancing tumor DNA delivery to DCs. Recently, Matsunaga and colleagues showed that carbon particle radiation coupled with DC-based immunotherapy resulted in control of primary tumors in immune competent mice (152). Further, these challenged animals developed long lasting CD8+ T-cell-mediated anti-tumor immunity, which prevented recurrence of the tumors. They could not replicate these results in immune-deficient nude mice, however, thus highlighting the importance of the immune system in controlling the tumor (152). In addition to priming DCs, RT also activates a specific subset of neutrophils, which induces oxidative stress on the tumor cells and eventually activates tumor-specific cytotoxic T cells (153). In addition to the adaptive immune response, studies have suggested that DNA repair and replication factors play a role in the innate immune response. For example, ATM-deficient cells were found to have increased cytosolic self-DNA, leading to increased inflammation (154). Similarly, MRE11, a DSB sensor protein, recognizes cytosolic DNA and initiates innate immune response signaling (155). Moreover, RPA2 and RAD51 were shown to protect the cytosol from the accumulation of self-DNA (156). Our recent study found a link between DNA replication, repair, and immune response after exposure to radiation (157). Our in vitro studies indicate that, in the absence of RAD51, exposure to radiation leads to increased replication stress, which results in the accumulation of cytosolic DNA, triggering a STING-mediated immune response. However, further in vivo studies are necessary to determine the efficacy of using small-molecule HRR inhibitors coupled with RT as an immunotherapeutic approach for killing tumor cells.
Current limitations and future direction
Abnormalities of the proteins involved in DDR and repair are common in cancers and may be induced by both genetic and epigenetic causes. Thus, the potential clinical utility of DNA repair inhibitors is encouraging. However, the greatest obstacle to developing inhibitors against DNA repair proteins is tumor heterogeneity, as levels of DNA repair proteins vary widely not only between different types of tumors, but also among patients with the same type of tumor. Registering the deregulated pathways and the cause of tumorigenesis in a given patient is necessary to determine the optimum treatment for that patient. However, analyzing the massive genomics and proteomics data to determine whether mutations in a particular gene or abnormal expression of a protein are clinically useful as a prognostic or predictive biomarker remains a daunting task. Also, both the specificity and the efficacy of small-molecule inhibitors should be taken into account when choosing the right course of treatment. Many DNA repair inhibitors that sensitize cancer cells to radiation are not specific to tumors, as these factors also affect normal tissue survival and function to some degree. Thus, applying small-molecule inhibitors with a low to moderate degree of specificity carries an inherent risk to clinical success. Therefore, utilizing our knowledge of synthetic lethal interactions, which make cancer cells overly dependent on one pathway in the absence of other functional pathways, and translating these cancer cell-specific molecular vulnerabilities into therapies can be pivotal for increasing the therapeutic ratio for radiation oncology. Even then, the evolution of resistance often limits the efficacy of the treatment. For example, BRCA1/2 mutant tumors treated with PARP1 inhibitors often generate a secondary mutation that makes them resistant to PARP inhibition (158–160). Additionally, many factors affect the tumor response to DNA damage and RT, including the tumor microenvironment. For instance, most cancer cells are hypoxic and show increased expression of HIF-1. Hypoxia is known to induce tumor radio resistance through the activation of HIF-1, which is entwined with molecules that are central to DDR and checkpoint control; thus, is rightly considered an attractive target molecule for cancer therapy (6).
In the last decade, research and clinical trials have proved the therapeutic importance of using DNA repair inhibitors can be beneficial for cancer treatment. The advent of databases like REPAIRtoire (http://repairtoire.genesilico.pl/) is a valuable resource, which lists known diseases correlated with mutations in genes encoding DNA repair proteins. Further understanding of the basic biology underlying the DDR and the mechanisms responsible for its dysregulation in cancer will provide exciting opportunities for new and effective cancer therapies. However, to take full advantage of the discoveries made in the laboratories, biomarker tests in the clinics need to be reliable, not cost prohibitive and able to be run with existing clinical facility in a timely manner. When combined with new discoveries of improved DNA repair inhibitors, patient specific biomarker detection will enable earlier diagnosis and better treatment of cancer, which is the ultimate goal of 21st century precision medicine.
Acknowledgements
We thank Dr. Jonathan Feinberg for the critical reading of this review.
Funding: The research in Asaithamby Aroumougame’s laboratory was partially supported by National Aeronautics and Space Administration (NNX13AD57G and NNX15AE06G), Cancer Prevention and Research Institute of Texas (RP160520), and National Institutes of Health (R01AG053341) grants.
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Bray F, Ren JS, Masuyer E, et al. Global estimates of cancer prevalence for 27 sites in the adult population in 2008. Int J Cancer 2013;132:1133–45. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646–74. [DOI] [PubMed] [Google Scholar]
- 3.Delaney G, Jacob S, Featherstone C, et al. The role of radiotherapy in cancer treatment: estimating optimal utilization from a review of evidence-based clinical guidelines. Cancer 2005;104:1129–37. [DOI] [PubMed] [Google Scholar]
- 4.Ringborg U, Bergqvist D, Brorsson B, et al. The Swedish Council on Technology Assessment in Health Care (SBU) systematic overview of radiotherapy for cancel-including a prospective survey of radiotherapy practice in Sweden 2001--summary and conclusions. Acta Oncol 2003;42:357–65. [DOI] [PubMed] [Google Scholar]
- 5.Hicks WL Jr, Kuriakose MA, Loree TR, et al. Surgery versus radiation therapy as single-modality treatment of tonsillar fossa carcinoma: the Roswell Park Cancel-Institute experience (1971-1991). Laryngoscope 1998;108:1014–9. [DOI] [PubMed] [Google Scholar]
- 6.Begg AC, Stewart FA, Vens C. Strategies to improve radiotherapy with targeted drugs. Nat Rev Cancer 2011;11:239–53. [DOI] [PubMed] [Google Scholar]
- 7.Barnett GC, West CM, Dunning AM, et al. Noimal tissue reactions to radiotherapy: towards tailoring treatment dose by genotype. Nat Rev Cancer 2009;9:134–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bhattachaiya S, Asaithamby A. Ionizing radiation and heart risks. Semin Cell Dev Biol 2016;58:14–25. [DOI] [PubMed] [Google Scholar]
- 9.Balentova S, Adamkov M. Molecular, Cellular and Functional Effects of Radiation-Induced Brain Injury: A Review. Int J Mol Sci 2015;16:27796–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Greene-Schloesser D, Robbins ME, Peiffer AM, et al. Radiation-induced brain injury: A review. Front Oncol 2012;2:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Plummer R Perspective on the pipeline of drugs being developed with modulation of DNA damage as a target. Clin Cancer Res 2010;16:4527–31. [DOI] [PubMed] [Google Scholar]
- 12.Kelley MR, Logsdon D, Fishel ML. Targeting DNA repair-pathways for cancer treatment: what’s new? Future Oncol 2014;10:1215–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bibault JE, Fumagalli I, Ferte C, et al. Personalized radiation therapy and biomarker-driven treatment strategies: a systematic review. Cancer Metastasis Rev 2013;32:479–92. [DOI] [PubMed] [Google Scholar]
- 14.Cadet J, Delatour T, Dould T, et al. Hydroxyl radicals and DNA base damage. Mutat Res 1999;424:9–21. [DOI] [PubMed] [Google Scholar]
- 15.Nikjoo H, Uehara S, Wilson WE, et al. Track structure in radiation biology: theory and applications. Int J Radiat Biol 1998;73:355–64. [DOI] [PubMed] [Google Scholar]
- 16.Lomax ME, Folkes LK, O’Neill P. Biological consequences of radiation-induced DNA damage: relevance to radiotherapy. Clin Oncol (R Coll Radiol) 2013;25:578–85. [DOI] [PubMed] [Google Scholar]
- 17.Asaithamby A, Chen DJ. Mechanism of cluster DNA damage repair in response to high-atomic number and energy particles radiation. Mutat Res 2011;711:87–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Asaithamby A, Hu B, Chen DJ. Unrepaired clustered DNA lesions induce chromosome breakage in human cells. Proc Natl Acad Sci U S A 2011;108:8293–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Asaithamby A, Uematsu N, Chatterjee A, et al. Repair of HZE-particle-induced DNA double-strand breaks in normal human fibroblasts. Radiat Res 2008;169:437–46. [DOI] [PubMed] [Google Scholar]
- 20.Caldecott KW. Single-strand break repair and genetic disease. Nat Rev Genet 2008;9:619–31. [DOI] [PubMed] [Google Scholar]
- 21.Jackson SP, Bartek J The DNA-damage response in human biology and disease. Nature 2009;461:1071–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Su F, Mukherjee S, Yang Y, et al. Nonenzymatic role for WRN in preserving nascent DNA strands after replication stress. Cell Rep 2014;9:1387–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mathew CG. Fanconi anaemia genes and susceptibility to cancer. Oncogene 2006;25:5875–84. [DOI] [PubMed] [Google Scholar]
- 24.Roy R, Chun J, Powell SN. BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat Rev Cancer 2011;12:68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shiloh Y ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer 2003;3:155–68. [DOI] [PubMed] [Google Scholar]
- 26.Su F, Bhattacharya S, Abdisalaam S, et al. Replication stress induced site-specific phosphorylation targets WRN to the ubiquitin-proteasome pathway. Oncotarget 2016;7:46–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jeggo PA, Pearl LH, Carr AM. DNA repair, genome stability and cancer: a historical perspective. Nat Rev Cancer 2016;16:35–42. [DOI] [PubMed] [Google Scholar]
- 28.O’Connor MJ. Targeting the DNA Damage Response in Cancer. Mol Cell 2015;60:547–60. [DOI] [PubMed] [Google Scholar]
- 29.Hickson I, Zhao Y, Richardson CJ, et al. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res 2004;64:9152–9. [DOI] [PubMed] [Google Scholar]
- 30.Rainey MD, Charlton ME, Stanton RV et al. Transient inhibition of ATM kinase is sufficient to enhance cellular sensitivity to ionizing radiation. Cancer Res 2008;68:7466–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dupre A, Boyer-Chatenet L, Sattler RM, et al. A forward chemical genetic screen reveals an inhibitor of the Mrel 1-Rad50-Nbsl complex. Nat Chem Biol 2008;4:119–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hashimoto M, Rao S, Tokuno O, et al. DNA-PK: the major target for wortmannin-mediated radiosensitization by the inhibition of DSB repair via NHEJ pathway. J Radiat Res 2003;44:151–9. [DOI] [PubMed] [Google Scholar]
- 33.Collis SJ, DeWeese TL, Jeggo PA, et al. The life and death of DNA-PK. Oncogene 2005;24:949–61. [DOI] [PubMed] [Google Scholar]
- 34.Willmore E, de Caux S, Sunter NJ, et al. A novel DNA-dependent protein kinase inhibitor, NTJ7026, potentiates the cytotoxicity of topoisomerase II poisons used in the treatment of leukemia. Blood 2004;103:4659–65. [DOI] [PubMed] [Google Scholar]
- 35.Zhou X, Zhang X, Xie Y, et al. DNA-PKcs inhibition sensitizes cancer cells to carbon-ion irradiation via telomere capping disruption. PLoS One 2013;8:e72641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hagner PR, Man HW Fontanillo C, et al. CC-122, a pleiotropic pathway modifier, mimics an interferon response and has antitumor activity in DLBCL. Blood 2015;126:779–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mukherjee B, Tomimatsu N, Amancherla K, et al. The dual PI3K/mTORinhibitorNVP-BEZ235 is a potent inhibitor of ATM- and DNA-PKCs-mediated DNA damage responses. Neoplasia 2012;14:34–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Srivastava M, Nambiar M, Sharma S, et al. An inhibitor of nonhomologous end-joining abrogates double-strand break repair and impedes cancer progression. Cell 2012;151:1474–87. [DOI] [PubMed] [Google Scholar]
- 39.Huang F, Motlekar NA, Burgwin CM, et al. Identification of specific inhibitors of human RAD51 recombinase using high-throughput screening. ACS Chem Biol 2011;6:628–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Budke B, Kalin JH, Pawlowski M, et al. An optimized RAD51 inhibitor that disrupts homologous recombination without requiring Michael acceptor reactivity. J Med Chem 2013;56:254–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Deakyne JS, Huang F, Negri J, et al. Analysis of the activities of RAD54, a SWI2/SNF2 protein, using a specific small-molecule inhibitor. J Biol Chem 2013;288:31567–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Frank AO, Feldkamp MD, Kennedy JP, et al. Discovery of a potent inhibitor of replication protein a protein-protein interactions using a fragment-linking approach. J Med Chem 2013;56:9242–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2 -deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005;434:913–7. [DOI] [PubMed] [Google Scholar]
- 44.Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005;434:917–21. [DOI] [PubMed] [Google Scholar]
- 45.Feng FY, de Bono JS, Rubin MA, et al. Chromatin to Clinic: The Molecular Rationale for PARP1 Inhibitor Function. Mol Cell 2015;58:925–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brown JS, Kaye SB, Yap TA. PARP inhibitors: the race is on. Br J Cancer 2016;114:713–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rottenberg S, Jaspers JE, Kersbergen A, et al. High sensitivity of BRCA1 -deficient mammary tumors to the PARP inhibitor AZD22 81 alone and in combination with platinum drugs. Proc Natl Acad Sci USA 2008;105:17079–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fong PC, Boss DS, Yap TA, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation earners. N Engl J Med 2009;361:123–34. [DOI] [PubMed] [Google Scholar]
- 49.DiNitto JP, Wu JC. Molecular mechanisms of drug resistance in tyrosine kinases cAbl and c Kit. Grit Rev Biochem Mol Biol 2011;46:295–309. [DOI] [PubMed] [Google Scholar]
- 50.Slupianek A, Schmutte C, Tombline G, et al. BCR/ABL regulates mammalian Rec A homologs, resulting in drug resistance. Mol Cell 2001;8:795–806. [DOI] [PubMed] [Google Scholar]
- 51.Zhu J, Zhou L, Wu G, et al. A novel small molecule RAD51 inactivator overcomes imatinib-resistance in chronic myeloid leukaemia. EMBO Mol Med 2013;5:353–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Noguchi M, Yu D, Hirayama R, et al. Inhibition of homologous recombination repair in irradiated tumor cells pretreated with Hsp90 inhibitor 17-allylamino-17-demethoxygeldanamycin. Biochem Biophys Res Commun 2006;351:658–63. [DOI] [PubMed] [Google Scholar]
- 53.Jhaveri K, Taldone T, Modi S, et al. Advances in the clinical development of heat shock protein 90 (Hsp90) inhibitors in cancers. Biochim Biophys Acta 2012;1823:742–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Reed AM, Fishel ML, Kelley MR. Small-molecule inhibitors of proteins involved in base excision repair potentiate the anti-tumorigenic effect of existing chemotherapeutics and irradiation. Future Oncol 2009;5:713–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Naidu MD, Agarwal R, Pena LA, et al. Lucanthone and its derivative hycanthone inhibit apurinic endonuclease-1 (APE1) by direct protein binding. PLoS One 2011;6:e23679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bapat A, Glass LS, Luo M, et al. Novel small-molecule inhibitor of apurinic/apyrimidinic endonuclease 1 blocks proliferation and reduces viability of glioblastoma cells. J Pharmacol Exp Ther 2010;334:988–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rai G, Vyjayanti VN, Dorjsuren D, et al. Small Molecule Inhibitors of the Human Apurinic/apyrimidinic Endonuclease 1 (APE1). Probe Reports from the NIH Molecular Libraries Program. Bethesda (MD): National Center for Biotechnology Information (US);2010. [PubMed] [Google Scholar]
- 58.Zou GM, Maitra A. Small-molecule inhibitor of the AP endonuclease 1/REF-l E3330 inhibits pancreatic cancer cell growth and migration. Mol Cancer Ther 2008;7:2012–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jaiswal AS, Banerjee S, Aneja R, et al. DNA polymerase beta as a novel target for chemotherapeutic intervention of colorectal cancer. PLoS One 2011;6: el6691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Srinivasan A, Gold B. Small-molecule inhibitors of DNA damage-repair pathways: an approach to overcome tumor resistance to alkylating anticancer drugs. Future Med Chem 2012;4:1093–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Turney LN, Bom D, Huck B, et al. The identification and optimization of a N-hydroxy urea series of flap endonuclease 1 inhibitors. Bioorg Med Chem Lett 2005;15:277–81. [DOI] [PubMed] [Google Scholar]
- 62.Garrett MD, Collins I. Anticancer therapy with checkpoint inhibitors: what, where and when? Trends Pharmacol Sci 2011;32:308–16. [DOI] [PubMed] [Google Scholar]
- 63.Zhao B, Bower MJ, McDevitt PJ, et al. Structural basis for Chkl inhibition by UCN-01. J Biol Chem 2002;277:46609–15. [DOI] [PubMed] [Google Scholar]
- 64.Riesterer O, Matsumoto F, Wang L, et al. A novel Chk inhibitor, XL-844, increases human cancer cell radiosensitivity through promotion of mitotic catastrophe. Invest New Drugs 2011. ;29:514–22. [DOI] [PubMed] [Google Scholar]
- 65.Zabludoff SD, Deng C, Grondine MR, et al. AZD7762, a novel checkpoint kinase inhibitor, drives checkpoint abrogation and potentiates DNA-targeted therapies. Mol Cancer Ther 2008;7:2955–66. [DOI] [PubMed] [Google Scholar]
- 66.Brenner AK, Reikvam H, Lavecchia A, et al. Therapeutic targeting the cell division cycle 25 (CDC25) phosphatases in human acute myeloid leukemia—the possibility to target several kinases through inhibition of the various CDC25 isoforms. Molecules 2014;19:18414–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lavecchia A, Di Giovanni C, Novellino E. CDC25 phosphatase inhibitors: an update. Mini Rev Med Chem 2012;12:62–73. [DOI] [PubMed] [Google Scholar]
- 68.Hartwell LH, Szankasi P, Roberts CJ, et al. Integrating genetic approaches into the discovery of anticancer drugs. Science 1997;278:1064–8. [DOI] [PubMed] [Google Scholar]
- 69.de Murcia JM, Niedergang C, Trucco C, et al. Requirement of poly(ADP-ribose) polymerase in recovery from DNA damage in mice and in cells. Proc Natl Acad Sci US A 1997;94:7303–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang ZQ, Stingl L, Morrison C, et al. PARP is important for genomic stability but dispensable in apoptosis. Genes Dev 1997;11:2347–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liu X, Holstege H, van der Gulden H, et al. Somatic loss of BRCA1 and p53 in mice induces mammary tumors with features of human BRCA1-mutated basal-like breast cancer. Proc Natl Acad Sci U S A2007;104:12111–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.O’Shaughnessy J, Osborne C, Pippen JE, et al. Iniparib plus chemotherapy in metastatic triple-negative breast cancer. N Engl J Med 2011;364:205–14. [DOI] [PubMed] [Google Scholar]
- 73.Guha M PARP inhibitors stumble in breast cancer. Nat Biotechnol 2011;29:373–4. [DOI] [PubMed] [Google Scholar]
- 74.Papeo G, Casale E, Montagnoli A, et al. PARP inhibitors in cancer therapy: an update. Expert Opin Ther Pat 2013;23:503–14. [DOI] [PubMed] [Google Scholar]
- 75.Kim YJ, Wilson DM 3rd. Overview of base excision repair biochemistiy. Curr Mol Pharmacol 2012;5:3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Robertson AB, Klungland A, Rognes T, et al. DNA repair in mammalian cells: Base excision repair: the long and short of it. Cell Mol Life Sci 2009;66:981–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Luo M, He H, Kelley MR, et al. Redox regulation of DNA repair: implications for human health and cancer therapeutic development. Antioxid Redox Signal 2010;12:1247–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Abbotts R, Madhusudan S. Human AP endonuclease 1 (APE1): from mechanistic insights to druggable target in cancer. Cancer Treat Rev 2010;36:425–35. [DOI] [PubMed] [Google Scholar]
- 79.Madhusudan S Evolving drug targets in DNA base excision repair for cancer therapy. Curr Mol Pharmacol 2012;5:1–2. [DOI] [PubMed] [Google Scholar]
- 80.Tell G, Wilson DM 3rd. Targeting DNA repair proteins for cancer treatment. Cell Mol Life Sci 2010;67:3569–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zou GM, Karikari C, Kabe Y, et al. The Ape-l/Ref-1 redox antagonist E3330 inhibits the growth of tumor endothelium and endothelial progenitor cells: therapeutic implications in tumor angiogenesis. J Cell Physiol 2009;219:209–18. [DOI] [PubMed] [Google Scholar]
- 82.Tell G, Quadrifoglio F, Tiribelli C, et al. The many functions of APEl/Ref-1: not only a DNA repair enzyme. Antioxid Redox Signal 2009;11:601–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fishel ML, Jiang Y, Rajeshkumar NVj et al. Impact of APEl/Ref-1 redox inhibition on pancreatic tumor growth. Mol Cancer Ther 2011;10:1698–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cardoso AA, Jiang Y, Luo M, et al. APEl/Ref-1 regulates STAT3 transcriptional activity and APE1/Ref-1-STAT3 dual-targeting effectively inhibits pancreatic cancer cell survival. PLoS One 2012;7:e47462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Srinivasan A, Wang L, Cline CJ, et al. Identification and characterization of human apurinic/apyrimidinic endonuclease-1 inhibitors. Biochemistiy 2012;51:6246–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Campalans A, Kortulewski T, Amouroux R, et al. Distinct spatiotemporal patterns and PARP dependence of XRCC1 recruitment to single-strand break and base excision repair. Nucleic Acids Res 2013;41:3115–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Helleday T, Petermann E, Lundin C, et al. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer 2008;8:193–204. [DOI] [PubMed] [Google Scholar]
- 88.Davis AJ, Chen DJ. DNA double strand break repair via non-homologous end-joining. Transl Cancer Res 2013;2:130–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Srivastava M, Raghavan SC. DNA double-strand break repair inhibitors as cancer therapeutics. Chem Biol 2015;22:17–29. [DOI] [PubMed] [Google Scholar]
- 90.Beskow C, Skikuniene J, Holgersson A, et al. Radioresistant cervical cancer shows up regulation of the NHEJ proteins DNA-PKcs, Ku70 and Ku86. Br J Cancer 2009;101:816–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Shintani S, Mihara M, Li C, et al. Up-regulation of DNA-dependent protein kinase correlates with radiation resistance in oral squamous cell carcinoma. Cancer Sci 2003;94:894–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sirzen F, Nilsson A, Zhivotovsky B, et al. DNA-dependent protein kinase content and activity in lung carcinoma cell lines: correlation with intrinsic radiosensitivity. Eur J Cancer 1999;35:111–6. [DOI] [PubMed] [Google Scholar]
- 93.Pyun BJ, Seo HR, Lee HJ, et al. Mutual regulation between DNA-PKcs and Snaill leads to increased genomic instability and aggressive tumor characteristics. Cell Death Dis 2013;4:e517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Goodwin JF, Kothari V, Drake JM, et al. DNA-PKcs-Mediated Transcriptional Regulation Drives Prostate Cancer Progression and Metastasis. Cancer Cell 2015;28:97–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kotula E, Berthault N, Agrario C, et al. DNA-PKcs plays role in cancer metastasis through regulation of secreted proteins involved in migration and invasion. Cell Cycle 2015;14:1961–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Groth P, Orta ML, Elvers I, et al. Homologous recombination repairs secondary replication induced DNA double-strand breaks after ionizing radiation. Nucleic Acids Res 2012;40:6585–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Harper JV, Anderson JA, O’Neill P. Radiation induced DNA DSBs: Contribution from stalled replication forks? DNA Repair (Amst) 2010;9:907–13. [DOI] [PubMed] [Google Scholar]
- 98.Hartlerode AJ, Scully R Mechanisms of double-strand break repair in somatic mammalian cells. Biochem J 2009;423:157–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mao Z, Bozzella M, Seluanov A, et al. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle 2008;7:2902–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hannay JA, Liu J, Zhu QS, et al. Rad51 overexpression contributes to chemoresistance in human soft tissue sarcoma cells: a role for p53/activator protein 2 transcriptional regulation. Mol Cancer Ther 2007;6:1650–60. [DOI] [PubMed] [Google Scholar]
- 101.Helleday T Homologous recombination in cancer development, treatment and development of drug resistance. Carcinogenesis 2010;31:955–60. [DOI] [PubMed] [Google Scholar]
- 102.Zafar F, Seidler SB, Rronenberg A, et al. Homologous recombination contributes to the repair of DNA double-strand breaks induced by high-energy iron ions. Radiat Res 2010;173:27–39. [DOI] [PubMed] [Google Scholar]
- 103.Gerelchuluun A, Manabe E, Ishikawa T, et al. The major DNA repair pathway after both proton and carbon-ion radiation is NHEJ, but the HR pathway is more relevant in carbon ions. Radiat Res 2015;183:345–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wang H, Wang X, Zhang P, et al. The Ku-dependent non-homologous end-joining but not other repair pathway is inhibited by high linear energy transfer ionizing radiation. DNA Repair (Amst) 2008;7:725–33. [DOI] [PubMed] [Google Scholar]
- 105.Budke B, Logan HL, Kalin JH, et al. RI-1: a chemical inhibitor of RAD51 that disrupts homologous recombination in human cells. Nucleic Acids Res 2012;40:7347–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lok BH, Powell SN. Molecular pathways: understanding the role of Rad52 in homologous recombination for therapeutic advancement. Clin Cancer Res 2012;18:6400–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Feng Z, Scott SP, Bussen W, et al. Rad52 inactivation is synthetically lethal with BRCA2 deficiency. Proc Natl Acad Sci US A 2011;108:686–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bucher N, Britten CD. G2 checkpoint abrogation and checkpoint kinase-1 targeting in the treatment of cancer. Br J Cancer 2008;98:523–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kam WW, Banati RB. Effects of ionizing radiation on mitochondria. Free Radic Biol Med 2013;65:607–19. [DOI] [PubMed] [Google Scholar]
- 110.Eker AP, Quayle C, Chaves I, et al. DNA repair in mammalian cells: Direct DNA damage reversal: elegant solutions for nasty problems. Cell Mol Life Sci 2009;66:968–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Fan CH, Liu WL, Cao H, et al. O6-methylguanine DNA methyl transferase as a promising target for the treatment of temozolomide-resistant gliomas. Cell Death Dis 2013;4:e876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Brown A, Suit H. The centenary of the discoveiy of the Bragg peak. Radiother Oncol 2004;73:265–8. [DOI] [PubMed] [Google Scholar]
- 113.Wambersie A RBE, reference RBE and clinical RBE: applications of these concepts in hadron therapy. Strahlenther Onkol 1999; 175 Suppl 2:39–43. [DOI] [PubMed] [Google Scholar]
- 114.Weyrather WK, Kraft G. RBE of carbon ions: experimental data and the strategy of RBE calculation for treatment planning. Radiother Oncol 2004;73 Suppl 2:S161–9. [DOI] [PubMed] [Google Scholar]
- 115.Cui X, Oonishi K, Tsujii H, et al. Effects of carbon ion beam on putative colon cancer stem cells and its comparison with X-rays. Cancer Res 2011;71:3676–87. [DOI] [PubMed] [Google Scholar]
- 116.Oonishi K, Cui X, Hirakawa H, et al. Different effects of carbon ion beams and X-rays on clonogenic survival and DNA repair in human pancreatic cancer stem-like cells. Radiother Oncol 2012;105:258–65. [DOI] [PubMed] [Google Scholar]
- 117.Quan Y, Wang W, Fu Q, et al. Accumulation efficiency of cancer stem-like cells post γ-ray and proton irradiation. Nuclear Instruments and Methods in Physics Research Section B: Beam Interactions with Materials and Atoms 2012;286:341–5. [Google Scholar]
- 118.Fu Q, Quan Y, Wang W, et al. Response of cancer stem-like cells and non-stem cancer cells to proton and γ-ray irradiation. Nuclear Instruments and Methods in Physics Research Section B: Beam Interactions with Materials and Atoms 2012;286:346–50. [Google Scholar]
- 119.Baumann M, Krause M, Hill R Exploring the role of cancer stem cells in radio resistance. Nat Rev Cancer 2008;8:545–54. [DOI] [PubMed] [Google Scholar]
- 120.Newhauser WD, Durante M. Assessing the risk of second malignancies after modem radiotherapy. Nat Rev Cancer 2011;11:438–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.De Ruysscher D, Mark Lodge M, Jones B, et al. Charged particles in radiotherapy: a 5-year update of a systematic review. Radiother Oncol 2012;103:5–7. [DOI] [PubMed] [Google Scholar]
- 122.Kuhlthau KA, Pulsifer MB, Yeap BY, et al. Prospective study of health-related quality of life for children with brain tumors treated with proton radiotherapy. J Clin Oncol 2012;30:2079–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hoppe BS, Flampouri S, Lynch J, et al. Improving the therapeutic ratio in Hodgkin lymphoma through the use of proton therapy. Oncology (Williston Park) 2012;26:456–9, 62-5. [PubMed] [Google Scholar]
- 124.Allen AM, Pawlicki T, Dong L, et al. An evidence based review of proton beam therapy: the report of ASTRO’s emerging technology committee. Radiother Oncol 2012;103:8–11. [DOI] [PubMed] [Google Scholar]
- 125.Ramaekers BL, Pijls-Johannesma M, Joore MA, et al. Systematic review and meta-analysis of radiotherapy in various head and neck cancers: comparing photons, carbon-ions and protons. Cancer Treat Rev 2011;37:185–201. [DOI] [PubMed] [Google Scholar]
- 126.Simone CB 2nd, Ly D, Dan TD, et al. Comparison of intensity-modulated radiotherapy, adaptive radiotherapy, proton radiotherapy, and adaptive proton radiotherapy for treatment of locally advanced head and neck cancer. Radiother Oncol 2011;101:376–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.van de Water TA, Bijl HP, Schilstra C, et al. The potential benefit of radiotherapy with protons in head and neck cancer with respect to noimal tissue sparing: a systematic review of literature. Oncologist 2011. ;16:366–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Bush DA, Slater JD, Garberoglio C, et al. Partial breast irradiation delivered with proton beam: results of a phase II trial. Clin Breast Cancer 2011;11:241–5. [DOI] [PubMed] [Google Scholar]
- 129.Schlaff CD, Krauze A, Belard A, et al. Bringing the heavy: carbon ion therapy in the radiobiological and clinical context. Radiat Oncol 2014;9:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Oike T, Sato H, Noda SE, et al. Translational Research to Improve the Efficacy of Carbon Ion Radiotherapy: Experience of Gunma University. Front Oncol 2016;6:139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Jeimann M Particle Therapy Statistics in 2014. International Journal of Particle Therapy 2015;2:50–4. [Google Scholar]
- 132.Ebner DK, Kamada T. The Emerging Role of Carbon-Ion Radiotherapy. Front Oncol 2016;6:140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Combs SE, Nikoghosyan A, Jaekel O, et al. Carbon ion radiotherapy for pediatric patients and young adults treated for tumors of the skull base. Cancer 2009;115:1348–55. [DOI] [PubMed] [Google Scholar]
- 134.Combs SE, Kessel KA, Herfarth K, et al. Treatment of pediatric patients and young adults with particle therapy at the Heidelberg Ion Therapy Center (HIT): establishment of workflow and initial clinical data. Radiat Oncol 2012;7:170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.McGovern SL, Mahajan A. Progress in radiotherapy for pediatric sarcomas. Curr Oncol Rep 2012;14:320–6. [DOI] [PubMed] [Google Scholar]
- 136.Matsunobu A, Imai R, Kamada T, et al. Impact of carbon ion radiotherapy for unresectable osteosarcoma of the trunk. Cancer 2012;118:4555–63. [DOI] [PubMed] [Google Scholar]
- 137.Imai R, Kamada T, Tsuji H, et al. Effect of carbon ion radiotherapy for sacral chordoma: results of Phase I-II and Phase II clinical trials. Int J Radiat Oncol Biol Phys 2010;77:1470–6. [DOI] [PubMed] [Google Scholar]
- 138.Nishida Y, Kamada T, Imai R, et al. Clinical outcome of sacral chordoma with carbon ion radiotherapy compared with surgery. Int J Radiat Oncol Biol Phys 2011;79:110–6. [DOI] [PubMed] [Google Scholar]
- 139.Schulz-Ertner D, Karger CP, Feuerhake A, et al. Effectiveness of carbon ion radiotherapy in the treatment of skull-base chordomas. Int J Radiat Oncol Biol Phys 2007;68:449–57. [DOI] [PubMed] [Google Scholar]
- 140.Tsujii H, Kamada T. A review of update clinical results of carbon ion radiotherapy. Jpn J Clin Oncol 2012;42:670–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Imada H, Kato H, Yasuda S, et al. Comparison of efficacy and toxicity of short-course carbon ion radiotherapy for hepatocellular carcinoma depending on their proximity to the porta hepatis. Radiother Oncol 2010;96:231–5. [DOI] [PubMed] [Google Scholar]
- 142.Shinoto M, Yamada S, Yasuda S, et al. Phase 1 trial of preoperative, short-course carbon-ion radiotherapy for patients with resectable pancreatic cancer. Cancer 2013;119:45–51. [DOI] [PubMed] [Google Scholar]
- 143.Yamada S, Kamada T, Ebner DK, et al. Carbon-Ion Radiation Therapy for Pelvic Recurrence of Rectal Cancer. International Journal of Radiation Oncology · Biology · Physics;96:93–101. [DOI] [PubMed] [Google Scholar]
- 144.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin 2013;63:11–30. [DOI] [PubMed] [Google Scholar]
- 145.Miyamoto T, Baba M, Sugane T, et al. Carbon ion radiotherapy for stage I non-small cell lung cancer using a regimen of four fractions during 1 week. J Thorac Oncol 2007;2:916–26. [DOI] [PubMed] [Google Scholar]
- 146.Ma H, Takahashi A, Yoshida Y, et al. Combining carbon ion irradiation and non-homologous end-joining repair inhibitor NTJ7026 efficiently kills cancer cells. Radiat Oncol 2015;10:225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Hamada N Recent insights into the biological action of heavy-ion radiation. J Radiat Res 2009;50:1–9. [DOI] [PubMed] [Google Scholar]
- 148.Le QT, Shirato H, Giaccia AJ, et al. Emerging Treatment Paradigms in Radiation Oncology. Clin Cancer Res 2015;21:3393–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Pilones KA, Aryankalayil J, Babb JS, et al. Invariant natural killer T cells regulate anti-tumor immunity by controlling the population of dendritic cells in tumor and draining lymph nodes. J Immunother Cancer 2014;2:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Woo SR, Fuertes MB, Corrales L, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 2014;41:830–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Deng L, Liang H, Xu M, et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 2014;41:843–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Matsunaga A, Ueda Y, Yamada S, et al. Carbon-ion beam treatment induces systemic antitumor immunity against murine squamous cell carcinoma. Cancer 2010;116:3740–8. [DOI] [PubMed] [Google Scholar]
- 153.Takeshima T, Pop LM, Laine A, et al. Key role for neutrophils in radiation-induced antitumor immune responses: Potentiation with G-CSF. Proc Natl Acad Sci U S A 2016;113:11300–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Hartlova A, Erttmann SF, Raffi FA, et al. DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity. Immunity 2015;42:332–43. [DOI] [PubMed] [Google Scholar]
- 155.Kondo T, Kobayashi J, Saitoh T, et al. DNA damage sensor MRE11 recognizes cytosolic double-stranded DNA and induces type I interferon by regulating STING trafficking. Proc Natl Acad Sci U S A2013;110:2969–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Wolf C, Rapp A, Berndt N, et al. RPA and Rad51 constitute a cell intrinsic mechanism to protect the cytosol from self DNA. Nat Commun 2016;7:11752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Bhattacharya S, Srinivasan K, Abdisalaam S, et al. RAD51 interconnects between DNA replication, DNA repair and immunity. Nucleic Acids Res 2017;45:4590–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Edwards SL, Brough R, Lord CJ, et al. Resistance to therapy caused by intragenic deletion in BRCA2. Nature 2008;451:1111–5. [DOI] [PubMed] [Google Scholar]
- 159.Swisher EM, Sakai W, Karlan BY, et al. Secondary BRCA1 mutations in BRCA1-mutated ovarian carcinomas with platinum resistance. Cancer Res 2008;68:2581–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Sakai W, Swisher EM, Karlan BY, et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 2008;451:1116–20. [DOI] [PMC free article] [PubMed] [Google Scholar]