

SUMMARY
Synthetically engineered DNA-encoded monoclonal antibodies (DMAbs) are an in vivo platform for evaluation and delivery of human mAb to control against infectious disease. Here, we engineer DMAbs encoding potent anti-Zaire ebolavirus (EBOV) glycoprotein (GP) mAbs isolated from Ebola virus disease survivors. We demonstrate the development of a human IgG1 DMAb platform for in vivo EBOV-GP mAb delivery and evaluation in a mouse model. Using this approach, we show that DMAb-11 and DMAb-34 exhibit functional and molecular profiles comparable to recombinant mAb, have a wide window of expression, and provide rapid protection against lethal mouse-adapted EBOV challenge. The DMAb platform represents a simple, rapid, and reproducible approach for evaluating the activity of mAb during clinical development. DMAbs have the potential to be a mAb delivery system, which may be advantageous for protection against highly pathogenic infectious diseases, like EBOV, in resource-limited and other challenging settings.
Graphical Abstract
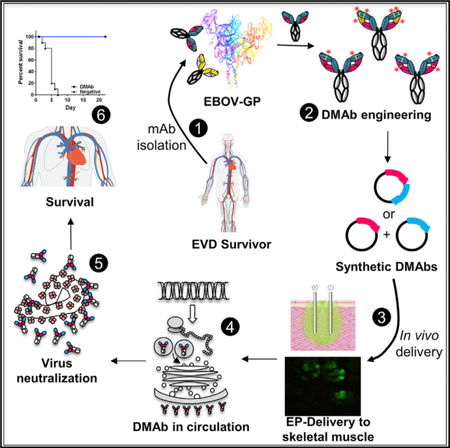
In Brief
Monoclonal antibodies are an important approach for emerging infectious disease prevention. Patel et al. demonstrate engineering and in vivo delivery of DNA-encoded monoclonal antibodies (DMAbs) targeting the Zaire ebolavirus (EBOV) glycoprotein. DMAbs protect against lethal mouse-adapted EBOV and are useful for rapid evaluation of fully human mAbs in live animal models.
INTRODUCTION
The 2013–2016 Zaire ebolavirus (EBOV) epidemic in West Africa was the most severe and devastating Ebola virus disease (EVD) epidemic reported to date. Several experimental treatments were administered to EBOV-infected individuals as part of compassionate-use protocols, including the ZMapp cocktail of monoclonal antibodies (mAbs), which demonstrated protection in non-human primates and promise in people (Davey et al., 2016; Petrosillo et al., 2015) (reviewed in Trad et al., 2017). ZMapp was originally developed as a cocktail of three mAb clones: 2G4, 4G7, and 13c6, which were isolated from vaccinated mice (Qiu et al., 2011; Wilson et al., 2000) and later developed into mouse/human chimeric immunoglobulin Gs (IgGs). Since the onset of the West Africa outbreak, several highly potent anti-Ebola virus mAbs targeting different regions of the glycoprotein (GP) have been identified from human survivors from the 1995 EVD outbreak (Corti et al., 2016) and 2014–2016 EVD epidemic (Bornholdt et al., 2016), and the 2007 Bundibugyo ebolavirus outbreak (Flyak et al., 2016).
The need for repeat, high-dose Ig infusions to overcome viral load during infection represents a hurdle for recombinant mAb therapeutics in pandemic outbreaks of highly infectious pathogens such as EBOV. However, further development of cell culture manufacturing technologies is necessary to fully realize bioprocessed IgG production to meet global demand for targeting infectious diseases and cost for world-wide availability in countries where such therapeutics are often most needed (Dumiak, 2014; Kunert and Reinhart, 2016; Samaranayake et al., 2009). We have developed an in vivo approach that can be used to rapidly develop and screen potentially important mAb candidates, independent of in vitro cell liabilities, which enables rapid evaluation of their properties in a live-model system.
We evaluated, optimized, and encoded 23 different fully human DNA-encoded monoclonal antibodies (DMAbs), which originated from EVD survivors, as well as the ZMapp antibodies. The DMAb strategy produces mAb in vivo, which allows for analysis that may otherwise be difficult to develop due to undesirable biophysical and biochemical sequence liabilities. Using this approach, 23 anti-GP DMAbs were engineered to express in vivo. From this analysis, we focused on two DMAbs with significant potency, targeting the fusion loop (DMAb-11) and heptad repeat 2 (HR2) region (DMAb-34). These clones demonstrated long-term expression and significant antiviral potency in vivo. The candidates were then evaluated for protection against lethal mouse-adapted EBOV (ma-EBOV), where both were highly effective. We further demonstrate that the top candidates can be co-administered as part of a cocktail, which can improve potency. Importantly, we present biological evidence that DMAbs and recombinant mAb bind to identical molecular epitopes, confirming equivalency and supporting the DMAb platform as an exciting approach for in vivo delivery of fully human mAb. Importantly, in vivo expression of DMAb is much longer than recombinant mAb expression. This in vivo strategy represents an important tool for the study and development of transient mAb delivery to prevent infectious diseases.
RESULTS
DMAbs Targeting Different EBOV-GP Regions Can Be Engineered and In Vivo-Delivered to Muscle
We selected 26 different mAb clones that target the EBOV-GP glycan cap, fusion loop, chalice base, HR2 region, membrane-proximal external region (MPER), and mucin-like domain for development into anti-EBOV-GP (anti-GP) DMAbs. The gene sequences of the human Ig heavy IgG1 and light chains were codon and RNA-optimized and encoded into a single modified-pVax1 DNA expression vector plasmid, separated by furin and P2A peptide cleavage sites (single plasmid) or encoded as two separate plasmid constructs (dual plasmid). Expression of all constructs was confirmed in vitro prior to administering anti-GP DMAb constructs in vivo (Table S1).
The DMAb single-plasmid or dual-plasmid (equal ratio of heavy and light chain plasmids) were administered to mice by in vivo intramuscular (IM) injection followed by facilitated CELLECTRA-3P electroporation (IM-EP). This resulted in DMAb expression and secretion directly into systemic circulation. Quadriceps muscle slices from mice injected with anti-GP DMAb or control pVax1 were harvested 2 days post-DMAb injection and stained for human IgG (Figure S1) to confirm expression in vivo in muscle cells (Figure S1A and S1B; 40× magnification) and within the fiber cross-section (Figure S1C and S1D; 40× magnification). A pseudocolor overlay was generated to demonstrate the intensity of anti-GP DMAb expression in comparison with control muscle (Figure S1E and S1F; 40× magnification).
DMAb Animal Model Development
In our initial DMAb studies, we observed a strong mouse antihuman antibody response in vivo to the foreign DMAb (Elliott et al., 2017; Patel et al., 2017). This observation is consistent with other groups and is the reason that many studies utilize fully immune-deficient RAG1-knockout (Limberis et al., 2016) or nude mouse models (Elliott et al., 2017; Muthumani et al., 2016; Patel et al., 2017) and deliver chimeric human/mouse mAbs (Andrews et al., 2017; Limberis et al., 2016). Although RAG is not required for natural killer (NK) cell development, studies have shown a role for RAG genes may contribute to NK cell fitness (Karo et al., 2014), and RAG-related immune deficiency may display a skewed NK cell profile that can directly impact antibody:Fc receptor interactions such as antibody-dependent cellular cytotoxicity. Therefore, we sought to develop a resource for evaluating the efficacy of fully human antibodies in a mouse model that would be immune competent at challenge. T cell depletion has been studied extensively in the immunology field and there are well-established in vivo anti-CD4 (clone GK1.5) and anti-CD8 (clone YTS169.4) antibodies are available for transient depletion in mouse models (Grcević et al., 2000). We first performed CD4+ and CD8+ T cell transient depletion studies, in BALB/c mice with anti-Pseudomonas aeruginosa DMAb-V2L2 (Patel et al., 2017), to identify which arm of the mouse immune system is involved with the anti-antibody responses. Anti-CD4 (GK1.5), anti-CD8 (YTS169.4), or both depleting antibodies were delivered immediately prior to DMAb administration (day 0). We observed that this anti-antibody response is driven primarily by CD4+ T cells in BALB/c mice (n = 5 mice/group) (Figure S2A) and is MHC class II dependent, as observed in an MHC class II knockout mouse on a C57BL6 background (n = 5 mice/ group) (Figure S2B). Complete CD4+ and CD8+ T cell depletion afforded the best transient suppression that lasts for 14–21 days, enabling long-term expression of DMAb in the mouse model long after both T cell populations fully recover (Figure S2C). Supported by this data, we performed a similar study depletion study with anti-GP DMAb-11 (Figure S2D; n = 5 mice/group), observing similar results. Non-depleted animals rapidly developed anti-antibody responses that completely shut down DMAb expression by day 14 (Figure S2E). Depleted animals do not develop an anti-antibody response (Figure S2F). DMAb-11 encoded as single-plasmid (400-µg dose) or dual-plasmid constructs (200-µg dose total DNA) was administered to BALB/c mice (n = 8–9 mice/group) and monitored expression for 365 days following administration (Figure S2G). Long-term expression at high levels was observed and administration of a single plasmid or dual plasmids had very similar expression kinetics in the T cell-depleted model. Taken together, this serves as an immune-competent model that can be utilized to evaluate fully human mAbs without generation of an anti-antibody response and that is also cost-effective for rapid screening of multiple potential DMAb constructs. T cell depletion was performed immediately prior to DMAb administration for all subsequent experiments.
Anti-GP DMAb In Vivo Expression Is Enhanced through Sequence and Formulation Optimization
It is well known that sequence liabilities of IgG can limit bio-processed mAb production, frequently leading to discarding of an otherwise highly potent mAb clone (Lauer et al., 2012; Sharma et al., 2014). The mAb genes for clones 4G7 (DMAb-4), 13c6 (DMAb-7), 5.6.1A2 (DMab-11), and 15784 (DMAb-34) were injected into mice by IM-EP to test expression in vivo (Figure S3; n = 5 mice/group). Clones 4G7 (mouse variable heavy chain [VH] 1–42, variable kappa chain [VK] 12–44) and 13c6 (mouse VH 8–8, VK 6–13) are two mAbs found in the ZMapp cocktail that were originally isolated from vaccinated mice and encoded into the DMAb platform as chimeric mouse/human IgG1. Clone 5.6.1A2 (human VH 3–53, VK 2–28) was isolated from a 2014 EVD survivor who was treated at Emory University. This clone was isolated from an EVD survivor at the 6-month time point post-treatment (C.W.D and R.A, unpublished data). Clone 15784 (human VH 1–18, VK 2–28) was isolated among hundreds of survivor-derived mAb clones from a different 2014 EVD survi-vor (Bornholdt et al., 2016).
We first hypothesized that nucleotide codon optimization would enhance DMAb in vivo expression (version 1). DMAbs were codon and RNA optimized for both mouse and human bias to increase expression in mammalian cells (Deml et al., 2001; Graf et al., 2004). The initial version 1 of DMAb-4 and DMAb-7 did not express well in vitro or in vivo (Figure S3; Table S1). We observed that the N terminus of both DMAbs is different than the germline sequences in the genome (Figures S3A and S3B). We next hypothesized that modification of these N terminus amino acids back to germline would improve antibody stability, as predicted by in silico methodologies (Dunbar et al., 2016). Following modification for DMAb-4 (E1Q and E6Q) and DMAb-7 (L2V), we observed a modest increase in DMAb in vivo expression (version 2; Figures S3A and S3B). The N terminus amino acid sequence of DMAb-11 and DMAb-34 were identical to germline; therefore, they were not modified. DMAb-34 was identified as a potential candidate later in the study and therefore benefited from the design discovery performed with the other anti-GP DMAbs.
To further increase DMAb expression in vivo, we evaluated the possibility of delivering the DMAb in combination with a hyaluronidase treatment formulation (version 3) as an approach to increase plasmid uptake (McMahon et al., 2001). Two different doses (50 and 200 µg) were delivered along with hyaluronidase treatment (200 U/mL) (Figures S3A–S3D). Overall, combination with hyaluronidase significantly increases DMAb expression. These collective optimizations were applied to all 26 anti-GP DMAbs (Figure 1). All of the data in Figure 1 are DMAb expression levels collected at day 7 post-administration in the T cell-depleted BALB/c mouse model.
Figure 1. Cmax Expression Levels for 26 Different Optimized DMAbs Targeting Various Regions of EBOV GP.
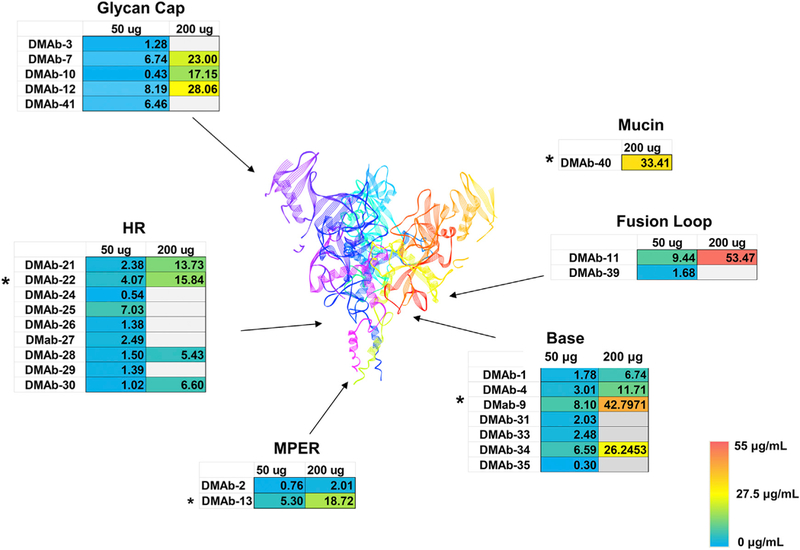
Optimized anti-GP DMAbs targeting the glycan cap, heptad repeat region 2 (HR2), membrane-proximal external region (MPER), base, fusion loop, and mucin-like domain were evaluated at 50 µg/mouse and 200 µg/mouse. Expression was assayed at day 7 post-DMAb administration. The gray box represents groups that were not evaluated for dose 2. Structure shown is based on Ebola virus GP PDB:5JQ3 (Zhao et al., 2016). A heatmap scale bar is included for colorimetric reference (0–55 µg/mL). Asterisks represent DMAbs that were optimized later during the study. The data represent the mean of n = 5 mice/group. Expression data for individual animals in each group are included in Supplemental Information.
In Vivo DMAb Expression Is Not Limited by Cell Culture Sequence Liabilities
Highly potent antibodies may be unsuited for manufacturing due to intrinsic biochemical and biophysical properties that could negatively impact production. Analysis of these parameters has been collectively termed the developability index (DI) (Lauer et al., 2012). This represents a tremendous challenge for difficult-to-treat infectious diseases and highly pathogenic viral infections such as EBOV where a potent mAb may be excluded in favor of another clone that is easier to manufacture but has a weaker potency profile. DI is predicted in silico utilizing proprietary and freely available algorithms that identify potential for antibody amino acid oxidation, deamination, or potential isomerization that could negatively impact antibody stability, aggregation, and clearance (Dunbar et al., 2016; Lauer et al., 2012). We calculated the predicted DIs for eight mAbs, based on their sequence information, utilizing in silico algorithms available in Biovia Discovery Studio (Accelyrs) and the freely available SAbPred algorithm (Dunbar et al., 2016) (http://opig.stats.ox.ac.uk/webapps/sabdab-sabpred/WelcomeSAbPred.php), and compared the output DIs (Table S2) with in vivo DMAb expression levels obtained from our in vivo experiments at day 7 post-DMAb administration. The Discovery Studio algorithm output ranked the antibody DI indexes from highest potential developability (rank 1) to lowest potential developability (rank 8). The SAbPred algorithm ranked the antibodies by low, moderate, or high liabilities. Taken together, this analysis showed that using the DMAb platform we can successfully deliver in vivo anti-GP mAb clones even with poor DI scores. Based on DI analysis, DMAb-11 and DMAb-34 scored in the middle of the ranking and are characterized with moderate to high negative biochemical features including Trp oxidation, Asn deamidation, Met oxidation, Asp isomerization, as well as aggregation scores that may be less favorable for bio-processed mAb production compared with other candidates. Even so, we observed strong in vivo DMAb expression of both DMAb-11 and DMAb-34, supporting a useful aspect of DMAb antibody production and illustrating that in vivo production can significantly differ from in vitro production systems.
Anti-GP DMAb Expression Can Be Screened In Vivo for Rapid Clinical Development
Rapid in vivo screening of potent mAb clones is one advantage of the DMAb platform. In total, we tested 26 different optimized anti-GP DMAbs in mice at 50 µg/mouse and/or 200 µg/mouse doses (Cmax expression at day 7 post-DMAb administration is shown in Figure 1; data from individual mice and SDs are listed in Table S3). Three DMAbs were mouse-human chimeras (DMAb-1, DMAb-4, DMAb7) and 23 were fully human IgG1 DMAbs. We demonstrated successful in vivo DMAb production and showed efficient expression of both chimeric and fully human clones. These DMAbs expressed clones with different VH and VL families including robust expression of the more frequently used human VH1, VH3, VH4, VК1, VК2, VК3, and Vλ3 families (Tables S3 and S4). Based on the strong expression in vivo, we focused on DMAb-11 and DMAb-34 for further characterization studies.
DMAb-11 and DMAb-34 Express for Weeks to Months and Produce Functional In Vivo-Delivered mAb
Long-term expression of different doses of DMAb-11 (dual plasmid; 25- to 100-µg total DNA; n = 5 mice/group) or DMAb-34 (dual plasmid; 50-µg total DNA; n = 5 mice/group) were monitored in parallel with single injection of different doses of recombinant 5.6.1A2 or 15784 (25 100 µg; n = 5 mice/group) (Figures 2A and 2B). Sera from mice administered DMAb-11 and DMAb-34 bound to 1976 EBOV-GP (strain Mayinga) comparably to recombinant mAb (Figures 2C and 2D; n = 10 mice/ group). The DMAb-containing sera neutralized live EBOV-GFP (strain Mayinga) virus (Figures 2E and 2F; n = 10 mice/group highlighting in vivo functional activity of DMAb). A second BSL-2 rVSV-EBOVGP-luciferase (strain Makona, performed on pooled sera; Figure S4) assay was performed to demonstrate that anti-GP DMAb functionality can also be monitored using this alternative assay.
Figure 2. Characterization of DMAb-11 and DMAb-34.
(A) Comparison of DMAb-11 expression kinetics with the equivalent recombinant mAb 5.6.1A2. Different doses of DMAb-11 (50–200 µg plasmid DNA IM-EP) and mAb 5.6.1A2 (25 µg-100 µg protein i.p.) were administered to mice, and serum human IgG1 levels were monitored over time (n = 5/group).
(B) Comparison of DMAb-34 expression kinetics with equivalent recombinant mAb 15784. A dose of 50-µg plasmid/mouse of DMAb-34 and different doses of mAb 15784 (25–100 µg protein) were administered to mice and serum human IgG1 levels were monitored over time (n = 5/group).
(C and D) Binding of sera from DMAb-11-administered mice to EBOV-GP in comparison with mAb 5.6.1A2 (C) and binding of DMAb-34 sera to EBOV-GP in comparison with mAb 15784 (D).
(E and F) Ebola virus neutralization IC50 for sera collected from mice administered DMAb-11 (E) or DMAb-34 (F). The neutralization assays were performed with EBOV (strain Mayinga) expressing GFP. Error bars represent the SD from the mean.
Anti-GP DMAbs Map to the Same Molecular Epitope as Recombinant mAb
To further address the question of in vivo-produced DMAb equivalency to recombinant mAb, shotgun mutagenesis epitope mapping (Davidson et al., 2015; Davidson and Doranz, 2014) was performed using HEK293 cells expressing EBOV-GP (1976-GP) with alanine (Ala) mutations at each position in the EBOV-GP. First, recombinant mAb 5.6.1A2 or recombinant mAb 15784 were run on the library to establish assay conditions and identify GP residues necessary for mAb binding. Pooled serum from mice administered DMAb-11 (dilution, 1:32) or DMAb-34 (dilution, 1:64) were then run using the same assay, with identical conditions. For mAb 5.6.1A2, mutations I527A and W531A, at residues in the GP fusion loop, were identified as critical (Figure 3A). The identical mutations were identified for DMAb-11. For mAb 15784, critical mutations W531A, Y534A, F535A, and T565A (in the fusion loop and GP base) were identified as critical for binding. The identical mutations were observed for DMAb-34 (Figure 3B). Three distinct positive control mAbs, A, B, and C (Davidson et al., 2015; Davidson and Doranz, 2014), were run in parallel. These epitope-mapping data indicate that the in vivo-produced DMAb and its respective mAb exhibit the identical binding characteristics at the molecular level.
Figure 3. Shotgun Mutagenesis Epitope Mapping by Alanine Scanning of a EBOV ΔMucin GP Library.
Recombinant mAb and pooled mouse serum were mapped on an EBOV Δmucin GP (1976 outbreak) alanine scan mutation library expressed in HEK293T cells and assayed by flow cytometry.
(A) Drop-out mutations that did not support antibody binding were identified for mAb 5.6.1A2 and pooled sera from mice administered DMAb-11 (dilution 1:32). mAb 5.6.1A2 and serum from BALB/c mice expressing DMAb-11 show reactivity with the fusion loop of EBOV GP2.
(B) Drop-out mutations that did not support antibody binding were identified for mAb 15784 and pooled serum from mice administered DMAb-34 (dilution, 1:64). mAb 15784 and serum from BALB/c mice expressing DMAb-34 show reactivity with the base/fusion loop of EBOV GP2. Residues identified as critical for DMAb-11 and DMAb-34 binding are shown mapped in green spheres on the GP monomer (left) and trimer structures (center and right) from EBOV GP crystal structure (PDB:5JQ3; Zhao et al., 2016). GP1 is shown as yellow, and GP2 as red. The right-hand figure shows the entire space-filled GP surface model.
Specific Anti-GP DMAbs Protect against Ebola Virus in a Mouse Challenge Model
Doses of DMAb-11, DMAb-13, and DMAb-34 were administered to BALB/c mice (n = 10 mice/group) 28 days prior to infection (day −28). On day −14, serum was harvested from animals before they were shipped to the biosafety level 4 (BSL4) containment laboratory at the Public Health Agency of Canada (PHAC) (Winnipeg, MB, Canada). DMAb-injected mice received 1,000 times the median lethal dose (1,000LD50) challenge of ma-EBOV on day 0 (Figure 4A). A negative control group (n = 10) and positive recombinant mAb 2G4 (n = 10) groups were included (Figure 4B). As expected, all of the negative controls succumbed to infection. Importantly, anti-GP DMAb-13 (100-µg dose) was not protective in mice, suggesting that not all EBOV-GP-specific DMAbs are protective Figure 4C. For DMAb-11 and DMAb-34, serum expression levels increased in a dose-dependent manner (Figures 4D and 4E). DMAb-11 was 100% protective at the 100-µg dose and 80% protective at the lower 50-µg dose. No signs of disease were observed in surviving animals (p < 0.001 in comparison with DMAb-13 and negative control). Full protection (100%) was observed with the 100-µg dose of DMAb-34 (p < 0.001 in comparison with DMAb-13 and negative control). A break in DMAb-34 protection was observed at the 50-µg dose, where only 40% of animals survive. This low-dose group still showed benefeit compared to the negative control and DMAb-13 group (Figure 4E).
Figure 4. In Vivo Protection by Anti-GP DMAb-11 and DMAb-34.
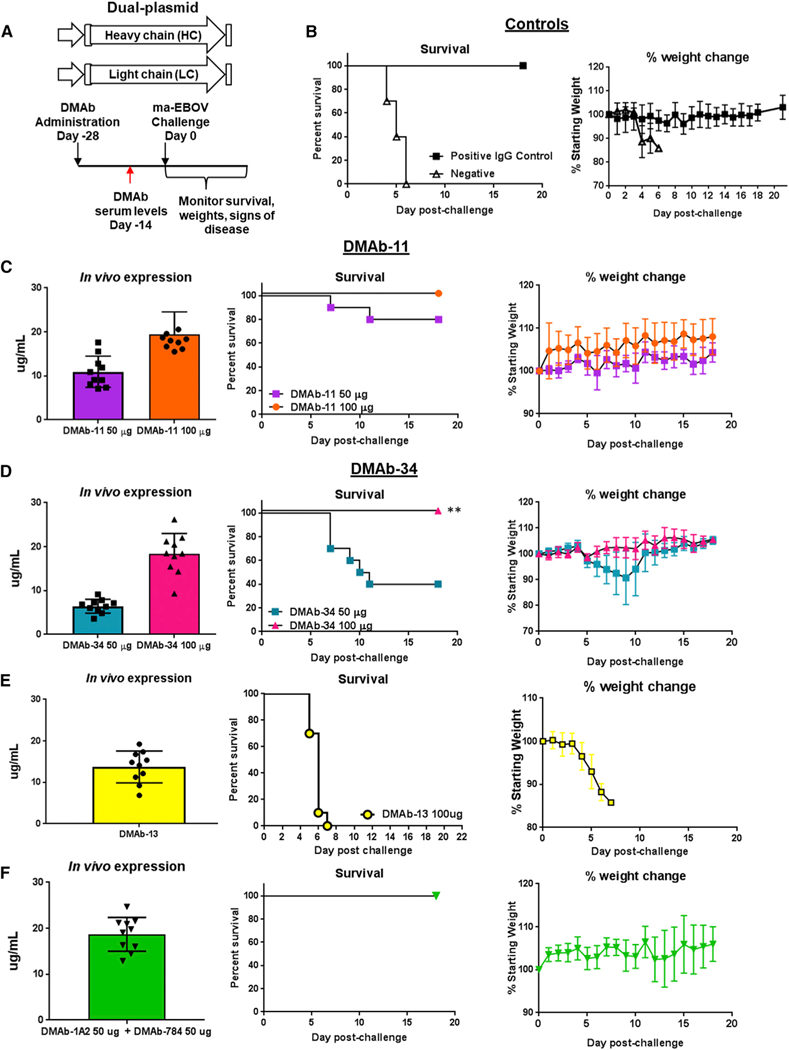
(A) Overview of the injection regimen. Individual DMAb-11 and DMAb-34 (n = 10 mice/group, 50 µg/mouse or 100 µg/mouse), or DMAb-13 (n = 10 mice/group 100 µg/mouse) were administered on day −28, and serum was collected on day −14 before lethal challenge with 1,000LD50 of ma-EBOV. DMAb-11 and DMAb-34 were also co-administered to BALB/c mice (n = 10 mice/group; 1 injection site/DMAb = 2 sites total; 50 µg/DMAb) Animals were monitored for 21 days post-challenge for signs of disease and weight loss.
(B) Survival and percentage weight change for positive control group receiving human 2G4 IgG1 (100 µg/mouse) and negative control group receiving DMAb empty vector pVax1.
(C) DMAb-11 expression at day −14 before challenge, survival, and weight change.
(D) DMab-34 expression at day −14 before challenge, survival, and weight change.
(E) DMAb-13 expression at day −14 before challenge, survival, and weight change for mice administered DMAb-13.
(F) Co-delivered DMAb-11 and DMAb-34 expression at day −14 before challenge, survival, and weight change. All controls and DMAb data in this figure were performed in the same experiment. Error bars represent the SD from the mean.
Co-delivered Anti-GP DMAb Protection against Lethal ma-EBOV Challenge
The potential for pathogen escape is a concern for anti-GP mAbs (Kugelman et al., 2015; Miller et al., 2016). One strategy is co-delivery of more than one antibody clone targeting different epitopes. Accordingly, we co-delivered DMAb-11 and DMAb-34 at separate injections sites on the mouse leg. Animals received 50 µg of DMAb-11 in one hindlimb and 50 µg of DMAb-34 in the opposite hindlimb (Figure 4) on day −28. Total IgG (both DMAb-11 and DMAb-34) was assayed (Figure 4F). Animals were challenged on day 0 with 1,000LD50 of ma-EBOV. Full protection was observed with no signs of disease (Figure 4E). One animal lost weight late during challenge; however, this animal fully recovered.
DMAb-11 Provides Rapid Protection against ma-EBOV Challenge
In all studies, DMAb-11 reliably expressed at high levels, and we observed consistent protection when administered −28 days before lethal challenge. To address the question of anti-GP DMAb protection at shorter time frames closer to lethal challenge, BALB/c mice (n = 10/group) were injected with 200 µg/mouse of DMAb-11 on days −14 and −8 before lethal challenge (Figure 5). Mice were challenged on day 0 with 1,000LD50 of ma-EBOV. The higher 200 µg/mouse dose was selected to observe optimal survival in this short-term experiment. We observed 90% and 80% protection in both groups, respectively, with signs of disease in only one surviving animals (p < 0.001). The other surviving animals did not have any signs of disease. The high protection levels support the hypothesis that the anti-GP DMAbs can rapidly deliver protective immunity and can serve as an important resource for rapid in vivo evaluation of mAb potency during viral challenge.
Figure 5. Rapid In Vivo Protection with DMAb-11.
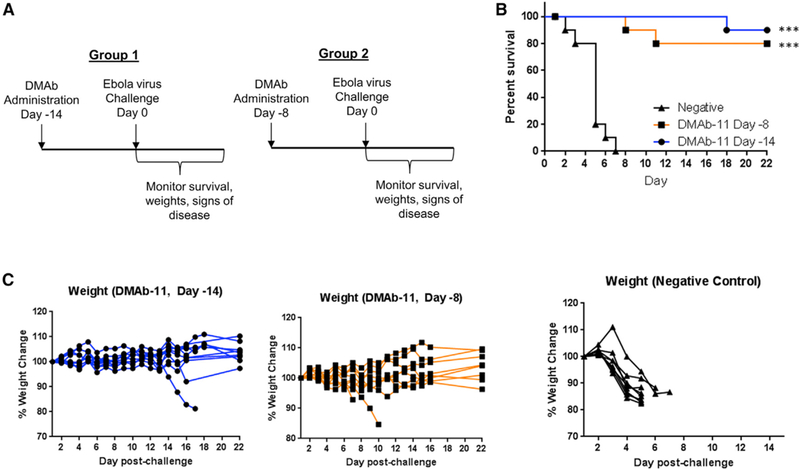
(A) Overview of the injection regimens. DMAbs were administered on day −14 or day −8 before lethal challenge with 1,000LD50 of ma-EBOV (n = 10 mice/group). Animals were monitored for 22 days post-challenge for signs of disease and weight loss.
(B and C) Survival (B) and percent weight change (C) in DMAb-11 groups receiving injection on day −14 or day −8 and the negative control. ***p < 0.001.
Single-Plasmid Delivery of DMAb-11 Provides Short-and Long-Term Protection against ma-EBOV Challenge
For potential DMAb clinical translation, it would be useful from a product perspective to encode both the HC and LC genes into a single plasmid. We encoded DMAb-11 as a single plasmid and performed a challenge experiment to confirm that this construct would be similarly protective. We administered the single-plasmid construct in different doses to BALB/c mice (Figure S5). Animals received 200, 300, or 400 µg of total DMAb-11 single-plasmid DNA. We observed high levels of protection (90%– 100% and no signs of morbidity) with each of the three doses (p < 0.001 compared to control).
To address whether a fully human anti-GP DMAb can provide long-term protection in this model, in one set of animals (n = 10), we administered DMAb-11 via the single-plasmid DMAb construct (400 µg/mouse) and challenged animals 82 days later. Serum mAb levels were monitored on day −26 before challenge, and animals were challenged on day 0 with 1,000LD50 of ma-EBOV. Based on our analyses of DMAb expression over time (Figure 3), it is likely that the animals had levels below 10 µg/mL at the time of challenge. Remarkably, we observed 40% survival in these animals, suggesting that DMAbs can afford long-term protection (Figure 6; p = 0.04). This would be particularly beneficial during a vaccination regimen that requires multiple boosts to achieve optimal efficacy and supports evaluation of a potential co-administration approach with DMAb and vaccine, which could provide rapid as well as long-term protection in a field setting.
Figure 6. Long-Term Protection with DMAb-11.
(A) Construct design.
(B) Overview of injection regimen. DMAbs were administered to BALB/c mice (n = 10/group) −82 days before lethal challenge, and serum was collected on day −56 before lethal challenge with 1,000LD50 of ma-EBOV. Animals were monitored for 21 days post-challenge administration.
(C) DMAb-11 expression in mouse serum. (D) Survival. A log-rank test was performed to compare the two groups. *p = 0.04.
(E) Percent weight change. The gray box represents the shipment and acclimatization period, so serum for these animals could not be tested during this period of time.
DISCUSSION
In these studies, we present in vivo delivery of fully human IgG anti-GP DMAbs that are derived from EVD survivors. We in vivo expressed 26 anti-GP DMAbs covering six different GP regions, representing neutralizing and non-neutralizing epitopes, demonstrating the consistency of this in vivo mAb delivery approach and its usefulness as a resource for rapid down-selection in a live-animal model. We previously showed that DMAbs provide interesting options for delivery of mAb targeting anti-microbial resistant Pseudomonas aeruginosa bacteria (Patel et al., 2017) and emerging viral pathogens including influenza A and B viruses (Elliott et al., 2017), chikungunya virus (Muthumani et al., 2016), and dengue virus (Flingai et al., 2015). In these studies, DMAbs were delivered 2–7 days before challenge. In the current manuscript, we demonstrate with this DMAb mouse model that fully human anti-GP DMAbs are 100% protective when administered at 8–28 days before lethal challenge and can express for months. This study confirms the equivalency of binding for in vivo-delivered DMAbs to recombinant mAb by epitope mapping at the molecular level. Consistent expression of DMAbs in vivo also benefits significantly from in silico sequence design, reductive antibody engineering, delivery, and formulation modifications to increase systemic human IgG expression. DMAb pharmacokinetic expression levels are not dependent on traditional in silico DI predictions, which are designed for conventional cell-based recombinant mAb manufacturing platforms but may not be relevant for in vivo production. Collectively, these studies demonstrate that fully human IgG anti-GP DMAbs can be rapidly evaluated in vivo, an important advance for mAb potency evaluation and translational studies for prevention of infectious diseases such as EBOV and potentially other diseases such as autoimmune disease and cancer.
Others have shown that DNA-encoded fully mouse IgG2a mAbs exhibit long-term expression in mouse models and protect against lethal ma-EBOV challenge (Andrews et al., 2017) and that mouse-human chimeric Ig and humanized mouse Fab VH and VL regions may significantly alter expression and binding, ultimately impacting protection against lethal ma-EBOV challenge (Limberis et al., 2016). This is not surprising given that altered antibody paratope binding and functionality have been observed with murine mAbs containing identical variable regions but different Fc isotypes (Janda et al., 2012, 2016), suggesting that the Fc domain may also place physical constraints on Fab allosteric cooperativity (Janda et al., 2016; Yang et al., 2017) with a potential impact on epitope specificity and virus neutralization (Tudor et al., 2012). Additionally, as our studies demonstrate, amino acid changes can have significant impact on in vivo expression levels (Figure S3) and conversion to a different Fc would likely have direct consequences on gene expression. Therefore, the anti-GP DMAb approach provides an important stepping-stone for evaluation of human DMAb expression and protective efficacy that will likely be enhanced in non-human primates (NHPs) and humans with better matched antibody-receptor interactions and functional responses.
Many fully human recombinant antibodies are well tolerated in people for significant periods of time with limited immunogenicity (Hwang and Foote, 2005) developing. However, some patients react to mAb therapy and are treated with combination approaches to limit anti-antibody responses. In this regard, additional preclinical models that will help evaluate potential candidates and understand this issue are important. In the current study, T cell-depleted DMAb model is fully immune competent at the time of challenge and therefore models immunological responses similar in aspects to a naive, undepleted mouse. The model allows full reconstitution of the immune response by normal thymopoiesis (Laky and Kruisbeek, 2016). As further support for the DMAb mouse model, it has been shown by others that human IgG1 can interact with mouse Fc gamma receptors (Bruhns, 2012; Dekkers et al., 2017), although less efficiently than mouse IgG2a Fc (Overdijk et al., 2012). Taken together, this supports the potential for this mouse model as a valuable resource to initially evaluate human DMAbs in a live model. Further development of the mouse model may benefit from DMAb evaluation in transgenic mice expressing human Fc gamma receptors (Smith et al., 2012) and studies in transgenic human neonatal Fc receptor mice (Proetzel and Roopenian, 2014).
During an outbreak, rapid isolation, evaluation, and delivery of protective mAbs directly from EVD survivors could be a very important approach for providing protection. DMAbs are an important platform for quick investigation of mAbs targeting Ebola virus and other infectious disease pathogens allowing rapid clinical translation, greatly expediting the simultaneous evaluation of multiple mAb clones in vivo and their delivery. Furthermore, DMAbs are simple to modify as additional, highly potent mAb clones are identified. In these studies, we developed the mouse model and optimized anti-GP DMAbs as soon as sequences became available through collaborators or in the public domain. Based on these developments, it is possible to engineer a panel of DMAb candidates for parallel in vivo testing of potency and efficacy. This approach is limited by the availability of potential mAb candidates that have at the least been characterized for binding and neutralization or functionality. DMAb in vivo studies can be performed in small parallel experiments to perform initial expression and characterization studies and down-selection for further studies in additional infectious disease models.
In this context, DMAb delivery fills an important gap between antibody production and in vivo administration, utilizing many of the advancements in mAb discovery and technology established through traditional mAb development. The field of bio-processed IgG production has developed highly sophisticated in silico analysis (Seeliger et al., 2015; Sharma et al., 2014), cell line-based large-scale bioreactors, and refined purification processes; however, their development is slow and it is usually associated with high cost and cold chain requirements for delivery. In vivo delivery strategies such as DMAb are potentially enabling for mAb administration utilizing a platform that is safe, non-integrating, and temperature stable in a diverse range of environments.
In these studies, we demonstrate that the window for protection with anti-GP DMAbs ranges from short-term expression to months of sustained levels, enabling potential administration with immunization campaigns to provide early protection during the time it takes to establish vaccine-induced memory responses. We previously demonstrated that an anti-chikungunya virus DMAb can be delivered in combination with a protective DNA vaccine in mice to provide both immediate and persistent protection without any negative impact on efficacy (Muthumani et al., 2016).
DMAbs have the potential to be administered to various demographic populations including deployable personnel, populations that are contraindicated for other treatments, and those living and working in resource-limited settings. The studies presented here represent a useful step supporting additional DMAb development and translation of in vivo-delivered mAbs to larger species. There have been significant advancements to plasmid DNA delivery technology for vaccine delivery (Amante et al., 2015; Broderick and Humeau, 2015). Rapid evaluation of infected individual repertoires by DMA technology in concert with rapid deployment into at-risk populations is work contemplation. Overall, the anti-GP DMAb approach provides a simple, transient in vivo delivery strategy for highly potent anti-EBOV mAb clones that can be applied to the engineering and screening of pan-filovirus and clones targeting diverse infectious diseases.
STAR★METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| unconjugated purified goat anti-human IgG-Fc | Bethyl | Cat#A-80–104; RRID:AB67060 |
| donkey anti-goat IgG (H+L) cross-adsorbed secondary antibody conjugated to Alexa Fluor 488 | Thermo Fisher Scientific | Cat#A-11055; RRID:AB_2534102 |
| Purified Human IgG/Kappa | Bethyl | P80–111 |
| anti-human Kappa light chain antibody conjugated to horseradish peroxidase | Bethyl | Cat#A80–115P; RRID:AB_67091 |
| anti-human IgG (H+L) conjugated to horseradish peroxidase | Sigma Aldrich | SAB3701359 |
|
Bacterial and Virus Strains | ||
| Mouse-adapted Zaire Ebolavirus (EBOV) | Public Health Agency of Canada | N/A |
|
Chemicals, Peptides, and Recombinant Proteins | ||
| GeneJammer | Agilent | 204130 |
| optimal cutting temperature (O.C.T) medium | Fisher Scientific | 23–730-571 |
| ProLong Gold Antifade reagent with DAPI | Thermo Fisher Scientific | P36931 |
| SIGMAFAST OPD | Sigma Aldrich | P9187 |
| Ebola virus Glycoprotein (strain Mayinga 1976) | Sino Biological | 40304-V08B1 |
| mAb 5.6.1A2 | Emory University | Dr. Rafi Ahmed |
| mAb 15784 | Bornholdt et al. (2) | KU602185 KU602186 |
|
Experimental Models: Cell Lines | ||
| Human embryonic kidney (HEK) 293 T cells | ATCC | Cat#ATCC #CRL-3216; RRID:CVCL_0063 |
| African green monkey Vero E6 cells | ATCC | Cat#ATCC #CRl-1586; RRID:CVCL_0574 |
|
Experimental Models: Organisms/Strains | ||
| BALB/c mouse | Charles River Labs | 028 |
|
Recombinant DNA | ||
| DMAb DNA | This paper | N/A |
|
Software and Algorithms | ||
| SPSS | IBM | N/A |
| GraphPad Prism 7.0 | GraphPad Software | N/A |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, David B. Weiner (dweiner@wistar.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
Human embryonic kidney (HEK) 293 T cells (ATCC #CRL-3216) and African green monkey Vero E6 cells (ATCC #CRl-1586) were maintained in Dulbecco’s Modified Eagles Medium (DMEM, GIBCO) at 37°C, 5% CO2. All cell lines were tested to be mycoplasma negative.
Viruses
All infectious work with Zaire ebolavirus was performed in the biosafety level 4 (BSL-4) facility at the National Microbiology Laboratory, Public Health Agency of Canada (NML/PHAC, Winnipeg, Manitoba, Canada). Zaire ebolavirus expressing enhanced green fluorescent protein (EBOV-GFP) stocks were titered on Vero E6 cells by plaque assay to determine the Plaque Forming Unit (PFU) using a final concentration of 0.7% Agarose (SeaPlaque, Lonza, Switzerland). Ma-EBOV virus stocks were originally obtained by serial passage in mice, as previously described (Bray et al., 1998) and titered using a focus-forming unit (FFU) assay.
In vivo animal studies
Female, six to eight-week old BALB/c mice were purchased from Charles River Laboratories (Malvern, PA) and housed in the animal facilities at the University of Pennsylvania, The Wistar Institute, and NML/PHAC. Female, six to eight-week old MHC Class II knockout mice (MHC II-) and the control parent C56BL6 mouse were obtained from Jackson Laboratories. (Bar Harbor, ME). All animal protocols were approved by the IACUC boards at the University of Pennsylvania (Protocol #: 805596) and Wistar (Protocol 112761) according to guidelines consistent with the Guide for the Care and Use of Laboratory Animals, 8th edition 2011 (the Guide), the Public Health Service Policy on Humane Care and Use of Laboratory Animals (PHS Policy revised 2015) and the Animal Welfare Act and Animal Welfare Regulations (AWRs). All animal research at Wistar adheres to the standards outlined in OLAW Assurance (A3432–01) and the University of Pennsylvania in OLAW Assurance # D16–00045 (A3079–01). All animal protocols at NML/PHAC were approved by the institutional Animal Care Committee at PHAC (Protocol # H15–007), in the guidelines maintained by the Canadian Council on Animal Care (CCAC), and consistent with the Containment Standards for Veterinary Facilities, Guide to the Care and Use of Experimental Animals. Further IACUC oversight was provided by The Animal Care and Use Review Office (ACURO).
Mice received intramuscular injections (50 µg/leg dual-plasmid, 25 μg heavy-chain plus 25 μg light chain or 100 μg/leg single-plasmid) in the tibialis anterior or quadriceps muscles of anti-GP DMAb DNA with hyalurondiase (200U/L, Sigma Aldrich, Saint Louis, MO), followed by electroporation (IM-EP) using the CELLECTRA® 3P adaptive constant current device (Inovio Pharmaceuticals, Ply-mouth Meeting, PA). BALB/c mice were transiently conditioned using T cell depleting antibodies to evaluate human IgG DMAb expression, unrestricted by the murine host immune system. Anti-CD4 (200μg/mouse, BioXcell clone GK1.5) and anti-CD8 (200μg/mouse, BioXcell clone YTS169.4) were administered by intraperitoneal injection immediately prior to DMAb administration. Full immune function is restored 14–21 days post-conditioning (Grcevic et al., 2000). Serum was collected longitudinally to monitor in vivo expression.
Mouse lethal challenge experiments were performed in the NML/PHAC BSL4 facility. Mice received bilateral IP injections at a total volume of 100 µl consisting of 1000 LD50 of ma-EBOV. The challenge stock titer is 1.29 ×107 FFU/mL and one LD50 is 0.01 FFU/ animal. Mice were weighed and scored for clinical signs daily for 21 days and animals were euthanized when their percent weight loss reached 75%.
METHOD DETAILS
In silico analysis
In silico analysis of mAb sequence liabilities was performed in Biovia Discovery Studio (Accelrys, San Diego, CA) and SAbPred (Dunbar et al., 2016). Further sequence analysis was performed using MEGA7.0 (Kumar et al., 2016) and germline protein display datasets obtained from the IMGT repertoire database (Lefranc, 2001; Scaviner et al., 1999). VH and VL family analysis was performed using the IMGT DomainGapAlign database (Ehrenmann et al., 2010; Ehrenmann and Lefranc, 2011).
DMAb construction
The sequences of twenty-six anti-GP monoclonal antibodies were obtained from collaborators at Emory University (Dr. Rafi Ahmed), the Public Health Agency of Canada (Dr. Xiangguo Qiu), Vanderbilt University (Dr. James E. Crowe) (Flyak et al., 2016), and from publically deposited sequences (Bornholdt et al., 2016). These clones bind to different regions of EBOV GP: glycan cap, HR2 region, fusion loop, chalice base, and the mucin domain. The nucleotide sequences for each heavy chain and light chain Fab and Fc regions were codon-optimized (mouse and human) to enhance transgene expression and RNA-optimized for improved stability (Deml et al., 2001; Graf et al., 2004). To further enhance expression, N terminus framework amino acid mutations were introduced for several anti-GP DMAbs. These amino acid changes were selected based on analysis of the germline Ig protein sequence available from the IMGT repertoire database (Lefranc, 2001; Scaviner et al., 1999). The optimized human IgG1 HC and LC were inserted into the pVax1 plasmid DNA expression vector, under the control of the human cytomegalovirus (hCMV) promoter and bovine grown hormone (BGH) polyA signal. The single-plasmid construct encoded both HC and LC genes in cis, separated by a furin cleavege site (RGRKRRS) and a porcine teschovirus-1 2A peptide (P2A). The dual-plasmid construct was encoded on separate plasmids.
DMAb-V2L2 is a single-plasmid construct that encodes an antibody binding to Pseudomonas aeruginosa PcrV protein (Patel et al., 2017).
DMAb expression in vitro
HEK293T cells were transfected with the DMAb DNA single-plasmid or equal mass of HC and LC plasmids (HC + LC) using GeneJammer (Agilent, Wilmington, DE) transfection reagent. Cell supernatants and cell lysates were harvested 40 hours post-transfection to be assayed for human IgG1 production.
Mouse muscle tissue immunofluorescence
BALB/c mice were injected with 50 µg of anti-GP DMAb dual-plasmid DNA by IM injection in the quad muscle followed by IM-EP. Muscles were harvest 2 days post-injection and embedded in optimal cutting temperature compound (Fisher Scientific, Waltham, MA) and snap-frozen on dryice. Muscles were sectioned and fixed with 100% methanol for ten minutes at −20°C. Slides were washed for three minutes with phosphate buffered saline (PBS) + 0.02% Tween 20 (PBST) and then placed in 0.03% Trixon-X100 in 0.05% PBST for fifteen minutes at room temperature. Slides were then washed three times for five minutes/wash with 0.05% PBST and blocked with 5% horse serum in 0.05% PBST for 1 hour. Following incubation, the serum was aspirated and 150 μL of unconjugated purified goat anti-human IgG-Fc (A-80–104, Bethyl, Montgomery, TX) was added to the slides (1:200 dilution in 10% BSA + 0.05% PBST) and incubated overnight at 4°C. The following day, slides were washed three times for five minutes/wash with 0.05% PBST and a donkey anti-goat IgG (H+L) cross-adsorbed secondary antibody conjugated to Alexa Fluor 488 (Thermo Fisher Scientific) was added (1:200 dilution in 0.05% PBST) for thirty minutes at room temperature. A final three washes for five minutes/wash was performed and slides were mounted with ProLong Gold Antifade reagent with DAPI (Thermo Fisher Scientific) before adding cover-slips. In vivo expression was imaged with a Nikon 80i upright fluorescent microscope at 40x magnification.
Human IgG quantification by ELISA
Ninety-six well, high-binding immunosorbent plates were coated with 1μg mL−1 purified anti-Human IgG-Fc (A-80–104A, Bethyl Lab-oratories, Montgomery, TX) and incubated overnight at 4°C. On the next day, plates were blocked with PBS containing 10% FBS for 1 hour at room temperature. Plates were washed with PBS containing 0.05% Tween-20 in between each incubation steps. Plates were incubated with a standard and samples for 1 hour at room temperature. Purified Human IgG/Kappa (P80–111, Bethyl Laboratories, Montgomery, TX) was used as a standard. Samples were diluted in PBS containing 1% FBS and 0.02% Tween-20. Following the incubation, samples were probed with anti-human Kappa light chain antibody conjugated to horseradish peroxidase (A80–115P, Bethyl Laboratories, Montgomery, TX) in 1:20,000 dilution and incubated for 1 hour at room temperature. After incubation, plates were developed with o-Phenylenediamine dihydrochloride (OPD) substrate (SIGMAFAST OPD, Sigma Aldrich, St. Louis, MO) for 25 minutes in the dark and stopped with 2N H2SO4. A BioTek Synergy2 plate reader (Biotek, Winooski, VT) was used to read plates at 450 nm wavelength.
Binding ELISA
Ninety-six well, high-binding immunosorbent plates were coated with 1μg mL−1 Ebola virus Glycoprotein (strain Mayinga 1976) (40304-V08B1, Sino Biological, Beijing, China) and incubated overnight at 4°C. On the next day, plates were blocked using PBS containing 5% non-fat milk and 0.02% Tween-20 for 90 minutes at 37°C. Plates were washed with PBS containing 0.05% Tween-20 in between each incubation steps. After being blocked, plates were incubated with samples in series of dilution for 1 hour at 37°C. Following incubation, samples were probed with anti-human IgG (H+L) conjugated to horseradish peroxidase (SAB3701359, Sigma Aldrich, St. Louis, MO) for 1 hour at 37°C. Plates were developed using OPD substrate for 25 minutes in the dark and stopped using 2N H2SO4. A BioTek Synergy2 plate reader was used to read plates at OD 450nm.
Neutralization assay
Neutralization assays were performed using live EBOV-GFP or rVSV-EBOV-GP. The day before the assay, Vero E6 cells were plated in ninety-six well black plates with a transparent bottom. Serum from DMAb-administered mice was heat inactivated at 56°C for 30 minutes and diluted 1 into 10 and then serially diluted two-fold in DMEM down a 96 well plate and incubated with 100 PFU of EBOV-GFP per well for one hour at 37°C, 5% CO2. The serum:virus mixture was then added to Vero E6 cells (85%–90% confluent) and incubated for one hour at 37°C, 5% CO2. After incubation, the mixture was removed and 100 ml of DMEM plus 2% Bovine Growth Serum (BGS, Hyclone, GE Healthcare Life Sciences, Mississauga, ON, Canada). Cells were then incubated at 37°C, 5% CO2 for up to 144 hours until the GFP signal became saturated. Plates were read for GFP fluorescence daily from the bottom using a Bio-Tek Synergy HT plate reader (Biotek, Winooski, VT).
Alternatively, a pre-titrated amount of rVSV-EBOV GP was incubated with antibody at 37°C for 1 hour before addition to confluent Vero monolayers in 96-well plates. Infection proceeded for 16–18 hr at 37°C in 5% CO2 before cells were fixed in 4% paraformaldehyde and stained with Hoescht. Cells were imaged using a CellInsight CX5 imager (Thermo Fisher) and infection was quantitated by automated enumeration of total cells and those expressing GFP. Infection was normalized to the percent cells infected with rVSV-EBOV GP incubated with a human IgG control antibody. Data are presented as the relative neutralization for each antibody concentration.
Shotgun mutagenesis epitope mapping
Shotgun Mutagenesis epitope mapping (Davidson and Doranz, 2014) on EBOV-GP was performed as described previously (Davidson et al., 2015). Briefly, alanine scanning mutagenesis was carried out on an expression construct for EBOV-GP (strain Mayinga-76; UniProt accession # Q05320) lacking the mucin-like domain (residues 311–461). Residues 33–310 and 462–676 of the EBOV delta (D) mucin GP were mutagenized to create a library of clones, each with an individual point mutant. Residues were changed to alanine, with alanine residues changed to serine. GP residues 1–32, which constitute the GP signal peptide, were not mutagenized. The resulting EBOV GP alanine-scan library covered 492 of 493 of target residues (99.9%). Each mutation was confirmed by DNA sequencing, and clones were arrayed into 384-well plates, one mutant per well. Each library plate also contained negative control wells with vector alone and positive control wells containing wild-type EBOV Δmucin GP.
Before epitope mapping on the mutation library, we confirmed that mAb 5.6.1A2 and 15784 and mouse DMAb-11 and DMAb-34 serum showed reactivity with EBOV-GP, and identified an appropriate mAb concentration and serum dilution for screening the mutation library. mAb 5.6.1A2 and 15784 and DMAb-11 and DMAb-34 mouse serum (each pooled from multiple mice) were tested for binding to wild-type EBOV Δmucin GP expressed in HEK293T cells. After addition of a fluorescent secondary antibody, the mean cellular fluorescence was detected using an Intellicyt flow cytometer. The entire EBOV Δmucin GP library expressed in HEK293T cells was screened for binding of mutant clones to mAb 5.6.1A2 and 15784, or to DMAb-11 and DMAb-34 mouse serum, by detecting mean cellular fluorescence. Mutations within clones were identified as critical to the mAb epitope if they did not support reactivity of the mAb, but did support reactivity of other conformation-dependent MAbs (Davidson et al., 2015; Davidson and Doranz, 2014). This counter-screen strategy facilitates the exclusion of GP mutants that are globally or locally misfolded or that have an expression defect. Validated critical residues represent amino acids whose side chains make the highest energetic contributions to the mAb-epitope interaction (Bogan and Thorn, 1998; Lo Conte et al., 1999).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistics
Statistical analyses were performed using GraphPad Prism 7.0 software (La Jolla, CA) or SPSS (IBM). For survival studies, we performed sample size calculations for two-independent proportions, alpha 0.05 and power 0.80. For DMAb expression studies, n = 5 mice/group and for challenge studies n = 10 mice/group was determined to be the number of animals need in order to achieve statistical significance. Protection study data were represented by a Kaplan-Meier survival curve and log-rank test analysis, followed by two-way ANOVA with correction for multiple comparisons. Samples and animal groups with a p value < 0.05 were considered statistically. All bar graphs and line graphs display individual animals or the mean value, and error bars represent the standard deviation. Supplementary tables display the results for individual mice. Statistical details can be found in the Results, figure legends, and on the figures.
Supplementary Material
Highlights.
DMAbs are an in vivo approach for mAb development/ delivery against Zaire ebolavirus
DMAbs can be functionally equivalent to recombinant mAb
Reproducible and cost-effective mouse model for in vivo mAb evaluation
Enables evaluation of fully human mAbs rapidly in vivo
ACKNOWLEDGMENTS
The authors would like to thank Jonathan Audet (PHAC) for analysis of the EBOV-GFP neutralization assay, Dr. Alfredo Perales-Puchalt (Wistar) for statis-tical analysis, and the Wistar Histotechnology Core Facility for tissue sectioning and staining. This research was funded by DARPA Grant W31P4Q-15-1-0003 awarded to Inovio Pharmaceuticals and NIH Contract HHSN272201400058C to B.J.D.
K.M. reports receiving grants from DARPA and Inovio, receiving consulting fees from Inovio related to DNA vaccine development, and a pending patent application (to Inovio) for delivery of DNA-encoded monoclonal antibodies. T.R.F.S., C.R., M.C.W., J.Y., J.C., K.E.B., L.H., and N.Y.S. are employees of Inovio Pharmaceuticals and as such receive salary and benefits, including ownership of stock and stock options, from the company. A.B., E.D., and B.J.D. are employees of Integral Molecular and as such receive salary and benefits, including ownership of stock and stock options, from the company. D.B.W. has received grant funding, participates in industry collaborations, has received speaking honoraria, and has received fees for consulting, including serving on scientific review committees and board services.
Footnotes
DECLARATION OF INTERESTS
Remuneration received by D.B.W. includes direct payments or stock or stock options, and in the interest of disclosure he notes potential conflicts associated with this work with Inovio and possibly others. In addition, he has a patent DNA vaccine delivery pending to Inovio.
REFERENCES
- Amante DH, Smith TR, Mendoza JM, Schultheis K, McCoy JR, Khan AS, Sardesai NY, and Broderick KE (2015). Skin transfection patterns and expression kinetics of electroporation-enhanced plasmid delivery using the CELLECTRA-3P, a portable next-generation dermal electroporation device. Hum. Gene Ther. Methods 26, 134–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews CD, Luo Y, Sun M, Yu J, Goff AJ, Glass PJ, Padte NN, Huang Y, and Ho DD (2017). In vivo production of monoclonal antibodies by gene transfer via electroporation protects against lethal influenza and Ebola infections. Mol. Ther. Methods Clin. Dev 7, 74–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogan AA, and Thorn KS (1998). Anatomy of hot spots in protein interfaces. J. Mol. Biol 280, 1–9. [DOI] [PubMed] [Google Scholar]
- Bornholdt ZA, Turner HL, Murin CD, Li W, Sok D, Souders CA, Piper AE, Goff A, Shamblin JD, Wollen SE, et al. (2016). Isolation of potent neutralizing antibodies from a survivor of the 2014 Ebola virus outbreak. Science 351, 1078–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray M, Davis K, Geisbert T, Schmaljohn C, and Huggins J (1998). A mouse model for evaluation of prophylaxis and therapy of Ebola hemorrhagic fever. J. Infect. Dis 178, 651–661. [DOI] [PubMed] [Google Scholar]
- Broderick KE, and Humeau LM (2015). Electroporation-enhanced delivery of nucleic acid vaccines. Expert Rev. Vaccines 14, 195–204. [DOI] [PubMed] [Google Scholar]
- Bruhns P (2012). Properties of mouse and human IgG receptors and their contribution to disease models. Blood 119, 5640–5649. [DOI] [PubMed] [Google Scholar]
- Corti D, Misasi J, Mulangu S, Stanley DA, Kanekiyo M, Wollen S, Ploquin A, Doria-Rose NA, Staupe RP, Bailey M, et al. (2016). Protective monotherapy against lethal Ebola virus infection by a potently neutralizing anti-body. Science 351, 1339–1342. [DOI] [PubMed] [Google Scholar]
- Davey RT Jr., Dodd L, Proschan MA, Neaton J, Neuhaus Nordwall J, Koopmeiners JS, Beigel J, Tierney J, Lane HC, Fauci AS, et al. ; PREVAIL II Writing Group; Multi-National PREVAIL II Study Team (2016). A randomized, controlled trial of ZMapp for Ebola virus infection. N. Engl. J. Med 375, 1448–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson E, and Doranz BJ (2014). A high-throughput shotgun mutagenesis approach to mapping B-cell antibody epitopes. Immunology 143, 13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson E, Bryan C, Fong RH, Barnes T, Pfaff JM, Mabila M, Rucker JB, and Doranz BJ (2015). Mechanism of binding to Ebola virus glycopro-tein by the ZMapp, ZMAb, and MB-003 cocktail antibodies. J. Virol 89, 10982–10992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekkers G, Bentlage AEH, Stegmann TC, Howie HL, Lissenberg-Thunnissen S, Zimring J, Rispens T, and Vidarsson G (2017). Affinity of human IgG subclasses to mouse Fc gamma receptors. MAbs 9, 767–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deml L, Bojak A, Steck S, Graf M, Wild J, Schirmbeck R, Wolf H, and Wagner R (2001). Multiple effects of codon usage optimization on expression and immunogenicity of DNA candidate vaccines encoding the human immunodeficiency virus type 1 Gag protein. J. Virol 75, 10991–11001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumiak M (2014). Making it to manufacturing. The potential success of broadly neutralizing monoclonal antibodies for HIV prevention, treatment, and possibly even a cure could come at a cost. IAVI Rep 18, 4–7, 17. [PubMed] [Google Scholar]
- Dunbar J, Krawczyk K, Leem J, Marks C, Nowak J, Regep C, Georges G, Kelm S, Popovic B, and Deane CM (2016). SAbPred: a structure-based antibody prediction server. Nucleic Acids Res 44 (W1), W474–W478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrenmann F, and Lefranc MP (2011). IMGT/DomainGapAlign: IMGT standardized analysis of amino acid sequences of variable, constant, and groove domains (IG, TR, MH, IgSF, MhSF). Cold Spring Harb. Protoc 2011, 737–749. [DOI] [PubMed] [Google Scholar]
- Ehrenmann F, Kaas Q, and Lefranc MP (2010). IMGT/3Dstructure-DB and IMGT/DomainGapAlign: a database and a tool for immunoglobulins or anti-bodies, T cell receptors, MHC, IgSF and MhcSF. Nucleic Acids Res 38, D301–D307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott STC, Kallewaard NL, Benjamin E, Wachter-Rosati L, McAuliffe JM, Patel A, Smith TRF, Schultheis K, Park DH, Flingai S, et al. (2017). DMAb inoculation of synthetic cross reactive antibodies protects against lethal influenza A and B infections. NPJ Vaccines 2, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flingai S, Plummer EM, Patel A, Shresta S, Mendoza JM, Broderick KE, Sardesai NY, Muthumani K, and Weiner DB (2015). Protection against dengue disease by synthetic nucleic acid antibody prophylaxis/immunotherapy. Sci. Rep 5, 12616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flyak AI, Shen X, Murin CD, Turner HL, David JA, Fusco ML, Lampley R, Kose N, Ilinykh PA, Kuzmina N, et al. (2016). Cross-reactive and potent neutralizing antibody responses in human survivors of natural Ebolavirus infection. Cell 164, 392–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graf M, Deml L, and Wagner R (2004). Codon-optimized genes that enable increased heterologous expression in mammalian cells and elicit efficient im-mune responses in mice after vaccination of naked DNA. Methods Mol. Med 94, 197–210. [DOI] [PubMed] [Google Scholar]
- Grcevic D, Lee SK, Marusic A, and Lorenzo JA (2000). Depletion of CD4 and CD8 T lymphocytes in mice in vivo enhances 1,25-dihydroxyvitamin D3-stimulated osteoclast-like cell formation in vitro by a mechanism that is dependent on prostaglandin synthesis. J. Immunol 165, 4231–4238. [DOI] [PubMed] [Google Scholar]
- Hwang WY, and Foote J (2005). Immunogenicity of engineered antibodies. Methods 36, 3–10. [DOI] [PubMed] [Google Scholar]
- Janda A, Eryilmaz E, Nakouzi A, Cowburn D, and Casadevall A (2012). Variable region identical immunoglobulins differing in isotype express different paratopes. J. Biol. Chem 287, 35409–35417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janda A, Bowen A, Greenspan NS, and Casadevall A (2016). Ig constant region effects on variable region structure and function. Front. Microbiol 7, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karo JM, Schatz DG, and Sun JC (2014). The RAG recombinase dictates functional heterogeneity and cellular fitness in natural killer cells. Cell 159, 94–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kugelman JR, Kugelman-Tonos J, Ladner JT, Pettit J, Keeton CM, Nagle ER, Garcia KY, Froude JW, Kuehne AI, Kuhn JH, et al. (2015). Emergence of Ebola virus escape variants in infected nonhuman primates treated with the MB-003 antibody cocktail. Cell Rep 12, 2111–2120. [DOI] [PubMed] [Google Scholar]
- Kumar S, Stecher G, and Tamura K (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol 33, 1870– 1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunert R, and Reinhart D (2016). Advances in recombinant antibody manufacturing. Appl. Microbiol. Biotechnol 100, 3451–3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laky K, and Kruisbeek AM (2016). In vivo depletion of T lymphocytes. Curr. Protoc. Immunol 113, 4.1.1–4.1.9. [DOI] [PubMed] [Google Scholar]
- Lauer TM, Agrawal NJ, Chennamsetty N, Egodage K, Helk B, and Trout BL (2012). Developability index: a rapid in silico tool for the screening of antibody aggregation propensity. J. Pharm. Sci 101, 102–115. [DOI] [PubMed] [Google Scholar]
- Lefranc MP (2001). IMGT, the international ImMunoGeneTics database. Nucleic Acids Res 29, 207–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limberis MP, Tretiakova A, Nambiar K, Wong G, Racine T, Crosariol M, Xiangguo Q, Kobinger G, and Wilson JM (2016). Adeno-associated virus serotype 9-expressed ZMapp in mice confers protection against systemic and airway-acquired Ebola virus infection. J. Infect. Dis 214, 1975– 1979. [DOI] [PubMed] [Google Scholar]
- Lo Conte L, Chothia C, and Janin J (1999). ). The atomic structure of protein-protein recognition sites. J. Mol. Biol 285, 2177–2198. [DOI] [PubMed] [Google Scholar]
- McMahon JM, Signori E, Wells KE, Fazio VM, and Wells DJ (2001). Optimisation of electrotransfer of plasmid into skeletal muscle by pretreatment with hyaluronidase – increased expression with reduced muscle damage. Gene Ther 8, 1264–1270. [DOI] [PubMed] [Google Scholar]
- Miller CR, Johnson EL, Burke AZ, Martin KP, Miura TA, Wichman HA, Brown CJ, and Ytreberg FM (2016). Initiating a watch list for Ebola virus antibody escape mutations. PeerJ 4, e1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthumani K, Block P, Flingai S, Muruganantham N, Chaaithanya IK, Tingey C, Wise M, Reuschel EL, Chung C, Muthumani A, et al. (2016). Rapid and long-term immunity elicited by DNA-encoded antibody prophylaxis and DNA vaccination against chikungunya virus. J. Infect. Dis 214, 369–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overdijk MB, Verploegen S, Ortiz Buijsse A, Vink T, Leusen JH, Bleeker WK, and Parren PW (2012). Crosstalk between human IgG iso-types and murine effector cells. J. Immunol 189, 3430–3438. [DOI] [PubMed] [Google Scholar]
- Patel A, DiGiandomenico A, Keller AE, Smith TRF, Park DH, Ramos S, Schultheis K, Elliott STC, Mendoza J, Broderick KE, et al. (2017). An engineered bispecific DNA-encoded IgG antibody protects against Pseudomonas aeruginosa in a pneumonia challenge model. Nat. Commun 8, 637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrosillo N, Nicastri E, Lanini S, Capobianchi MR, Di Caro A, Antonini M, Puro V, Lauria FN, Shindo N, Magrini N, et al. ; INMI EBOV Team (2015). Ebola virus disease complicated with viral interstitial pneumonia: a case report. BMC Infect. Dis 15, 432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proetzel G, and Roopenian DC (2014). Humanized FcRn mouse models for evaluating pharmacokinetics of human IgG antibodies. Methods 65, 148–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X, Alimonti JB, Melito PL, Fernando L, Stro¨her U, and Jones SM (2011). Characterization of Zaire ebolavirus glycoprotein-specific monoclonal antibodies. Clin. Immunol 141, 218–227. [DOI] [PubMed] [Google Scholar]
- Samaranayake H, Wirth T, Schenkwein D, Ra¨ty JK, and Yla¨-Herttuala S (2009). Challenges in monoclonal antibody-based therapies. Ann. Med 41, 322–331. [DOI] [PubMed] [Google Scholar]
- Scaviner D, Barbie V, Ruiz M, and Lefranc MP (1999). Protein displays of the human immunoglobulin heavy, kappa and lambda variable and joining re-gions. Exp. Clin. Immunogenet 16, 234–240. [DOI] [PubMed] [Google Scholar]
- Seeliger D, Schulz P, Litzenburger T, Spitz J, Hoerer S, Blech M, Enenkel B, Studts JM, Garidel P, and Karow AR (2015). Boosting antibody developability through rational sequence optimization. MAbs 7, 505–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma VK, Patapoff TW, Kabakoff B, Pai S, Hilario E, Zhang B, Li C, Borisov O, Kelley RF, Chorny I, et al. (2014). In silico selection of therapeutic antibodies for development: viscosity, clearance, and chemical stability. Proc. Natl. Acad. Sci. USA 111, 18601–18606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith P, DiLillo DJ, Bournazos S, Li F, and Ravetch JV (2012). Mouse model recapitulating human Fcg receptor structural and functional diversity. Proc. Natl. Acad. Sci. USA 109, 6181–6186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trad MA, Naughton W, Yeung A, Mazlin L, O’Sullivan M, Gilroy N, Fisher DA, and Stuart RL (2017). Ebola virus disease: an update on current prevention and management strategies. J. Clin. Virol 86, 5–13. [DOI] [PubMed] [Google Scholar]
- Tudor D, Yu H, Maupetit J, Drillet AS, Bouceba T, Schwartz-Cornil I, Lopalco L, Tuffery P, and Bomsel M (2012). Isotype modulates epitope specificity, affinity, and antiviral activities of anti-HIV-1 human broadly neutralizing 2F5 antibody. Proc. Natl. Acad. Sci. USA 109, 12680–12685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson JA, Hevey M, Bakken R, Guest S, Bray M, Schmaljohn AL, and Hart MK (2000). Epitopes involved in antibody-mediated protection from Ebola virus. Science 287, 1664–1666. [DOI] [PubMed] [Google Scholar]
- Yang D, Kroe-Barrett R, Singh S, Roberts CJ, and Laue TM (2017). IgG cooperativity—is there allostery? Implications for antibody functions and ther-apeutic antibody development. MAbs 9, 1231–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Ren J, Harlos K, Jones DM, Zeltina A, Bowden TA, Padilla-Parra S, Fry EE, and Stuart DI (2016). Toremifene interacts with and de-stabilizes the Ebola virus glycoprotein. Nature 535, 169–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.