

Abstract
Background:
Congenital heart disease (CHD) is one of the most common birth defects; however, the mechanisms underlying its development are poorly understood. Recently, heritable genetic factors, including copy number variations (CNVs) and single nucleotide polymorphisms (SNPs), have been implicated in its etiology. The aim of this study was to investigate the utility of a SNP array for the prenatal diagnosis of CHD and the improvement of prenatal genetic counseling and to compare this approach to traditional chromosome analysis.
Methods:
One hundred and fortysix cases of CHD detected by prenatal echocardiography were analyzed. Of these, 110 were isolated CHD and 36 were of CHD with extracardiac defects. SNP analysis was performed using the Affymetrix CytoScan HD platform, which was followed by karyotype analysis. All annotated CNVs were validated by fluorescence in situ hybridization.
Results:
Karyotype analysis identified chromosomal abnormalities in 19 of 146 cases. In addition to the 15 chromosomal abnormalities that were consistent with the results of karyotype analysis, the SNP array identified abnormal CNVs in an additional 15.2% (22/145) cases; of these, 15 were pathogenic CNVs, three were variations of uncertain clinical significance, and four were benign CNVs. The rates at which the SNP array detected pathogenic CNVs differed significantly between cases of isolated CHD and CHD with extracardiac defects (13.6% vs. 72.2%, P = .001). The results of the SNP array also affected the rate of pregnancy termination.
Conclusion:
Combining SNP array with cytogenetic analyses is particularly effective for identifying chromosomal abnormalities in CNVs in fetuses with CHD, which also affects obstetrical outcomes.
Keywords: congenital heart disease, fetal, SNP array
1. Introduction
Congenital heart disease (CHD) is one of the most common birth defects, affecting up to 8 of every 1000 babies born in China.[1] It is characterized by defective heart structure, which results from the malformation of the heart wall and veins during embryogenesis, and is considered the main noninfectious cause of fetal death. Currently, diagnosis relies on medical imaging and CHDs are classified into 2 types based on their association with other congenital defects as follows: isolated CHD, without other congenital defects, and syndromic CHD associated with urinary or nervous system malformations.[2]
Although environmental and genetic factors or a combination thereof are considered the main causes of CHD,[3] the mechanism underlying its development is poorly understood. Recent studies showed that heritable genetic factors play important roles in its etiology.[4,5] These include chromosomal variations and mutations, as well as copy number variations (CNVs), which comprise one of the main causes.[6] Chromosomal microarray analysis (CMA) entails the use of microarray-based comparative genomic hybridization and a single nucleotide polymorphism (SNP) array, both of which can detect microdeletions and microduplications in the genome. In addition to CNVs, SNPs can be used to detect uniparental disomy and chimeras.
In this study, we performed a whole-genome scan using SNP array technology and karyotyping to analyze 146 CHD cases assessed by ultrasonic cardiography to identify the genetic factors responsible for fetal CHDs and explore the clinical utility of SNPs in diagnosing this disease.
2. Methods
2.1. Patient data
We conducted a retrospective study on CHD cases diagnosed prenatally using ultrasound data from January 2016 to April 2018 at the Prenatal Diagnosis Center of the Fujian Provincial Maternal and Children Health Hospital. Fetal samples were collected by amniocentesis (n = 76) and cord blood sampling (n = 70) according to gestational age. Amniotic fluid was collected by amniocentesis at 16 to 24 gestational weeks, and fetal blood was collected by cordocentesis after the 24th gestational week. The fetuses underwent routine ultrasonic scans, and fetal biometry was assessed at a median gestational age of 25 + 5 weeks (range, 18 + 3–33 + 4 weeks). This study was approved by the ethics committee of the Fujian Provincial Maternal and Child Health Hospital, and informed consent was obtained from the parents for invasive prenatal diagnosis. Approximately 2.0 mL of peripheral venous blood was collected from the parents once fetal pathogenic CNVs were confirmed. DNA was extracted using a Gentra Puregene Blood Kit (Qiagen, Hilden, Germany).
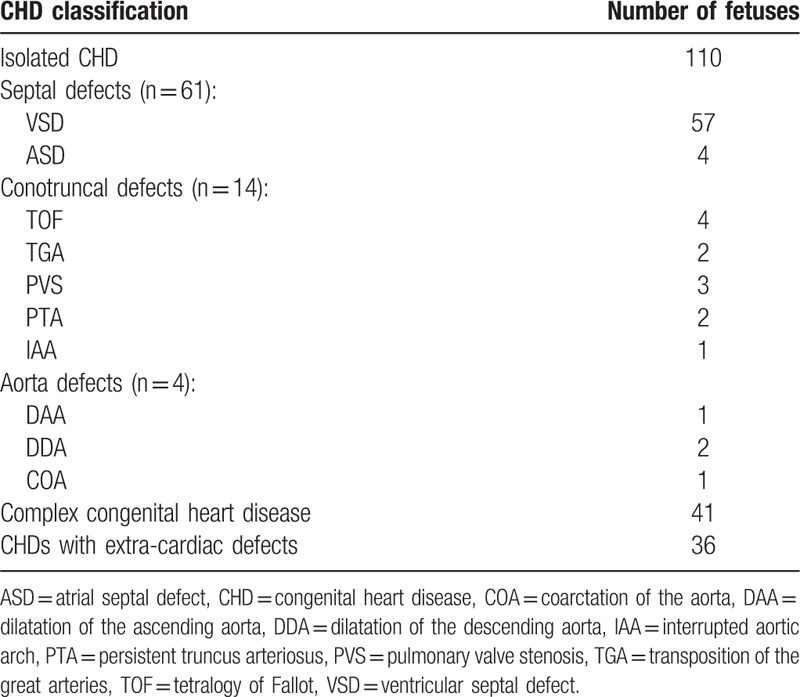
In total, 146 CHD cases were included, among which 110 (75.3%) were of isolated CHD and 36 (24.7%) were of CHD with extra-cardiac defects. Among the 110 isolated CHD fetuses, 61 were cases of septal defects (57 cases with ventricular septal defects and 4 with atrial septal defects), 14 of conotruncal defects (4 with tetralogy of Fallot, 2 with transposition of great arteries, 3 with pulmonary valve stenosis, 2 with persistent truncus arteriosus, and 1 with interrupted aortic arch), 4 of aortal defects (one with dilatation of the ascending aorta, 2 with dilatation of the descending aorta, and 1 with coarctation of the aorta), and 41 of complex congenital heart diseases (detailed information in Table 1).
Table 1.
Phenotypic characteristics of 146 fetuses with congenital heart diseases.
2.2. Karyotype analysis
Cultured amniocytes or lymphocytes were analyzed by regular cytogenetic analysis using Giemsa banding at a resolution of 450 to 550 bands.
2.3. SNP array
Genomic DNA was extracted directly from uncultured amniotic fluid and cord blood samples using a QIAamp DNA Blood Mini Kit (Qiagen). DNA was quantified using a NanoDrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer's instructions. The DNA quality was assessed using agarose gel electrophoresis. The CytoScan HD genome-wide high-resolution SNP array (Affymetrix, Santa Clara, CA), which includes both SNPs and oligonucleotide probes, was used.
DNA (250 ng) was amplified, labeled, and hybridized according to the manufacturer's instructions. Procedures for DNA digestion, ligation, polymerase chain reaction (PCR), fragmentation, labeling, and array hybridization were performed according to the manufacturer's protocol. The CNV-reporting filter was set at > 100 kb, with a minimum set of 50 marker counts. The results obtained by scanning the CytoScan arrays were analyzed using the Chromosome Analysis Suite software (Affymetrix, Inc., Santa Clara, CA, USA) and annotated based on GRCh37 (hg19). The CNVs were classified as benign, pathogenic, or variants of uncertain clinical significance (VOUS) according to the American College of Medical Genetics (ACMG) guidelines.[7] Parental testing was performed for fetuses with CHD that had abnormal SNP array results to determine the inheritance pattern of the deletions and/or duplications. All annotated CNVs were experimentally validated using fluorescence in situ hybridization (FISH).
2.4. Statistical analysis
SPSS Statistics 20 software (IBM, Armonk, NY) was used for statistical analysis. The detection rates of pathogenic results were compared between the karyotype cytogenetic analyses and SNP array groups of the fetuses with isolated CHD and fetuses with CHD with extra-cardiac defects. P < .05 indicated statistical significance.
3. Results
3.1. Karyotype analysis
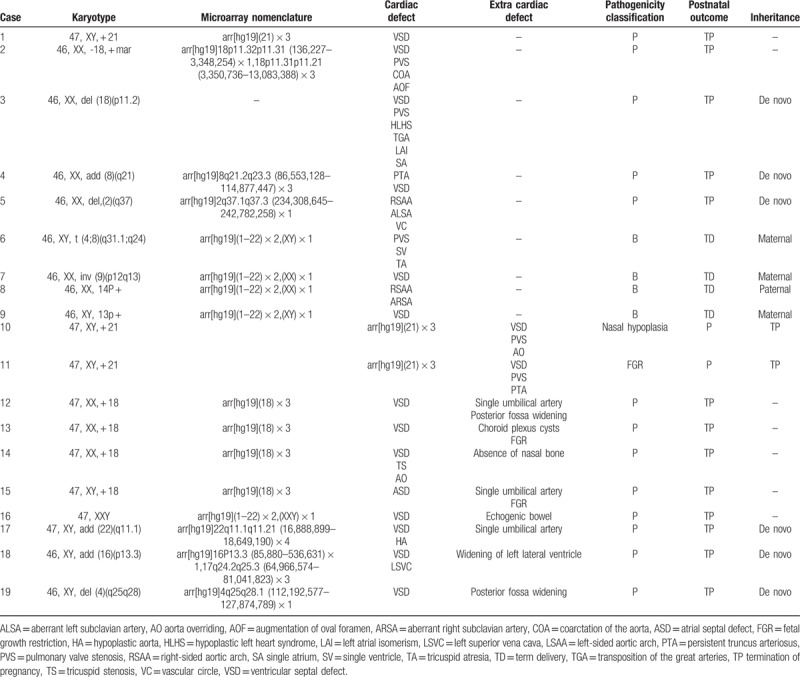
Karyotype analysis identified chromosomal abnormalities in 19 of 146 cases of CHD. Among the 110 fetuses with isolated CHD, karyotype cytogenetic analysis identified 9 cases (8.2%) of clinically significant chromosomal abnormalities, including one case each of trisomy 21, deletions of 2q37 and 18p11.2, duplication of 8q21, and an unusual partial aneuploidy that involved monosomy 18 and trisomy 18, with one chromosome exhibiting a 18p11.32p11.31 microdeletion and another a 18p11.31p11.21 duplication, respectively. Furthermore, we detected one balanced translocation t (4;8) (q31.1;q24), one pericentric (9) (p12q13) inversion, one 14p + , and one 13p + that were not detected by the SNP array. Among the 36 fetuses exhibiting CHD with extra-cardiac defects, karyotype analysis identified chromosomal abnormalities in 10 (27.8%) as follows: trisomy 21 (n = 2), trisomy 18 (n = 4), Klinefelter syndrome (n = 1), deletion of 4q25q28 (n = 1), and duplications of 22q11.2 (n = 1) and 6p13.3 (n = 1; Table 2).
Table 2.
Results of chromosomal microarray analysis of fetuses with congenital heart diseases and abnormal karyotypes.
3.2. Detection rates of abnormal karyotypes using the SNP array
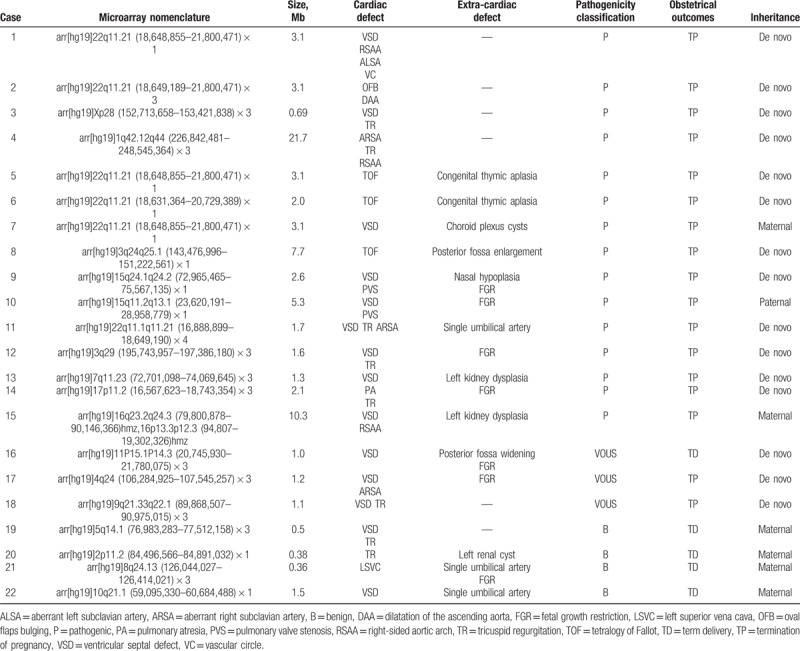
SNP array hybridization was performed for 145 of 146 fetuses with CHD (one of the parents refused to participate in the SNP array analysis). The results of this analysis differed from that of karyotype analysis in 24.8% of cases (36/145). In addition to the 15 cases of chromosome abnormalities that were consistent with the results of the karyotype analysis, the SNP array identified abnormal CNVs in 22 (15.2%) cases; of these CNVs, 15 were pathogenic, 3 were VOUS, and 4 were benign. Among the 15 cases with pathogenic CNVs, 4 were isolated CHD. In these 15 cases, the SNP array identified deletions of 3q24q25.1, 15q11.2q13.1, 15q24.1q24.2, and 22q11.21; duplications of 1q42.12q44, 3q29, 7q11.23, 9q21.33q22.1, 17p11.2, 22q11.21, 22q11.1q11.21, and Xp28; and losses of heterozygosity (LOHs) in 16q23.2q24.3 and 16p13.3p12.3. Eight of the identified CNVs were related to known chromosomal disorders, namely, DiGeorge (n = 4), cat eye (n = 2), Prader–Willi (n = 1), and Potocki–Lupski syndromes (n = 1).
3.3. Comparison of pathogenic CNV detection rates
Overall, the rate at which pathogenic CNVs were detected by the SNP array was significantly higher than that of standard karyotype analysis (P < .05). Of 146 fetuses with CHD that underwent karyotype cytogenetic and SNP array analyses, 110 (75.3%) harbored isolated cardiac defects, whereas 36 (24.7%) possessed extra-cardiac defects. The detection rates of pathogenic CNVs using the SNP array differed significantly between cases of isolated CHD and CHD with extra-cardiac defects (P < .05) (Table 3).
Table 3.
Comparison of the results of chromosomal microarray analysis and normal karyotype analysis.
3.4. Inheritance analysis and obstetrical outcomes
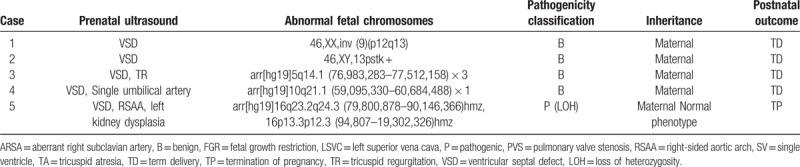
We next screened inheritance information of 32 families with abnormal karyotype results (excluding those with chromosome aneuploidies) and abnormal CNVs. Eleven fetuses inherited abnormal CNVs from unaffected parents, whereas 21 were de novo CNVs. Chromosomal abnormalities (n = 15), pathogenic CNVs (n = 15), and VOUS CNVs (n = 2) accounted for the termination of respective pregnancies. Furthermore, we observed that 18.2% (20/110) of isolated CHD cases and 72.2% (26/36) of CHD cases with extra-cardiac defects resulted in the termination of pregnancy (Table 4).
Table 4.
Fetuses with congenital heart diseases inherited from unaffected parents.
4. Discussion
In this study, 146 pregnant women with fetuses with CHD consented to undergo karyotyping and SNP array testing after fetal anatomy scans and echocardiography. We obtained clinically significant results in 21.2% (31/146) of fetuses. Karyotype cytogenetic analysis identified 7 chromosomal aneuploidies associated with trisomy 21, trisomy 18, and Klinefelter syndrome. In the prenatal setting, the incidence of chromosomal anomalies in fetuses with CHD is reportedly as high as 18% to 22%, with most anomalies being trisomy 21, trisomy 18, and 22q11 microdeletions.[8–10] Moreover, cytogenetic karyotype analysis identified one balanced translocation, one pericentric inversion, and duplications in the short arms of 2 chromosomes that were not detected by the SNP array.
According to Wapner et al,[11] SNP arrays cannot replace karyotype analysis because of their inability to detect chromosomal translocations or inversions. However, in one case where amniotic fluid cell culture was unsuccessful, the SNP array was able to detect an abnormal pathogenic CNV. As such, the SNP array is superior to karyotype analysis because of its ability to detect CNVs with high accuracy and resolution without the need for amniotic fluid cell culture.
According to the SNP array performed for 145 CHD fetuses, 24.8% (36/145) of cases had abnormalities. Several studies have reported that the diagnostic yield of SNP array testing used in prenatal evaluation ranges from 6.6% to 19.2%.[8,12–14] Thus, the actual clinical detection rates in our cohort were slightly higher than those in previous studies on CHD. These results demonstrate that variations in the detection rates of pathogenic CNVs might occur based on CHD type, clinical differences among cases, and differences in the scales of array probes.
Among the 15 pathogenic CNVs identified, 4 were cases of isolated CHD, specifically, one 22q11.2 deletion and one each of duplication in 22q11.2, 1q42, and Xq28. Both duplications and deletions of 22q11.2 are associated with cardiac abnormalities in approximately 75% and 15% of cases, respectively.[15] Some patients with 1q42–44 duplications presented with cardiac abnormalities. Although a correlation between Xq28 duplications and heart malformations has not been reported, this fragment is important for intellectual development and is a known pathogenic CNV.[16] A CHD is more likely to be related to genetic disorders if it is detected in the presence of other structural anomalies.[17] We also identified a set of 10 different CNVs associated with rare chromosomal diseases as follows: 6 microdeletions, namely, three 22q11.21 (DiGeorge syndrome[15]), one 3q24q25.1, one 15q11.2q13.1 (Prader–Willi syndrome[18]), and one 15q24.1q24.2, and 4 microduplications, namely, 22q11.1q11.21 (cat eye syndrome[19]), 17p11.2 (Potocki–Lupski syndrome[20]), 7q11.23, and 3q29. These syndromes are associated with a range of physical and mental disabilities, as well as congenital organ malformations, including CHD. Syndromic CHD has various etiologies, such as microscopic and submicroscopic chromosomal abnormalities, monogenic syndromes, and epigenetic and environmental factors.[21] We also detected one case of CHD with LOH in 16q23.2q24.3 and 16p13.3p12.3.
VOUS was detected at a rate of 2.1% (3/145) in our analysis. This is lower than, but not substantially different from, the rates reported previously.[22] Among the 145 cases tested by chromosomal microarray, 4 were considered benign based on current knowledge and would not have been reported otherwise. Recently, a systematic meta-analysis compared the rates at which imbalances are detected by CMA to those detected by conventional karyotyping and 22q11 microdeletion analysis by FISH in fetuses with cardiac malformations. CMA yielded additional clinically valuable information for 7.0% of fetal CHD cases. Subgroup analysis showed an increased detection rate of 3.4% and 9.3% in isolated and nonisolated CHD cases, respectively.[22–23] In this study, the detection rates of pathogenic CNVs by SNP array differed significantly between cases with isolated CHD and those with CHD with extra-cardiac defects (13.6% vs 72.2% P < .001).
We detected 4 CHD cases with 22q11 deletion syndrome. Genetic analysis of the parents of these 4 fetuses using SNP arrays showed that one of the cases arose from a maternal deletion of chromosome 22, wherein the mother's elder child, who also suffered from CHD, showed an identical genetic abnormality. Clinical manifestations of the 22q11 deletion syndrome differ considerably; further, the phenotype, which varied significantly even within a single family tree, is not definitively associated with the genotype. In another case, the phenotype of the mother was normal, whereas an ultrasonic cardiogram indicated a ventricular septal defect and choroid plexus cysts in the fetus. The mother's elder child had syndromic CHD. The parents were informed that the baby might show disease symptoms after birth and decided to terminate the pregnancy.
The SNP array cannot only detect CNVs but can also detect uniparental disomy. In this study, 2 CHD fetuses harbored uniparental disomy as follows: one had Prader–Willi syndrome and the other had an LOH at q23.2q24 and p13.3p12.3 on chromosome 16. SNP array analysis of the parents revealed maternal uniparental disomy, which was classified as a pathogenic variation. Therefore, these parents also decided to terminate the pregnancy.
We observed that 18.2% (20/110) of isolated CHD cases and 72.2% (26/36) of cases of CHD with extra-cardiac defects resulted in pregnancy termination. The reasons for pregnancy terminations were chromosome abnormalities (n = 15) and the presence of pathogenic CNVs (n = 15). We also observed that 10% (11/110) of families associated with isolated CHD and 66.7% (2/3) of those associated with VOUS still terminated their respective pregnancies. Therefore, genetic counseling should be improved, psychological support should be provided, and public awareness regarding CHDs should be increased to reduce the unnecessary termination of pregnancies.
Our study had some limitations. First, the number of cases included was small. A study with a larger sample size is required to reduce the large confidence interval for the detection rates of abnormal results. Second, we did not have access to information regarding CNVs for parents of all fetuses. Therefore, only some genetic information related to CNVs was obtained. In addition, an important component of prenatal consultation for the patients is the long-term physical, language, and speech development associated with CHD, as well as clinical symptoms, which could not be obtained from this study.
5. Conclusion
SNP array testing accurately detects cases of prenatal CHD, including those with isolated heart malformations and extra-cardiac defects. Our study shows that SNP array analysis combined with cytogenetic analysis is particularly effective for identifying chromosomal abnormalities and CNVs in fetuses with CHDs, which affects obstetrical outcomes. Based on this and other studies,[9] chromosomal abnormalities and CNVs form the genetic basis for CHDs and extra-cardiac abnormalities in only approximately 20% of CHD fetuses. Thus, in this study, factors contributing to 78.8% (115/146) of the cases remained elusive, suggesting that different mutations or factors might be involved in CHD etiology.
Author contributions
Data curation: Gang An, ying li.
Formal analysis: Linjuan Su.
Funding acquisition: Hailong Huang, Yuan Lin.
Investigation: Xiaoqing Wu.
Project administration: Liangpu Xu.
Software: Xiaorui Xie.
Supervision: Na Lin.
Writing – original draft: Meiying Cai.
Footnotes
Abbreviations: CHD = congenital heart disease, CMA = chromosomal microarray analysis, CNV = copy number variation, FISH = fluorescence in situ hybridization, LOH = loss of heterozygosity, PCR = polymerase chain reaction, SNP = single nucleotide polymorphism, VOUS = variation of uncertain clinical significance.
This work was supported by the Key Special Projects of Fujian Provincial Department of Science and Technology (no. 2013YZ0002-1), the Key Clinical Specialty Discipline Construction Program of Fujian (no. 20121589), and the Fujian Provincial Natural Science Foundation (2017J01238).
The authors have no conflicts of interest to disclose.
References
- [1].Lin KY, D’Alessandro LC, Goldmuntz E. Genetic testing in congenital heart disease: ethical considerations. World J Pediatr Congenit Heart Surg 2013;4:53–7. [DOI] [PubMed] [Google Scholar]
- [2].Derwińska K, Barnik M, Wiśniowiecka-Kowalnik B, et al. Assessment of the role of copy-number variants in 150 patients with congenital heart defects. Med Wieku Rozwoj 2012;16:175–82. [PubMed] [Google Scholar]
- [3].Hinton RB. Genetic and environmental factors contributing to cardiovascular malformation: a unified approach to risk. J Am Heart Assoc 2014;3:e000292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Bruneau BG, Srivastava D. Congenital heart disease: entering a new era of human genetics. Circ Res 2014;114:598–9. [DOI] [PubMed] [Google Scholar]
- [5].Edwards JJ, Gelb BD. Genetics of congenital heart disease. Curr Opin Cardiol 2016;31:235–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Fahed AC, Gelb BD, Seidman JG, et al. Genetics of congenital heart disease: the glass half empty. Circ Res 2013;112:707–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hanemaaijer NM, Sikkema-Raddatz B, van der Vries G, et al. Practical guidelines for interpreting copy number gains detected by high-resolution array in routine diagnostics. Eur J Hum Genet 2012;20:161–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Mademont-Soler I, Morales C, Soler A, et al. Prenatal diagnosis of chromosomal abnormalities in fetuses with abnormal cardiac ultrasound findings: evaluation of chromosomal microarray-based analysis. Ultrasound Obstet Gynecol 2013;41:375–82. [DOI] [PubMed] [Google Scholar]
- [9].Song MS, Hu A, Dyamenahalli U, et al. Extracardiac lesions and chromosomal abnormalities associated with major fetal heart defects: comparison of intrauterine, postnatal and postmortem diagnoses. Ultrasound Obstet Gynecol 2009;33:552–9. [DOI] [PubMed] [Google Scholar]
- [10].Chaoui R, Körner H, Bommer C, et al. Prenatal diagnosis of heart defects and associated chromosomal aberrations [German]. Ultraschall Med 1999;20:177–84. [DOI] [PubMed] [Google Scholar]
- [11].Wapner RJ, Martin CL, Levy B, et al. Chromosomal microarray versus karyotyping for prenatal diagnosis. N Engl J Med 2012;367:2175–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Liao C, Li R, Fu F, et al. Prenatal diagnosis of congenital heart defect by genome-wide high-resolution SNP array. Prenat Diagn 2014;34:858–63. [DOI] [PubMed] [Google Scholar]
- [13].Yan Y, Wu Q, Zhang L, et al. Detection of submicroscopic chromosomal aberrations by array-based comparative genomic hybridization in fetuses with congenital heart disease. Ultrasound Obstet Gynecol 2014;43:404–12. [DOI] [PubMed] [Google Scholar]
- [14].Zhu X, Li J, Ru T, et al. Identification of copy number variations associated with congenital heart disease by chromosomal microarray analysis and next-generation sequencing. Prenat Diagn 2016;36:321–7. [DOI] [PubMed] [Google Scholar]
- [15].Wilson DI, Goodship JA, Burn J, et al. Deletions within chromosome 22q11 in familial congenital heart disease. Lancet 1992;340:573–5. [DOI] [PubMed] [Google Scholar]
- [16].Amir RE, Ib VDV, Wan M, et al. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet 1999;23:185–8. [DOI] [PubMed] [Google Scholar]
- [17].Donnelly JC, Platt LD, Rebarber A, et al. Association of copy number variants with specific ultrasonographically detected fetal anomalies. Obstet Gynecol 2014;124:83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Driscoll DJ, Miller JL, Schwartz S, et al. Am Fam Physician 2008;72:827–30. [Google Scholar]
- [19].Johnson A, Minoshima S, Asakawa S, et al. A 1.5-Mb contig within the cat eye syndrome critical region at human chromosome 22q11.2. Genomics 1999;57:306–9. [DOI] [PubMed] [Google Scholar]
- [20].Potocki L, Bi W, Treadwell-Deering D, et al. Characterization of Potocki–Lupski syndrome (dup (17)(p11.2p11 2)) and delineation of a dosage-sensitive critical interval that can convey an autism phenotype. Am J Hum Genet 2007;80:633–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Sukenik-Halevy R, Sukenik S, Koifman A, et al. Clinical aspects of prenatally detected congenital heart malformations and the yield of chromosomal microarray analysis. Prenat Diagn 2016;36:1185–91. [DOI] [PubMed] [Google Scholar]
- [22].Jansen FA, Blumenfeld YJ, Fisher A, et al. Array comparative genomic hybridization and fetal congenital heart defects: a systematic review and meta-analysis. Ultrasound Obstet Gynecol 2015;45:27–35. [DOI] [PubMed] [Google Scholar]
- [23].Oneda B, Rauch A. Microarrays in prenatal diagnosis. Best Pract Res Clin Obstet Gynaecol 2017;42:53–63. [DOI] [PubMed] [Google Scholar]