

Supplemental Digital Content is available in the text
Keywords: Dravet syndrome, genetic modifier, SCN1A mutation, targeted-exome sequencing
Abstract
Dravet syndrome is considered to be one of the most severe types of genetic epilepsy. Mutations in SCN1A gene have been found to be responsible for at least 80% of patients with Dravet syndrome, and 90% of these mutations arise de novo. The variable clinical phenotype is commonly observed among these patients with SCN1A mutations, suggesting that genetic modifiers may influence the phenotypic expression of Dravet syndrome. In the present study, we described the clinical, pathological, and molecular characteristics of 13 Han Chinese pedigrees clinically diagnosed with Dravet syndrome. By targeted-exome sequencing, bioinformatics analysis and Sanger sequencing verification, 11 variants were identified in SCN1A gene among 11 pedigrees including 7 missense mutations, 2 splice site mutations, and 2 frameshift mutations (9 novel variants and 2 reported mutations). Particularly, 2 of these Dravet syndrome patients with SCN1A variants also harbored SCN9A, KCNQ2, or SLC6A8 variants. In addition, 2 subjects were failed to detect any pathogenic mutations in SCN1A and other epilepsy-related genes. These data suggested that SCN1A variants account for about 84.6% of Dravet syndrome in our cohort. This study expanded the mutational spectrum for the SCN1A gene, and also provided clinical and genetic evidence for the hypothesis that genetic modifiers may contribute to the variable manifestation of Dravet syndrome patients with SCN1A mutations. Thus, targeted-exome sequencing will make it possible to detect the interactions of epilepsy-related genes and reveal their modification on the severity of SCN1A mutation-related Dravet syndrome.
1. Introduction
Dravet syndrome (DS) is an infrequent progressive epileptic encephalopathy with an incidence between 1:20,000 and 1:60,000.[1–3] It was firstly reported by Dravet in 1978, and initially called severe myoclonic epilepsy in infancy (SMEI). In 2001, the name was officially changed into DS by the International League Against Epilepsy. DS is one of the most severe types of genetic epilepsy characterized by febrile seizures and psychomotor developmental delay after seizures onset. Mutations in SCN1A gene, which encodes the voltage-gated sodium channel αl-subunit, have been shown to be responsible for at least 80% of patients with DS, and 90% of these mutations arise de novo.[4] Furthermore, DS rarely involves PCDH19, STXBP1, GABRA1, GABARG2, SCN1B, and SCN2A mutations.[5–8] In addition, individuals with SCN1A mutations exhibit widely different seizure severities ranges from intractable seizures of DS, through comparatively benign febrile seizure, to completely normal family members in some cases. It has been reported that other genetic susceptibility factors may exist and act as a partner with SCN1A mutations, leading to the various clinical phenotype of patients with DS.[9,10] Genetic modifiers including SCN2A, KCNQ2, and SCN8A have been showed to influence the clinical presentation of human GEFS+ (generalized epilepsy with febrile seizures plus) associated SCN1A mutation by mouse models.[11] Furthermore, Singh et al[12] found that variants in SCN9A may act as a genetic modifier that exacerbated mild SCN1A mutation-associated GEFS+ and as a susceptibility gene for DS. However, more definitive clinical and genetic evidence is lacking. Here, the clinical and molecular characteristics of 13 DS cases were analyzed by targeted-exome sequencing to identify the novel variants in SCN1A gene among Han Chinese children and investigate the possibility of other epilepsy-related genes contributing to the variable manifestation of DS associated with SCN1A mutations.
2. Materials and methods
2.1. Subjects
Peripheral blood samples from 13 DS pediatric probands hospitalized in the Department of Neurology, Children's Hospital of School of Medicine, Zhejiang University from January 2014 to September 2017, as well as their families, were collected. Furthermore, a comprehensive history and physical examination were performed on these subjects to fulfill the clinical registration forms including name, sex, date of birth, age at onset, inducing factor, clinical manifestations during onset, perinatal situation, previous history, family history, auxiliary examination results, treatment, and prognosis. The study has been approved by Ethic Committee of Zhejiang University and written informed consents were obtained from all participants or their guardians (in the case of children).
2.2. Methods
2.2.1. Diagnostic criteria
According to Classification criteria of epilepsy and epilepsy syndrome (ILAE, 2010),[13] the detailed diagnostic criteria for DS were as follows: onset age within 1 year with febrile seizures; normal mental and motor development before onset, but retrogression in the second year; longstanding generalized or lateral seizures or generalized tonic-clonic seizures, often induced by fever; multi-seizure types such as myoclonic seizures, partial seizures, and atypical absence seizures shown at 1 to 4-year-old, often presented as status epilepticus; onset characterized by heat sensitivity; mental and motor retardation after onset combined with ataxia and pyramidal signs; poor therapeutic efficacy of anti-epileptic drugs.
2.2.2. Mutational screening of epilepsy-related genes
Blood samples from subjects were collected (5 mL, EDTA anticoagulated), and FlexiGene DNA Kit (Qiagen, Germany) was used for whole genomic DNA extraction. Exon target capture and second-generation sequencing were used in combination to screen mutations in 308 epilepsy-related genes (Table S1). Agilent SureDesign was employed to design target capture probes on target gene exons and flanking sequences (± 10 bp) (total 60,605 with 1.961 Mbp). Then, Agilent SureSelect Target Enrichment System was combined with Illumina NextSeq 500 to perform high-throughput sequencing. Data analysis included fundamental assessment of original sequences and bioinformatics evaluation. After analysis by the RTA software (real-time analysis, Illumina San Diego, California), the CASAVA software v1.8.2 (Illumina), BWA and GATK, sequencing quality was assessed, and single nucleotide variation (SNV) reports were obtained. Furthermore, ANNOVAR, PolyPhen-2, SIFT, HGMD database, dbSNP database, and 1000 Genome database were used to conduct variation annotation and analysis to confirm candidate pathogenic mutations. To identify putative pathogenic mutation, variants associated with DS were evaluated using following criteria: recorded or reported as pathogenic mutation by HGMD and PubMed databases (reported mutation); minor allele frequency <0.01 in dbSNP and genotype frequency <0.005 in 1000 Genome databases; predicted as detrimental mutation by PolyPhen-2 and SIFT; alignment by National Center for Biotechnology Information (NCBI) BLAST Link showing highly conservation in evolution. Finally, the PCR-Sanger method was used to assess probands and their family members, as well as normal controls to confirm the pathogenic mutations.
3. Results
3.1. Mutational analysis of SCN1A gene in DS patients
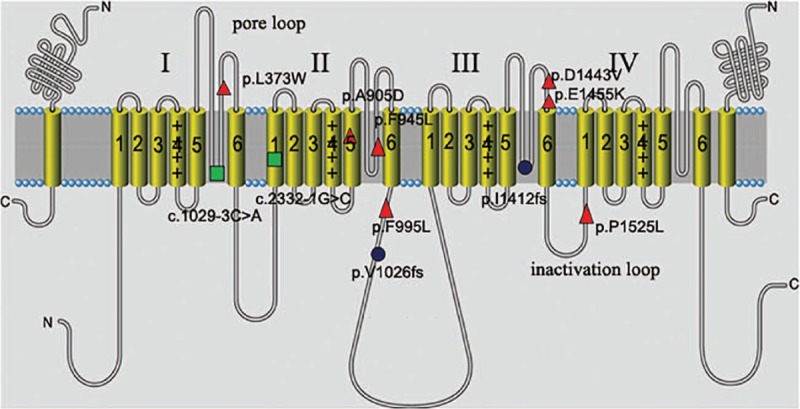
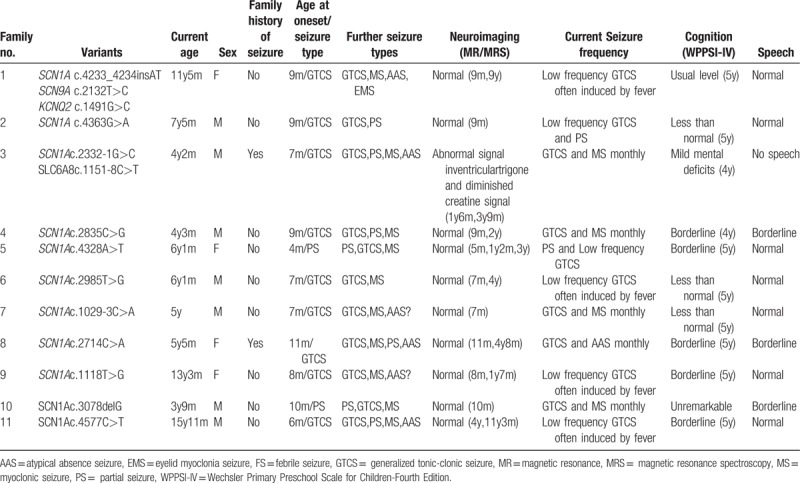
As shown in Table 1, targeted-exome sequencing, bioinformatic analysis, and Sanger sequencing verification revealed that 11 out of 13 (84.6%) pediatric patients harbored SCN1A putative pathogenic variants in heterozygous state, including 7 missense mutations, 2 splice site mutations, and 2 frameshift mutations (9 novel variants and 2 reported mutations). Furthermore, the localization of these variants in the type I voltage-gated sodium channel (Nav1.1) alpha subunit was exhibited in Fig. 1. Among these missense variants, p.L373W, p.F945L, p.D1443V, and p.E1455K variants reside at the S5 to S6 pore loop regions, p.P1525L variant is located at inactivation loop (connecting DIII and DIV), p.A905D variant occurs in S5 segment (DIII), and p.F995L variant resides at the loop between DII and DIII. The parental origin analyses for these variants by Sanger sequencings showed that 10 SCN1A variants in these DS pediatric patients were de novo, except the SCN1A c.2714C > A (p.A905D) variant. As shown in Fig. 2, the variant was inherited from his mother who displayed febrile seizures in Family 8, and the younger brother with DS phenotype also carried SCN1A c.2714C > A (p.A905D) variant. Additionally, 2 patients in our cohort were failed to detect any pathogenic mutations in 308 epilepsy-related genes.
Table 1.
Molecular analysis of epilepsy-related genes in patients with Dravet syndrome.
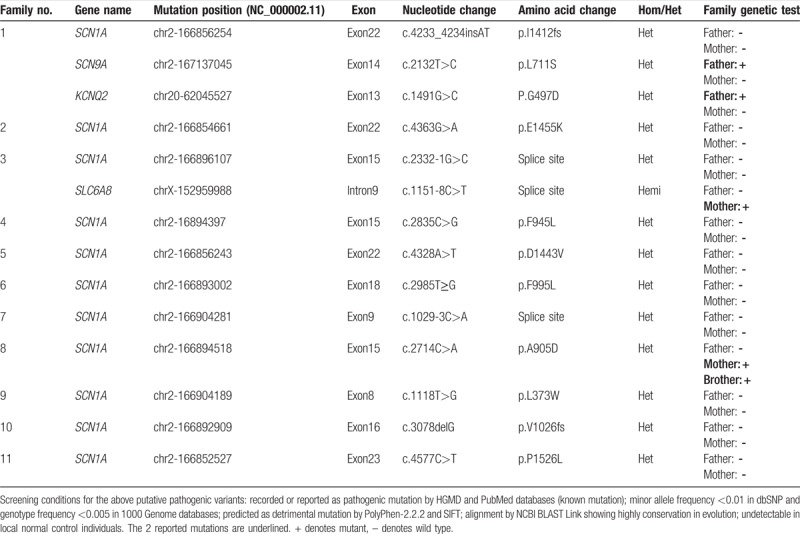
Figure 1.
The schematic topology of the α-subunit of the sodium channel and the distribution of variants identified in our study. The α-subunit is composed of 4 homologous domains (DI–DIV) of 6 transmembrane segments each (S1–S6). S4 is the voltage zone. S5, S6, and the ring between them is the gate zone. Seven missense mutations were denoted by red triangles (p.L373W, p.A905D, p.F945L, p.F995L, p.D1443V, p.E1445K, and p.P1526L), 2 splice site mutations were indicated by green rectangles (c.2332-1G>C and c.1029-3C>A), and 2 frameshift mutations were in deep blue ellipses (p.l1412fs and p.V1026fs).
Figure 2.
The SCN1A (c.2714C>A, p.A905D) variant was identified in Family 8. The variant was found in heterozygous state in mother with febrile seizures and 2 brothers with DS. The arrow denotes the position of the variant. DS = Dravet syndrome.
3.2. Mutational analysis of other epilepsy-related genes in DS patients
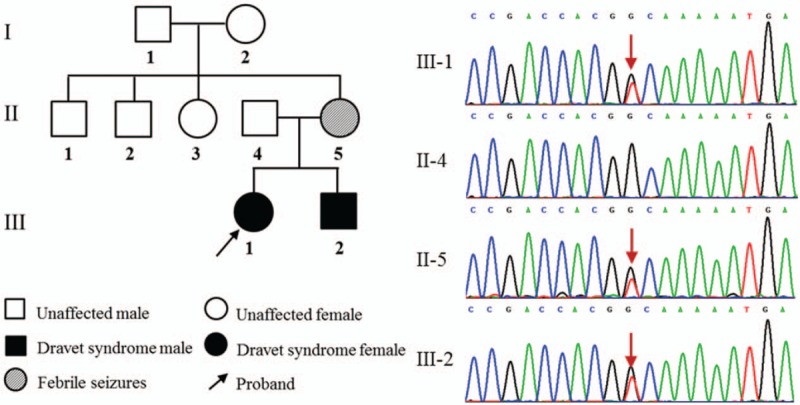
In addition to the SCN1A variants mentioned above, putative pathogenic mutations in other epilepsy-related genes associated with DS were also evaluated (Fig. 3). In this cohort, 2 of these DS subjects with SCN1A variants were found to be in combination with the other epilepsy-related variants. Particularly, the pediatric patient from Family 1 carried SCN9A (c.2132T > C, p.L711S) and KCNQ2 (c.1491G > C, P.G497D) variants along with SCN1A (c.4233_4234insAT, p.I1412Mfs37) variant. The variant p.L711S is located at the inactivation loop connecting DI and DII of voltage-gated sodium channel NaV1.7 which encoded by SCN9A and shared same overall structural motif as NaV1.1. The resulting P.G497D amino-acid change which occurred in the C-terminal of the potassium-channel subunit Kv7.2 (encoded by KCNQ2), was located in an evolutionarily conserved region. Furthermore, the proband (boy) in Family 3 with SCN1A c.2332-1G > C variant also harbored SLC6A8 c.1151-8C > T variant, which may affect the process of precursor mRNA into mature mRNA and lead to the production of abnormal protein. In fact, neither of 2 variants was detected in the 100 unrelated population-matched controls by Sanger sequencing. The 2 missense mutations all localized at highly conserved amino acids of proteins and may cause potential structural and functional alterations. High probability of damaging effect by PolyPhen-2 and SIFT prediction of these variants further supported their pathogenic nature, and may contribute to the variable DS phenotype associated with SCN1A mutations.
Figure 3.
Genetic modifier was identified in Family 3 with SCN1A variant. The SCN1A c.2332-1G>C variant was de novo, and SLC6A8 c.1151-8C>T variant was inherited from his mother in Family 3. The arrow denotes the position of the variant.
3.3. Clinical and genetic characterization of SCN1A mutated patients with additional modifier variations
A genotype–phenotype correlation analysis was carried out among 2 SCN1A mutated patients carrying additional modifier variations (Table 2). In Family 1, the proband carried SCN1A variant along with SCN9A and KCNQ2 variants has an unusually favorable cognitive and behavioral development. At admission to primary school, no obvious decline in cognitive level was observed. In Family 3, the proband with SCN1A and SLC6A8 variants exhibited more serious of speech deficits, developmental delay, and intellectual deficiency. He is currently 5 years old, and has no speech and limited language comprehension. The interictal electroencephalogram (EEG) showed slow background activity and multifocal spike slow waves. The brain magnetic resonance imaging (MRI) suggested abnormal signal in ventricular trigone. Long echo time (288 ms) proton magnetic resonance spectroscopy (MRS) reveals diminished creatine signal which was acquired within frontal white matter. Furthermore, the patient's uncle from mother's side suffered seizures, mental, and motor retardation.
Table 2.
Summary of clinical and molecular data for 11 DS patients with puptative pathogenic variants on epilepsy-related genes.
4. Discussion
Mutations in SCN1A gene have been found to be responsible for at least 80% of patients with Dravet syndrome, and 90% of these mutations arise de novo. In the present study, we detected 11 variants (including 10 de novo variants) in SCN1A gene among 11 pedigrees, accounting for about 84.6% (11/13) of DS in our cohort. Claes et al[14] reported SCN1A gene mutation resulting in DS for the first time in 2001. Marini et al[15] indicated that SCN1A mutation accounts for ∼80% of all DS cases, while mutations in other genes only account for a small part. It has been reported that a total of 1727 SCN1A gene variants have been reported, including 1448 mutations associated with the DS phenotype. Among these, 617 variants were missense mutations, and there was no hot spot mutation.[4] In this study, SCN1A c.4233_4234insAT variant from Family 1 and SCN1A c.2985T > G variant from Family 6 has been previously reported[16,17], while the remaining 9 variants were novel. These findings expanded the mutational spectrum for the SCN1A gene in Chinese Han population with DS.
A total of 11 SCN1A putative pathogenic variants were identified among 11 pedigrees including 2 frameshift mutations, 2 splice site mutations, and 7 missense mutations. SCN1A encodes the α1 subunit of the sodium channel, and is located on chromosome 2q24. The α-subunit includes 4 homologous regions with 6 transmembrane fragments (S1–S6) each. Among them, S4 is the voltage zone. S5, S6, and the ring between them is the gate zone. The SCN1A protein coding region, especially mutations in the gate zone can lead to permeability and electro-conductibility changes of the sodium channel that results in increased cell excitability and neuronal discharge with tiny stimulations, causing epilepsy. The c.4233_4234insAT variant in Family 1 and c.3078delG variant in family 10 caused an altered open reading frame, resulting in a completely different translation and dysfunction of Nav1.1. Two splice site mutations (c.2332–1G>C and c.1029–3C>A) which located at the acceptor-sites or recognition sites, leading to the abnormal processing of mRNA, were identified in Family 3 and Family 7, respectively. Amino acid synthesis and arrangement are greatly influenced by such mutations, and the function of the product is significantly changed, resulting in severe clinical manifestations. The remaining 7 missense mutations in SCN1A gene, including p.L373W, p.F945L, p.P1443V, and p.E1445K (S5–S6 pore loop regions), p.A905D (S5), p.F995L (intracellular DII and DIII connecting ring), and p.L373W (intracellular DIII and DIV connecting ring). In our study, 5 missense mutations occurred in the gate zone (71.4%, 5/7), suggesting the SCN1A missense mutation associated with DS mostly located at important functional region. Our finding was consistent with previous studies by Zuberi et al[18] and Meng et al.[4]
SCN1A mutations in DS pediatric patients were mostly de novo, and their clinical phenotypes were greatly different.[19] In our study, 10 of these SCN1A variants in DS patients were de novo, except the SCN1A c.2714C > A (p.A905D) variant. Sanger sequencing showed that this variant was inherited from his mother who exhibited febrile seizure before 6 years of age. In addition, his younger brother with DS phenotype also carried this variant. Several studies have reported that some DS subjects inherited SCN1A mutations from asymptomatic parents or mildly affected adult patients. Actually, somatic and germline SCN1A mosaicism has been reported in a small number of cases.[20,21] Somatic mosaicism may account for variable expressivity of SCN1A mutations in SMEI families.
Genetic interaction between sodium channel mutations and variants in other channels have been described both in mouse models and affected patients.[11] These results suggest that ion channel variants may contribute to the clinical variation observed in subjects with monogenic epilepsy. Particularly, interactions between genetic variations of SCN1A and KCNQ2 observed in the mouse and mutations of SCN1A and SCN9A described in DS patients provide important models for potential genetic modifier effects of DS.[12] In our cohort, SCN1A mutations were found to be combined with other epilepsy-related mutations (SCN9A and KCNQ2 variants in Family 1 and SLC6A8 variant in Family 3) that may contribute to the variable phenotype of SCN1A-related DS. In Family 1, segregation analysis showed that the SCN9A p.L711S and KCNQ2 p.G497D variations were inherited from the asymptomatic father without a family history of seizures. It has been reported that SCN9A variations themselves alone might be asymptomatic or cause infrequent febrile seizures due to incomplete penetrance and variable expressivity, but likely contribute to the modification of the severity of SCN1A related DS. There is indeed that a subset of DS patients commonly with mutations in SCN1A gene also harbor SCN9A mutations.[22] In addition, KCNQ2 mutation has been considered to be as a cause of seizures benign neonatal and epileptic encephalopathy early infantile.[23] Previous study have demonstrated that KCNQ2 mutation could exhibit increased susceptibility to induced seizures, and can dramatically influence the phenotype of mice carrying the SCN1A mutation.[24] However, it is reassuring that the proband in Family 1 has undergone an unusually favorable cognitive and behavioral development in comparison with other DS subjects. Furthermore, mutations in SLC6A8 has been reported to be associated with X-linked cerebral creatine deficiency syndrome1 (CCDS1), and speech difficulty is considered as a hallmark of CCDS.[25] The defect of SLC6A8 impaired the transport of creatine into brain and resulted in creatine transporter deficiency, thereby producing the phenotype of CCDS1. The SCL6A8 c.1151–8C>T variant that identified in proband 3 has been reported to be pathogenic in X-linked recessive inherited model.[26] Clinical evaluation showed that his speech deficits, developmental delay and intellectual deficiency are more serious than other DS patients, MRS reveals diminished creatine signal. Thus, these findings suggested that SCN9A, KCNQ2, and SCL6A8 may act as modifiers influenced the phenotypic manifestation of the DS associated SCN1A mutations. Furthermore, these modifying alleles may preferentially found in DS patients with SCN1A mutations that are less deleterious when compared with complete heterozygous loss of function mutations. Additional functional data in mouse models to evaluate the interaction between the variants in genetic modifiers and SCN1A gene will be required to test if the modification is valid in certain DS subjects.
In conclusion, these data suggested that SCN1A putative pathogenic variants account for about 84.6% of DS in our cohort. The study expanded the mutational spectrum for the SCN1A gene, and also provided clinical and genetic evidence for the hypothesis that genetic modifiers may contribute to the variable manifestation of DS patients with SCN1A mutations. Thus, targeted-exome sequencing will make it possible to detect the interactions of epilepsy-related genes and reveal their modification on the severity of SCN1A-related DS. The hypothesis of genetic modifiers on SCN1A-related variable phenotype should require more evidences from clinical patients and mouse models. Additionally, the whole exome or genome screening would be helpful to explain the molecular pathogenesis of the 2 patients who failed to detect any pathogenic mutations in 308 epilepsy-related genes.
Author contributions
Conceptualization: Tiejia Jiang, Feng Gao.
Data curation: Tiejia Jiang, Yaping Shen, Feng Gao, Huai Chen.
Formal analysis: Tiejia Jiang, Zhefeng Yuan, Shanshan Mao.
Investigation: Yaping Shen, Huai Chen, TieJia Jiang, Zhefeng Yuan, Shanshan Mao, Feng Gao.
Methodology: Tiejia Jiang.
Project administration: TieJia Jiang, Feng Gao
Resources: Huai Chen, Shanshan Mao, Feng Gao.
Supervision: Feng Gao, TieJia Jiang, Zhefeng Yuan.
Writing – original draft: Tiejia Jiang.
Writing – review & editing: Zhefeng Yuan, Shanshan Mao, Feng Gao.
Supplementary Material
Footnotes
Abbreviations: CCDS1= cerebral creatine deficiency syndrome1, DS = Dravet syndrome, GEFS+ = generalized epilepsy with febrile seizures plus, MRS= magnetic resonance spectroscopy, Nav1.1 = type I voltage-gated sodium channel, SMEI = severe myoclonic epilepsy in infancy, SNV = single nucleotide variation.
The authors have no conflicts of interest to disclose.
Supplemental Digital Content is available for this article.
References
- [1].Wu] YW, Sullivan J, McDaniel SS, et al. Incidence of Dravet syndrome in a US population. Pediatrics 2015;136:E1310–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Rosander C, Hallbook T. Dravet syndrome in Sweden: a population-based study. Dev Med Child Neurol 2015;57:628–33. [DOI] [PubMed] [Google Scholar]
- [3].Bayat A, Hjalgrim H, Moller RS. The incidence of SCN1A-related Dravet syndrome in Denmark is 1: 22,000: a population-based study from 2004 to 2009. Epilepsia 2015;56:E36–9. [DOI] [PubMed] [Google Scholar]
- [4].Meng H, Xu HQ, Yu L, et al. The SCN1A mutation database: updating information and analysis of the relationships among genotype, functional alteration, and phenotype. Hum Mutat 2015;36:573–80. [DOI] [PubMed] [Google Scholar]
- [5].Ishii A, Kanaumi T, Sohda M, et al. Association of nonsense mutation in GABRG2 with abnormal trafficking of GABA (A) receptors in severe epilepsy. Epilepsy Res 2014;108:420–32. [DOI] [PubMed] [Google Scholar]
- [6].Carvill GL, Weckhuysen S, McMahon JM, et al. GABRA1 and STXBP1: novel genetic causes of Dravet syndrome. Neurology 2014;82:1245–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Shi XY, Yasumoto S, Nakagawa E, et al. Missense mutation of the sodium channel gene SCN2A causes Dravet syndrome. Brain Dev 2009;31:758–62. [DOI] [PubMed] [Google Scholar]
- [8].Depienne C, Bouteiller D, Keren B, et al. Sporadic infantile epileptic encephalopathy caused by mutations in PCDH19 resembles Dravet syndrome but mainly affects females. PLoS Genet 2009;5:e1000381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Gaily E, Anttonen AK, Valanne L, et al. Dravet syndrome: new potential genetic modifiers, imaging abnormalities, and ictal findings. Epilepsia 2013;54:1577–85. [DOI] [PubMed] [Google Scholar]
- [10].Meisler MH, O’Brien JE, Sharkey LM. Sodium channel gene family: epilepsy mutations, gene interactions and modifier effects. J Physiol 2010;588(pt 11):1841–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Hawkins NA, Martin MS, Frankel WN, et al. Neuronal voltage-gated ion channels are genetic modifiers of generalized epilepsy with febrile seizures plus. Neurobiol Dis 2011;41:655–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Singh NA, Pappas C, Dahle EJ, et al. A role of SCN9A in human epilepsies, as a cause of febrile seizures and as a potential modifier of Dravet syndrome. PLoS Genet 2009;5:e1000649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Berg AT, Berkovic SF, Brodie MJ, et al. Revised terminology and concepts for organization of seizures and epilepsies: Report of the ILAE Commission on Classification and Terminology, 2005-2009. Epilepsia 2010;51:676–85. [DOI] [PubMed] [Google Scholar]
- [14].Claes L, Del-Favero J, Ceulemans B, et al. De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J Hum Genet 2001;68:1327–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Marini C, Scheffer IE, Nabbout R, et al. The genetics of Dravet syndrome. Epilepsia 2011;52:24–9. [DOI] [PubMed] [Google Scholar]
- [16].Xu XJ, Zhang YH, Sun HH, et al. Early clinical features and diagnosis of Dravet syndrome in 138 Chinese patients with SCN1A mutations. Brain Dev 2014;36:676–81. [DOI] [PubMed] [Google Scholar]
- [17].Jiang P, Shen J, Yu Y, et al. Dravet syndrome with favourable cognitive and behavioral development due to a novel SCN1A frameshift mutation. Clin Neurol Neurosurg 2016;146:144–6. [DOI] [PubMed] [Google Scholar]
- [18].Zuberi SM, Brunklaus A, Birch R, et al. Genotype-phenotype associations in SCN1A-related epilepsies. Neurology 2011;76:594–600. [DOI] [PubMed] [Google Scholar]
- [19].Mancardi MM, Striano P, Gennaro E, et al. Familial occurrence of febrile seizures and epilepsy in severe myoclonic epilepsy of infancy (SMEI) patients with SCN1A mutations. Epilepsia 2006;47:1629–35. [DOI] [PubMed] [Google Scholar]
- [20].Depienne C, Trouillard O, Gourfinkel-An I, et al. Mechanisms for variable expressivity of inherited SCN1A mutations causing Dravet syndrome. J Med Genet 2010;47:404–10. [DOI] [PubMed] [Google Scholar]
- [21].Tuncer FN, Gormez Z, Calik M, et al. A clinical variant in SCN1A inherited from a mosaic father cosegregates with a novel variant to cause Dravet syndrome in a consanguineous family. Epilepsy Res 2015;113:5–10. [DOI] [PubMed] [Google Scholar]
- [22].Singh NA, Pappas C, Dahle EJ, et al. A role of SCN9A in human epilepsies, as a cause of febrile seizures and as a potential modifier of Dravet Syndrome. PLoS Genet 2009;5:e1000649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Weckhuysen S, Mandelstam S, Suls A, et al. KCNQ2 encephalopathy: emerging phenotype of a neonatal epileptic encephalopathy. Ann Neurol 2012;71:15–25. [DOI] [PubMed] [Google Scholar]
- [24].Makinson CD, Dutt K, Lin F, et al. An Scn1a epilepsy mutation in Scn8a alters seizure susceptibility and behavior. Exp Neurol 2016;275:46–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hahn KA, Salomons GS, Tackels-Horne D, et al. X-linked mental retardation with seizures and carrier manifestations is caused by a mutation in the creatine-transporter gene (SLC6A8) located in Xq28. Am J Hum Genet 2002;70:1349–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Betsalel OT, Pop A, Rosenberg EH, et al. Detection of variants in SLC6A8 and functional analysis of unclassified missense variants. Mol Genet Metab 2012;105:596–601. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.