

Abstract
Lysine acetyltransferases (KATs) are critical regulators of signaling in many diseases, including cancer. A major challenge in establishing the targetable functions of KATs in disease is a lack of well-characterized, cell-active KAT inhibitors. To confront this challenge, here we report a microfluidic mobility shift platform for the discovery and characterization of small molecule KAT inhibitors. Novel fluorescent peptide substrates were developed for four well-known KAT enzymes (p300, Crebbp, Morf, and Gcn5). Enzyme-catalyzed acetylation alters the electrophoretic mobility of these peptides in a microfluidic chip, allowing facile and direct monitoring of KAT activity. A pilot screen was used to demonstrate the utility of microfluidic mobility shift profiling to identify known and novel modulators of KAT activity. Real-time kinetic monitoring of KAT activity revealed that garcinol, a natural product KAT inhibitor used in cellular studies, exhibits time-dependent and detergent-sensitive inhibition, consistent with an aggregation-based mechanism. In contrast, the cell-permeable bisubstrate inhibitor Tat-CoA exhibited potent and time-independent KAT inhibition, highlighting its potential utility as a cellular inhibitor of KAT activity. These studies define microfluidic mobility shift profiling as a powerful platform for the discovery and characterization of small molecule inhibitors of KAT activity, and provide mechanistic insights potentially important for the application of KAT inhibitors in cellular contexts.
Graphical Abstract
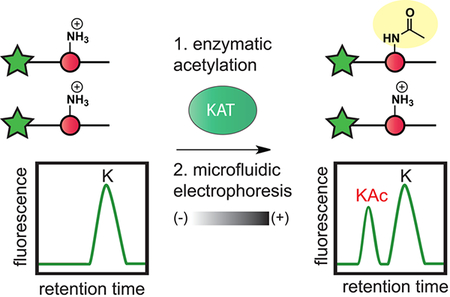
Lysine acetyltransferases (KATs) catalyze lysine acetylation, a reversible protein modification that plays a key role in the regulation of genome function.1 Lysine acetylation alters chromatin accessibility by modulating electrostatic histone–DNA interactions and can facilitate transcriptional elongation by providing high affinity binding sites for acetyl-lysine binding protein motifs, such as bromodomains.2 Beyond histones, acetylation has also been shown to directly influence the stability, localization, and DNA-binding affinity of many transcription factors, including c-Myc, p53, and PGC-1α.3 The biological significance of these modifications has spurred efforts to target the cellular acetylation machinery for therapeutic benefit. Recent preclinical studies of bromodomain inhibitors have provided strong validation for the therapeutic targeting of acetylation-dependent nuclear signaling in cancer.4–6 Small molecule KAT inhibitors could possibly augment or extend this paradigm by allowing the inhibition of specific enzymatic programs of acetylation. However, our ability to test this hypothesis is hindered by a lack of small molecules capable of probing KAT activity in cells.
Unlike many epigenetic enzyme families, few potent and well-characterized inhibitors of KATs are known. Molecules commonly applied to probe KAT activity in cells include cell-penetrating CoA-based bisubstrate inhibitors (p300/CBP or Gcn5/pCAF targeting),7,8 the polyphenolic natural product garcinol (p300/pCAF inhibitor),9 and the synthetic molecule C646 (p300/CBP inhibitor).10 While novel KAT inhibitors would be powerful tools, one challenge in their discovery is a lack of methods for the rapid and direct assay of KAT activity. Existing methods for the direct assay of KAT-catalyzed acetylation are based on radiation or mass spectrometry, which can be challenging to adapt to library screening.11–13 In contrast, KAT assays that have been shown to be amenable to high-throughput screening rely on detection of the reaction’s CoA byproduct,14,15 which can make them prone to false positives. For example, a recent screen of the fungal KAT Rtt109 used released CoA as a surrogate for KAT activity and found the vast majority of “hits” were caused by the ability of thiol-reactive compounds to consume CoA directly, rather than inhibit KAT activity.16 Such studies emphasize the need for high-throughput analytical tools capable of directly profiling KAT activity.
These considerations led us to explore the development of a microfluidic mobility shift platform for profiling of lysine acetylation (Figure 1). This approach utilizes quantitative microfluidic capillary electrophoresis to separate acetylated and nonacetylated fluorescent histone peptides on the basis of their different charge-to-mass ratios, with the ratio of substrate-to-product peak height providing a measure of conversion. Commercial microfluidic chip readers use only nanoliter-sized aliquots per measurement and can monitor up to 12 reactions simultaneously. Thus, rapid cycling between sample loading and fast electrophoretic separation enables high-throughput, kinetically resolved measurements of enzyme activity. Studies of histone deacetylases have demonstrated the ability of micro-fluidic mobility shift platforms to monitor acetylation-dependent changes in the charge of fluorescent peptide substrates.17 However, the similar application of this method to study KAT enzymes is limited by a lack of separable, fluorescent KAT substrates.17,18
Figure 1.

Microfluidic mobility shift profiling of lysine acetyltransferase activity. (a) Assay principle. Fluorescent KAT substrate peptides are incubated with KAT and acetyl-CoA. Acetylation of substrate alters its charge-to-mass ratio, enabling electrophoretic separation and direct measurement of KAT enzyme activity. (b) Schematic and net charges of histone H4/H3 KAT substrate peptides used in this study. All peptides were modified with an N-terminal aminohexanoic acid-FITC to facilitate detection. Sequences of each fluorescent substrate are provided in Table S1. (c) Charge-dependent separation of acetylated (“KAc”) and nonacetylated (“K”) fluorescent histone H4 substrates. SR = separation resolution.
Here, we report the development of a microfluidic mobility shift platform for the screening and mechanistic analysis of KAT inhibitors. Literature analysis and systematic truncation were applied to design new fluorescent KAT substrates based on the histone H3 and H4 tails. When combined with microfluidic capillary electrophoresis, these novel substrates enable the sensitive, separation-based assay of four different KAT enzymes. A hydroxylamine-based quenching protocol was developed to facilitate the screening of large compound libraries, and we demonstrate in a pilot screen that this approach can be used to identify known modulators of KAT activity. The real-time kinetic aspect of our platform enabled the surprising discovery that garcinol, a natural product commonly cited and used as a pan-KAT inhibitor, exhibits time- and detergent-sensitive inhibition consistent with an aggregation-based mechanism. In contrast, the cell-permeable bisubstrate inhibitor Tat-CoA demonstrates an inhibition profile indicative of a reversible inhibitor and also antagonizes acetylation in cells. These studies integrate KAT substrate design and microfluidic capillary electrophoresis to generate a powerful tool for the screening and mechanistic analysis of KAT inhibitors and provide unexpected insights that will guide the use of small molecules to probe cellular KAT activity.
Definition of a Minimal Histone H4 Fluorescent Acetylation Substrate.
Many cellular KATs modify the N-terminal tail of histone H4. These include the prototypical KAT family member p300, whose overactivity has been implicated in many diseases.3,19,20 Since mobility shift assays for p300 and other H4 KATs have not previously been described, we focused our initial efforts on identifying a minimal fluorescent substrate suitable for their separation-based analysis. The KAT activity of p300 is most commonly assayed with peptides based on histone H4 (1–21), which possesses a charge of +7 at physiological pH (Figure 1b, Table S1).21,22 This lies far outside the range of net charge ideal for commercial microfluidic capillary electrophoresis instruments, which are designed to separate peptides with charges ranging from +3 to −3. However, early studies of p300 by Thompson et al. reported that the truncated histone H4 substrates were also turned over by p300, albeit with reduced catalytic efficiency.21 This inspired us to synthesize and evaluate a series of fluorescent H4 peptides, toward the goal of identifying an electrophoretically separable p300 substrate.
Solid-phase peptide synthesis was used to synthesize a series of peptides based on the canonical H4 (1–21) p300 substrate (Scheme S1). Each peptide contains an N-terminal FITC to facilitate fluorescence detection, as well as an aminohexanoic acid linker, which separates the fluorophore from the peptide and minimizes any potential deleterious effects it may have on KAT recognition. In each construct, we also maintained K8, which has been determined by kinetic and mutational analysis to be a major site of p300 acetylation (for full sequences, see Table S1).21,23 The first peptide tested was FITC-H4 (1–19; net charge: +6), a minimally truncated peptide in which only the two C-terminal residues (lysine and valine) were removed from the canonical p300 substrate. Incubation of FITC-H4 (1–19) with p300 and acetyl-CoA led to turnover and clear formation of a product peak (Figure 1c), confirmed to be the acetylated peptide by LC-MS (Figure S1). However, peaks for the starting material and product were only modestly separable (separation resolution [SR] = 0.9), and LC-MS analysis indicated the formation of a bis-acetylated product that could also not be separated. By comparison, the less basic FITC-H4 (3–14) substrate (net charge: + 3) revealed an improved baseline separation of the acetylated product from the nonacetylated substrate (SR = 1.8; Figure 1c). This improved resolution also enabled the visualization of a separable third peak corresponding to the bis-acetylated product (Figure S1). Removing an additional C-terminal charge yielded FITC-H4 (3–11) (net charge: + 2), which exhibited near identical resolution but ~60% less turnover than H4 (3–14), suggesting a strong contribution of the K12 residue to p300 substrate recognition. Consistent with this, further truncated substrates FITC-H4 (4–11) and FITC-H4 (6–11) (net charge: + 1 and 0, respectively) showed little or no turnover with p300 (Figure 1). These results illustrate the balance that must be struck between turnover and capillary electrophoretic resolution for fluorescent KAT substrates. Furthermore, they specify FITCH4 (3–14) as an exemplary peptide for the separation-based assay of H4 KAT activity.
Fluorescent Substrates Enable the Kinetic Profiling of Diverse KAT Enzymes.
Next, we sought to apply these insights to expand the utility of the microfluidic mobility shift assay to profile diverse KAT enzyme activities. CREB-binding protein (Crebbp) is an H4 acetyltransferase that is functionally distinct from p300 but shares an 87% identical KAT catalytic domain. Hypothesizing that it may also use FITC-H4 (3–14) as a fluorescent substrate, we performed microfluidic mobility shift analysis of FITC-H4 (3–14) following incubation with Crebbp and acetyl-CoA and observed clear, time-dependent formation of a product peak (Figure 2, Figure S2). Next, we assessed the utility of FITC-H4 (3–14) in analyzing the mechanistically distinct MYST family of KATs, many of which catalyze H4 acetylation.24 Focusing on Morf, a MYST family member whose pathologic activity results from chromosomal translocations in cancer,25 we observed similar time-dependent acetylation. Thus, FITC-H4 (3–14) can be applied as a sensitive and versatile reporter of H4 acetyltransferase activity.
Figure 2.

Microfluidic mobility shift analysis enabling real-time monitoring of diverse KAT activities. (a) p300 turnover of FITC-H4 (3–14). (b) Crebbp turnover of FITC-H4 (3–14). (c) Morf turnover of FITC-H4 (3–14). (d) Gcn5 turnover of FITC-H3 (5–20). Model separations for each substrate/product pair are provided as Supporting Information (Figure S2).
A third major class of KAT enzymes is the GCN5 family, composed of two well-characterized members: Gcn5 and pCAF.3 Gcn5 was previously analyzed using a separation-based assay by Fanslau and co-workers, who demonstrated that a highly charged FITC-histone H3 peptide (residues 5–23; net charge +6) could be used to monitor KAT activity.26 However, this study also reported a nonbaseline separation of fluorescent substrate and product, as well as a small interfering contaminant that could not be removed. Therefore, to expand the scope of our microfluidic mobility shift platform, we sought to apply insights from our studies of H4 KATs to improve methods for the analysis of Gcn5. In addition to H3 (5–23), literature analysis highlighted H3 (1–20) as another substrate commonly applied in Gcn5 biochemical assays. This prompted us to synthesize and examine the separative properties of FITCH3 (5–20), a peptide that contains the overlapping segments of the H3 (5–23) and H3 (1–20) substrates but possesses a reduced net charge (+4; Figure 1b). Validating this design, the acetylated form of FITC-H3 (5–20) demonstrates baseline separation from the parent peptide when incubated with Gcn5 and acetyl-CoA (Figure S2). Kinetic analysis by an orthogonal assay indicated that the H3 5–20 peptide was processed with a similar catalytic efficiency as the canonical substrate H3 (5–23) (Table S2).15 Furthermore, the H3 KAT substrates were found to display linear fluorescence at concentrations ranging from 1 to 50 μM, enabling reagent usage and balanced assay conditions to be tailored for individual applications (Figure S3). These studies demonstrate that microfluidic mobility shift can be used to profile the activity of diverse KAT enzymes and highlight fluorescent H3/H4 peptides of net charges from +3 to +4 as ideal substrates for the separation-based analysis of KAT activity.
Development of an End Point Microfluidic KAT Assay for Library Screening.
Having established the ability of our microfluidic assay to monitor diverse KAT activities, we next focused on its adaptation for the screening of large chemical libraries. In order to establish uniform KAT assay conditions, we first required a method to efficiently quench the enzymatic reaction. KATs lack a general inhibitor and are commonly quenched using denaturing reagents such as urea, SDS, or organic cosolvents.14 Unfortunately, each of these had deleterious effects on the resolution of acetylated and nonacetylated peptides in our microfluidic assay. Searching for alternatives, we were inspired by the seminal studies of Lipmann et al., who used hydroxylamine as a reagent to quench acetyl-CoA dependent enzymes via cleavage of the acylthioester bond.27,28 Indeed, we found that KAT enzymatic reactions quenched with hydroxylamine (70 mM) were amenable to microfluidic analysis and showed time-dependent turnover that was identical to that of reactions analyzed in real time (Figure S4). Applying the hydroxylamine-quenched KAT end point assay in 384-well format revealed that both the H3 and H4 KAT assays demonstrate excellent well-to-well consistency, affording Z′-values of 0.97 (p300) and 0.98 (Gcn5), respectively (Figure S5).
To validate our assay for screening, we assembled a small library of 145 known epigenetic modulators and assessed the ability of our platform to discriminate known KAT inhibitors from the background (Figure 3). Compounds were initially screened in unicate against p300 and Gcn5, using FITC-H4 (3–14) and FITC-H3 (5–20) substrates, respectively. Following preincubation with inhibitor (40 μM), KAT reactions were initiated with acetyl-CoA (1 μM) and quenched with hydroxylamine at a time point where the kinetics of KAT turnover were linear (<15% peptide acetylation). Of the 145 compounds screened, only the bisubstrate inhibitors Lys-CoA and Tat-CoA showed greater than 40% inhibition of both KATs (Figure 3, Table S3). Consistent with the literature, Lys-CoA inhibited p300 considerably more strongly than Gcn5, while Tat-CoA is less selective, inhibiting p300 and Gcn5 to similar extents. Selective inhibition of p300 was observed by the known KAT inhibitors C646 and garcinol. Several compounds activated p300 (Table S3), including mitoxantrone, whose selective p300 activation we validated (Figure S6). Small molecule activation of p300 has been previously observed.13,29 Focusing on Gcn5, we found that MG-149, a previously identified Tip60 inhibitor, and the natural product gossypol each selectively inhibited Gcn5 activity (>40% inhibition at 40 μM; Figure 3). These studies demonstrate the utility of applying our microfluidic assay to identify KAT inhibitors from small molecule libraries.
Figure 3.

(a) Pilot screen of p300 and Gcn5 using an epigenetic-targeted inhibitor library. Percent activity represents relative KAT-catalyzed turnover of FITC-peptide in the presence of compound compared to vehicle DMSO control. All reactions were quenched and analyzed under conditions where enzyme velocity was linear. (b) Structures of selected compounds which showed >40% inhibition of p300 or Gcn5 at 40 μM. Color code for highlighted compounds: yellow, p300 inhibitors; green, Gcn5 inhibitors; orange, inhibitor of both p300 and Gcn5.
Kinetic Analysis Reveals Garcinol Is Time- and Detergent-Sensitive KAT Inhibitor.
A major advantage of the microfluidic assay platform is its versatility, which enables both screening as well as real-time kinetic measurements of KAT activity. We reasoned this kinetic mode could be exploited to rapidly assess the time dependence of inhibitors and provide critical insights into their mechanism of action. To demonstrate this, we assessed the kinetics of Gcn5 inhibition by three screening hits: Tat-CoA, gossypol, and garcinol (Figure 4). Tat-CoA is a cell permeable bisubstrate inhibitor first synthesized by Cole and co-workers, which has been reported to inhibit both GCN5 and P300-family KATs with micromolar potency.7 Gossypol is a polyphenol natural product that demonstrates diverse biological activities, including inhibition of the steroid coactivator (SRC) family of KAT enzymes;30 however, gossypol is also a known frequent hitter in biochemical assays, due to its redox and aggregation properties.31 Garcinol is a polyisoprenylated benzophenone natural product that shares a structurally conserved catechol with gossypol and has been applied in several cellular studies as a general antagonist of KAT activity.32–34 In addition to its structural similarity to gossypol, garcinol is of interest because it has been reported to exhibit broad-spectrum inhibition of KATs in biochemical assays9 but did not manifest as a Gcn5 hit in our model screen. Notably, the time-dependent inhibition of KATs by any of these small molecules has not been studied.
Figure 4.

Real-time kinetic monitoring of histone acetylation provides insights into the mechanism of KAT inhibitors. (a) Kinetic monitoring of Gcn5-catalyzed acetylation in the presence or absence of inhibitors.(b) Ratio of acetylation rates at early (0–25 min) and late (65–90 min) time points. High ratios indicate reduced reaction rates at later time points, suggestive of time-dependent inhibition. (c) Garcinol inhibition of Gcn5 and p300 is sensitive to the concentration of the detergent Triton-X. (d) Tat-CoA inhibition of p300 and Gcn5 is not sensitive to detergent. (f) Tat-CoA inhibits acetylation in cells.
To assess time dependence, we monitored KAT reactions in the presence or absence of each inhibitor for 100 min, and then compared the rate of acetylated peptide formation over the first 25 and last 25 min (Figure 4a). The uninhibited enzyme serves as the control, with a time dependence value (the ratio of rates at early/late intervals) set equal to 1. Our hypothesis was that at a fixed concentration, reversible inhibitors would impact the rate of acetylation similarly at early and late time intervals (time dependence ≤1), reflecting a constant level of enzyme activity. In contrast, irreversible inactivators of Gcn5 would exhibit lower rates of acetylation at late compared to early time points (time dependence >1; Figure 4b), reflecting a decrease in the concentration of enzyme available for catalysis over time. The ability of microfluidic capillary electrophoresis instruments to monitor up to 12 KAT reactions in parallel allowed us to examine simultaneously two concentrations of each inhibitor, as well as controls containing no enzyme, no inhibitor, and the known reversible inhibitor CoA. Tat-CoA inhibits Gcn5 but exhibits a time dependence value of <1, indicative of a stabilizing effect on Gcn5 activity (Figure 4a,b). This is consistent with previous studies that found that Gcn5 is stabilized by molecules binding the cofactor binding site such as the feedback inhibitor CoA, an observation we can recapitulate (Figure 4b).35 In contrast, the inhibition of Gcn5 by garcinol and gossypol exhibits a time dependence >1. This indicates time-dependent inhibition by these two agents, a property suggestive of an irreversible mechanism.
The time-dependent inhibition of Gcn5 by garcinol was unexpected and has not been previously reported. Seeking to explain this phenomenon, we were intrigued by the structural similarities between garcinol and gossypol, which has been shown to inhibit many enzymes by aggregation-based mechanisms.31 While our assays were performed at a constant detergent concentration of 0.05% Triton-X, Shoichet and coworkers have observed that some aggregation-prone molecules display inhibitory activity even in the presence of low levels of detergent.36 Thus, to better understand garcinol’s mechanism of inhibition, we analyzed the dose and detergent dependence of Gcn5 inhibition by garcinol. Consistent with an aggregation-based mechanism, garcinol exhibited significantly greater inhibitory activity in Gcn5 and p300 reactions performed at low (0.01%) compared to high (0.05%) detergent concentrations (Figure 4c, Figure S7). Gossypol also displayed detergent-sensitive inhibition of Gcn5 (Figure S8). Notably, the IC50 of garcinol for p300 observed at low detergent concentrations correlates well with the literature value initially reported by Kundu et al., whose KAT assay did not report the use of any solublizing detergent.9 While we were able to observe effects of garcinol on acetylation at concentrations as low as ~25 μM in HepG2 cells (Figure S9), the IC50 of garcinol for p300 in the presence of 0.05% Triton-X is ~60 μM, concentrations at which garcinol is toxic. These studies highlight the ability of our microfluidic assay to provide new insights into KAT inhibitor mechanisms and suggest the natural products garcinol and gossypol inhibit KATs in vitro by promiscuous, aggregation-based mechanisms.
Tat-CoA Is a Reversible Cell-Active KAT Inhibitor.
Finally, we examined the cell-permeable bisubstrate inhibitor Tat-CoA in greater detail. As mentioned above, kinetic analyses revealed Tat-CoA did not display time-dependent inhibition of Gcn5. To rule out an aggregation-based inhibitory mechanism, we performed similar dose- and detergent-sensitivity profiling as above. Tat-CoA was a potent inhibitor of p300 (IC50 = 0.9 μM) as well as Gcn5 (IC50 = 19 μM) and demonstrated a lack of detergent sensitivity (Figure 4d, Figure S7). Since Tat-CoA has not been previously analyzed in biological settings, we tested its ability to antagonize acetylation in cells. We found Tat-CoA reduced the acetylation of histone H4 in cells in a dose-dependent manner (Figure 4e) at concentrations that did not affect cell viability. This inhibitory effect is consistent with cellular and in vivo studies suggesting H4 is the major cellular target of p300-catalyzed acetylation.37 Notably, concentrations of Tat-CoA above 10 μM were required to see substantial effects on cellular acetylation. This concentration is far higher than the IC50 of Tat-CoA for p300 in the KAT biochemical assay, suggesting delivery of Tat-CoA to the nucleus may be a limiting determinant for activity. While a full characterization of Tat-CoA’s cellular effects is beyond the scope of this study, these findings suggest Tat-CoA may have utility as a reversible probe of KAT activity in cellular systems.
Here, we have developed an analytical platform for microfluidic mobility shift profiling of KAT activity. Synthetic optimization of highly charged histone peptides facilitated the generation of fluorescent substrates that are efficiently recognized and acetylated by KAT enzymes. Microfluidic electrophoresis and fluorescence detection was then applied to separate and directly visualize acetylated and nonacetylated peptides. Detailed analysis of a p300 substrate revealed that a net charge of +3 to +4 facilitated optimal resolution of acetylated and deacetylated peptides; more highly charged substrates hindered electrophoretic separation, while truncation of substrates below a + 3 net charge substantially reduced their enzymatic acetylation. While this study focused on the well-known KATs p300, Crebbp, Morf, and Gcn5, we expect these insights to be of general utility to the design of microfluidic mobility shift assays for any KAT capable of modifying a peptide substrate, including less well-studied “orphan” KATs.38 Development of assays for these enzymes should be greatly facilitated by peptide array technology, which has already proven a powerful tool for the discovery and mechanistic study of KAT substrates.11
A major advantage of our microfluidic assay is that each data point requires only 10 nL of a given biochemical reaction, enabling repeated sampling and real-time monitoring of acetylation. Thus, in contrast to radiation and mass spectrometry-based methods, microfluidic profiling enables both direct detection as well as kinetic analysis of KAT activity. To adapt our assay for chemical library screening, we developed a novel hydroxylamine protocol to quench the KAT reaction and examined a small library of molecules for inhibition of the prototypical KATs p300 and Gcn5. This single-point mobility shift screen identified several known inhibitors of KAT activity, including Lys-CoA, Tat-CoA, C646, MG-149, and garcinol, as well the novel modulators mitoxantrone (Figure S6) and gossypol. Mechanistic analysis using the assay’s kinetic mode revealed garcinol and gossypol were time-dependent, detergent-sensitive inhibitors of Gcn5 and p300. These properties are consistent with inhibition due to aggregation, rather than direct molecular recognition of KAT enzymes. The detergent sensitivity of garcinol has not been previously reported and is notable, given that this molecule is commonly invoked as a generic KAT inhibitor,32–34 and the initial publication reporting garcinol’s KAT inhibitory properties has been cited over 200 times.9 While the precise molecular mechanisms responsible for the cellular antiacetylation activity of garcinol remain to be determined, our studies suggest caution may be warranted when interpreting biological results based on the use of this molecule as a specific pan-KAT inhibitor. In addition, the finding that garcinol retained a degree of KAT inhibitory activity even at 0.05% Triton-X reiterates the principle that aggregation-based effects should not be ruled out even when pursuing leads discovered from screens performed in the presence of detergent36 and suggests KATs may be especially susceptible to aggregation-based inhibition.
Broad-spectrum inhibitors, including bromosporine,39 DZNep,40 nicotinamide,41 and SAHA,42 have proven valuable tools for probing the dependence of biological processes on specific epigenetic enzyme families. However, similar reagents do not exist for the study of KATs. Intrigued by a previous report indicating the bisubstrate Tat-CoA may function as a class-wide inhibitor of KAT activity,7 we included it in our screen, verified its KAT inhibitory activity, and performed biochemical and cellular analyses of its effects. Tat-CoA was found to be a potent inhibitor of p300 and also inhibited Gcn5, albeit ~20-fold less potently. Furthermore, we found that Tat-CoA is an active antagonist of histone acetylation in cells. CoA-based bisubstrate probes have several advantages relative to electrophilic and aggregating KAT inhibitors, including a well-defined mechanism of reversible inhibition,21 facile synthesis via solid-phase methods,7 and the ability to characterize targets via chemoproteomic methods.43,44 While p300-selective cell permeable KAT bisubstrates have been previously applied in several studies, our results are the first to indicate that less selective inhibitors such as Tat-CoA are also active biologically and therefore may have use as broad-spectrum probes of KAT-dependent processes. Future studies will be required to determine whether Tat-CoA, or alternative cell-permeable bisubstrates,45 are capable of functioning as true pan-KAT inhibitors. Such molecules will provide additional useful stopgaps to enable the study of KAT biology while the discovery of reversible small molecule KAT inhibitors remains an active goal, one whose achievement may be considerably facilitated by the application of the microfluidic mobility shift strategy reported herein.
METHODS
Compounds, Enzymes, and Materials.
Recombinant p300 (1195–1662) and Gcn5 (497–662) were expressed and purified from E. coli as N-terminal, hexahistidine-tagged constructs. Crebbp (1319–1710) and Morf (431–2073) were obtained from SignalChem. Labchip EZ-Reader 12-sipper chip (#760404) and ProfilerPro Separation Buffer (#760367) were purchased from PerkinElmer. A library of 148 molecules previously characterized as inhibitors of KATs, lysine deacetylases, protein methyltransferases, lysine demethylases, bromodomains, and kinases were assembled, subjected to quality control, and obtained from NCATS. Lys-CoA and Tat-CoA were synthesized according to a previously reported procedure.43 Materials and methods for the synthesis of FITC-labeled KAT substrate peptides are provided in the Supporting Information.
General Procedure for Microfluidic Mobility Shift Assay.
FITC-H4 (3–14, FITC-Ahx-RGKGGKGLGKGG [Ahx = 6-amino-hexanoic acid]) and FITC-H3 (5–20; FITC-Ahx-TARKSTGGKAPRKQL) were used for all microfluidic KAT assays unless otherwise specified. KAT assays consisting of reaction buffer (50 mM HEPES, pH 7.5, 50 mM NaCl, 2 mM EDTA, 2 mM DTT, 0.05% Triton-X-100) with KAT (p300 [50 nM], Crebbp [150 nM], Morf [200 nM], Gcn5 [100 nM]) and FITC-peptide (FITC-H4 [2 μM ] for p300, FITC-H4 [2 μM] for Crebbp and Morf, FITC-H3 [2 μM] for Gcn5) were plated in 384-well plates and allowed to equilibrate at RT for 10 min. Reactions were initiated by the addition of acetyl-CoA (final concentration = 1 μM), bringing the final assay volume to 30 μL. End-point assays were quenched at appropriate time points (<15% product accumulation) by the addition of 5 μL of 0.5 M neutral hydroxylamine. Reactions were then transferred to a PerkinElmer Lab-Chip EZ-Reader instrument for analysis by microfluidic electrophoresis. Optimized separation conditions were as follows: downstream voltage of −500 V, upstream voltage of −2500 V, and a pressure of −1.5 psi for FITC-H4 (3–14) and FITC-H3 (5–20). Separation resolution (SR) in Figure 1 was calculated using the formula SR = ΔL/2(σ1 + σ2), where ΔL, σ1, and σ2 are the distance between peaks, standard deviation of the first peak, and standard deviation of the second peak, respectively. Percent conversion is calculated by ratiometric measurement of substrate/product peak heights. Model screen for p300 inhibitors was performed using a compound concentration of 40 μM (1.5% DMSO). Percent activity represents the percent conversion of KAT reactions treated with inhibitors relative to untreated control KAT reactions and corrected for nonenzymatic acetylation. Dose–response analysis of p300 and Gcn5 were performed in triplicate and analyzed by nonlinear least-squares regression fit to y = 100/(1 + 10^(log IC50 − x)*H), where H = Hill slope (variable). IC50 values represent the concentration that inhibits 50% of KAT activity. All calculations were performed using Prism 6 (GraphPad) software.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Dr. Masoud Vedadi and Taraneh Hajian (University of Toronto-Structural Genomics Consortium) for providing recombinant p300 and Gcn5, Dr. Jay Schneekloth (NCI) for access to the PerkinElmer EZ-Reader instrument and Prof. Cheryl Arrowsmith (University of Toronto-Structural Genomics Consortium), Dr. Anton Simenov (NCATS), and Dr. David Maloney (NCATS) for many helpful discussions. This work was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research (ZIA BC011488–02), and National Center for Advancing Translational Sciences, National Institutes of Health.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.5b00709.
Tables S1–S3, Figures S1–S9, and supporting materials and methods (PDF)
Table of structures, SMILES, and inhibition data for p300 and Gcn5 (XLSX)
The authors declare no competing financial interest.
REFERENCES
- (1).Verdin E, and Ott M (2015) 50 years of protein acetylation: from gene regulation to epigenetics, metabolism and beyond. Nat. Rev. Mol. Cell Biol. 16, 258–264. [DOI] [PubMed] [Google Scholar]
- (2).Dhalluin C, Carlson JE, Zeng L, He C, Aggarwal AK, and Zhou MM (1999) Structure and ligand of a histone acetyltransferase bromodomain. Nature 399, 491–496. [DOI] [PubMed] [Google Scholar]
- (3).Farria A, Li W, and Dent SY (2015) KATs in cancer: functions and therapies. Oncogene 34, 4901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, Philpott M, Munro S, McKeown MR, Wang Y, Christie AL, West N, Cameron MJ, Schwartz B, Heightman TD, La Thangue N, French CA, Wiest O, Kung AL, Knapp S, and Bradner JE (2010) Selective inhibition of BET bromodomains. Nature 468, 1067–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, Chesi M, Schinzel AC, McKeown MR, Heffernan TP, Vakoc CR, Bergsagel PL, Ghobrial IM, Richardson PG, Young RA, Hahn WC, Anderson KC, Kung AL, Bradner JE, and Mitsiades CS (2011) BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 146, 904–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Nicodeme E, Jeffrey KL, Schaefer U, Beinke S, Dewell S, Chung CW, Chandwani R, Marazzi I, Wilson P, Coste H, White J, Kirilovsky J, Rice CM, Lora JM, Prinjha RK, Lee K, and Tarakhovsky A (2010) Suppression of inflammation by a synthetic histone mimic. Nature 468, 1119–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Zheng Y, Balasubramanyam K, Cebrat M, Buck D, Guidez F, Zelent A, Alani RM, and Cole PA (2005) Synthesis and evaluation of a potent and selective cell-permeable p300 histone acetyltransferase inhibitor. J. Am. Chem. Soc 127, 17182–17183. [DOI] [PubMed] [Google Scholar]
- (8).Guidez F, Howell L, Isalan M, Cebrat M, Alani RM, Ivins S, Hormaeche I, McConnell MJ, Pierce S, Cole PA, Licht J, and Zelent A (2005) Histone acetyltransferase activity of p300 is required for transcriptional repression by the promyelocytic leukemia zinc finger protein. Mol. Cell. Biol 25, 5552–5566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Balasubramanyam K, Altaf M, Varier RA, Swaminathan V, Ravindran A, Sadhale PP, and Kundu TK (2004) Polyisoprenylated benzophenone, garcinol, a natural histone acetyltransferase inhibitor, represses chromatin transcription and alters global gene expression. J. Biol. Chem 279, 33716–33726. [DOI] [PubMed] [Google Scholar]
- (10).Stimson L, Rowlands MG, Newbatt YM, Smith NF, Raynaud FI, Rogers P, Bavetsias V, Gorsuch S, Jarman M, Bannister A, Kouzarides T, McDonald E, Workman P, and Aherne GW (2005) Isothiazolones as inhibitors of PCAF and p300 histone acetyltransferase activity. Mol. Cancer Ther. 4, 1521–1532. [DOI] [PubMed] [Google Scholar]
- (11).Kornacki JR, Stuparu AD, and Mrksich M (2015) Acetyltransferase p300/CBP associated Factor (PCAF) regulates crosstalk-dependent acetylation of histone H3 by distal site recognition. ACS Chem. Biol 10, 157–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Berndsen CE, and Denu JM (2005) Assays for mechanistic investigations of protein/histone acetyltransferases. Methods 36, 321–331. [DOI] [PubMed] [Google Scholar]
- (13).Henry RA, Kuo YM, Bhattacharjee V, Yen TJ, and Andrews AJ (2015) Changing the selectivity of p300 by acetyl-CoA modulation of histone acetylation. ACS Chem. Biol 10, 146–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Trievel RC, Li FY, and Marmorstein R (2000) Application of a fluorescent histone acetyltransferase assay to probe the substrate specificity of the human p300/CBP-associated factor. Anal. Biochem 287, 319–328. [DOI] [PubMed] [Google Scholar]
- (15).Kim Y, Tanner KG, and Denu JM (2000) A continuous, nonradioactive assay for histone acetyltransferases. Anal. Biochem 280, 308–314. [DOI] [PubMed] [Google Scholar]
- (16).Dahlin JL, Nissink JW, Strasser JM, Francis S, Higgins L, Zhou H, Zhang Z, and Walters MA (2015) PAINS in the Assay: Chemical Mechanisms of Assay Interference and Promiscuous Enzymatic Inhibition Observed during a Sulfhydryl-Scavenging HTS. J. Med. Chem 58, 2091–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Liu Y, Gerber R, Wu J, Tsuruda T, and McCarter JD (2008) High-throughput assays for sirtuin enzymes: a microfluidic mobility shift assay and a bioluminescence assay. Anal. Biochem 378, 53–59. [DOI] [PubMed] [Google Scholar]
- (18).Wigle TJ, Provencher LM, Norris JL, Jin J, Brown PJ, Frye SV, and Janzen WP (2010) Accessing protein methyltransferase and demethylase enzymology using microfluidic capillary electrophoresis. Chem. Biol 17, 695–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Min SW, Cho SH, Zhou Y, Schroeder S, Haroutunian V, Seeley WW, Huang EJ, Shen Y, Masliah E, Mukherjee C, Meyers D, Cole PA, Ott M, and Gan L (2010) Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 67, 953–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Ott M, Schnolzer M, Garnica J, Fischle W, Emiliani S, Rackwitz HR, and Verdin E (1999) Acetylation of the HIV-1 Tat protein by p300 is important for its transcriptional activity. Curr. Biol 9, 1489–1492. [DOI] [PubMed] [Google Scholar]
- (21).Thompson PR, Kurooka H, Nakatani Y, and Cole PA (2001) Transcriptional coactivator protein p300. Kinetic characterization of its histone acetyltransferase activity. J. Biol. Chem 276, 33721–33729. [DOI] [PubMed] [Google Scholar]
- (22).Wu J, and Zheng YG (2008) Fluorescent reporters of the histone acetyltransferase. Anal. Biochem 380, 106–110. [DOI] [PubMed] [Google Scholar]
- (23).Liu X, Wang L, Zhao K, Thompson PR, Hwang Y, Marmorstein R, and Cole PA (2008) The structural basis of protein acetylation by the p300/CBP transcriptional coactivator. Nature 451, 846–850. [DOI] [PubMed] [Google Scholar]
- (24).Avvakumov N, and Cote J (2007) The MYST family of histone acetyltransferases and their intimate links to cancer. Oncogene 26, 5395–5407. [DOI] [PubMed] [Google Scholar]
- (25).Panagopoulos I, Fioretos T, Isaksson M, Samuelsson U, Billstrom R, Strombeck B, Mitelman F, and Johansson B (2001) Fusion of the MORF and CBP genes in acute myeloid leukemia with the t(10;16)(q22;p13). Hum. Mol. Genet 10, 395–404. [DOI] [PubMed] [Google Scholar]
- (26).Fanslau C, Pedicord D, Nagulapalli S, Gray H, Pang S, Jayaraman L, Lippy J, and Blat Y (2010) An electrophoretic mobility shift assay for the identification and kinetic analysis of acetyl transferase inhibitors. Anal. Biochem 402, 65–68. [DOI] [PubMed] [Google Scholar]
- (27).Lipmann F, and Tuttle LC (1945) The detection of activated carboxyl groups with hydroxylamine as interceptor. J. Biol. Chem 161, 415. [PubMed] [Google Scholar]
- (28).Chou TC, and Lipmann F (1952) Separation of acetyl transfer enzymes in pigeon liver extract. J. Biol. Chem 196, 89–103. [PubMed] [Google Scholar]
- (29).Balasubramanyam K, Swaminathan V, Ranganathan A, and Kundu TK (2003) Small molecule modulators of histone acetyltransferase p300. J. Biol. Chem 278, 19134–19140. [DOI] [PubMed] [Google Scholar]
- (30).Wang Y, Lonard DM, Yu Y, Chow DC, Palzkill TG, and O’Malley BW (2011) Small molecule inhibition of the steroid receptor coactivators, SRC-3 and SRC-1. Mol. Endocrinol 25, 2041–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Jadhav A, Ferreira RS, Klumpp C, Mott BT, Austin CP, Inglese J, Thomas CJ, Maloney DJ, Shoichet BK, and Simeonov A (2010) Quantitative analyses of aggregation, autofluorescence, and reactivity artifacts in a screen for inhibitors of a thiol protease. J. Med. Chem 53, 37–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Sethi G, Chatterjee S, Rajendran P, Li F, Shanmugam MK, Wong KF, Kumar AP, Senapati P, Behera AK, Hui KM, Basha J, Natesh N, Luk JM, and Kundu TK (2014) Inhibition of STAT3 dimerization and acetylation by garcinol suppresses the growth of human hepatocellular carcinoma in vitro and in vivo. Mol. Cancer 13, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Inuzuka H, Gao D, Finley LW, Yang W, Wan L, Fukushima H, Chin YR, Zhai B, Shaik S, Lau AW, Wang Z, Gygi SP, Nakayama K, Teruya-Feldstein J, Toker A, Haigis MC, Pandolfi PP, and Wei W (2012) Acetylation-dependent regulation of Skp2 function. Cell 150, 179–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Kuhn AN, van Santen MA, Schwienhorst A, Urlaub H, and Luhrmann R (2009) Stalling of spliceosome assembly at distinct stages by small-molecule inhibitors of protein acetylation and deacetylation. RNA 15, 153–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Herrera JE, Bergel M, Yang XJ, Nakatani Y, and Bustin M (1997) The histone acetyltransferase activity of human GCN5 and PCAF is stabilized by coenzymes. J. Biol. Chem 272, 27253–27258. [DOI] [PubMed] [Google Scholar]
- (36).Feng BY, and Shoichet BK (2006) A detergent-based assay for the detection of promiscuous inhibitors. Nat. Protoc 1, 550–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Ogryzko VV, Schiltz RL, Russanova V, Howard BH, and Nakatani Y (1996) The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell 87, 953–959. [DOI] [PubMed] [Google Scholar]
- (38).Montgomery DC, Sorum AW, and Meier JL (2015) Defining the Orphan Functions of Lysine Acetyltransferases. ACS Chem. Biol 10, 85–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Gallenkamp D, Gelato KA, Haendler B, and Weinmann H (2014) Bromodomains and their pharmacological inhibitors. Chem-MedChem 9, 438–464. [DOI] [PubMed] [Google Scholar]
- (40).Glazer RI, Hartman KD, Knode MC, Richard MM, Chiang PK, Tseng CK, and Marquez VE (1986) 3-Deazaneplanocin: a new and potent inhibitor of S-adenosylhomocysteine hydrolase and its effects on human promyelocytic leukemia cell line HL-60. Biochem. Biophys. Res. Commun 135, 688–694. [DOI] [PubMed] [Google Scholar]
- (41).Bitterman KJ, Anderson RM, Cohen HY, Latorre-Esteves M, and Sinclair DA (2002) Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J. Biol. Chem 277, 45099–45107. [DOI] [PubMed] [Google Scholar]
- (42).Richon VM, Webb Y, Merger R, Sheppard T, Jursic B, Ngo L, Civoli F, Breslow R, Rifkind RA, and Marks PA (1996) Second generation hybrid polar compounds are potent inducers of transformed cell differentiation. Proc. Natl. Acad. Sci. U. S. A 93, 5705–5708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Montgomery DC, Sorum AW, and Meier JL (2014) Chemoproteomic Profiling of Lysine Acetyltransferases Highlights an Expanded Landscape of Catalytic Acetylation. J. Am. Chem. Soc 136, 8669–8676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Montgomery DC, Sorum AW, Guasch L, Nicklaus MC, and Meier JL (2015) Metabolic Regulation of Histone Acetyltransferases by Endogenous Acyl-CoA Cofactors. Chem. Biol 22, 1030–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Bandyopadhyay K, Baneres JL, Martin A, Blonski C, Parello J, and Gjerset RA (2009) Spermidinyl-CoA-based HAT inhibitors block DNA repair and provide cancer-specific chemo- and radiosensitization. Cell Cycle 8, 2779–2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.