

Abstract
The kinase-inducible domain (KIX) of the transcriptional coactivator CBP binds multiple transcriptional regulators through two allosterically-connected sites. Establishing a method for observing activator-specific KIX conformations would facilitate the discovery of drug-like molecules that capture specific conformations and further elucidate how distinct activator-KIX complexes produce differential transcriptional effects. However, the transient and low to moderate affinity interactions between activators and KIX are difficult to capture using traditional biophysical assays. Here, we describe a collision induced unfolding-based approach that produces unique fingerprints for peptides bound to each of the two available sites within KIX, as well as a third fingerprint for ternary KIX complexes. Furthermore, we evaluate the analytical utility of unfolding fingerprints for KIX complexes using CIUSuite, and conclude by speculating as to the structural origins of the conformational families created from KIX:peptide complexes following collisional activation.
Keywords: transcriptional coactivator, Protein:Protein Interaction, Inhibitor, Ion Mobility, Native Mass Spectrometry
INTRODUCTION
The master coactivator CREB Binding Protein (CBP) contains a kinase-inducible (KIX) domain that interacts with over a dozen activators through two allosterically connected sites to regulate diverse processes including hematopoiesis, metabolism, and cognitive function [1]. DNA-bound activators bind to KIX through transcriptional activation domains (TADs). These TADs (~8 to 45 amino acids) are intrinsically disordered and adopt helical structures upon binding to KIX [2-5]. KIX consists of a three-helix bundle with two well-defined interfaces that are often referred to as the mixed-lineage leukemia (MLL)- and phosphorylated kinase-inducible domain (pKID)- binding sites (Figure 1 A). Although KIX is a conformationally dynamic protein, NMR data for binary and ternary KIX complexes reveal that the domain is composed of three alpha-helices (α1, α2, α3) and two 310 helices [6]. The MLL-binding site is formed by a hydrophobic groove between α2, α3 and one of the 310 helices (Figure 1A, red), while pKID binds to the opposite face of KIX in a shallow groove between α1 and α3, with the pKID peptide adopting a conformation that wraps around the third alpha helix (Figure 1 A, blue).
Figure 1.

Structure of KIX with its peptide binding partners and corresponding mass spectra, (A) NMR structures of KIX (gray) with four peptides: MLL (red), E2A (pink), c-Myb (light blue), and pKID (blue). (B) Mass spectra of (from top to bottom) apo KIX, KIX:MLL, KIX:pKID, and KIX:MLL:pKID. The apo peaks are in red, the KIX+MLL peaks in red, KIX+pKID in blue, and the ternary KIX+MLL+pKID in green.
While the activators described above bind canonically to either the MLL- or pKID-binding site within KIX, their effects extend beyond their local interactions. Distal residues shift between these sites to mediate specific activator-coactivator interactions. This is exemplified by KIX bound to the activator, MLL; where the affinity of the complex for the phosphorylated form of CREB increases by two-fold at a remote second site [7, 8]. As an inducible activator, pKID interacts more strongly than its constitutive counterpart, c-Myb, leading to alterations in gene expression [9, 10]. Both the c-Myb and pKID regions share distinct, but overlapping binding sites, with pKID exhibiting stronger binding compared to c-Myb, thus leading to a key regulation mechanism for gene expression. The MLL binding site is also shared by other activators, such as the E-protein activation domain 1 (E2A) [11]. Overall, these structure-function relationships are difficult to capture because they are short-lived, obscuring the functions of specific transcriptional complexes [12]. Purified KIX complexes often exist as ensembles, defined by relatively weak (Kd < 10 μM) peptide binding affinities in solution [2, 12, 13]. Using high peptide concentrations forces the generation of more homogenous KIX:peptide complexes, but mixed conformational ensembles remain in solution [14-16]. This fact raises the question of whether it is possible to rapidly identify distinguishable conformations of KIX that are related to its binding partner.
While many have worked to identify ligands and mutants that modulate KIX structure and function [17-19], it remains difficult to rapidly predict the structural and functional impacts of KIX ligand binding. For instance, functional readouts such as competitive fluorescence polarization assays screen for inhibitors against defined partners such as the MLL-KIX complex [17], but do not reveal alterations in KIX structure or interactions at remote sites. While standard mass spectrometry (MS)-based screens can detect target-ligand binding, the functional and structural consequences of such binding is not often available [20, 21]. Therefore, novel and high-throughput techniques that are sensitive to both ligand binding and protein structure would aid analysis of these protein-protein interactions (PPIs) and identify novel ligands based on their ability to produce distinct conformations within KIX.
KIX is particularly amenable to native MS studies given its surprising stability in the gas phase and network of electrostatic interactions [22, 23]. Here, we describe an ion mobility-mass spectrometry (IM-MS) based method capable of detecting peptide binding and assigning the location of that binding uniquely to either of the two available sites within KIX. In general, IM-MS can measure both protein mass and size within complex mixtures, requiring only small amounts (<1 ng) of unlabeled analyte. In an IM-MS experiment, intact protein:inhibitor complexes are ionized by nano-electrospray ionization (nESI) prior to separation by size and charge by IM and mass analysis by a time-of-flight mass analyzer. While protein complexes that are >3% different in total global size (collision cross section, CCS) can be readily separated by IM alone, proteins that undergo smaller conformation changes induced by ligand binding can be analyzed by a gas-phase activation technique termed collision induced unfolding (CIU) [24], which is analogous to a gas-phase differential scanning calorimetry (DSC) experiment. Additionally, collision induced dissociation (CID) can be used to measure the ejection of ligands from protein:ligand complexes [25]. CIU has been used to distinguish between modes of ATP-competitive kinase inhibitors [26], in biosimilar analysis [27, 28], to assess biotherapeutics [29], and to probe the binding of specific lipids to membrane protein complexes [30-34]. In this report, we develop a method using CIU to differentiate between KIX:peptide(MLL, E2A, pKID, and c-Myb) complexes. Through a detailed quantitative analysis of the CIU data presented [35], we speculate on the origins of the conformational transitions observed and their broader analytical utility in KIX inhibitor discovery efforts.
MATERIALS AND METHODS
Protein Expression and Purification.
The DNA sequence encoding the KIX domain from mouse CBP (residues 56-672) was cloned into the bacterial expression pRSETB vector with an additional hexahistadine tag and a short polar linker fused to the N-terminus of KIX resulting in protein with the following sequence (tag and linker residues are shown in lower case)[36]: mrgshhhhhhgmasGVRKGWHEHVTQDLRSHLVHKLVQAIFPTPDPAALKDRRMENLVAYAKKVEGDMYESANSRDEYYHLLAEKIYKIQKELEEKRRSRL (molecular weight (MW)=11.9 kDa).
The pRSETB KIX plasmid was transformed into Rosetta 2(DE3) pLysS Escherichia coli (Novagen, Merck KGaA, Darmstadt, Germany) competent. A colony was used to inoculate 50 mL LB starter cultures containing 0.1 mg/mL ampicillin and 0.34 μg/mL chloramphenicol and allowed to shake overnight at 250 rpm, 37 °C. The overnight culture was added to 1 L of sterile terrific broth (12 g tryptone, 24 g yeast extract, 4% (v/v) glycerol, 900 mL water, 100 mM 1 M potassium phosphate buffer) at a ratio of 10 mL overnight to 1 L media. Cells were grown to an OD600nm of approximately 0.8-1.0 (37 °C, 250 rpm) and induced with 0.25 mM IPTG (25 °C, 18 hours). The cells were harvested by centrifugation for 15 minutes at 8,000xg and the cell pellet was stored at −80 °C. From a 1 L expression the pellet was thawed on ice and re-suspended in 20 mL of lysis buffer (50 mM sodium phosphate, 300 mM NaCl, 10 mM imidazole, pH 7.2 10 mM β-ME). Cells were lysed by sonication and the lysate was separated from cellular debris by centrifugation at 9,000xg for 30 minutes at 4 °C.
KIX was purified using a standard batch nickel affinity protocol, as previously described using Ni-NTA agarose resin (QIAGEN GmbH, Hilden, Germany) according to the protocol provided by the manufacturer. The protein was eluted using a high concentration of imidazole (400 mM), and then was further isolated from contaminants by cation-exchange chromatography with Source 15S column (GE Healthcare Bio-Sciences Corp., Marlborough, MA) using the AKTA FPLC system using buffer A (50 mM sodium phosphate pH 7.2 1 mM DTT) and buffer B (50 mM sodium phosphate, 1 M NaCl, pH 7.2 1 mM DTT). After the protein was applied to the column, unbound sample was washed for two column volumes. For the first segment, 60% of buffer B was reached for 5 column volumes. For the second, 100% of buffer B ran through for two column volumes. Purified protein was buffer exchanged into 10 mM sodium phosphate, 100 mM NaCl, pH 6.8 using a PD-10 column (GE Healthcare Bio-Sciences Corp., Marlborough, MA). The protein concentration was determined by ultraviolet light with a wavelength at 280 nm using the extinction coefficient of 12950 cm−1 M−1 [37]. Final protein purity was assessed by electrophoresis, and the mass of the protein was verified by Q-ToF LC-MS (Agilent Technologies, Santa Clara, CA).
Peptide synthesis.
MLL (GNILPSDIMDFVLKNTPSMQALGESPES, MW=2991 Da) and E2A (Y-βAla-GTDKELSDLLDFSAMFS, MW=2110 Da) were synthesized on CLEAR (Cross-linked Ethoxylate Acrylate Resins) amide resin (Peptides International Inc., Louisville KY) by standard HBTU/HOBT/DIEA and N-9 Fluorenylmethoxycarbonyl (Fmoc) solid phase synthesis methods as previously described and purified by HPLC [38]. Dr. Ningkun Wang provided the c-Myb25mer (KEKRIKELELLLMSTENELKGQQAL, MW=2942 Da) and pKID29mer (AcβA-TDSQKRREILSRRPS(Phos)YRKILNDLSSDAPG, MW=3553 Da) peptide.
Ion Mobility-Mass Spectrometry.
Peptides were added to KIX in a 10:1 molar ratio and incubated at room temperature for 30 minutes. Samples were then buffer exchanged into 100 mM ammonium acetate at pH 7 using Micro Bio-Spin 6 columns (Bio-Rad, Hercules, CA) and prepared to a final concentration of 25 μM. After buffer exchange, all samples were moved to ice before analysis. 5-7 μL of sample was analyzed on a Q-ion mobility-ToF mass spectrometer (Q-IM-TOF) instrument (Synapt G2 HDMS, Waters, Milford, MA). Samples were ionized using a nESI source as previously described [39, 40]. Capillary voltages ranged from 1.5-1.7 V, with the sample cone set at 10 V and the extraction cone set to 1 V in positive ion mode. The quadrupole was operated at a range of 500-5000 m/z and set to scan automatically. The trap traveling-wave ion guide was pressurized using a gas flow rate of 2 mL/min, with the ion mobility separation cell operating with a wave height of 25 V and a wave velocity of 600 m/s. The ToF was operated at a pressure of 8.70×10−7 mbar. The protein:peptide ratios were kept sufficiently low to avoid the formation of ESI artifact complexes [25]. All mass spectra were externally calibrated using a solution of cesium iodide (20 mg/mL) and processed using the Masslynx 4.1 software (Waters, UK). CCS measurements were calibrated externally using a database of proteins and peptides with known helium CCS values that bracketed the CCS and ion mobility values of KIX and KIX:peptide complexes [41].
For CIU experiments, ions were activated in the trap traveling-wave ion guide prior to IM separation. Each ion was mass-selected in the quadrupole prior to activation by increasing the collision voltage from 4 V to 50 V in steps of 2 V. After 50 V, the signal-to-noise (S/N) ratios for KIX:peptide complexes typically decreased to values less than 3. The corresponding drift times for the 8+ ions were extracted, normalized and processed using the CIUSuite software package as described previously [35]. The 8+ charge state was chosen as the focus of our KIX CIU assay development for its relative compactness, high signal relative intensity in comparison to other protein charge states observed under native-like buffer conditions and generated a relatively large number of CIU transitions following activation. Specifically, CIU fingerprints recorded for 7+ KIX ions exhibited fewer structural transitions upon activation, and 9+ ions exhibited larger CCS values in comparison to lower charge states prior to CIU (data not shown). Similar criteria have been used previously to select charge states for CIU assay development [26, 27, 42].
Some datasets shown are analyzed using a scaled deviation score (SDS), as described previously [26, 35]. Briefly, the SDS uses a scaled z-score to quantify a level of deviation (or difference) observed in a CIU dataset in comparison to an average CIU fingerprint, plotted as a function of the collision voltage used to achieve the indicated level of CIU and normalized to the total intensity values found within the fingerprint at the indicated voltage. Error bars reported for CIU stability values represent a single standard deviation generated from replicate measurements.
RESULTS AND DISCUSSION
KIX:Peptide Complexes Generate Unique CIU Fingerprints
Signals for unbound (apo) KIX, along with those associated with KIX:peptide complexes, are observed in our native IM-MS experiments (Figure 1B). In the presence of two peptides that bind to different sites within KIX, we observe signals from each binary KIX:peptide complex, as well as signals corresponding to the intact ternary KIX:peptide:peptide complex. Native IM-MS allows for structural information to be extracted from each of these signals, whereas most bulk solution-phase technologies provide signatures averaged over all complex stoichiometries present in a sample. When CCS values are extracted from these data, the KIX:pKID 8+ charge state exhibits a 1397 ± 20 Å2 and the KIX:MLL 8+ charge state exhibits a value of 1382 ± 19 Å2. These CCS values differ by less than 2%, making the complexes essentially indistinguishable by our IM measurements alone. Therefore, in order to differentiate these KIX complexes, we implemented a CIU strategy aimed at identifying differences in the unfolding patterns of the assemblies.
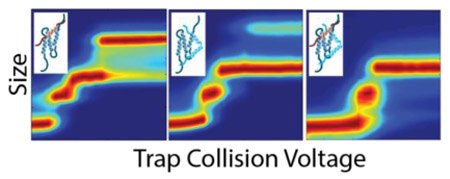
Since the ground state CCS values for unbound KIX, 1:1 and 1:2 KIX and KIX:peptide complexes vary primarily upon the number of peptide partners involved, and such differences reveal little information on protein tertiary structure (see above), CIU fingerprints shown here are plotted on a normalized IM drift time axis (Δdrift time) in order to facilitate their direct comparisons (Figure 2). For binary KIX:peptide complexes, we observe four CIU features having Δdrift time centroid values of ~0.00 ms, 2.26 ms, 3.75 ms, and 5.06 ms (Figure 2B-E), while ternary KIX:peptide complexes have Δdrift time centroid values of 0.21 ms, 2.30 ms and 3.93 ms (Figure 2F,G). A cursory examination of data shown in Figure 2 indicates that MLL or E2A bound KIX ions appear to follow similar unfolding pathways in the gas-phase (Figure 2B, C), whereas pKID and c-Myb KIX complexes exhibit similar CIU data (Figure 2D, E) that is substantially different from those recorded for MLL and E2A KIX complexes. For CIU fingerprints generated from ternary complexes formed by KIX:MLL:pKID and KIX:E2A:c-Myb, only three features are observed (Figure 2F,G), and the first feature appears to be highly stabilized, generating drift time features that span 24 V versus 5 V for apo KIX (Figure 2A). It is also apparent that relative intensity values for the CIU features observed vary significantly for each class of KIX complex studied here. For example, feature four maintains ~50% relative intensity for pKID-like binders (Figure 2D, E), whereas this same feature achieves 100% relative intensity in CIU data recorded for MLL-like binders (Figure 2B, C). In addition, the first three CIU features in Δdrift time are stabilized relative to the apo KIX CIU fingerprint (Figure 2A). While we observe small differences in the Δdrift time centroid values between CIU fingerprints, the main driver for the differences observed between KIX complexes in these experiments are the number and stability of the CIU features recorded.
Figure 2.

CIU fingerprints for the 8+ ions of the following KIX-associated proteins and complexes: (A) Apo KIX (B) KIX:MLL (C) KIX:E2A (D) KIX:pKID (E) KIX:c-Myb (F) KIX:MLL:pKID, and (G) KIX:E2A:c-Myb. Each of these CIU fingerprints is displayed on a Δdrift time axis in order to facilitate comparisons across complexes having disparate starting sizes.
We computed pair-wise root-mean-square deviation (RMSD) values in order to quantify the CIU differences described qualitatively above (Figure 3) [35]. The RMSD values computed by comparing the three different classes of KIX complex (Figure 3E,F,H) range between 23 and 27. These values are 2.3-3x higher than the RMSD values we compute for replicates of KIX:pKID and KIX:MLL (Figure 3C,D) complex CIU fingerprints, which are 9 and 10, respectively. The RMSD values we compute for CIU data recorded within each KIX complex class (Figure 3A,B,G) range from 13 to 25. Interestingly, the comparison of the CIU data recorded for c-Myb and pKID bound KIX have the greatest RMSD value within a given ligand class, producing an RMSD value of 25. We attribute this relatively large RMSD value to the slightly different binding locations of these two peptides on the KIX surface [10, 43] In general, KIX complexes follow a trend of low RMSD values (9 to10) for replicate measurements, larger RMSD values when comparing CIU data within a KIX binding class (13 to 25), with the largest RMSD values observed when comparing CIU data across KIX binding classes (23 to 27). Furthermore, when cross-comparing RMSD values computed between CIU fingerprints recorded for KIX complexes (Figure 4A), the lowest RMSD values are obtained for data acquired for complexes comprised of similar peptide binders.
Figure 3.

RMSD values and CIU difference plots, computed using CIUSuite, for binary comparison across the data shown in Figure 2. Each difference map indicates where the intensity values for the two subtracted CIU fingerprints are greatest with a blue/red color code, indicated above each panel. The comparisons made are between: (A) MLL and E2A KIX:peptide complexes, (B) pKID and c-Myb KIX:peptide complexes, (C) replicate KIX:pKID CIU fingerprints, (D) replicate KIX:MLL CIU fingerprints, (E) the ternary KIX:MLL:pKID complex and the KIX:pKID complex, (F) the ternary KIX:MLL:pKID complex and KIX:MLL the complex, the (G) the KIX:E2A:c-Myb and KIX:MLL:pKID complexes, and (H) the KIX:pKID and KIX:MLL complexes.
Figure 4.

Histograms of the RMSD values and stabilities of the CIU features observed in Figure 2. (A) RMSD values from Figure 3 in histogram form. (B) A histogram of the CIU stability values recorded of each of the features observed in Figure 2.
Differences in CIU Stabilities Further Discriminate KIX:Peptide Complexes
The CIU datasets were compared by using stability values which are represented by the amount of accelerating voltage necessary to initiate unfolding in gas-phase KIX:peptide complexes (Figure 4A), using a simple first derivative test to identify features within CIU fingerprints (Figure 4B)[35]. We found that the feature appearing at lowest activation energy in apo KIX possessed a stability (defined as a range of voltage values over which the feature is observed) of 5 ± 1 V, compared to stability values of 7 ± 1 V and 6 ± 0 V for KIX:MLL and KIX:E2A, stability values of 12 ± 1 V and 9 ± 1 V for KIX:pKID and KIX:c-Myb, and 21 ± 3 V and 15 ± 1 V for KIX:MLL:pKID and KIX:E2A:c-Myb (n=3 for each CIU fingerprint). While little additional stability is imparted through peptide binding on the second CIU feature observed, the third feature is significantly stabilized upon KIX binding pKID-like ligands, generating stability values of 28 ± 0 V and 32 ± 0 V for KIX:pKID and KIX:c-Myb respectively, as compared to 4 ± 0 V, 8 ± 0 V, 10 ± 0 V, 22 ± 0 V and 19 ± 1 V for apo KIX, KIX:MLL, KIX:E2A, KIX:MLL:pKID, and KIX:MLL:E2A. Notably, a fourth feature is absent in ternary complexes, and the stabilities of this feature scales in stability from pKID-like binders (19 ± 1 V and 13 ± 1 V for KIX:pKID and KIX;MLL) to the MLL-like binders (22 ± 0 V for both KIX:MLL and KIX:E2A), to apo KIX with a stability of 30 ± 0 V.
The differences in stabilities for the CIU features observed in our experiments can be used to theorize as to their origins. Recently, we discovered a strong correlation between the number of CIU transitions and the number of domains for multi-protein domain systems [42]. While the KIX structure is well-described as a single domain, it possesses three α-helices, a number which correlates well to our observations of four CIU transitions for our apo and binary complex CIU fingerprints. Furthermore, the CIU feature appearing at highest activation voltage values dramatically decreases in intensity upon pKID and c-Myb binding and is completely absent for CIU data acquired for ternary KIX complexes. These observations, in combination with knowledge of the binding locations of the ligands studied here on the KIX surface, leads us to speculate that the highest energy CIU transition is likely related to the unfolding of α3. Solution structures of the KIX:peptide complexes reveal that all four peptides contact this helix when bound to the KIX surface [6, 8, 11, 43, 44]. Such binding would likely lead to significant stabilization of this region of the protein complex, leading to the absence of the CIU feature appearing at highest collision energies in our data.
Previous studies have used electron capture dissociation (ECD) to reveal that KIX is stabilized in the gas phase through primarily salt bridges and hydrogen bonds. In particular, ECD of 7+ KIX ions revealed that the observed c and z• fragment ions mainly originated from the backbone near the protein termini, and not from the 3-helix bundle [22]. Further ECD and CID experiments found that the propensity for KIX folding is determined by the intramolecular distribution of charges on the protein surface, rather than its net charge, leading ultimately to produce the surprising stability of KIX in the gas-phase [23]. Together, these experiments showed the gas-phase stabilities of the KIX helices to be α3>α2>α1 [22], and in particular, for the 8+ charge state, α1 appears to be the least stable [23]. This rank ordering agrees well with our CIU interpretation discussed above, and future work will focus on further assigning the precise origins of the structural transitions observed in our KIX:peptide CIU data.
Using CIU to Localize Peptide Binding on the KIX Surface
In order to better project the clustering of each peptide type, the z-scores for each peptide against an MLL-like, pKID-like, or ternary-like complex were computed to describe how far away each individual complex was from the average of each of the three types of complex. (Table 1). Against the average MLL-like score, MLL-like binders had an average score of 0.1±1.1, while pKID-like peptides exhibited a score of −2.1±3.1 and the ternary complexes had a score of −5.3±2.4. Against the pKID-like score, MLL-like binders possessed a score of −1.1±0.6, pKID-like binders had a score of 1.7×10−10±1.1, and ternary binders a score of −0.4±1.5. Against a ternary average, pKID-, MLL-, and ternary-like peptides had scores of −4.0±1.9, −5.7±1.9, and 0.2±1.1, respectively. These scores indicate that, if complete CIU fingerprints are analyzed, MLL-like and pKID-like complexes are challenging to differentiate with confidence, while ternary complexes are readily discriminated.
Table 1.
Z-scores computed for KIX:peptide complexes analyzed by CIU. Values shown in the shaded-boxes represent z-scores computed for replicate datasets.
| MLL-Like | pKID-like | Ternary-like | |
|---|---|---|---|
| MLL-like score | 0.1 ± 1.1 | −2.1 ± 3.1 | −5.3 ± 2.4 |
| pKID-like | −1.1 ± 0.6 | 1.7×10−10 ± 1.1 | −0.4 ± 1.5 |
| Ternary-like | −4.0 ± 1.9 | −5.7 ± 1.9 | 0.2 ± 1.1 |
Previously, we identified regions within CIU fingerprints where differences between different classes of ligands are maximized and can be used to screen unknown binding agents and assign their interactions with a target protein [26]. In order to replicate such results for KIX, we plotted average SDS values [26, 35] for the two classes of peptide binders against collision voltage. The CIU fingerprints collected for all KIX:peptide complexes were compared against averaged CIU fingerprints for MLL-like, pKID-like, or ternary-complexes (Figure 5). Distinct collision voltage ranges are identified that differentiate ternary (Figure 5A), MLL-like (Figure 5B) and pKID-like (Figure 5C) complexes from all others tested. Despite this result, pKID-like complexes proved more challenging to separate from all other KIX complexes tested, exhibiting widely-varying, non-zero SDS scores generated for transitions at every collision voltage value. Overall, CIU transitions appearing at 30 V appear to possess the greatest potential for differentiation of pKID-like binders from other KIX complexes. In contrast, our analysis of MLL-like (Figure 5C) and ternary (Figure 5A) KIX complexes produce a large number of collision voltage values where SDS values for the targeted assemblies were near zero, and those computed for all other complexes were significantly larger, with tight variance values. We note that our SDS score analysis reveals that the areas between 4 and 8 V, 30 V, and 12-16 V appeared most promising for future screening experiments as diagnostic regions for ternary, pKID-like, and MLL-like KIX complexes respectively (shaded in beige), while noting narrower differences in SDS scores for higher voltage values. While this result may seem surprising given our feature analysis and discussion above (Figure 4), it is important to note that the absolute signal intensities recorded for complexes at higher energies is considerably smaller than those observed at lower collision voltages due to CID of the KIX complexes measured here. This decreased signal intensity results in a reduced weighting for feature 4 and other CIU features that appear at larger collision voltage values in our SDS scoring structure.
Figure 5.

A plot of SDS vs collision voltage for peptides compared to an (A) ternary complex average, (B) a pKID-like average, or a (C) MLL-like average. The blue circles represent the average values computed for all other peptides, and the red circles represent the average values computed for the complex targeted for the analysis. Color coded error bars indicate standard deviation values for the analysis produced across replicate CIU experiments (n =3). (D) Z-scores for each KIX:peptide complex plotted against an MLL-like average, pKID-like average, and ternary complex average. Data for the pKID-like complexes are represented by blue circles, the MLL-like complexes by red circles, and the ternary complexes by green circles. The ellipses around the points are added to guide the eye and illustrate the clustering of the different complex classes.
CONCLUSIONS
Here, we present an assay based on IM-MS that rapidly identifies the binding site of a peptide to KIX by using CIU. This is a rare example of using CIU to identify unique fingerprints for different binding sites within a single protein. Specifically, we identified a region between 12-16 V in our CIU data that provides the best differentiation between KIX complex types. This region could be used to build a high-throughput CIU screen capable of rapidly identifying the binding modes of potential PPI inhibitors. Such a screening approach would necessarily rely upon a discrete relationship between CIU fingerprints and KIX-peptide binding modes adopted by the complexes observed. More broadly, through detailed comparisons of CIU fingerprints, quantified using RMSD values, our CIU experiments reveal that the differences in the observed CIU features arise from the altered stabilities of the complexes imparted through peptide binding. We hypothesize that KIX unfolds in a linear α-helix 1-3 fashion in the gas-phase, which correlates well with previous ECD and CID experiments [22, 23]. Such a detailed understanding of the KIX CIU process, linking CIU features to specific regions of the KIX structure, may enable the development of improved PPI inhibitor binding assays.
Despite the progress summarized above, neither native MS nor CIU data were able to detect evidence of allostery between the pKID- and MLL-binding sites. We propose that further MS-based studies could be used examine the allosteric effect by determining the dissociation constants, Kds,[45] for the different complexes by carefully titrating the concentration of one peptide, and measuring the amount of complex formation targeting both both apoKIX and a pre-existing KIX complexes where the alternative binding site is occupied. Such experiments would need to be conducted carefully, taking into account potential differences in the ionization efficiencies of the KIX:peptide complexes studied. These Kd measurements could later be built into a full CIU-MS screen to provide both allostery and ligand localization information for high-throughput screening.
ACKNOWLEDGMENT
The authors would like to acknowledge Daniel Polasky for his assistance with the in-house data extraction software
Funding Sources
The development of CIU technologies in the Ruotolo lab is supported by the National Science Foundation (CAREER, 1253384). JML was supported by NIH F31GM113561 and NIH 3RO1 GM65530 (to A.K.M.).
ABBREVIATIONS
- KIX
kinase inducible domain
- DBD
DNA-binding domain
- TAD
transcriptional activation domain
- PPI
protein-protein interaction
- CBP
CREB-binding protein
- CREB
cAMP response element binding protein
- E2A
E-protein activation domain 1
- HTVL-1
human immunodeficiency virus type 1
- IM-MS
ion mobility-mass spectrometry
- nESI
nano-electrospray ionization
- CCS
collision cross section
- CIU
collision induced unfolding
- CID
collision induced dissociation
- TCV
trap collision voltage
REFERENCES
- 1.Thakur JK, Yadav A, Yadav G: Molecular recognition by the KIX domain and its role in gene regulation. Nucleic Acids Res 42, 2112–2125 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shammas SL, Travis AJ, Clarke J: Allostery within a transcription coactivator is predominantly mediated through dissociation rate constants. Proc Natl Acad Sci U S A. 111, 12055–12060 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rowe SP, Mapp AK: Assessing the permissiveness of transcriptional activator binding sites. Biopolymers. 89, 578–581 (2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Babu MM, van der Lee R, de Groot NS, Gsponer J: Intrinsically disordered proteins: regulation and disease. Current opinion in structural biology. 21, 432–440 (2011) [DOI] [PubMed] [Google Scholar]
- 5.Sugase K, Dyson HJ, Wright PE: Mechanism of coupled folding and binding of an intrinsically disordered protein. Nature. 447, 1021–1025 (2007) [DOI] [PubMed] [Google Scholar]
- 6.Radhakrishnan I, Pérez-Alvarado GC, Parker D, Dyson HJ, Montminy MR, Wright PE: Solution structure of the KIX domain of CBP bound to the transactivation domain of CREB: a model for activator: coactivator interactions. Cell. 91, 741–752 (1997) [DOI] [PubMed] [Google Scholar]
- 7.Radhakrishnan I, Perez-Alvarado GC, Parker D, Dyson HJ, Montminy MR, Wright PE: Solution structure of the KIX domain of CBP bound to the transactivation domain of CREB: a model for activator:coactivator interactions. Cell. 91, 741–752 (1997) [DOI] [PubMed] [Google Scholar]
- 8.Brüschweiler S, Konrat R, Tollinger M: Allosteric Communication in the KIX Domain Proceeds through Dynamic Repacking of the Hydrophobic Core. ACS Chem Biol 8, 1600–1610 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mayr B, Montminy M: Transcriptional regulation by the phosphorylation-dependent factor CREB. Nature reviews Molecular cell biology. 2, 599–609 (2001) [DOI] [PubMed] [Google Scholar]
- 10.Radhakrishnan I, Pérez-Alvarado GC, Dyson HJ, Wright PE: Conformational preferences in the Ser 133-phosphorylated and non-phosphorylated forms of the kinase inducible transactivation domain of CREB. FEBS letters. 430, 317–322 (1998) [DOI] [PubMed] [Google Scholar]
- 11.Denis CM, Chitayat S, Plevin MJ, Wang F, Thompson P, Liu S, Spencer HL, Ikura M, LeBrun DP, Smith SP: Structural basis of CBP/p300 recruitment in leukemia induction by E2A-PBX1. Blood. 120, 3968–3977 (2012) [DOI] [PubMed] [Google Scholar]
- 12.Wands AM, Wang N, Lum JK, Hsieh J, Fierke CA, Mapp AK: Transient-state Kinetic Analysis of Transcriptional Activator·DNA Complexes Interacting with a Key Coactivator. Journal of Biological Chemistry. 286, 16238–16245 (2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shammas SL, Travis AJ, Clarke J: Remarkably Fast Coupled Folding and Binding of the Intrinsically Disordered Transactivation Domain of cMyb to CBP KIX. J Phys Chem B. 117, 13346–13356 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Palazzesi F, Barducci A, Tollinger M, Parrinello M: The allosteric communication pathways in KIX domain of CBP. Proc Natl Acad Sci U S A. 110, 14237–14242 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brüschweiler S, Schanda P, Kloiber K, Brutscher B, Kontaxis G, Konrat R, Tollinger M: Direct Observation of the Dynamic Process Underlying Allosteric Signal Transmission. J Am Chem Soc 131, 3063–3068 (2009) [DOI] [PubMed] [Google Scholar]
- 16.Clark MD, Kumar GS, Marcum R, Luo Q, Zhang Y, Radhakrishnan I: Molecular Basis for the Mechanism of Constitutive CBP/p300 Coactivator Recruitment by CRTC1-MAML2 and Its Implications in cAMP Signaling. Biochemistry. 54, 5439–5446 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Majmudar CY, Højfeldt JW, Arevang CJ, Pomerantz WC, Gagnon JK, Schultz PJ, Cesa LC, Doss CH, Rowe SP, Vásquez V, Tamayo-Castillo G, Cierpicki T, Brooks CL, Sherman DH, Mapp AK: Sekikaic Acid and Lobaric Acid Target a Dynamic Interface of the Coactivator CBP/p300. Angew Chem 51, 11258–11262 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gee CT, Koleski EJ, Pomerantz WCK: Fragment Screening and Druggability Assessment for the CBP/p300 KIX Domain through Protein-Observed 19F NMR Spectroscopy. Angew Chem 54, 3735–3739 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Best JL, Amezcua CA, Mayr B, Flechner L, Murawsky CM, Emerson B, Zor T, Gardner KH, Montminy M: Identification of small-molecule antagonists that inhibit an activator: coactivator interaction. Proc Natl Acad Sci U S A. 101, 17622–17627 (2004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Erlanson DA, Wells JA, Braisted AC: Tethering: fragment-based drug discovery. Annu. Rev. Biophys. Biomol. Struct 33, 199–223 (2004) [DOI] [PubMed] [Google Scholar]
- 21.Wang N, Majmudar CY, Pomerantz WC, Gagnon JK, Sadowsky JD, Meagher JL, Johnson TK, Stuckey JA, Brooks CL III, Wells JA: Ordering a dynamic protein via a small-molecule stabilizer. Journal of the American Chemical Society. 135, 3363–3366 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Breuker K, Brüschweiler S, Tollinger M: Electrostatic stabilization of a native protein structure in the gas phase. Angewandte Chemie International Edition. 50, 873–877 (2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schennach M, Schneeberger E-M, Breuker K: Unfolding and folding of the three-helix bundle protein KIX in the absence of solvent. Journal of The American Society for Mass Spectrometry. 27, 1079–1088 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hyung S-J, Robinson CV, Ruotolo BT: Gas-phase unfolding and disassembly reveals stability differences in ligand-bound multiprotein complexes. Chemistry & biology. 16, 382–390 (2009) [DOI] [PubMed] [Google Scholar]
- 25.Niu S, Rabuck JN, Ruotolo BT: Ion mobility-mass spectrometry of intact protein–ligand complexes for pharmaceutical drug discovery and development. Current opinion in chemical biology. 17, 809–817 (2013) [DOI] [PubMed] [Google Scholar]
- 26.Rabuck JN, Hyung S-J, Ko KS, Fox CC, Soellner MB, Ruotolo BT: Activation state-selective kinase inhibitor assay based on ion mobility-mass spectrometry. Analytical chemistry. 85, 6995–7002 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tian Y, Han L, Buckner AC, Ruotolo BT: Collision induced unfolding of intact antibodies: rapid characterization of disulfide bonding patterns, glycosylation, and structures. Analytical chemistry. 87, 11509–11515 (2015) [DOI] [PubMed] [Google Scholar]
- 28.Beck A, Debaene F, Diemer H, Wagner-Rousset E, Colas O, Dorsselaer AV, Cianférani S: Cutting- edge mass spectrometry characterization of originator, biosimilar and biobetter antibodies. Journal of Mass Spectrometry. 50, 285–297 (2015) [DOI] [PubMed] [Google Scholar]
- 29.Dixit SM, Polasky DA, Ruotolo BT: Collision induced unfolding of isolated proteins in the gas phase: past, present, and future. Current Opinion in Chemical Biology. 42, 93–100 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saikusa K, Kuwabara N, Kokabu Y, Inoue Y, Sato M, Iwasaki H, Shimizu T, Ikeguchi M, Akashi S: Characterisation of an intrinsically disordered protein complex of Swi5–Sfr1 by ion mobility mass spectrometry and small-angle X-ray scattering. Analyst. 138, 1441–1449 (2013) [DOI] [PubMed] [Google Scholar]
- 31.Pagel K, Natan E, Hall Z, Fersht AR, Robinson CV: Intrinsically disordered p53 and its complexes populate compact conformations in the gas phase. Angewandte Chemie International Edition. 52, 361–365 (2013) [DOI] [PubMed] [Google Scholar]
- 32.Vahidi S, Stocks BB, Konermann L: Partially disordered proteins studied by ion mobility-mass spectrometry: implications for the preservation of solution phase structure in the gas phase. Analytical chemistry. 85, 10471–10478 (2013) [DOI] [PubMed] [Google Scholar]
- 33.Dickinson ER, Jurneczko E, Nicholson J, Hupp TR, Zawacka-Pankau J, Selivanova G, Barran PE: The use of ion mobility mass spectrometry to probe modulation of the structure of p53 and of MDM2 by small molecule inhibitors. Frontiers in molecular biosciences. 2, (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.D’Urzo A, Konijnenberg A, Rossetti G, Habchi J, Li J, Carloni P, Sobott F, Longhi S, Grandori R: Molecular basis for structural heterogeneity of an intrinsically disordered protein bound to a partner by combined ESI-IM-MS and modeling. Journal of The American Society for Mass Spectrometry. 26, 472–481 (2015) [DOI] [PubMed] [Google Scholar]
- 35.Eschweiler JD, Rabuck-Gibbons JN, Tian Y, Ruotolo BT: CIUSuite: A Quantitative Analysis Package for Collision Induced Unfolding Measurements of Gas-Phase Protein Ions. Analytical chemistry. 87, 11516–11522 (2015) [DOI] [PubMed] [Google Scholar]
- 36.Buhrlage SJ, Bates CA, Rowe SP, Minter AR, Brennan BB, Majmudar CY, Wemmer DE, Al-Hashimi H, Mapp AK: Amphipathic small molecules mimic the binding mode and function of endogenous transcription factors. ACS chemical biology 4, 335–344 (2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pace CN, Vajdos F, Fee L, Grimsley G, Gray T: How to measure and predict the molar absorption coefficient of a protein. Protein Sci 4, 2411–2423 (1995) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pomerantz WC, Wang N, Lipinski AK, Wang R, Cierpicki T, Mapp AK: Profiling the dynamic interfaces of fluorinated transcription complexes for ligand discovery and characterization. ACS chemical biology. 7, 1345–1350 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhong Y, Hyung S-J, Ruotolo BT: Characterizing the resolution and accuracy of a second-generation traveling-wave ion mobility separator for biomolecular ions. Analyst. 136, 3534–3541 (2011) [DOI] [PubMed] [Google Scholar]
- 40.Hernández H, Robinson CV: Determining the stoichiometry and interactions of macromolecular assemblies from mass spectrometry. Nature protocols. 2, 715–726 (2007) [DOI] [PubMed] [Google Scholar]
- 41.Bush MF, Hall Z, Giles K, Hoyes J, Robinson CV, Ruotolo BT: Collision cross sections of proteins and their complexes: a calibration framework and database for gas-phase structural biology. Analytical chemistry. 82, 9557–9565 (2010) [DOI] [PubMed] [Google Scholar]
- 42.Zhong Y, Han L, Ruotolo BT: Collisional and Coulombic Unfolding of Gas-Phase Proteins: High Correlation to Their Domain Structures in Solution. Angewandte Chemie 126, 9363–9366 (2014) [DOI] [PubMed] [Google Scholar]
- 43.Zor T, De Guzman RN, Dyson HJ, Wright PE: Solution structure of the KIX domain of CBP bound to the transactivation domain of c-Myb. Journal of molecular biology. 337, 521–534 (2004) [DOI] [PubMed] [Google Scholar]
- 44.De Guzman RN, Goto NK, Dyson HJ, Wright PE: Structural basis for cooperative transcription factor binding to the CBP coactivator. J Mol Biol 355, 1005–1013 (2006) [DOI] [PubMed] [Google Scholar]
- 45.Jecklin MC, Touboul D, Bovet C, Wortmann A, Zenobi R: Which electrospray-based ionization method best reflects protein-ligand interactions found in solution? A comparison of ESI, nanoESI, and ESSI for the determination of dissociation constants with mass spectrometry. Journal of the American Society for Mass Spectrometry. 19, 332–343 (2008) [DOI] [PubMed] [Google Scholar]