

Abstract
Background:
Neuroendocrine prostate cancer (NEPC) is an aggressive variant of prostate cancer that may develop de novo or as a mechanism of treatment resistance. N-myc is capable of driving NEPC progression. Alisertib inhibits the interaction between N-myc and its stabilizing factor Aurora-A, inhibiting N-myc signaling, and suppressing tumor growth.
Experimental Design:
Sixty men were treated with alisertib 50mg twice daily for 7 days every 21-days. Eligibility included metastatic prostate cancer and at least one: small cell neuroendocrine morphology; ≥50% neuroendocrine marker expression; new liver metastases without PSA progression; elevated serum neuroendocrine markers. The primary endpoint was six-month radiographic progression free survival (rPFS). Pre-treatment biopsies were evaluated by whole exome and RNA-seq and patient-derived organoids were developed.
Results:
Median PSA was 1.13 ng/ml (0.01–514.2), number of prior therapies was three, and 68% had visceral metastases. Genomic alterations involved RB1 (55%), TP53 (46%), PTEN (29%), BRCA2 (29%), AR (27%), and there was a range of androgen receptor signaling and NEPC marker expression. Six-month rPFS was 13.4% and median overall survival was 9.5 months (7.3–13). Four exceptional responders were identified, including complete resolution of liver metastases and prolonged stable disease, with tumors suggestive of N-myc and Aurora-A overactivity. Patient organoids exhibited concordant responses to alisertib and allowed for the dynamic testing of Aurora-N-myc complex disruption.
Conclusions:
Although the study did not meet its primary endpoint, a subset of patients with advanced prostate cancer and molecular features supporting Aurora-A and N-myc activation achieved significant clinical benefit from single-agent alisertib.
Introduction
Prostate cancer arises as an androgen driven disease, and systemic therapies targeting the androgen receptor (AR) are a mainstay of treatment for patients at all stages of the disease. Most tumors that develop resistance to AR-therapies remain dependent on AR signaling through AR mutation, overexpression, or other means (1). However in recent years, with the earlier and more potent targeting of the AR with newer drugs, non-AR-driven prostate cancer has emerged as a clinically relevant problem(2,3). This has been associated with low AR-signaling, loss of luminal prostate markers, and the development of small cell neuroendocrine features through a process of lineage plasticity(3–6). Patients with AR-independent neuroendocrine prostate cancer (NEPC) often present with metastatic disease to visceral sites in the setting of a low or modestly rising serum prostate specific antigen(7). NEPC may share morphologic, clinical, and molecular (eg., RB1 and TP53 loss) features with small cell carcinoma of the lung(8), and patients are often treated with similar platinum-based chemotherapy regimens(9–11). Prognosis is poor and new therapies are urgently needed(3,12).
We previously identified the oncogenic transcriptional factor N-Myc and the cell cycle kinase Aurora kinase A as overexpressed in the majority of metastatic neuroendocrine prostate tumors and a subset of castration resistant prostate adenocarcinomas(13). N-myc is capable of suppressing AR-signaling and driving lineage plasticity, tumor aggressiveness, and AR-independent progression in prostate cancer preclinical models(14,15). Aurora-A stabilizes N-myc and prevents N-myc protein degradation in human neuroblastoma and in prostate cancer(13–16). The Aurora kinase A catalytic inhibitor alisertib also disrupts the N-myc-Aurora A protein complex(17), thereby inhibiting N-myc signaling and tumor growth.
In this study, we aimed to investigate the clinical efficacy of alisertib in patients with the NEPC phenotype. As NEPC is not well-defined clinically, we established distinct eligibility criteria to capture the range of clinical and pathologic definitions within the “NEPC syndrome” with shared biologic features. A mandatory pre-treatment biopsy was performed in all patients to investigate the relationship between biopsy pathology and molecular features and clinical characteristics. Our objective of this trial was to identify the clinical characteristics and molecular profiles of responding patients to define distinct cohorts most likely to benefit.
Patients and Methods
In this single arm, multi-institutional open label phase 2 trial of the Department of Defense Prostate Cancer Clinical Trials Consortium (NCT01799278), patients with metastatic prostate cancer and at least one of the following key eligibility criteria were enrolled across nine centers between 2013–2016: 1) small cell NEPC morphology (determined by the enrolling center) based on any current or prior tissue sample, 2) prostate adenocarcinoma with greater than 50% immunohistochemical (IHC) staining for neuroendocrine markers (e.g., chromogranin, synaptophysin), 3) development of liver metastases in the absence of PSA progression defined by Prostate Cancer Working Group 2 criteria(18); 4) serum chromogranin A level ≥5X upper limit of normal (ULN) and/or serum neuron specific enolase (NSE) ≥2X ULN. Institutional review board approval was obtained (see trial protocol, supplement) and all patients gave written informed consent. The study was conducted in accordance with ethical principles founded in the Declaration of Helsinki.
A new metastatic pre-treatment biopsy was sent centrally for pathology review and molecular analysis. Patients were treated with alisertib (supplied by Millennium Pharmaceuticals) at the recommended phase 2 dose of 50 mg twice daily for 7 days every 21-days and followed with radiographic evaluation every 3 cycles. Concomitant proton pump inhibitor medication was not allowed on study (see protocol, supplement). Dose reductions were specified based on adverse events (AEs). Therapy was continued until disease progression, unacceptable toxicity, or withdrawal of consent. The primary endpoint was six-month radiographic progression free survival. Secondary endpoints included response rate, overall survival (OS), and toxicity.
Whole exome sequencing (WES) and RNA-seq and analysis were performed using protocols previously described(4) with the addition of Excavator(19) for copy number segmentation. Targeted mRNA gene expression (nanostring) was performed in cases without sufficient frozen tissue using a published assay(20). AURKA and MYCN dual-color fluorescence in situ hybridization (FISH) amplification was defined by the presence of ≥4 copies per nucleus with >100 nuclei evaluated per section (13,21). Patient-derived organoids were developed from fresh tissues using published protocols (22,23). Organoids were treated with alisertib and viability determined using CellTiter-Glo (Promega). A proximity ligation assay of N-Myc/Aurora A interaction was applied to human tissues using protocols described in Dardenne et al(14).
Statistical Methods
Based on the phase II trial of danusertib(24), less than 15% of unselected patients with metastatic castration resistant prostate cancer (CRPC) would be expected to respond to single agent aurora kinase inhibition. Radiographic response rate was initially selected as the primary endpoint. The trial was amended after an interim analysis of the first 19 patients demonstrating limited responses but with clinical improvement and stable disease in this patient population with aggressive disease and limited treatment options. Based on recommendations of the steering committee, the primary endpoint was modified to six-month radiographic progression free survival. This was an endpoint also used in other recent clinical studies including Rathkopf et al (25). Secondary endpoints included response rate and OS. The null hypothesis (H0) was that ≤15% of patients would be radiographic progression-free at 6 months and the alternative hypothesis (Ha) was ≥30%. A sample size of 48 patients was determined according to Ahern’s exact single-stage phase II design(26) (5% significance, 80% power). Requiring at least 20% to have histologic entry criteria to assess sufficient NEPC patients, we planned to treat a total 60 patients. All patients who received at least one dose of alisertib were included in the safety analysis. Toxicities were graded according to CTCAEv.4.0. Analysis for organoid treatment was performed on three independent biological replicates using a non-linear regression curve fit (three parameters) method in Prism 6. All other analyses were performed in R 3.2.3.
Results
75 patients screened and 60 patients were treated. At the end of the study, 48 patients (80%) had died and median follow-up among patients not known to be deceased was 9.7 months (1.1–14.9). Median age was 67 years (45–87), median PSA 1.13 ng/ml (0.01–514.2) (Table 1). Thirty-seven patients (61%) received prior local therapy for prostate cancer and the median number of prior systemic therapies was three (40% enzalutamide or abiraterone, 32% docetaxel, 58% platinum chemotherapy). The majority of patients had adverse prognostic risk factors, including visceral metastases (68%) and elevated LDH (53%; median 233, 109–1428). By central labs, serum neuroendocrine markers were elevated above the upper limit of normal in 46/55 (84%) evaluable patients (chromogranin 71%, NSE 47%; concordance 51%). Carcinoembryonic antigen (CEA) was elevated in 18/31(58%) evaluable patients (median 18.1 [2.3,204.5]).
Table 1:
Baseline clinical characteristic. A total of 75 patients were screened and 60 patients were treated.
|
Patient Characteristics |
|
|---|---|
| Number of patients treated | 60 |
| Median Age (years) | 67 years (45–87) |
| Prior Local Therapy (surgery or radiation) | 37 (61%) |
| Gleason Grade 6 7 8–10 |
8 6 (13%) 15 (33%) 26 (59%) |
| Median PSA (ng/ml) | 1.07 (0.01–514.2) |
| Sites of Metastases Bone Lymph Node Lung Liver Any visceral |
47 (78%) 45 (75%) 22 (37%) 36 (60%) 41 (68%) |
| Elevated LDH | 29/57 (51%) |
| Median number of prior systemic therapies for CRPC Platinum Enzalutamide or abiraterone Docetaxel Cabazitaxel Radium-223 |
3 35 (58%) 24 (40%) 19 (32%) 6 (10%) 1(2%) |
| Histologic diagnosis of NEPC by local review (inclusion criteria 1) |
45 (75%) |
| Prostate adenocarcinoma plus ≥50% immunohistochemical staining for neuroendocrine markers (inclusion criteria 2) |
11 (18%) |
| Development of liver metastases in the absence of PSA progression (inclusion criteria 3) |
15 (25%) |
| Serum chromogranin >5X ULN and/or NSE>2X ULN (inclusion criteria 4) |
18 (30%) |
| Median time from prostate cancer diagnosis to dose (months) | 44.23 (3.9–1313) |
Seventy-five percent (45) of treated patients had pathologic features of NEPC at screening. Of the others with adenocarcinoma, eligibility was based on IHC (5), serum neuroendocrine markers (6), liver metastases without PSA progression (6). Overall, 35% (21) met more than one key eligibility criteria (Table S1). All patients underwent a new pre-treatment metastatic biopsy, of which 56/60 were evaluable by central review, and tumors were classified as NEPC based on published morphologic criteria(27) or castration resistant prostate adenocarcinoma (CRPC-Adeno). NEPC pathology concordance rate between study biopsy and screening tissue was 75% (42/56) (Table S1) with differences likely reflecting heterogeneity, temporal differences, and/or possibly inter-reader variability between centers. By central review, 54% of evaluable patients (30/56) were classified as NEPC based on morphologic criteria(27); of these, 73%(22) were small cell carcinoma, 23%(7) neuroendocrine differentiation, and one mixed small cell carcinoma and adenocarcinoma. Eleven patients had a de novo presentation of small cell carcinoma of the prostate (ie, no known history of prior prostate adenocarcinoma).
The median duration of drug exposure was 7.0 weeks (IQR 5.3 – 14.1 weeks). For the primary endpoint, eight patients (13.4%) were progression free at six months (16.7% NEPC; 5.3% CRPC-Adeno). Eighteen patients on study (30%) had stable disease or better at cycle 3 scans, all of whom had demonstrated progression prior to starting alisertib; the majority (89%) of them had NEPC histology (clinical features summarized in Table S2). Overall median PFS was 2.2 months (95% CI 2.0– 2.6; 2.3 months, NEPC, 2.0 months, CRPC-Adeno) (Figures 1A, S1) and objective response rate was 3.3%. Exceptional responders were identified (Figures S2-5) and described below including two with complete resolution of liver metastases on therapy and two with prolonged stable disease (14 months and 3.8 years).
Figure 1:
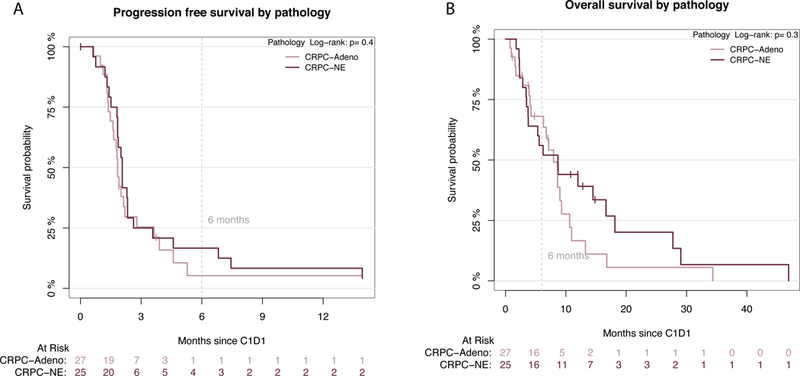
(A) Progression free survival and (B) Overall survival Kaplan Meir Curves based on pathology subtype by central pathology review (CRPC-Adeno= red; NEPC= blue). PFS and OS for the entire cohort is shown in Supplementary Figure 5.
Median OS was 9.5 months (95% CI 7.4–13.0) with no significant differences between NEPC and CRPC-Adeno (9.5 vs 8.6 months, respectively, p=0.28) (Figures 1B, S1). Forty-eight deaths were recorded on-study, most due to progressive disease. Median OS of patients with lymph node and/or bone-only metastases was 12.1 months compared with 9.4 months for those with visceral metastases (p=0.08).
Reasons for treatment discontinuation were disease progression (88%), adverse event (8%), or unrelated medical condition (3%). The toxicity profile was as expected based of prior trials of alisertib (Tables S3-4) with most common AEs being fatigue (78%), anorexia (45%), nausea (38%). Grade 3–4 neutropenia occurred in 13%, febrile neutropenia in 6.6%, grade 3–4 thrombocytopenia in 5%. The most common grade ≥3 non-hematologic toxicities were fatigue (10%), GI (10%), dehydration (5%). Dose reductions and/or delays were necessary in 37% patients. A similar proportion of NEPC and CRPC-Adeno patients reported treatment-related AEs.
Molecular analyses
Forty-nine patients had pre-treatment metastatic biopsies sufficient for whole exome sequencing (WES), 20 for both WES and RNA-seq, 25 for AURKA and MYCN FISH. Genomic analysis including purity and ploidy determined by CLONET(28), genomic burden, mutation rate, and differences between NEPC and CRPC-Adeno are shown in Figures 2and S6. Recurrent mutations and somatic copy number alterations included RB1 deletion (55%), TP53 mutation or deletion (46%), PTEN deletion (29%), BRCA2 mutation or deletion (29%). There was a lower frequency of AR amplification and mutations (29%) in this cohort compared to what has been reported in CRPC(29) and consistent with what we have previously reported in NEPC(4). As expected, expression of AR and AR-signaling genes was lower in NEPC and expression of NEPC-associated genes was higher in NEPC, but there was a spectrum within both pathologic subgroups (Figure S7). Overall 24% of evaluable cases harbored AURKA or MYCN amplification (16% AURKA (5), 16% MYCN (6), 8% concurrent (2)), lower than observed in prior studies of treatment related NEPC (21). Aurora kinase A (AURKA) amplification was associated with improved OS (p=0.05) but no difference in PFS (p=0.4) on alisertib (Figure S8, log-rank test). While N-myc (MYCN) amplification was not associated with outcomes across the cohort, responders were identified that demonstrated molecular features suggestive of MYCN overactivity.
Figure 2:
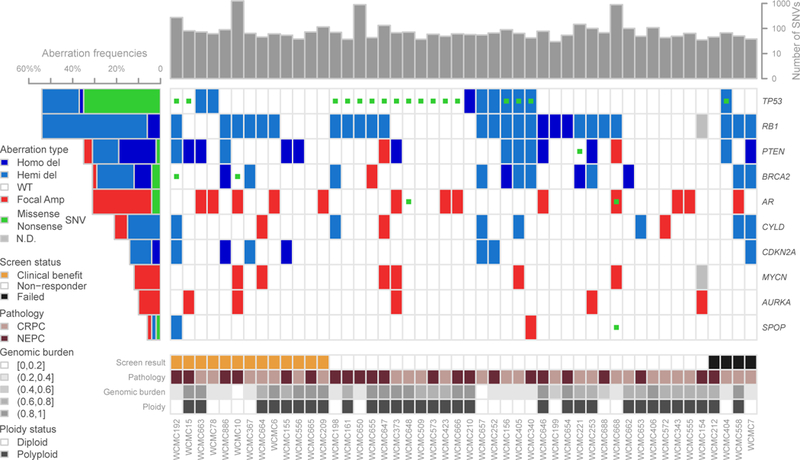
Whole exome sequencing analysis of the cohort annotated by pathology subtype (CRPC-Adeno= pink; NEPC= purple) using the same methodologies described in Beltran et al(4). Plot shows total number of SNVs, aberrations in relevant genes, including non-sense and missense SNVs, copy number deletions and focal amplifications. Copy number calls derive from log2 ratio (tumor/normal) adjusted by ploidy and tumor purity. Deletions and amplifications are defined using the thresholds on log2 ratio.
Patient 10 developed progressive bulky retroperitoneal and pelvic nodal (up to 7.5 cm) and new bone metastases within six months on primary androgen deprivation therapy (ADT) for metastatic prostate cancer, serum PSA 3ng/ml and chromogranin 1379 ng/ml (upper limit, 95). Lymph node biopsy revealed neuroendocrine morphology with diffuse staining for chromogranin and synaptophysin (Figure S1). The patient was treated with alisertib, which he tolerated well and was maintained on therapy with no evidence of progression at 3.8 years. His tumor demonstrated hyper-mutated phenotype, mutation of MSH6, as well as concurrent genomic amplification of both MYCN and AURKA (Figure S1) possibly contributing to his long-term durable response.
Patient 886 initially presented with de novo small cell carcinoma of the prostate with local invasion and extensive liver and lytic bone metastases, serum PSA 5.2 ng/ml. He was treated with carboplatin and etoposide without response (primary refractory) including progression in liver. He was started on alisertib and had complete resolution of metastases within three months (Figure S2). The patient was maintained on alisertib for 8.3 months in near CR. He subsequently developed parenchymal brain metastases without evidence of systemic relapse. His pre-treatment liver biopsy had demonstrated small cell carcinoma (Figure S2) with focal deletion of CDKN2A, CDKN2B, RB1, BRCA2, low AR signaling, elevated neuroendocrine markers (Figure S2). Although AURKA was not amplified and only 20% of tumor cells harbored MYCN amplification by FISH (Figure S2), there was significant AURKA and MYCN overexpression (Figure S3).
Patient 155 developed innumerable lung and liver metastases after twelve months of primary ADT for metastatic prostate cancer. Serum PSA was 0.2 ng/ml, chromogranin A was elevated at 1157 ng/ml, NSE 61.2 ug/L, CEA 128 ng/ml. Liver biopsy revealed small cell NEPC. He was treated with alisertib and within 12 weeks had improvement in symptoms and complete resolution of liver and lung metastases (Figure 3A). His CEA and serum neuroendocrine markers rapidly normalized (Figure 3B). Radiographic CR was maintained for 14 months. Therapy was then held for an intervening illness (gallstone cholecystitis); during this time, he developed rapid progression including brain metastases. WES of his pre-treatment and progression tumor biopsies as well as pre-treatment circulating tumor DNA (ctDNA) showed high degree of genomic similarity between the three samples (Figure 3C) suggesting minimal intra-patient tumoral heterogeneity in this patient. All three samples harbored focal loss of PTEN and CDKN2A. AURKA and MYCN were not amplified but were overexpressed. Large scale deletions involving FANCA, SMARCB1, and FBXW7 were identified. FBXW7 encodes the Fbox protein that regulates degradation of both Aurora A and N-myc by targeting them for ubiquitization(16,30). There was a similar clonality of mutations observed in his progression tumor, high NEPC score and low AR signaling, suggesting that after therapeutic release from alisertib, the tumor retained its original molecular profile (Figure 3D).
Figure 3:
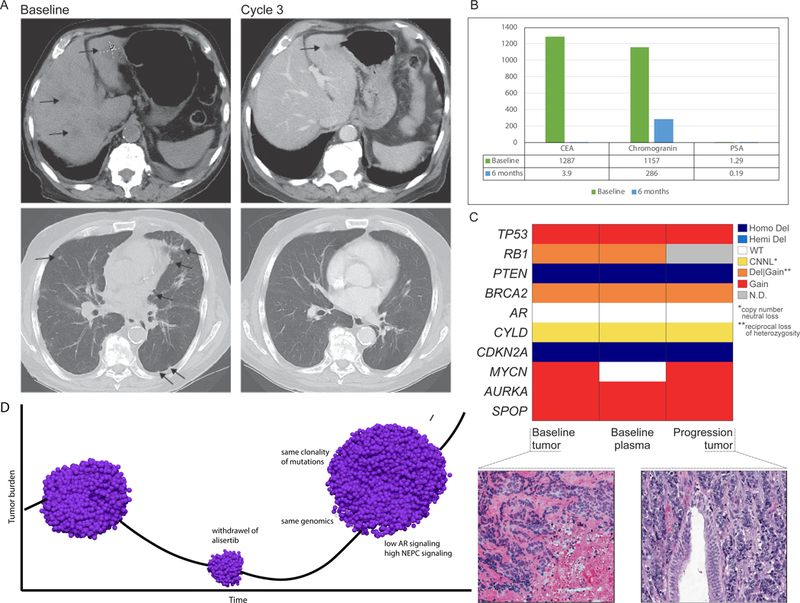
Exceptional responder patient 155 (A) CT scan at baseline (pre-treatment) and after cycle 3; (B) Serum chromogranin, CEA, PSA at baseline and after six months on alisertib; (C) Comparison of copy number of representative genes (determined by WES) showing concordance between baseline biopsy, baseline ctDNA, and progression biopsy; colors represent allele specific copy number states; hematoxylin and eosin (H&E) images showing pre-treatment baseline tumor biopsy and post-treated (progression) tumor both showing similar morphology (small cell carcinoma); (D) Schematic of patient 155 tumor response and progression after therapeutic release from alisertib (drug was held 17 days). pre-treatment biopsy: AR signaling score 0.04; NEPC score 0.5; progression tumor: AR signaling score 0.05, NEPC score 0.47.
A patient-derived organoid was developed from the pre-treatment biopsy of exceptional responder patient 155 and compared with another organoid (Figure 4A) developed from a bone biopsy from another patient on this alisertib trial (patient 154), who presented with small cell NEPC after prior ADT and platinum-etoposide with progressive bone, lung, and liver metastases; he did not respond to alisertib (progressed within 3 months). Both organoids were characterized histologically and molecularly(23), and were concordant with their matched patient tumor and AR negative by IHC (Figure 4A). The organoids were treated with alisertib in vitro and demonstrated directionally similar responses as observed in the clinic by their corresponding patients (Figures 4B, S9). Alisertib consistently disrupted the N-myc–Aurora A complex in control cell lines (Figure 4C, S10)(14) but was not capable of disrupting their allosteric interaction in 154 organoids (the non-responder patient) despite high baseline levels of N-myc-Aurora A. These data point to the potential utility of the N-myc –Aurora A proximal ligation assay as a functional readout of alisertib on–target effect and sensitivity.
Figure 4:
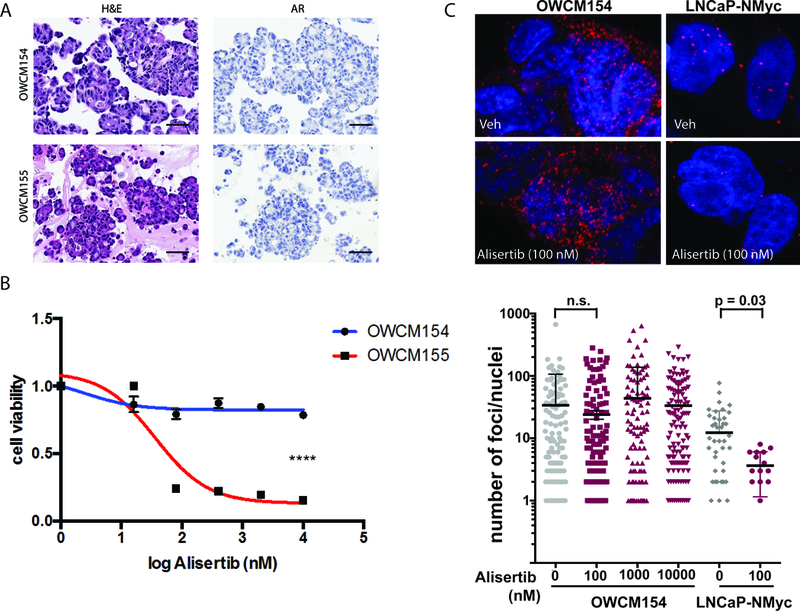
(A) Representative hematoxylin and eosin (H&E) images of tumor organoids derived from pre-treatment biopsies of patients 154 (liver) and 155 (bone) both showing small cell carcinoma, AR negative by immunohistochemistry (B) Cell viability graph using NEPC patient-derived organoids. A significant effect of alisertib is observed in OWCM155 (red) (IC50= 35.98nM), pvalue<0.0001 **** compared to the resistant NEPC organoid OWCM154 (blue). Organoids were treated with increasing doses of alisertib (range 16nM-10uM) for 6 days and viability was determined using CellTiter Glo (Promega). Statistical analysis was performed on three independent biological replicates using a non-linear regression curve fit (three parameters) method in Prism 6 for Mac Os X. (C) Proximal ligation assay (PLA) to measure the interaction between N-myc and Aurora A and quantify the amount of complex present in organoids using in situ probes(14). Shown here are representative confocal images of the N-Myc/Aurora A complex PLA visualized by discreet red dots in OWCM154 and LNCaP-N-Myc cells treated with 100 nM of alisertib or vehicle for 24 hours or 72 hours, respectively (top). Fixed cells were incubated with antibodies against N-Myc (dilution: 1/200; sc53993 (B8.4.B, N-Myc) and Aurora-A (dilution: 1/200; cell signaling 4718S), and interactions were revealed using secondary antibodies coupled to PLA DNA probes that hybridized and enzymatically joined when in close proximity. After rolling circle amplification, each interaction generated a fluorescent spot. (D) Confocal microscopy quantifications were carried out using BlobFinder software(33) using minimum nucleus size: 50 pixel2, Blob threshold: 12. Shown is a summary of the quantification of number of PLA dots calculated from OWCM154 and cells (number of nuclei evaluation per dose: n = 115, 0 nM; n = 164, 100 NM; n = 135, 1,000 nM; n = 137; 10,000 uM) or LNCaP-NMyc cells (number of nuclei evaluation per dose: n = 49, 0 nM; n = 16, 100 nM).
Discussion
It has become increasingly recognized that a subset of advanced prostate tumors evolve to become less dependent on the AR(2–4). AR-independence is challenging to recognize clinically and neuroendocrine features may not always be present on tumor biopsy. Recent evidence suggests that NEPC arises clonally from prostate adenocarcinoma cells during the course of therapy through the acquisition of genomic and epigenomic alterations(4,31), yet the clinical and molecular features and timing of these events are not well defined. In this trial, we sought to address some of these challenges by establishing specific inclusion criteria to stratify the heterogeneously defined entity of NEPC from both a clinical and pathologic standpoint. While we did not expect all patients with clinical criteria to demonstrate pathologic features of NEPC, we posited that these aggressive tumors would share molecular features and therapy responsiveness with NEPC. The goal of establishing multiple inclusion cohorts was in line with PCWG3 objectives to identify which patient features are most likely to benefit by any criteria (eg., clinical, radiologic, biologic) as a strategy to inform future trials(32).
Collectively, the trial enrolled patients with aggressive disease including those previously treated with platinum chemotherapy. Median PSA on this study was low at 1.13 ng/ml supportive of less AR-driven disease. We learned that the genomic profiles, range of AR signaling, and expression of neuroendocrine markers varied, which may reflect limitations of single site biopsy and/or a biologic spectrum of CRPC, with initial retention of the AR and expression of luminal prostate markers even in tumors that start to lose AR- dependence(5,6,14). Although the study did not meet its primary endpoint, significant responders were identified with molecular alterations potentially contributing to response. The overall lack of strong signal of alisertib response across the cohort may reflect challenges of underlying heterogeneity, context with other drivers of CRPC, and/or the mechanism of action of alisertib that may not be inhibiting N-myc sufficiently.
Responders on the study included those with extensive visceral metastases and platinum refractory disease. Notably, one of the highlighted exceptional responders to alisertib had a de novo presentation of small cell prostate cancer with a clinical presentation of visceral and lytic bone metastases. These data suggest that there may be similarities between this subgroup and the more common treatment related NEPC, and further study of the role of N-myc and Aurora A in de novo small cell prostate cancer is warranted. Alisertib has also been investigated in other de novo neuroendocrine tumors including small cell lung cancer and neuroblastoma with the cooperation of Aurora A and Myc family transcription factors(8).
The results from exceptional responder patient 155 highlight the impact of intra-patient tumor consistency (or lack of heterogeneity) in mediating therapeutic response to a targeted therapy. The considerable similarity of the whole exome sequence of pre-treatment and progression biopsies and circulating tumor DNA suggests shared vulnerabilities across tumor metastases potentially contributing toward the patient’s dramatic clinical response. Upon release of the therapeutic pressure of alisertib, the patient developed rapid progression, with tumor transcriptome changes associated with NEPC progression and a maintained genomic profile, suggesting that that the patient may have responded to re-treatment had he been well enough to do so.
Given the heterogeneity within this clinically enriched trial subgroup, future trials for non-AR driven disease should be aimed at molecularly-defined inclusion criteria. This is especially important as distinct subclasses of prostate cancer are biologically characterized, including not only NEPC but also other AR-independent subtypes such as double negative (AR-negative, NE-negative) CRPC(2). Understanding the spectrum and chronology of the molecular events underlying disease progression will allow us to identify patient subsets based on their underlying resistance patterns and will pave the way for non-invasive testing (eg., circulating tumor DNA or circulating tumor cell profiles), as metastatic biopsies are invasive and remain challenging to perform serially in patients.
The development of patient-derived organoids from patients on early phase clinical trials represents a unique approach for patient interrogation. We have demonstrated how organoid models maintain the genomic profiles of their corresponding tumor biopsies(23) and here show how they recapitulate the drug responses of the patients, and therefore may be useful tools as patient ‘avatars’ for co-clinical trials and as a resource to inform drug and biomarker development. In prostate cancer, this is especially relevant as there are few cell line models and only one bona-fide NEPC cell line (the NCI-H660 cell line) widely available for preclinical studies. Such models may be used to study dynamic biomarkers to evaluate pharmacodynamic effects of drug such as the PLA assay described in this study, which could be applicable for future preclinical studies and trials using alisertib or other inhibitors of the AURKA/MYCN complex to define the most appropriate patients moving forward.
Cancers may adopt different means to evade therapy and evolve in the face of pressure from a targeted therapy. Further investigation of targeting N-myc and the Aurora A- N-myc complex for patients with AR-independent NEPC is warranted but biomarker selection will be critical.
Supplementary Material
Statement of Translational Relevance:
A subset of prostate cancers evade androgen receptor (AR)- targeted therapies through the development of lineage plasticity, neuroendocrine features, and loss of AR-signaling dependence, with prior translational data pointing to N-myc and Aurora kinase A as key targets. In this Phase 2 trial, clinically enriched for patients with AR-independent and neuroendocrine prostate cancer, we investigated the efficacy of alisertib, a drug that inhibits the allosteric interaction between N-myc and Aurora-A. Exceptional responders were identified and characterized, suggesting that some but not all patients with NEPC do benefit from alisertib. Pre-treatment biopsies were performed in all patients to understand the clinical, pathologic, and molecular features underlying response to alisertib and to characterize the molecular characteristics of this aggressive subgroup of prostate cancer.
Acknowledgements:
We are grateful to our patients and their families for participation in this study. We appreciate all the work of the site coordinators, data managers, and investigators. In particular, we would like to acknowledge Irene Karpenko, Gillian Hodes, Olivera Calukovic, Michael Sigouros, Danielle Pancirer, Jessica Padilla, Theresa MacDonald, and Terra McNary for their help with regulatory, tissue, and data coordination for this study. This work was supported by Millennium Pharmaceuticals, Inc and the Prostate Cancer Foundation. Additional funding sources include the Department of Defense PC131961 (D.M.N., S.T.T., H.B.), PC121341 (H.B.), PC160264 (H.B., D.R.), PC121111 (H.I.S.), PC131984 (H.I.S); NIH/NCI SPORE in Prostate Cancer P50-CA211024 (H.B., J.M.M., M.A.R., D.R.) and P50-CA92629 (H.I.S.); NIH/NCI Cancer Center Support Grant P30-CA008748 (H.I.S.); and the Ann and William Bresnan Foundation (H.B., D.M.N). The molecular data for this study is accessible at dbGaP phs001666.v1.p1
Footnotes
Disclosures: Weill Cornell Medicine received funding from Millennium Pharmaceuticals, Inc to conduct this study. The authors declare no other conflicts of interest.
References
- 1.Watson PA, Arora VK, Sawyers CL. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat Rev Cancer 2015;15(12):701–11 doi 10.1038/nrc4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bluemn EG, Coleman IM, Lucas JM, Coleman RT, Hernandez-Lopez S, Tharakan R, et al. Androgen Receptor Pathway-Independent Prostate Cancer Is Sustained through FGF Signaling. Cancer Cell 2017;32(4):474–89.e6 doi 10.1016/j.ccell.2017.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aggarwal R, Huang J, Alumkal JJ, Zhang L, Feng FY, Thomas GV, et al. Clinical and Genomic Characterization of Treatment-Emergent Small-Cell Neuroendocrine Prostate Cancer: A Multi-institutional Prospective Study. J Clin Oncol 2018:JCO2017776880 doi 10.1200/JCO.2017.77.6880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med 2016;22(3):298–305 doi 10.1038/nm.4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mu P, Zhang Z, Benelli M, Karthaus WR, Hoover E, Chen CC, et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 2017;355(6320):84–8 doi 10.1126/science.aah4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ku SY, Rosario S, Wang Y, Mu P, Seshadri M, Goodrich ZW, et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 2017;355(6320):78–83 doi 10.1126/science.aah4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beltran H, Tagawa ST, Park K, MacDonald T, Milowsky MI, Mosquera JM, et al. Challenges in recognizing treatment-related neuroendocrine prostate cancer. J Clin Oncol 2012;30(36):e386–9 doi 10.1200/JCO.2011.41.5166. [DOI] [PubMed] [Google Scholar]
- 8.Rickman DS, Beltran H, Demichelis F, Rubin MA. Biology and evolution of poorly differentiated neuroendocrine tumors. Nat Med 2017;23(6):1–10 doi 10.1038/nm.4341. [DOI] [PubMed] [Google Scholar]
- 9.Aparicio AM, Harzstark AL, Corn PG, Wen S, Araujo JC, Tu SM, et al. Platinum-based chemotherapy for variant castrate-resistant prostate cancer. Clin Cancer Res 2013;19(13):3621–30 doi 10.1158/1078-0432.CCR-12-3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aparicio AM, Shen L, Tapia EL, Lu JF, Chen HC, Zhang J, et al. Combined Tumor Suppressor Defects Characterize Clinically Defined Aggressive Variant Prostate Cancers. Clin Cancer Res 2016;22(6):1520–30 doi 10.1158/1078-0432.CCR-15-1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beltran H, Tomlins S, Aparicio A, Arora V, Rickman D, Ayala G, et al. Aggressive variants of castration-resistant prostate cancer. Clin Cancer Res 2014;20(11):2846–50 doi 10.1158/1078-0432.CCR-13-3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang HT, Yao YH, Li BG, Tang Y, Chang JW, Zhang J. Neuroendocrine Prostate Cancer (NEPC) progressing from conventional prostatic adenocarcinoma: factors associated with time to development of NEPC and survival from NEPC diagnosis-a systematic review and pooled analysis. J Clin Oncol 2014;32(30):3383–90 doi 10.1200/JCO.2013.54.3553. [DOI] [PubMed] [Google Scholar]
- 13.Beltran H, Rickman DS, Park K, Chae SS, Sboner A, MacDonald TY, et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov 2011;1(6):487–95 doi 10.1158/2159-8290.CD-11-0130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dardenne E, Beltran H, Benelli M, Gayvert K, Berger A, Puca L, et al. N-Myc Induces an EZH2-Mediated Transcriptional Program Driving Neuroendocrine Prostate Cancer. Cancer Cell 2016;30(4):563–77 doi 10.1016/j.ccell.2016.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee JK, Phillips JW, Smith BA, Park JW, Stoyanova T, McCaffrey EF, et al. N-Myc Drives Neuroendocrine Prostate Cancer Initiated from Human Prostate Epithelial Cells. Cancer Cell 2016;29(4):536–47 doi 10.1016/j.ccell.2016.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Otto T, Horn S, Brockmann M, Eilers U, Schüttrumpf L, Popov N, et al. Stabilization of N-Myc is a critical function of Aurora A in human neuroblastoma. Cancer Cell 2009;15(1):67–78 doi 10.1016/j.ccr.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 17.Brockmann M, Poon E, Berry T, Carstensen A, Deubzer HE, Rycak L, et al. Small molecule inhibitors of aurora-a induce proteasomal degradation of N-myc in childhood neuroblastoma. Cancer Cell 2013;24(1):75–89 doi 10.1016/j.ccr.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scher HI, Halabi S, Tannock I, Morris M, Sternberg CN, Carducci MA, et al. Design and end points of clinical trials for patients with progressive prostate cancer and castrate levels of testosterone: recommendations of the Prostate Cancer Clinical Trials Working Group. J Clin Oncol 2008;26(7):1148–59 doi 10.1200/JCO.2007.12.4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Magi A, Tattini L, Cifola I, D’Aurizio R, Benelli M, Mangano E, et al. EXCAVATOR: detecting copy number variants from whole-exome sequencing data. Genome Biol 2013;14(10):R120 doi 10.1186/gb-2013-14-10-r120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beltran H, Wyatt AW, Chedgy EC, Donoghue A, Annala M, Warner EW, et al. Impact of Therapy on Genomics and Transcriptomics in High-Risk Prostate Cancer Treated with Neoadjuvant Docetaxel and Androgen Deprivation Therapy. Clin Cancer Res 2017;23(22):6802–11 doi 10.1158/1078-0432.CCR-17-1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mosquera JM, Beltran H, Park K, MacDonald TY, Robinson BD, Tagawa ST, et al. Concurrent AURKA and MYCN gene amplifications are harbingers of lethal treatment-related neuroendocrine prostate cancer. Neoplasia 2013;15(1):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gao D, Vela I, Sboner A, Iaquinta PJ, Karthaus WR, Gopalan A, et al. Organoid cultures derived from patients with advanced prostate cancer. Cell 2014;159(1):176–87 doi 10.1016/j.cell.2014.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Puca L, Bareja R, Prandi D, Shaw R, Benelli M, Karthaus WR, et al. Patient derived organoids to model rare prostate cancer phenotypes. Nat Commun 2018;9(1):2404 doi 10.1038/s41467-018-04495-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meulenbeld HJ, Bleuse JP, Vinci EM, Raymond E, Vitali G, Santoro A, et al. Randomized phase II study of danusertib in patients with metastatic castration-resistant prostate cancer after docetaxel failure. BJU Int 2013;111(1):44–52 doi 10.1111/j.1464-410X.2012.11404.x. [DOI] [PubMed] [Google Scholar]
- 25.Rathkopf DE, Antonarakis ES, Shore ND, Tutrone RF, Alumkal JJ, Ryan CJ, et al. Safety and Antitumor Activity of Apalutamide (ARN-509) in Metastatic Castration-Resistant Prostate Cancer with and without Prior Abiraterone Acetate and Prednisone. Clin Cancer Res 2017;23(14):3544–51 doi 10.1158/1078-0432.CCR-16-2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.A’Hern RP. Sample size tables for exact single-stage phase II designs. Stat Med 2001;20(6):859–66 doi 10.1002/sim.721. [DOI] [PubMed] [Google Scholar]
- 27.Epstein JI, Amin MB, Beltran H, Lotan TL, Mosquera JM, Reuter VE, et al. Proposed morphologic classification of prostate cancer with neuroendocrine differentiation. Am J Surg Pathol 2014;38(6):756–67 doi 10.1097/PAS.0000000000000208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Prandi D, Baca SC, Romanel A, Barbieri CE, Mosquera JM, Fontugne J, et al. Unraveling the clonal hierarchy of somatic genomic aberrations. Genome Biol 2014;15(8):439 doi 10.1186/s13059-014-0439-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015;161(5):1215–28 doi 10.1016/j.cell.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kwon YW, Kim IJ, Wu D, Lu J, Stock WA, Liu Y, et al. Pten regulates Aurora-A and cooperates with Fbxw7 in modulating radiation-induced tumor development. Mol Cancer Res 2012;10(6):834–44 doi 10.1158/1541-7786.MCR-12-0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zou M, Toivanen R, Mitrofanova A, Floch N, Hayati S, Sun Y, et al. Transdifferentiation as a Mechanism of Treatment Resistance in a Mouse Model of Castration-Resistant Prostate Cancer. Cancer Discov 2017;7(7):736–49 doi 10.1158/2159-8290.CD-16-1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scher HI, Morris MJ, Stadler WM, Higano C, Basch E, Fizazi K, et al. Trial Design and Objectives for Castration-Resistant Prostate Cancer: Updated Recommendations From the Prostate Cancer Clinical Trials Working Group 3. J Clin Oncol 2016;34(12):1402–18 doi 10.1200/JCO.2015.64.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Allalou A, Wählby C. BlobFinder, a tool for fluorescence microscopy image cytometry. Comput Methods Programs Biomed 2009;94(1):58–65 doi 10.1016/j.cmpb.2008.08.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.