

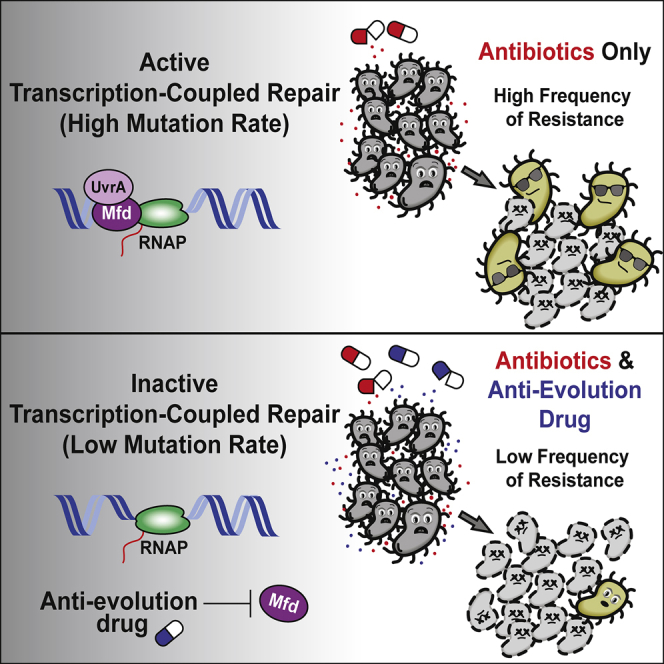
Summary
Efforts to battle antimicrobial resistance (AMR) are generally focused on developing novel antibiotics. However, history shows that resistance arises regardless of the nature or potency of new drugs. Here, we propose and provide evidence for an alternate strategy to resolve this problem: inhibiting evolution. We determined that the DNA translocase Mfd is an “evolvability factor” that promotes mutagenesis and is required for rapid resistance development to all antibiotics tested across highly divergent bacterial species. Importantly, hypermutator alleles that accelerate AMR development did not arise without Mfd, at least during evolution of trimethoprim resistance. We also show that Mfd’s role in AMR development depends on its interactions with the RNA polymerase subunit RpoB and the nucleotide excision repair protein UvrA. Our findings suggest that AMR development can be inhibited through inactivation of evolvability factors (potentially with “anti-evolution” drugs)—in particular, Mfd—providing an unexplored route toward battling the AMR crisis.
Keywords: antimicrobial resistance, antibiotic resistance, transcription-coupled repair, Mfd, evolution, Mycobacteria, hypermutator, anti-evolution
Graphical Abstract
Highlights
-
•
The bacterial transcription-coupled repair (TCR) factor Mfd promotes mutagenesis
-
•
Mfd-driven mutagenesis accelerates the evolution of antimicrobial resistance (AMR)
-
•
The rapid evolution of AMR requires Mfd’s interaction with RpoB and UvrA
-
•
Mfd may be an ideal target for “anti-evolution” drugs that inhibit AMR development
Bacterial evolution drives antimicrobial resistance (AMR) development. We identify the protein Mfd as a highly conserved “evolvability factor” that increases mutagenesis and the capacity of bacteria to evolve antibiotic resistance. We propose inhibiting the activity of evolvability factors through “anti-evolution” drugs during antibiotic treatment to ameliorate the global AMR crisis.
Introduction
The battle between antimicrobial-resistant pathogens and antibiotic therapy is an evolutionary arms race—one that we are currently losing. Consequently, antimicrobial resistance (AMR)-related deaths have reached alarming rates throughout the world. Estimates suggest that at least 700,000 people die annually from drug-resistance infections; this number could rise to 10 million by 2050, far surpassing cancer as the major cause of death worldwide (O’Neill, 2014). Most efforts to resolve AMR are geared toward the development of novel antibiotics, yet resistance has arisen to every antibiotic used in the clinic. Innovative strategies to reduce the rise of drug-resistant pathogens are therefore a necessary public health concern.
For many pathogens and antibiotic classes, de novo mutations play a critical role in AMR development. For example, in the case of Mycobacterium tuberculosis (Mtb), the causative agent of tuberculosis (TB), AMR acquisition arises exclusively through chromosomal mutations (Almeida Da Silva and Palomino, 2011). Given the alarming global burden of TB drug resistance in addition to the rise of chromosomally acquired AMR in many other pathogens, reducing the mutational capacity of organisms could significantly inhibit their ability to develop AMR. This approach requires the identification and subsequent inhibition of active factors that increase mutation rates. We term these proteins “evolvability factors” given that they can promote evolution by increasing mutation rates (either directly or indirectly).
The DNA translocase protein Mfd is highly conserved across bacterial phyla, suggesting that it plays an important physiological role in cells. Like its functional analog CSB in humans, Mfd’s main function has long been thought to be in the initiation of nucleotide excision repair (NER—which repairs bulky lesions on DNA) at sites of stalled RNA polymerases (RNAP) (Hanawalt and Spivak, 2008). This mechanism is referred to as transcription-coupled repair (TCR). Comprehensive biochemical studies have provided insight into the various functions of Mfd, including its role in the recruitment of NER proteins to regions of stalled RNAP. Curiously though, cells lacking Mfd do not display increased sensitivity to DNA-damaging agents (Cohen et al., 2010, Epshtein et al., 2014, Kamarthapu et al., 2016, Witkin, 1966b, Witkin, 1969; Figure S1). Furthermore, overexpression of Mfd sensitizes cells to DNA damage (Kamarthapu et al., 2016). Moreover, even though Mfd is canonically known to promote DNA repair, it paradoxically increases mutagenesis in certain contexts, such as at regions of replication-transcription conflicts and in stationary-phase mutagenesis (Han et al., 2008, Lee et al., 2009, Million-Weaver et al., 2015, Martin et al., 2011, Ross et al., 2006, Wimberly et al., 2013). These findings can be interpreted in at least three different (but not mutually exclusive) ways: (1) redundant TCR mechanisms exist (also proposed by Kamarthapu et al., 2016), (2) Mfd may actually inhibit DNA repair in some contexts (also proposed by Pani and Nudler, 2017), and (3) Mfd may promote DNA repair, but this repair is mutagenic in the absence of exogenous DNA damage (e.g., Million-Weaver et al., 2015).
Mfd may have additional functions outside of TCR. Recently, Mfd was found to associate with RNAP even in the absence of exogenous DNA damage (Ho et al., 2018, Le et al., 2018), suggesting that it may play a more general housekeeping role during transcription elongation. Furthermore, Mfd acts as an RNAP anti-backtracking factor and therefore could be critical for RNAP processivity. Mfd’s anti-backtracking activity also alleviates genomic instability caused by collisions between replication and transcription elongation complexes (Dutta et al., 2011).
Here, we identify Mfd as an evolvability factor, the absence of which hinders antibiotic resistance development. We show that Mfd promotes mutagenesis in bacteria both during laboratory growth and during infection of eukaryotic cells. Our experiments show that the Mfd-dependent increase in mutagenesis accelerates AMR development and that this holds true for multiple classes of antibiotics. We also find that Mfd promotes the evolution of hypermutation, one important mechanism known to lead to rapid AMR development. Importantly, our findings show that the role of Mfd in AMR development is highly conserved across bacteria, including several clinically relevant pathogens. Finally, we pinpoint critical regions of Mfd that are required for its evolvability function. Specifically, we show that the interactions of Mfd with the RNA polymerase beta subunit RpoB as well as the NER protein UvrA are required for its role in the rapid evolution of resistance to several classes of antibiotics. Altogether, these results provide evidence that blocking evolvability factors—in particular, Mfd—can inhibit resistance development in a diverse array of bacterial pathogens.
Results
Mfd Is a Mutagenic Factor in Divergent Bacterial Species
The role of Mfd in DNA repair has remained controversial: cells lacking Mfd are not sensitive to DNA-damaging agents and previous work hints at a mutagenic role for Mfd in specific contexts. We decided to thoroughly examine Mfd’s role in mutagenesis, specifically in the absence of exogenous DNA damage. We measured mutation rates with and without Mfd in divergent bacterial species using Luria-Delbrück fluctuation analysis (Luria and Delbrück, 1943). We observed that strains lacking Mfd had a 2- to 5-fold decrease in mutation rates as measured by rifampicin resistance compared to wild-type (WT) strains (Figure 1). This decreased mutation rate was conserved between Gram-negative and Gram-positive species, including Bacillus subtilis and clinical isolates of Salmonella typhimurium (Hayden et al., 2016) and Pseudomonas aeruginosa (Wolfgang et al., 2003; Figure 1A). These results are in contrast to Mfd’s previously published anti-mutagenic properties during UV exposure (Selby and Sancar, 1993, Witkin, 1966a).
Figure 1.

Mfd Promotes Mutagenesis in Diverse Bacterial Species, Related to Figures S1 and S5
(A) Mutation rates of WT (black) and Δmfd (gray) strains to rifampicin for three indicated species (Bs, B. subtilis HM1; Pa, P. aeruginosa CF127; St, S. typhimurium ST19). Number of replicates for Bs = 75, Pa = 42, St = 36. Error bars are 95% confidence intervals.
(B) Mutation rates of Mtb (H37Rv) to three different antibiotics for WT (black) and Δmfd (gray). Number of replicates for Mtb = 33–48. Error bars are 95% confidence intervals. ∗Ciprofloxacin y-axis is mutations per 108 cells per generation.
(C) Mutation frequency of S. typhimurium in culture tubes and during infection of CACO-2 cells. Frequency was measured by plating on M9 glycerol with 5-flourocytosine for CFU enumeration. Error bars are standard error of the mean. Two-tailed Student’s t test determined statistical significance (∗∗p value < 0.01, ∗∗∗p value < 0.001).
(D) CFU enumeration of WT and Δmfd S. typhimurium strains upon infection of CACO-2 cells.
Chromosomal mutations are the sole means by which AMR develops in Mtb (Almeida Da Silva and Palomino, 2011). Differences in mutation rates between clinical isolates of Mtb are thought to underlie AMR development (Ford et al., 2013). Therefore, if conserved, Mfd-driven mutagenesis could have a significant impact on the development of AMR in Mtb. Indeed, when we deleted the gene encoding Mfd, mutation rates in Mtb were reduced by roughly 2- to 3-fold (Figure 1B), as measured by resistance to three different antimicrobials frequently used to treat tuberculosis: rifampicin, ethambutol, and ciprofloxacin. This suggests that Mfd promotes mutagenesis across different resistance loci in Mtb and is likely critical for the development of antibiotic resistance.
Mfd’s Mutagenic Function Is Conserved during Infection of Eukaryotic Cells
We wanted to determine if the mutagenic effects of Mfd are conserved in an infection model of drug resistance. For these experiments, we infected CACO-2 epithelial cells with a clinical isolate of S. typhimurium and subsequently measured mutation frequency using resistance to 5-fluorocytosine (Richardson et al., 2009). Interestingly, compared to the ∼2- to 4-fold decrease observed during laboratory growth (Figure 1C, left), we see a ∼5-fold decrease in mutagenesis in the absence of Mfd upon host cell infection (Figure 1C, right). These differences are not related to growth defects during infection of host cells, as there is no change in the number of colony-forming units following infection (Figure 1D). Therefore, the effect of Mfd-mediated mutagenesis is both conserved and potentially enhanced during growth and replication in the host.
Mfd Accelerates AMR Development
We next assessed the impact of Mfd on both the kinetics and the levels of AMR development in short-term evolution experiments in the Gram-negative pathogen S. typhimurium. Given that the differences in mutation rates between WT and cells lacking Mfd were modest (2- to 5-fold), we wondered if these differences could impact the kinetics and evolution of resistance in a meaningful way. To test this model, we developed an assay that measures both metrics over roughly 35 to 70 generations in the presence of antibiotics (ranging from sub-inhibitory to ∼16x MIC for our first time course). We monitored the evolution of S. typhimurium resistance to a panel of clinically relevant antibiotics (rifampicin, phosphomycin, trimethoprim, kanamycin, and vancomycin), which act through different mechanisms and have different resistance loci. We found that resistance to all the antibiotics tested arose significantly faster and to higher levels in WT compared to the Δmfd strain (Figures 2A–2E). The difference in the median resistance levels between the two strains at the end of the S. typhimurium evolution experiments was 6-to 21-fold greater in WT than in Δmfd. These results were not specific to S. typhimurium: we found a ∼32 fold difference in median antibiotic concentration tolerated by WT compared to Δmfd cells in the highly divergent, Gram-positive bacterium B. subtilis (Figure 2F). These findings show that Mfd promotes resistance development in diverse bacterial species.
Figure 2.

Mfd Promotes Evolution to Various Classes of Antibiotics, Related to Figure S2, S3, and Table S1
Evolution of S. typhimurium ST19 to (A) rifampicin, (B) phosphomycin, (C) trimethoprim, (D) kanamycin, and (E) vancomycin; evolution of B. subtilis HM1 to (F) rifampicin. Heatmaps and line plots show median antibiotic concentration for WT and Δmfd strains at each sampled time point. Black bars represent median growth greater than highest concentration shown on the scale. Concentrations for all antibiotics are in μg/mL. Statistical significance was determined using a two-tailed Mann-Whitney U test (∗p value < 0.05, ∗∗p value < 0.01, ∗∗∗p value < 0.001). Number of replicates for each strain and antibiotic of St and Bs are 12–30.
Mfd Is Critical for the Development of AMR in Mtb
Mtb is arguably the most difficult-to-treat pathogen due to AMR development. Therefore, we were interested in determining whether Mfd is responsible for the evolution of resistance in this pathogen. We adapted our short-term evolution assays to the unique culture conditions of Mtb and performed the evolution experiments using rifampicin as a representative antibiotic. The difference in median resistance to rifampicin between the two strains at the end of the experiment was striking: the median resistance level to rifampicin in WT was in some experiments up to 1,000-fold greater than Δmfd strains (Figure 3A). This difference is significantly greater than that observed for S. typhimurium or B. subtilis. Additionally, we find that by the end of our evolution assays, roughly 2/3 of our evolved WT strains were above the clinical MIC breakpoint of Mtb to rifampicin (1 mg/L) (Schon et al., 2009), whereas none of the Δmfd strains reached this threshold. These data suggest that, as observed in other species, Mfd is critical in the development of AMR in Mtb—a finding with potential clinical implications.
Figure 3.

Mfd Promotes Evolution to Antibiotics in Mtb
(A) Evolution of Mtb H37Rv to rifampicin. Heatmaps and line plots showing median rifampicin concentration for WT and Δmfd strains at each sampled passage from a representative experiment are shown. Black bars represent median growth greater than highest concentration shown on the scale. Concentrations are in ng/mL. Statistical significance was determined using a two-tailed Mann-Whitney U test (∗p value < 0.10). Number of replicates for each strain of Mtb is 6.
(B) Mtb Mfd and ST19 RpoB interact. Mtb Mfd RNAP interacting domain (RID) and S. typhimurium ST19 RpoB N-terminal domain were cloned into a luciferase-based bacterial 2-hybrid system. Interactions between these respective protein domains were measured by luminescence and normalized to OD600. Results are from three independent experiments, and error bars indicate standard error of the mean. Statistical significance was determined using two-tailed Student’s t test (∗∗p value < 0.01).
(C) Mutation rate analyses were performed with indicated strains of S. typhimurium to rifampicin as in Figure 1. Number of replicates is 36–96.
(D) Evolution of indicated strains of S. typhimurium to rifampicin. Plots and statistical testing for evolution assays were performed as described in Figure 2. Number of replicates per strain is 12–24. ∗p value < 0.05 between WT and Δmfd strains and ∗∗p value < 0.01 between Δmfd::Mtb-mfd and Δmfd strains.
The Evolvability Function of Mfd Can Be Cross-complemented between Divergent Species
The data presented above suggest that Mfd’s role in AMR development is conserved across species. To test the degree of conservation, we first performed bacterial 2-hybrid assays to determine if Mfd’s well-documented interaction with RpoB can be detected between S. typhimurium and Mtb proteins. We chose to test these species for our experiments because they are highly divergent. Furthermore, although minimal, these two species have the biggest difference in the Mfd sequence at the amino acid level. We found that S. typhimurium RpoB interacts with the Mtb Mfd-RNAP interaction domain (RID) (Figure 3B). These data suggest that the mechanism by which Mfd promotes the evolution of AMR could be conserved across these species.
We then performed cross-complementation experiments using S. typhimurium and Mtb as models (Figures 3C and 3D). We introduced a copy of the Mtb mfd gene into S. typhimurium strains lacking mfd and performed both mutation rate and evolution assays to rifampicin. Strikingly, the Mtb mfd gene fully complemented the reduced mutation rates (Figure 3C) as well as the delayed evolution of Δmfd S. typhimurium resistance to rifampicin (Figure 3D). These results indicate that the mechanism facilitating the evolvability function of Mfd is highly conserved across bacterial species.
Mfd Promotes Evolution by Increasing Mutagenesis
To determine if Mfd promotes AMR development through its mutagenic properties, we used Sanger sequencing to identify mutations that arose within the known rifampicin and trimethoprim resistance genes (rpoB [Brandis et al., 2015] and folA [Watson et al., 2007], respectively) during our evolution experiments. Analysis of the sequences obtained from every time point for 12 different replicates in both S. typhimurium WT and Δmfd strains revealed several resistance mutations. However, the Δmfd replicates consistently accumulated roughly 1/2 of the number of mutations in rpoB and 1/3 of the number of mutations in folA compared to WT (Figures S2A and S2B). Importantly, we observed a significant delay in the acquisition of mutations in the Δmfd strains compared to those in WT and rarely observed additional second and third mutations in the Δmfd strains (Figures S2A and S2B). These data strongly suggest that Mfd promotes the evolution of resistance to antibiotics through its pro-mutagenic function and that it may be critical for the acquisition of multiple mutations.
Mfd Promotes the Rise of Hypermutators
To determine if there were any mutations outside of the resistance loci in WT compared to Δmfd strains, we performed whole-genome sequencing (WGS) of six randomly chosen replicates from our rifampicin and trimethoprim evolution experiments. WGS of six WT and Δmfd S. typhimurium isolates from every time point of our rifampicin and trimethoprim evolution assays confirmed that, compared to our WT strain, Δmfd strains accumulated significantly fewer mutations in the rpoB locus, as we had observed using Sanger sequencing (Figures S2A and S2B). We did not find any additional mutations outside of the rpoB gene in any of the evolved rifampicin-resistant strains that we sequenced. In contrast, our WGS of strains evolved in trimethoprim revealed the presence of additional mutations outside of the coding region, within the putative promoter region of folA. All six sequenced WT strains contained one of two putative promoter mutations (either 35 or 61 base pairs upstream of the folA coding sequence), while only one of our sequenced Δmfd isolates carried one of these mutations (Table S1).
Interestingly, we found that three out of six WT sequenced trimethoprim-evolved strains contained a point mutation in the dnaQ gene (all strains had the same dnaQ(I33N) mutation), while none of the Δmfd strains contained any mutations in the dnaQ gene (Table S1). Mutations in dnaQ are known to generate hypermutator phenotypes (Echols et al., 1983), so, to determine if this new allele indeed conferred a hypermutator phenotype, we performed Luria-Delbrück fluctuation analysis of an evolved WT strain before and after gaining the identified dnaQ mutation. We found that the mutation rate upon gaining this dnaQ allele was ∼1,000-fold higher than the ancestor strain (Figure S3). We subsequently performed Sanger sequencing of the dnaQ allele on four additional WT and Δmfd strains and found that two out of four WT strains contained the same dnaQ mutation, while none of the four Δmfd strains contained this mutation. Overall, we can estimate that roughly 50% of WT strains developed hypermutator alleles during the evolution of trimethoprim resistance, while strains lacking Mfd are restrained in developing this phenotype (we did not find a hypermutator Δmfd isolate).
As expected, we found that WT S. typhimurium isolates carrying the dnaQ hypermutator allele accumulated a high number of mutations across the genome (up to 600 in some of our evolved isolates), including mutations that may confer an adaptive advantage in the presence of trimethoprim. These mutations should be examined further to discern true adaptive mutations from hitchhiker mutations. These potentially adaptive mutations are in genes previously implicated in promoting trimethoprim resistance (Baym et al., 2016), such as aroK (involved in the shimikate pathway for folate synthesis) (Table S1). We also found putative adaptive mutations in genes not previously associated with trimethoprim resistance. These mutations arose in genes such as fis (DNA binding and regulator of replication initiation and global transcription), pyrG (CTP synthetase), ygdP (RNA pyrophosphohydrolase), and ybgC (Acyl-CoA thioester hydrolase), among many others (Table S1). Mutations in many of these genes arose in independent lineages, suggesting that they may confer adaption to trimethoprim. These findings show that, in our evolution assays (as in clinical settings), the generation of hypermutation may offer an adaptive strategy to evolve high-level antibiotic resistance and that Mfd promotes this phenomenon.
Mfd-Mediated Evolution Requires Its Interaction with RNAP and UvrA
To test whether the evolvability function of Mfd depended on transcription and its conserved interaction with RpoB, we constructed an L499R mutation in the RNAP interaction domain (RID) of S. typhimurium Mfd. The Mfd L499R mutant was previously characterized in E. coli and was shown to alter Mfd-RpoB interaction without affecting Mfd’s DNA binding and ATPase activity (Deaconescu et al., 2006). Our bacterial 2-hybrid assays confirmed that disrupting this residue in S. typhimurium Mfd abrogates its binding to RpoB (Figure 4A). We found that WT Mfd fully complements the decreased evolvability of Δmfd strains, whereas the L499R point mutant cannot complement the evolution of resistance to rifampicin (Figures 4B and 4C), trimethoprim, or phosphomycin (Figure S4). Therefore, the interaction between Mfd and RpoB is essential for Mfd’s mutagenesis and subsequent evolvability function.
Figure 4.

Mfd-RpoB and Mfd-UvrA Interactions Are Essential for Mfd-Driven Mutagenesis and Evolution to Antibiotics, Related to Figure S4
(A) Mutation of Mfd L499R and R165A residues abrogates RNAP and UvrA interactions, respectively. Relevant domains of the RpoB, Mfd, and UvrA proteins of S. typhimurium ST19 were cloned into a luciferase-based bacterial 2-hybrid system. Interactions between the respective protein domains were measured as in Figure 3. Results are from three independent experiments, and error bars indicate standard error of the mean. Statistical significance was determined using two-tailed Student’s t test (∗∗∗p value < 0.001).
(B) Mutation rate analysis of indicated strains of S. typhimurium to rifampicin. Complement and point mutant (L499R and R165A) strains were expressed episomally. WT and Δmfd strains contain pUC19 empty vector control (see Tables S2 and S3). Number of replicates per strain is 36–112. Errors bars are 95% CI.
(C) Evolution of indicated S. typhimurium strains to rifampicin. Complement and point mutant strains (L499R and R165A) were expressed episomally. WT and Δmfd strains contain pMMB67EH empty vector controls. Strains were grown in 50 μg/mL carbenicillin to maintain selection of episomes. Plots and statistical testing for evolution assays were performed as described in Figure 2. Number of replicates per strain is 12–24. ∗∗p value < 0.01 between WT and Δmfd strains and ∗p value < 0.05 between WT and Δmfd::mfd(L499R) and WT and Δmfd::mfd(R165A) strains.
We next sought to determine whether Mfd’s interaction with its other known binding partner, UvrA, was also necessary for its evolvability function. In order to test this, we constructed a point mutation in the D2 domain (part of the UvrB homology module) of the Mfd protein, known to mediate binding to UvrA (Deaconescu et al., 2006). Bacterial 2-hybrid assays confirmed that a point mutation in the UvrA interaction domain of S. typhimurium Mfd (R165A) successfully abrogated its binding to UvrA (Figure 4A). We expressed S. typhimurium Mfd containing the R165A point mutation in the Δmfd background and tested the impact of this mutant on mutation rates and evolution of resistance to rifampicin or phosphomycin. The Mfd R165A mutant was unable to complement any of the phenotypes (lowered mutagenesis or restricted evolution of resistance) associated with the lack of Mfd (Figures 4B–4C and S4A). The D2 domain of Mfd is also thought to form an interface with the C terminal D7 domain as a means of restricting Mfd-UvrA interactions in the absence of RNAP stalling (Deaconescu et al., 2006). While we cannot exclude the possibility that the R165A mutation alters this interaction, it is unlikely that we can attribute a disruption of the interface between D2 and D7 to our observed results. The major effects of abrogating D7 activity are thought to be tighter binding of Mfd to UvrA (Deaconescu et al., 2006, Savery, 2007, Selby and Sancar, 1995), yet the phenotypes of this mutant mimic those of Δmfd strains. Overall, our data suggest that Mfd promotes mutagenesis and evolution of drug resistance through its interactions with both RpoB and UvrA.
Discussion
In this work, we assign a novel function to Mfd as a general evolvability factor and demonstrate that it accelerates AMR development. We show that Mfd’s evolvability function requires its evolutionarily conserved interactions with both RpoB and UvrA. Arguably, the ability to evolve is critical for bacterial survival under ever-changing environmental conditions. This is especially important in the context of pathogenesis, where escaping host immunity is essential and requires constant adaptation. Therefore, our model of Mfd as an evolvability factor could explain its high degree of conservation across phyla, especially in pathogens, which are unlikely to experience TCR activation through UV exposure. This model is consistent with data showing that a deletion of Mfd has minor effects on DNA damage tolerance—especially compared to cells missing NER (Figure S1; Witkin, 1966b, Witkin, 1969)—and that bacteria lacking Mfd are insensitive to the DNA-damaging environment within macrophages (Figure S5). These observations suggest that Mfd is largely dispensable for the coordination of DNA repair. TCR is still an important and effective method of lesion detection and repair; however, Mfd may not be the main driver of this DNA repair mechanism. Recently, a new TCR pathway (Cohen et al., 2010) driven by the helicase UvrD (Epshtein et al., 2014, Kamarthapu et al., 2016) was discovered, indicating that cells also harbor Mfd-independent TCR mechanisms.
How Mfd promotes antimicrobial resistance and mutagenesis is unclear. One possible explanation is that Mfd promotes mutagenic DNA repair through error-prone gap filling at sites of NER activity, as previously suggested (Million-Weaver et al., 2015). Mfd may also promote DNA repair at sites that do not contain damaged DNA, given that Mfd can associate with RNAP in the absence of exogenous DNA damage (Ho et al., 2018). In vitro data showing that NER can promote gratuitous repair of undamaged DNA leading to recurrent DNA re-synthesis, which could consequently promote mutagenesis, is also consistent with this model (Branum et al., 2001). Alternatively, Mfd may promote mutagenesis by inhibiting the activation of other DNA repair pathways, at least under “normal” growth conditions—e.g., absence of UV damage. These pathways may include Mfd-independent TCR or global NER. Given our data with the UvrA interaction mutant of Mfd, we would predict that such inhibition would be through sequestration of UvrA.
Mfd-mediated evolution may be critical in the context of host infection. During infections, bacterial replication is reduced (Gill et al., 2009, Helaine et al., 2010), consequently reducing replication fork errors and possibly enhancing the relative contribution of non-replicative mutations (Gao et al., 2016). Given that transcription is still active under these conditions, Mfd may play a critical role in promoting bacterial mutagenesis during infections. This may explain the exaggerated effects of Mfd that we observed in our infection model. Additionally, our evolution assays, which mimic the variable antibiotic concentrations seen during clinical infections, suggest that Mfd is required for developing high levels of drug resistance upon primary exposure to sub-inhibitory concentrations of antibiotics, which may be critical in the context of AMR development (Wistrand-Yuen et al., 2018).
Lastly, Mfd may be even more important when multiple mutations are necessary to confer resistance, such as in the context of multi-drug resistance acquisition or in the context of compensatory mutations. Our sequencing data are consistent with this prediction, given that the occurrence of second and third mutations was quite rare in the absence of Mfd. Additionally, we observe that the rise of dnaQ hypermutator strains is inhibited in the absence of Mfd. Hypermutation is a key strategy that bacteria use to evolve resistance in the context of infections (Blázquez, 2003, Oliver et al., 2000). However, strains specifically containing the dnaQ hypermutator alleles have not been identified in clinical settings. This could simply be due to the fact that few (if any) isolates from trimethoprim-treated patients have been sequenced. Therefore, it is still possible that the dnaQ hypermutator allele is relevant in clinical settings. This question should be investigated by WGS of pathogens isolated from trimethoprim-treated patients.
Given our findings, we propose that blocking evolvability factors, and in particular Mfd, could be a revolutionary strategy to address the AMR crisis. A new class of “anti-evolution” drugs that target Mfd or other evolvability factors that promote mutagenesis may complement new antimicrobials and alleviate the problem of chromosomally acquired mutations that promote AMR. For example, LexA, which induces the SOS response upon exposure to DNA damage, has been suggested to promote AMR development, likely through trans-lesion synthesis (TLS) at replication forks (Cirz et al., 2005, Cirz et al., 2006, Mo et al., 2016). This mechanism could also be a good target for the inhibition of AMR development. However, SOS-mediated AMR development may be distinct from the transcription-dependent evolvability function of Mfd, which (in addition to replicating cells) could be relevant in infections in which pathogens are not replicating and/or have not been exposed to extensive DNA damage but are transcriptionally active. Therefore, in principle, drugs that target Mfd (or key SOS factors) could be co-administered with antibiotics during treatment of infections, reducing the likelihood of resistance development at the onset of treatment. Overall, efforts to understand and target the evolutionary capacity of cells could also have wide-ranging implications outside of AMR development, from reducing cancer evolution to limiting pathogenic diversity in the context of host immunity.
The ideas discussed here deliver a second message regarding drug discovery and therapeutics. Although drug discovery efforts are generally geared toward targeting essential proteins, the effectiveness of this approach may be limited. Supplemental drugs that target non-essential proteins (e.g., Mfd) during the treatment of infections (or various diseases such as cancer) have the potential to significantly improve the efficiency and/or potency of current treatment regimens. Therefore, development of novel therapeutics targeting non-essential proteins could expand the arsenal of drugs available to combat AMR and potentially other diseases.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| BamHI-HF | NEB | R3136S |
| HindIII-HF | NEB | R3104S |
| NotI-HF | NEB | R3189S |
| BglII | NEB | R0144S |
| NheI | NEB | R0131S |
| XhoI | NEB | R0146S |
| 4-Nitroquinoline N-oxide | Sigma | N8141 |
| Gibson Assembly Master Mix | NEB | E2611S |
| Phusion High-Fidelity PCR Master Mix | Thermo | F531S |
| Nano-Glo Luciferase Assay System | Promega | N1130 |
| Critical Commercial Assays | ||
| Nextera XT DNA Library Preparation Kit | Illumina | (FC-131-1024) |
| GeneJET PCR Purification Kit | Thermo | K0701 |
| GeneJET Genomic DNA purification Kit | Thermo | K0722 |
| MasterPure complete DNA and RNA Purification Kit | Epicenter | MC85200 |
| QIAquick PCR Purification Kit | QIAGEN | 28106 |
| Deposited Data | ||
| All fastq files and descriptions uploaded to Sequence Read Archive | NCBI SRA SUB4542953 |
Bioproject: PRJNA492467 |
| Experimental Models: Cell Lines | ||
| Human: Caco-2 epithelial cell line | ATCC | HTB-37; RRID:CVCL_0025 |
| Harvested Bone Marrow Macrophages from BALB/c mice | N/A | Miller Lab |
| Experimental Models: Organisms/Strains | ||
| B. subtilis trpC2 pheA1 | Brehm et al., 1973 | HM1 |
| B. subtilis trpC2 uvrA::mls | Bacillus stock center | HM713 |
| B. subtilis trpC2 mfd::mls | Bacillus stock center | HM1720 |
| S. typhimurium ST19 | Hayden et al., 2016 | HM1996 |
| P. aeruginosa CF127 | Wolfgang et al., 2003 | HM2212 |
| P. aeruginosa CF127 Δmfd | This study | HM2260 |
| E. coli F’(kan) placOL2-62-lacZ | Dove et al., 1997 | HM2295 |
| B. subtilis mfd::mls, trpC2 pheA1 | Million-Weaver et al., 2015 | HM2521 |
| E. coli F’(kan) placOL2-62, pSIM27(tet) | This study | HM2602 |
| B. subtilis uvrA::mls, trpC2 pheA1 | Million-Weaver et al., 2015 | HM2633 |
| E. coli FW102 OL2-62-Nanoluc(hyg), pSIM27 | This study | HM2747 |
| E. coli FW102 OL2-62-Nanoluc(hyg), pACλCI, pBRα Plac-a-ST19rpoB(19-142) | This study | HM2838 |
| E. coli FW102 OL2-62-Nanoluc(hyg), pBRα, pACλCI Plac-CI-ST19mfd(1-450) | This study | HM2875 |
| S. typhimurium ST19, pUC19 | This study | HM2880 |
| S. typhimurium ST19 mfd::cat, pUC19 | This study | HM2881 |
| S. typhimurium ST19 mfd::cat, pUC19-ST19mfd | This study | HM2882 |
| S. typhimurium ST19 mfd::cat, pUC19-ST19mfdL499R | This study | HM2886 |
| E. coli FW102 OL2-62-Nanoluc(hyg), pACλCI Plac-CI-ST19mfd(1-450), pBRα Plac-a-ST19UvrA(88-505) | This study | HM2913 |
| E. coli FW102 OL2-62-Nanoluc(hyg), pACλCI Plac-CI-ST19rpoB(19-142), pBRα Plac-a-ST19mfd (RID) | This study | HM2920 |
| E. coli FW102 OL2-62-Nanoluc(hyg), pACλCI Plac-CI-ST19rpoB(19-142), pBRα Plac-a-ST19mfd (RID) L499R | This study | HM2921 |
| E. coli FW102 OL2-62-Nanoluc(hyg), pACλCI Plac-CI-ST19rpoB(19-142), pBRα | This study | HM2925 |
| E. coli FW102 OL2-62-Nanoluc(hyg), pACλCI, pBRα Plac-a-ST19mfd (RID) | This study | HM2926 |
| E. coli FW102 OL2-62-Nanoluc(hyg), pACλCI, pBRα Plac-a-ST19UvrA(88-505) | This study | HM2949 |
| E. coli FW102 OL2-62-Nanoluc(hyg), pACλCI Plac-CI-ST19mfdR165A, pBRα Plac-a-ST19UvrA(88-505) | This study | HM2962 |
| S. typhimurium ST19 mfd::cat, pUC19-Mtbmfd | This study | HM3134 |
| E. coli FW102 OL2-62-Nanoluc(hyg), pACλCI Plac-CI-Mtbmfd(334-651), pBRα | This study | HM3193 |
| E. coli FW102 OL2-62-Nanoluc(hyg), pACλCI Plac-CI-Mtbmfd(334-651), pBRα Plac-a-ST19rpoB(19-142) | This study | HM3225 |
| S. typhimurium ST19 uvrA::kan | This study | HM3245 |
| S. typhimurium ST19 ST19mfd::Mtbmfd-kan | This study | HM3406 |
| S. typhimurium ST19 mfd::cat | This study | HM3429 |
| S. typhimurium ST19, pMMB67EH | This study | HM3585 |
| S. typhimurium ST19 mfd::cat, pMMB67EH | This study | HM3586 |
| S. typhimurium ST19 mfd::cat, pMMB67EH-ST19mfd | This study | HM3590 |
| S. typhimurium ST19 mfd::cat, pMMB67EH-ST19mfdR165A | This study | HM3666 |
| S. typhimurium ST19 mfd::cat, pMMB67EH-ST19mfdL499R | This study | HM3667 |
| M. tuberculosis H37Rv | ATCC | ATCC 27294 |
| M. tuberculosis H37Rv mfd::hyg | This study | MR02 |
| Oligonucleotides | ||
| Primer sequences provided in Table S3 | N/A | N/A |
| Recombinant DNA | ||
| pHM443 | This study | pHM443 |
| pHM453 | This study | pHM453 |
| pHM457 | This study | pHM457 |
| pHM458 | This study | pHM458 |
| pHM474 | This study | pHM474 |
| pHM480 | This study | pHM480 |
| pHM481 | This study | pHM481 |
| pHM484 | This study | pHM484 |
| pHM494 | This study | pHM494 |
| pHM499 | This study | pHM499 |
| pHM550 | This study | pHM550 |
| pHM566 | This study | pHM566 |
| pHM629 | This study | pHM629 |
| pHM649 | This study | pHM649 |
| pHM650 | This study | pHM650 |
| pHM651 | This study | pHM651 |
| pHM661 | This study | pHM661 |
| pHM662 | This study | pHM662 |
| pBRα | Addgene | Addgene 53731 |
| pBRα-β-flap | Addgene | Addgene 53734 |
| pACλCI | Addgene | Addgene 53730 |
| pACλCI-β-flap | Addgene | Addgene 53733 |
| pEX18 | Gift from Dr. Matthew Parsek | pEX18 |
| pKD3 | Addgene | Addgene 45604 |
| pKD13 | Miller Lab Stock | pKD13 |
| pKD46 | Miller Lab Stock | pKD46 |
| pMMBEH67 | Miller Lab Stock | pMMBEH67 |
| pNIT | Sherman Lab Stock | pNIT |
| pNL1.1 | Promega | Promega N1441 |
| pSIM27 | Gift from Dr. Don Court | pSIM27 |
| pUC19 | Addgene | Addgene 50005 |
| Software and Algorithms | ||
| SAMtools | Li et al., 2009 | N/A |
| Bowtie 2 | Langmead and Salzberg, 2012 | N/A |
| breseq | Deatherage and Barrick, 2014 | N/A |
| Prism 7 Graphpad | N/A | N/A |
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to Lead Contact, Houra Merrikh (merrikh@uw.edu).
Experimental Model and Subject Details
Strains for the following species were built as described: deletions in B. subtilis were built with transformation of marked genomic DNA into the appropriate background strain. Deletions in S. typhimurium were built using the λ-red recombineering (Datsenko and Wanner, 2000) and all plasmids used were transformed by electroporation. The mfd deletion in P. aeruginosa was built using the pEX18 suicide plasmid (with homology regions to mfd) as previously described (Hoang et al., 1998), and the mfd deletion in Mtb was built using recombineering (van Kessel and Hatfull, 2007). E. coli strains for the bacterial 2-hybrid assay were built via electroporation of the designated plasmid into the appropriate strain background. E. coli DH5α was used to propagate recombinant DNA vectors. Transformations were done using heat shock of competent E. coli. E. coli cultures were grown at 37°C with shaking (260 RPM) in LB supplemented with antibiotics where appropriate. All plasmid vectors were purified using a commercially available plasmid extraction kit (Thermo). Specific details of strains and plasmid constructed used in this work, including primers used, are listed in Table S2 and S3. All strain modifications were confirmed by PCR and sequencing.
Method details
Strain constructions
Details of bacterial strains and recombinant plasmids built in this study are described in Table S2.
Luria-Delbruck fluctuation analysis
For B. subtilis, cultures were grown from single colonies at 37°C with aeration in LB media (10 g Tryptone, 5g yeast extract and 5g NaCl per liter). Exponential phase cultures (OD600 = 0.3) were diluted back to OD600 = 0.0005 in parallel cultures containing LB, and plated following 4.5 hours of growth at 37°C with aeration. Cultures were plated on 50 μg/mL rifampicin to quantify the number of mutants and serially diluted and plated on LB to quantify total viable cells. Colonies were quantified after overnight incubation at 37°C (for rifampicin plates) and 30°C (for LB plates).
S. typhimurium and P. aeruginosa mutation rates were measured by growing overnight cultures from single colonies and subsequently back diluting parallel cultures to an OD600 = 0.0005 in LB. Cultures were grown to an OD600 = 0.8-1.0 (OD600 = 1.0 for P. aeruginosa) at 37°C with aeration. Cultures were plated as described for B. subtilis. For mutation rate analysis of WT-pUC19, Δmfd-pUC19, ST19 mfd complementation, Mtb mfd complementation, and the ST19 mfd point mutant (L499R and R165A) strains of S. typhimurium, overnight cultures were grown to saturation in 50 μg/mL carbenicillin to maintain plasmid selection. Cultures were back diluted to an OD600 = 0.0005, grown in LB only to OD600 = 0.8-1.0 and plated as previously described.
For Mtb, experiments were performed as previously described (Ford et al., 2013). Briefly, cultures were grown in 7H9 mycobacterial media + ADC to saturation. Multiple, independent cultures were back diluted to final OD600 = 0.0001 and grown at 37°C to OD600 = 0.8-1.2. Cells were plated on 7H10 mycobacterial agar + OADC and 2μg/mL rifampicin, 5μg/mL ethambutol or 1.5μg/mL of ciprofloxacin to quantify resistant mutants and on 7H10 + OADC for CFU enumeration. Plates were incubated at 37°C for approximately 10 days for CFU enumeration and 25-30 days for antibiotic plates. Mutation rates for all species were calculated using the Ma-Sandri-Sarkar Maximum Likelihood method (Hall et al., 2009).
Mutagenesis measurements post epithelial cell infection
Colorectal adenocarcinoma cells line CACO-2 were cultured in DMEM medium with 20% heat-inactivated FBS at 37°C in 5% CO2. Approximately 106 CACO-2 cells were plated overnight in 6-well plates at 37°C in 5% CO2 for infection. A single S. typhimurium colony was picked and grown overnight at 37°C in LB, diluted back to an OD600 = 0.05 the following day and grown at 37°C in LB until cultures reached OD600 = 0.5. Cells were washed 2x with 1X PBS resuspended in DMEM +20% FBS and inoculated at 100:1 multiplicity of infection with CACO-2 cells at 37°C in 5% CO2 for one hour. Cells were then washed 2x with 1X PBS and DMEM + 20% FBS + 50 μg/mL gentamicin was added to plates to kill extracellular bacteria. After 6 hours of infection, cells were washed in 1X PBS and lysed in 1X PBS + 0.1% Triton X-100. Cells were plated on M9 minimal + 0.4% glycerol agar for CFU enumeration and M9 minimal + 0.4% glycerol agar containing 100μg/mL 5-fluorocytosine (5FC) and grown at 37°C to determine mutation frequency. Mutation frequency was determined by taking the ratio of 5FC colonies to the viable cell count for each sample. For experiments measuring cell viability over multiple time points, S. typhimurium and CACO-2 cells were grown as described and bacterial cells were harvested for CFU enumeration at defined time points.
Antibiotic evolution assays
Evolution experiments were performed for the indicated strains. For S. typhimurium, overnight cultures, started from a single colony, were back diluted to OD600 = 0.005 and used to inoculate a 96-well plate. Cells were grown for either 12 or 24 hours with agitation, at 37°C, in LB with a gradient of concentrations of the indicated antibiotic to select for resistance. ODs were subsequently measured in an Epoch/2 microplate spectrophotometer (Bio-Tek). Cultures that grew (defined by at least 50% growth relative to LB only) at the highest concentration of antibiotic were passaged into fresh LB +antibiotic in a subsequent plate. A total of 5-8 serial passages were performed depending on the antibiotic used. Evolution experiments with WT-pMMB67EH, Δmfd –pMMB67EH, complementation and point mutant (L499R and R165) strains of S. typhimurium were grown identical to WT and Δmfd strains except with the addition of 50 μg/mL carbenicillin to maintain selection of episomes. For B. subtilis, cultures were started from a single colony were grown for 4-5 hours until they reached OD600 = 1.0. Cultures were back diluted to OD600 = 0.005, inoculated into a 96-well plate and grown for 12 hours at 37°C in LB in an Epoch/2 microplate spectrophotometer (Bio-Tek) for 9 serial passages, with a gradient of concentrations of rifampicin to select for resistance. For Mtb, saturated cultures were back diluted to OD600 = 0.05, inoculated into a 96-well plate and grown in 7H9 +ADC in a 37°C incubator without aeration. Strains were serially passaged when the density of no antibiotic control wells reached approximately OD600 = 1.5-2.0 (approximately 15-20 days). Cultures that grew (defined by at least 50% growth relative to 7H9+ADC) at the highest concentration of rifampicin were passaged into a fresh 7H9 + ADC+ rifampicin in a subsequent plate, and a total of 6 serial passages were performed. For all species, antibiotics were diluted 2-fold down each given row in a 96 well plate.
Sequencing of antibiotic evolution assays
Genomic DNA was harvested from evolved strains of S. typhimurium and purified using either the MasterPure complete DNA and RNA Purification Kit (Epicenter) or GeneJet Genomic DNA Purification Kit (ThermoFisher) in accordance with manufacturer instructions. For WGS experiments, gDNA samples were processed for sequencing using the Nextera XT DNA Library Preparation Kit (Illumina). Paired-end libraries were sequenced on an Illumina NextSeq sequencing platform yielding an average of 40X coverage sequencing depth. The resulting FASTQ reads were trimmed for quality using the FASTX quality filter such that 95% of bases were required to have a Phred score of 30 or higher (Available at http://hannonlab.cshl.edu/fastx_toolkit/index.html). SNPs against the S. typhimurium ST19 genome (available from the Prokaryotic Genome Analysis Tool http://tools.uwgenomics.org/pgat/), a derivative of S. enterica Typhimurium LT2 (GenBank: NC_003197.2) were then identified using BreSeq (Deatherage and Barrick, 2014). For Sanger sequencing, amplification of rpoB and folA loci was performed using Phusion DNA polymerase (ThermoFisher). PCR products were purified using the QIAquick PCR Purification Kit (QIAGEN) and sequencing was subsequently performed to identify mutations.
Bacterial 2-hybrid assays
Bacterial 2-hybrid assays were performed as previously described (Dove et al., 1997). Briefly, domains from the genes of interest were fused to the Lambda repressor (cI) and the N-terminal domain of E. coli RNA polymerase’s alpha subunit (α-NTD) using the plasmids pACλCI and pBRα, respectively. These fusion constructs were transformed into E. coli containing the Lambda operator sequence inserted upstream of a luciferase reporter (NanoLuc, Promega) using an F’ episome. For expression of fusion constructs, cells were grown overnight in LB + 20μM IPTG at 30°C and diluted 1:100 intro fresh LB + 20μM IPTG at 30°C the next morning and were grown until OD600 = 2. For relative light unit measurements, Nano-glo substrate (Promega) was added to cultures according to the manufacturer’s instructions and luminescence was measured in a SpectraMax M3 96-well plate reader.
DNA damage survival assays
For both S. typhimurium and B. subtilis, cultures were started from single colonies and harvested at exponential growth (OD600 = 0.3-0.6). To determine 4-Nitro-Quinolone Oxide (4-NQO) survival, cell dilutions were spotted onto LB agar plates (for CFU enumeration) and LB agar plates containing either 0.2 μM (B. subtilis) or 4 μM (S. typhimurium) 4-NQO. To determine UV sensitivity, cells were spotted onto LB agar plates and exposed to the indicated intensity of UV light using a Mineralight XX 15V UV light source (UVP). Surviving colonies were enumerated after overnight incubation at 30°C.
Bone Marrow-Derived Macrophage (BMM) infections
BMMs were derived from BALB/c mice as previously described (Weischenfeldt and Porse, 2008). All protocols for harvesting BMMs were reviewed and approved by the Institutional Animal Care and Use Committee at the University of Washington. BMMs were cultured in RPMI media with 10% heat-inactivated FBS at 37°C in 5% CO2. Approximately 106 BMMs were plated overnight in 24-well plates at 37°C in 5% CO2. For infections, a single colony of S. typhimurium was picked and grown overnight at 37°C in LB, diluted back to an OD600 = 0.05 the following day and grown at 37°C in LB until cultures reached OD600 = 0.5. Bacteria were then washed 2x with 1X PBS and resuspended in RPMI +10% FBS and inoculated at 10:1 multiplicity of infection with BMMs at 37°C in 5% CO2 for 30 min. Plates were then washed 2x with 1X PBS and RPMI +10% FBS + 50 μg/mL gentamicin was added for killing of extracellular bacteria. Infected macrophages were lysed at indicated time points with 1X PBS + 0.1% Triton X-100 and plated on LB for CFU enumeration.
Quantification and Statistical Analysis
The definition of all data points, variance measurement, and statistical tests used are included in each figure legends. The number of replicates for each experiment are also described in each figure legend. Statistical measurements were performed in Prism 7.0 (Graphpad).
Data and Software Availability
The accession number for the data reported in this paper is NCBI SRA: PRJNA492467, SRA:SUB4542953.
Acknowledgments
We acknowledge the work of others that have contributed to our understanding of this topic and members of the Merrikh Lab for helpful discussions. We thank Brandon Gallaher for construction of pUC19-mfd strains. We thank Erik Peterson for harvesting bone marrow macrophages and for help with S. typhimurium infections. We thank the labs of Joshua Woodward (for the CACO-2 cells), Matt Parsek (for the Pseudomonas CF127 isolate and pEX18 plasmid), and Don Court (for the pSIM27 plasmid) for their donations. We thank Ann Hochschild for the bacterial 2-hybrid system and for troubleshooting assistance. We thank Andrew Frando and Jessica Winkler for help with strain construction in Mtb. A special thank you to Jay Shendure and Choli Lee for assistance in whole-genome sequencing experiments. We would like to also thank the anonymous reviewers for helping improve this manuscript. Support for this project was provided by the Bill & Melinda Gates Foundation OPP1154551 to H.M. and D.R.S. as well as the University of Washington Innovation Award and the National Institute of General Medical Sciences Award DP2GM110773 to H.M.
Author Contributions
H.M., M.N.R., M.K.T., A.K., S.I.M., and D.R.S. designed experiments and provided technical assistance. M.N.R., M.K.T., C.H., C.N.M., P.N., J.G., A.N.S., and A.K. performed experiments and analyzed data. H.M., M.N.R., and M.K.T. wrote the paper.
Declaration of Interests
The authors declare no competing interests.
Published: November 15, 2018
Footnotes
Supplemental Information includes five figures and three tables and can be found with this article online at https://doi.org/10.1016/j.molcel.2018.10.015.
Supplemental Information
Catalog of mutations for each replicate (6 WT and 6 Δmfd) at each time course of trimethoprim evolution (7 total). Highlighted in yellow are folA resistance conferring mutations (including putative promoter mutations), and highlighted in blue are dnaQ hypermutator mutations. For each mutation, the genomic location, nucleotide change, amino acid change, and gene are listed. Each individual replicate sequenced is cross-referenced with Figure S2, and isolates used to measure mutation rate in Figure S3 are indicated.
References
- Almeida Da Silva P.E., Palomino J.C. Molecular basis and mechanisms of drug resistance in Mycobacterium tuberculosis: classical and new drugs. J. Antimicrob. Chemother. 2011;66:1417–1430. doi: 10.1093/jac/dkr173. [DOI] [PubMed] [Google Scholar]
- Baym M., Lieberman T.D., Kelsic E.D., Chait R., Gross R., Yelin I., Kishony R. Spatiotemporal microbial evolution on antibiotic landscapes. Science. 2016;353:1147–1151. doi: 10.1126/science.aag0822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blázquez J. Hypermutation as a factor contributing to the acquisition of antimicrobial resistance. Clin. Infect. Dis. 2003;37:1201–1209. doi: 10.1086/378810. [DOI] [PubMed] [Google Scholar]
- Brandis G., Pietsch F., Alemayehu R., Hughes D. Comprehensive phenotypic characterization of rifampicin resistance mutations in Salmonella provides insight into the evolution of resistance in Mycobacterium tuberculosis. J. Antimicrob. Chemother. 2015;70:680–685. doi: 10.1093/jac/dku434. [DOI] [PubMed] [Google Scholar]
- Branum M.E., Reardon J.T., Sancar A. DNA repair excision nuclease attacks undamaged DNA. A potential source of spontaneous mutations. J. Biol. Chem. 2001;276:25421–25426. doi: 10.1074/jbc.M101032200. [DOI] [PubMed] [Google Scholar]
- Brehm S.P., Staal S.P., Hoch J.A. Phenotypes of pleiotropic-negative sporulation mutants of Bacillus subtilis. J. Bacteriol. 1973;115:1063–1070. doi: 10.1128/jb.115.3.1063-1070.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirz R.T., Chin J.K., Andes D.R., de Crécy-Lagard V., Craig W.A., Romesberg F.E. Inhibition of mutation and combating the evolution of antibiotic resistance. PLoS Biol. 2005;3:e176. doi: 10.1371/journal.pbio.0030176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirz R.T., Gingles N., Romesberg F.E. Side effects may include evolution. Nat. Med. 2006;12:890–891. doi: 10.1038/nm0806-890. [DOI] [PubMed] [Google Scholar]
- Cohen S.E., Lewis C.A., Mooney R.A., Kohanski M.A., Collins J.J., Landick R., Walker G.C. Roles for the transcription elongation factor NusA in both DNA repair and damage tolerance pathways in Escherichia coli. Proc. Natl. Acad. Sci. USA. 2010;107:15517–15522. doi: 10.1073/pnas.1005203107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko K.A., Wanner B.L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deaconescu A.M., Chambers A.L., Smith A.J., Nickels B.E., Hochschild A., Savery N.J., Darst S.A. Structural basis for bacterial transcription-coupled DNA repair. Cell. 2006;124:507–520. doi: 10.1016/j.cell.2005.11.045. [DOI] [PubMed] [Google Scholar]
- Deatherage D.E., Barrick J.E. Identification of mutations in laboratory-evolved microbes from next-generation sequencing data using breseq. Methods Mol. Biol. 2014;1151:165–188. doi: 10.1007/978-1-4939-0554-6_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dove S.L., Joung J.K., Hochschild A. Activation of prokaryotic transcription through arbitrary protein-protein contacts. Nature. 1997;386:627–630. doi: 10.1038/386627a0. [DOI] [PubMed] [Google Scholar]
- Dutta D., Shatalin K., Epshtein V., Gottesman M.E., Nudler E. Linking RNA polymerase backtracking to genome instability in E. coli. Cell. 2011;146:533–543. doi: 10.1016/j.cell.2011.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echols H., Lu C., Burgers P.M. Mutator strains of Escherichia coli, mutD and dnaQ, with defective exonucleolytic editing by DNA polymerase III holoenzyme. Proc. Natl. Acad. Sci. USA. 1983;80:2189–2192. doi: 10.1073/pnas.80.8.2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epshtein V., Kamarthapu V., McGary K., Svetlov V., Ueberheide B., Proshkin S., Mironov A., Nudler E. UvrD facilitates DNA repair by pulling RNA polymerase backwards. Nature. 2014;505:372–377. doi: 10.1038/nature12928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford C.B., Shah R.R., Maeda M.K., Gagneux S., Murray M.B., Cohen T., Johnston J.C., Gardy J., Lipsitch M., Fortune S.M. Mycobacterium tuberculosis mutation rate estimates from different lineages predict substantial differences in the emergence of drug-resistant tuberculosis. Nat. Genet. 2013;45:784–790. doi: 10.1038/ng.2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z., Wyman M.J., Sella G., Przeworski M. Interpreting the Dependence of Mutation Rates on Age and Time. PLoS Biol. 2016;14:e1002355. doi: 10.1371/journal.pbio.1002355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill W.P., Harik N.S., Whiddon M.R., Liao R.P., Mittler J.E., Sherman D.R. A replication clock for Mycobacterium tuberculosis. Nat. Med. 2009;15:211–214. doi: 10.1038/nm.1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall B.M., Ma C.X., Liang P., Singh K.K. Fluctuation analysis CalculatOR: a web tool for the determination of mutation rate using Luria-Delbruck fluctuation analysis. Bioinformatics. 2009;25:1564–1565. doi: 10.1093/bioinformatics/btp253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J., Sahin O., Barton Y.W., Zhang Q. Key role of Mfd in the development of fluoroquinolone resistance in Campylobacter jejuni. PLoS Pathog. 2008;4:e1000083. doi: 10.1371/journal.ppat.1000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanawalt P.C., Spivak G. Transcription-coupled DNA repair: two decades of progress and surprises. Nat. Rev. Mol. Cell Biol. 2008;9:958–970. doi: 10.1038/nrm2549. [DOI] [PubMed] [Google Scholar]
- Hayden H.S., Matamouros S., Hager K.R., Brittnacher M.J., Rohmer L., Radey M.C., Weiss E.J., Kim K.B., Jacobs M.A., Sims-Day E.H. Genomic analysis of Salmonella enterica serovar Typhimurium characterizes strain diversity for recent U.S. salmonellosis cases and identifies mutations linked to loss of fitness under nitrosative and oxidative stress. MBio. 2016;7 doi: 10.1128/mBio.00154-16. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4810482/ e00154–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helaine S., Thompson J.A., Watson K.G., Liu M., Boyle C., Holden D.W. Dynamics of intracellular bacterial replication at the single cell level. Proc. Natl. Acad. Sci. USA. 2010;107:3746–3751. doi: 10.1073/pnas.1000041107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho H.N., van Oijen A.M., Ghodke H. The transcription-repair coupling factor Mfd associates with RNA polymerase in the absence of exogenous damage. Nat. Commun. 2018;9:1570. doi: 10.1038/s41467-018-03790-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang T.T., Karkhoff-Schweizer R.R., Kutchma A.J., Schweizer H.P. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene. 1998;212:77–86. doi: 10.1016/s0378-1119(98)00130-9. [DOI] [PubMed] [Google Scholar]
- Kamarthapu V., Epshtein V., Benjamin B., Proshkin S., Mironov A., Cashel M., Nudler E. ppGpp couples transcription to DNA repair in E. coli. Science. 2016;352:993–996. doi: 10.1126/science.aad6945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le T.T., Yang Y., Tan C., Suhanovsky M.M., Fulbright R.M., Jr., Inman J.T., Li M., Lee J., Perelman S., Roberts J.W. Mfd Dynamically Regulates Transcription via a Release and Catch-Up Mechanism. Cell. 2018;173:1823. doi: 10.1016/j.cell.2018.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee G.H., Jeong J.Y., Chung J.W., Nam W.H., Lee S.M., Pak J.H., Choi K.D., Song H.J., Jung H.Y., Kim J.H. The Helicobacter pylori Mfd protein is important for antibiotic resistance and DNA repair. Diagn. Microbiol. Infect. Dis. 2009;65:454–456. doi: 10.1016/j.diagmicrobio.2009.08.007. [DOI] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luria S.E., Delbrück M. Mutations of Bacteria from Virus Sensitivity to Virus Resistance. Genetics. 1943;28:491–511. doi: 10.1093/genetics/28.6.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin H.A., Pedraza-Reyes M., Yasbin R.E., Robleto E.A. Transcriptional de-repression and Mfd are mutagenic in stressed Bacillus subtilis cells. J. Mol. Microbiol. Biotechnol. 2011;21:45–58. doi: 10.1159/000332751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Million-Weaver S., Samadpour A.N., Moreno-Habel D.A., Nugent P., Brittnacher M.J., Weiss E., Hayden H.S., Miller S.I., Liachko I., Merrikh H. An underlying mechanism for the increased mutagenesis of lagging-strand genes in Bacillus subtilis. Proc. Natl. Acad. Sci. USA. 2015;112:E1096–E1105. doi: 10.1073/pnas.1416651112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo C.Y., Manning S.A., Roggiani M., Culyba M.J., Samuels A.N., Sniegowski P.D., Goulian M., Kohli R.M. Systematically Altering Bacterial SOS Activity under Stress Reveals Therapeutic Strategies for Potentiating Antibiotics. MSphere. 2016;1:1–15. doi: 10.1128/mSphere.00163-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill, J. (2014). Antimicrobial Resistance: Tackling a Crisis for the Health and Wealth of Nations. The Review on Antimicrobial Resistance, 4–16. https://amr-review.org/sites/default/files/AMR%20Review%20Paper%20-%20Tackling%20a%20crisis%20for%20the%20health%20and%20wealth%20of%20nations_1.pdf.
- Oliver A., Cantón R., Campo P., Baquero F., Blázquez J. High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science. 2000;288:1251–1254. doi: 10.1126/science.288.5469.1251. [DOI] [PubMed] [Google Scholar]
- Pani B., Nudler E. Mechanistic insights into transcription coupled DNA repair. DNA Repair (Amst.) 2017;56:42–50. doi: 10.1016/j.dnarep.2017.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson A.R., Soliven K.C., Castor M.E., Barnes P.D., Libby S.J., Fang F.C. The Base Excision Repair system of Salmonella enterica serovar typhimurium counteracts DNA damage by host nitric oxide. PLoS Pathog. 2009;5:e1000451. doi: 10.1371/journal.ppat.1000451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross C., Pybus C., Pedraza-Reyes M., Sung H.M., Yasbin R.E., Robleto E. Novel role of mfd: effects on stationary-phase mutagenesis in Bacillus subtilis. J. Bacteriol. 2006;188:7512–7520. doi: 10.1128/JB.00980-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savery N.J. The molecular mechanism of transcription-coupled DNA repair. Trends Microbiol. 2007;15:326–333. doi: 10.1016/j.tim.2007.05.005. [DOI] [PubMed] [Google Scholar]
- Schon T., Jureen P., Giske C.G., Chryssanthou E., Sturegard E., Werngren J., Kahlmeter G., Hoffner S.E., Angeby K.A. Evaluation of wild-type MIC distributions as a tool for determination of clinical breakpoints for Mycobacterium tuberculosis. J. Antimcrobial Chemother. 2009:786–793. doi: 10.1093/jac/dkp262. [DOI] [PubMed] [Google Scholar]
- Selby C.P., Sancar A. Transcription-repair coupling and mutation frequency decline. J. Bacteriol. 1993;175:7509–7514. doi: 10.1128/jb.175.23.7509-7514.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selby C.P., Sancar A. Structure and function of transcription-repair coupling factor. I. Structural domains and binding properties. J Biol Chem. 1995;270:4882–4889. doi: 10.1074/jbc.270.9.4882. https://www.ncbi.nlm.nih.gov/pubmed/7876261 [DOI] [PubMed] [Google Scholar]
- van Kessel J.C., Hatfull G.F. Recombineering in Mycobacterium tuberculosis. Nat. Methods. 2007;4:147–152. doi: 10.1038/nmeth996. [DOI] [PubMed] [Google Scholar]
- Watson M., Liu J.-W., Ollis D. Directed evolution of trimethoprim resistance in Escherichia coli. FEBS J. 2007;274:2661–2671. doi: 10.1111/j.1742-4658.2007.05801.x. [DOI] [PubMed] [Google Scholar]
- Weischenfeldt J., Porse B. Bone marrow-derived macrophages (BMM): Isolation and applications. CSH Protoc. 2008;2008:t5080. doi: 10.1101/pdb.prot5080. [DOI] [PubMed] [Google Scholar]
- Wimberly H., Shee C., Thornton P.C., Sivaramakrishnan P., Rosenberg S.M., Hastings P.J. R-loops and nicks initiate DNA breakage and genome instability in non-growing Escherichia coli. Nat. Commun. 2013;4:2115. doi: 10.1038/ncomms3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wistrand-Yuen E., Knopp M., Hjort K., Koskiniemi S., Berg O.G., Andersson D.I. Evolution of high-level resistance during low-level antibiotic exposure. Nat. Commun. 2018;9:1599. doi: 10.1038/s41467-018-04059-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witkin E.M. Mutation and the repair of radiation damage in bacteria. Radiat. Res. 1966;6:6. 30–53. [PubMed] [Google Scholar]
- Witkin E.M. Radiation-induced mutations and their repair. Science. 1966;152:1345–1353. doi: 10.1126/science.152.3727.1345. [DOI] [PubMed] [Google Scholar]
- Witkin E.M. Ultraviolet-induced mutation and DNA repair. Annu. Rev. Microbiol. 1969;23:487–514. doi: 10.1146/annurev.mi.23.100169.002415. [DOI] [PubMed] [Google Scholar]
- Wolfgang M.C., Kulasekara B.R., Liang X., Boyd D., Wu K., Yang Q., Miyada C.G., Lory S. Conservation of genome content and virulence determinants among clinical and environmental isolates of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA. 2003;100:8484–8489. doi: 10.1073/pnas.0832438100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Catalog of mutations for each replicate (6 WT and 6 Δmfd) at each time course of trimethoprim evolution (7 total). Highlighted in yellow are folA resistance conferring mutations (including putative promoter mutations), and highlighted in blue are dnaQ hypermutator mutations. For each mutation, the genomic location, nucleotide change, amino acid change, and gene are listed. Each individual replicate sequenced is cross-referenced with Figure S2, and isolates used to measure mutation rate in Figure S3 are indicated.