

Abstract
Metabolic syndrome is a cluster of inherited metabolic traits, which centers around obesity and insulin resistance and is a major contributor to the growing prevalence of cardiovascular disease. The factors that underlie the association of metabolic traits in this syndrome are poorly understood due to disease heterogeneity and complexity. Genetic studies of kindreds with severe manifestation of metabolic syndrome has led to the identification of casual rare mutations in the LDL receptor-related protein 6 (LRP6), which serves as a co-receptor with frizzled protein receptors for Wnt signaling ligands. Extensive investigations have since unraveled the significance of the Wnt pathways in regulating body mass, glucose metabolism, de-novo lipogenesis, low density lipoprotein (LDL) clearance, vascular smooth muscle plasticity, liver fat and liver inflammation. The impaired canonical Wnt signaling observed in the R611C mutation carriers and the ensuing activation of non-canonical Wnt signaling constitute the underlying mechanism for these cardiometabolic abnormalities. Transcription factor 7-like 2 (TCF7L2) is a key transcription factor activated through LRP6 canonical Wnt and reciprocally inhibited by the non-canonical pathway. TC7L2 increases insulin receptor expression, decreases LDL and triglyceride synthesis, and inhibits vascular smooth muscle proliferation. Canonical Wnt also inhibits noncanonical protein kinase C (PKC), Ras homolog gene family member A (RhoA), Rho associated coiled-coil containing protein kinase 2 (ROCK2) activation thus inhibiting steatohepatitis and transforming growth factor β (TGF-β) mediated extracellular matrix deposition and hepatic fibrosis. Therefore, dysregulation of the highly conserved Wnt signaling pathway underlies the pleiotropy of metabolic traits of the metabolic syndrome and the subsequent end organ complications.
Keywords: Atherosclerosis, Wnt, LRP6, Metabolic syndrome, early onset coronary artery disease
1.Introduction
Obesity is a major trait of metabolic syndrome (MetS) that confers high risk for cardiovascular disease. Obesity has become a public health threat with a prevalence that has doubled over the last four decades and now affects approximately 40% of the adult population in the United States [1]. It is estimated that 40–70% of the variation in body mass index (BMI), a surrogate marker for abdominal obesity, is heritable [2, 3]. Rare monogenic disorders of obesity involve mutations affecting neuro-hormonal circuits that control food intake and energy expenditure [4]. Conversely, common polymorphisms in many more genes involving a variety of pathways have also been discovered in association with obesity albeit with smaller effects. While obesity can be measured with different metrics such as body mass index or waist circumference, it is clear that fat distribution is a major determinant of the severity of metabolic derangements in obese patients. The extreme spectrum of fat distribution is manifested in the hereditary lipodystrophies, inherited disorders of fat tissue associated with severe metabolic and atherosclerotic disease. A full review on the genetics of obesity and metabolic syndrome can be found elsewhere [4].
Other components of MetS include hypertension, insulin resistance, atherogenic dyslipidemia, a prothrombotic state, and a proinflammatory state. Interestingly, these risk factors cluster together more than expected by random chance and an element of heritability has been repeatedly identified in several familial studies [4]. Furthermore, MetS significantly increases the risk for cardiovascular disease after adjusting for the individual risk imparted by its component traits [5, 6]. Therefore, major scientific efforts have focused on the underlying molecular mechanism of MetS as a whole that could unify the association between the various components. In this review, we will focus on our laboratory’s discoveries linking Wnt signaling defects to metabolic disease.
2.Wnt signaling and metabolic syndrome
The Wnt signaling pathway is a ubiquitous signaling cascade that regulates a wide range of physiologic processes from stem cell pluripotency to cell fate determination during embryonic development. There are 3 distinct Wnt signaling pathways; the canonical pathway, the non-canonical cell polarity pathway, and the non-canonical calcium signaling pathway. In the canonical pathway, Wnt ligands bind to the frizzled receptor and LRP-5 or 6 co-receptors, resulting in the accumulation of β-catenin in the nucleus leading to cell cycle activation and transcriptional regulation [7]. In the non-canonical cell polarity pathway, the Wnt ligands bind to the frizzled receptor and the RYK/ROR co-receptors thus activating the Rho and Rac signaling cascades [7]. In the calcium signaling non-canonical pathway different Wnt ligands bind to the frizzled receptor and activate phospholipase C (PLC), thus leading to intracellular calcium release [8].
During adult life, Wnt signaling maintains its role in stem cells within tissues that require constant renewal such as gut epithelia and hair follicles. In humans, Wnt derangements have been implicated in cancer [9, 10], developmental disorders [11, 12], pulmonary disease [13], renal disease [14], cardiovascular disease and others [15]. The Wnt receptors belong to the Frizzled (Fz) receptor family [16] and Wnt co-receptors include LDL receptor-related proteins (LRP) 5 and 6, the Derailed/receptor tyrosine kinase (RYK) [17], and receptor tyrosine kinase-like orphan receptor (ROR) trans-membrane tyrosine kinases [18]. LRP 5 and 6 are co-receptors for canonical Wnt ligands and when phosphorylated lead to activation of canonical Wnt signaling and inactivation of non-canonical Wnt, whereas the other ligand co-receptor combinations activate the non-canonical cascade.
Our group was the first to implicate altered Wnt signaling in metabolic syndrome [19]. Genetic linkage analysis and positional cloning in a large outlier kindred with high prevalence of early onset coronary artery disease (CAD) and MetS led to discovery of novel nonconservative LRP6 mutations [19]. The index patient presented with myocardial infarction at 48 years of age and had a history of hypertension, hyperlipidemia and diabetes but not obesity. The mutation precisely cosegregated with MetS and resulted in substitution of Cysteine for Arginine at amino-acid 611 in the second epidermal growth factor (EGF)–like domain. The crystal structure of LRP6 revealed that this substitution disrupts a salt bridge between Arginine at 611 and Glutamate at 477 in the E2 propeller domain, relaxing the relative orientation between YWTD and EGF domains [20]. Many other LRP6 mutations similarly result in substitution of arginine by another amino acid [21].
2.1 LRP6 and plasma LDL clearance
Common variations in LRP6 had been linked to mild increase of LDL cholesterol in the general population [22]. Strikingly, patients with the LRP6-R611C mutation had blood cholesterol levels similar to heterozygote familial hypercholesterolemia manifesting after mid-thirties [19]. LRP6 is a lipid raft receptor protein that consists of LDLR-like binding domains, a cytoplasmic YXXL motif, and undergoes coated-pit-mediated internalization after Wnt stimulation in a Cav-1-mediated process [23, 24]. While LRP6 was known to mediate Wnt signaling [25] and not lipid homeostasis, other members of the family such as LRP1 and 2 were known to play important roles in apolipoprotein E (apoE) binding and cholesterol uptake by cells [26].
The function of LRP6 in LDL clearance was investigated in cells overexpressing LRP6-R611C or wild type LRP6 receptor. Cells overexpressing the wild type receptor showed higher LDL uptake compared to the cells transfected with empty vector or the LRP6-R611C vector. Similarly, LDL uptake in lymphoblastoid cells and native lymphocytes from heterozygote R611C mutation carriers were significantly lower compared to those of unaffected family members [27]. This effect was independent of LDL receptor mRNA and protein levels. Intracellular colocalization of LRP6 with tagged-LDL differed significantly between wild type and mutant cells. The R611C mutant cells had reduced cell surface expression of LRP6 with significant accumulation of LRP6 in late endocytic compartments compared to wild type where it was mainly present at the cell surface and early endocytic compartments [27].
Solid-phase binding assays showed low binding affinity of LRP6 and apolipoprotein-B (apoB) and no affinity for ApoE. However, the LRP6-R611C had higher affinity for apoB than the wild type receptor, which means that the reduced LDL uptake in R611C cells is not related to binding affinity with ApoB. Fluorescence-activated cell sorting (FACS) surface analysis showed lower cell surface expression of LRP6-R611C compared to wild type LRP6. In the presence of high LDL content in the medium, the expression level of wild type LRP6 was decreased. However, this phenomenon was reversible only with the wild type LRP6 and not LRP6-R611C cells. The enhanced ligand binding affinity of the mutant LRP6 in acidic pH and the continuous decline of its membrane expression in LDL-rich medium imply that impaired membrane recycling of LRP6 is a mechanism for the impaired LDL cholesterol clearance and hyperlipidemia in LRP6 mutation carriers.
In order to study the effect of LRP6 on LDL receptor (LDLR) function, CHO-ldlA7 cells lacking the LDLR were transfected with LRP6 [28]. Cells overexpressing LRP6 showed significantly higher LDL binding and uptake compared to those transfected with empty vector. Knock down of LRP6 by RNA interference in CHO-ldlA7 cells reduced endogenous LRP6 protein by 80% and caused significant reduction in LDL binding and uptake compared to controls, which means that LRP6 mediated LDL uptake is independent of the LDLR [28].
However, the mild impact of LRP6 on LDL uptake wouldn’t account for the severe hyperlipidemia in patients with loss of function LRP6 mutations. Hence, CHO cells (CHO-k1) that express the LDLR were transfected with hemagglutinin (HA) tagged LRP6 plasmid. At baseline, LDL uptake was significantly higher in the CHO-k1 cell-line compared to the CHO-ldlA7 cell-line. Overexpression of LRP6 significantly increased LDL uptake to a greater extent compared to CHO-ldlA7 cells overexpressing LRP6. Likewise, LRP6 knockdown in CHO-k1 cells caused a greater decrease in LDL binding and uptake in CHO-k1cells versus CHO-ldlA7 cells. The decline in LDL uptake was gradual and steady unlike that observed with CHO-ldlA7 cells, which means that the LDLR function was compromised in the absence of LRP6 thus implicating LRP6 in LDLR mediated LDL uptake [28].
Given the importance of lipoprotein lipase (LPL) in facilitating LDL uptake in an LDLR-dependent manner [29], the effect of LRP6 on LPL was tested. CHO-k1 overexpressing LRP6 doubled the LDL uptake in the presence of LPL compared to control. This effect was not observed in CHO-ldlA7 cells that lack the LDLR, which further confirmed that LRP6 plays an important role in LDLR-dependent LDL uptake.
The rate of LDLR internalization in the presence of LDL was diminished after LRP6 knock down in CHO-k1 cells, which means that LRP6 is important for LDLR internalization. Sucrose gradient fractionation demonstrated that LRP6 is present in lipid rafts containing clathrin and LDLR. LRP6 co-immunoprecipitated with the LDLR and colocalized at the plasma membrane and juxtanuclear region within the cytoplasm. Furthermore, the weak LRP6 and clathrin association increased in the presence of LDL. Although Caveolin 1 (Cav1) had been implicated in LRP6 internalization in response to Wnt3a stimulation [23], it had no effect on LRP6 internalization in response to LDL. However, LRP6 internalization was significantly impaired after clathrin knock down. Both clathrin and LRP6 colocalized in the cell membrane and the juxtanuclear region suggesting clathrin-mediated LRP6 vesicular internalization in response to LDL [28].
The overexpression of LRP6-R611C in both CHO-k1and CHO-ldlA7 did not increase LDL binding and uptake. Of note, LDLR internalization in response to LDL was significantly impaired in LRP6-R611C compared to wild type. The LDLR-clathrin complex was mainly present on cell membrane in the LRP6-R611C cells with minimal distribution in the juxtanuclear region compared to wild type cells. This confirmed that the damaging LRP6 mutation compromised LDLR internalization and LDL uptake and is likely the significant contributor to hypercholesterolemia phenotype in the LRP6-R611C mutation carriers.
2.2 LRP6, de novo lipogenesis, and LDL synthesis
LRP6 facilitates LDL clearance via LDL receptor (LDLR)-dependent vesicular endocytosis by forming a complex with LDLR and clathrin. Elevated LDL in patients with the LRP6-R611C mutation is, partially, due to decreased receptor-mediated uptake [27, 28, 30].
In vivo experimentation, 3 months mice carrying the LRP6 R611C mutation (heterozygous) on chow diet had significantly higher LDL, cholesterol and triglycerides compared to wild type. Furthermore, these levels were even higher in R611C homozygous mice. The hyperlipidemia phenotype was exacerbated by placing the mice on a high cholesterol/high fat diet with evidence of high VLDL triglyceride content and higher non-HDL cholesterol content compared to wild type.
In a mouse model that is deficient of the LDLR, introduction of the LRP6-R611C mutation resulted in significant increase of hypertriglyceridemia associated with increased very low-density lipoprotein (VLDL) particles, in addition to LDL and total cholesterol. Given that the effect of the R611C mutation on LDL binding and uptake has been shown to be mild, the major differences in plasma lipoproteins levels were attributed to increased LDL and VLDL synthesis and ApoB secretion [31].
While VLDL clearance was similar in LRP6-R611C homozygous mice compared to control, VLDL-apoB production was significantly increased in the LRP6 mutant mice. Furthermore, there was a 7-fold increase in de novo lipogenesis in the LRP6 mutant mice concordant with upregulation of SREBP1 and LXRα [31]. In parallel the expression of Insig1 and Insig2 were reduced. Interestingly, TCF7L2 binds the 3′ transcription start site of Insig1 [32] and TCF7L2 is a transcription factor downstream of LRP6. Hence, the reduced expression of Insig1 mRNA and protein are consistent with impaired canonical Wnt signaling in the LRP6-R611C homozygous mice.
LDLR negative and LRP6 mutant mice also had significantly higher blood LDL levels compared to LDLR negative mice. The majority of this increase was attributed to increased hepatic cholesterol production in the setting of increased hepatic mRNA and protein expression of HMG-CoA reductase. The expression and activities of enzymes regulating de novo lipogenesis, triglyceride and cholesterol synthesis (Sp1, IGF1, mTOR, ACC1, FASN, SCD1, HMGCR, apoB, MTP, SREBP1 and SREBP2) decreased significantly when mouse hepatocytes were treated with recombinant mouse Wnt3a (rmWnt3a). Furthermore, systemic in vivo administration of Wnt3a to LRP6-R611C homozygous mice significantly reduced the plasma levels of triglycerides, LDL and total cholesterol.
2.3 LRP6 and non-alcoholic fatty liver disease (NAFLD)
Interestingly, patients with the LRP6-R611C mutation were diagnosed with NAFLD and non-alcoholic steatohepatitis (NASH) based on computed tomography imaging and laboratory analysis with increased plasma aminotransferases. LRP6-R611C homozygous mice on high fat diet also developed hepatic insulin resistance and severe fatty liver disease with increased transaminase levels, increased total hepatic triglyceride and cholesterol content compared to controls [31].
The LRP6 mutant mice exhibited increased mTORC1 activity a key regulator of metabolism [33], as evidenced by increased phosphorylation of S6K, S6, and 4E-BP1 compared to wild type. Hepatic AKT/PI3K/mTOR can be activated by insulin growth factor (IGF1) [34] and this was significantly higher in plasma and the liver of LRP6-R611C homozygous mice compared to wild type. Hepatic IGF1R was also overexpressed in mutant mice compared to wild type. However, IRS1 phosphorylation was decreased in the mutant mice, which means that AKT/mTOR activation in these mice is independent of IRS-1 [31]. The increased IGF1R expression in the setting of impaired LRP6 is due to decreased ubiquitination and enhanced sumoylation [35]. Furthermore, impaired canonical Wnt in LRP6-R611C mice caused downregulation of Sp5 that suppresses Sp1. As a result, increased Sp1 transcriptional activity upregulated IGF1 [31].
Further analysis of the liver tissue of the LRP6 mutant mice showed significant infiltration with inflammatory cells aside from the greater fat content compared to controls [36]. These mice also went on to develop liver fibrosis. Protein levels of inflammatory cell markers CD68, F4/80, IL-6, and MPO were also considerably elevated in the liver. The expression of PNPLA3, a membrane-bound SREBP1c-regulated triacylglycerol lipase that is associated with NAFLD [37], was also increased in the LRP6-R611C mice.
The expression and phosphorylation of RhoA and ROCK2 were significantly increased in LRP6-R611C mouse liver and in cells with LRP6 knockdown [36]. Both enzymes are downstream effectors of non-canonical Wnt that are usually inhibited by canonical Wnt signaling. PKC is another enzyme in the non-canonical pathway that displayed increased phosphoactivation in the mutant mice with significantly increased activity in its downstream effector transforming growth factor (TGF)-β. In turn TGF-β triggers hepatic fibrosis by increasing expression of extracellular matrix proteins. Of note non-canonical Wnt has been implicated in inflammation [38, 39] and in the setting of LRP6-R611C mutation the activation of the non-canonical pathway seems to underlie hepatic steatosis, inflammation and steatofibrosis in affected patients that is rescued by administration of Wnt3a administration [36].
2.4 LRP6 and vascular smooth muscle proliferation
Traditionally, atherosclerotic arterial disease is attributed to arterial wall plaque formation with lipid laden macrophages. There is an increasing recognition for a disease entity known as plaque erosion. This type of coronary artery disease and cause of myocardial infarction was first discovered in pathological specimens of patients with myocardial infarction. The disease is more prevalent in women and young individuals than plaque rupture from atherosclerosis. The hallmark of plaque erosion is absence or reduced inflammatory cells and proliferation of vascular smooth muscle cells (VSMC) [40, 41] . The role of vascular smooth muscle cell (VSMC) proliferation in pathogenesis of CAD is also noted in recent genome-wide association (GWAS) studies of myocardial infarction and CAD. Common variants in TCF21 and Anril locus are linked to VSMC proliferation [42, 43]. In fact, patients with rare mutations in the alpha actin gene develop coronary artery occlusions due to excessive proliferation of VSMC [44].
VSMC proliferation, activated by platelet derived growth factor (PDGF) in response to endothelial injury, contributes to atherosclerosis [45, 46]. Inhibition of PDGF VSMC proliferation is known to diminish the atherosclerotic burden [47, 48]. Examination of coronary arteries from patients with atherosclerosis and healthy individuals showed significantly increased expression of LRP6 and PDGF receptor (PDGFR)-β in the subintima and muscularis layer of all atherosclerotic arteries compared to normal. Furthermore, LRP6 and PDGFR-β co-localized and immunoprecipitated together [49].
Although canonical Wnt stimulation with Wnt3a causes significant increase in smooth muscle cell proliferation, this effect was blunted in cells expressing LRP6-R611C. Therefore, the atherosclerosis in LRP6-R611C patients is not explained by increased Wnt-dependent smooth muscle proliferation. Surprisingly, the proliferative effect of PDGF-β stimulation was significantly enhanced in smooth muscle cells overexpressing LRP6-R611C compared to wild type LRP6 and this was concomitant with cyclin D1 levels. While PDGFR-β (receptor) was downregulated after exposure to PDGF-β in wild type LRP6 cells, there was no downregulation in the LRP6-R611C cells. Interestingly, wild type LRP6 caused increased ubiquitination of PDGFR-β and degradation through the lysosomal pathway. The antiproliferative effect of LRP6 also resulted from decreased phosphoactivation of ERK1/2 and the JAK–STAT pathway. This effect was impaired in the LRP6-R611C mutation carriers [49].
Aortic walls from mice homozygous for LRP6-R611C mutation on a 3-month chow diet revealed increased medial thickening, VSMC hyperplasia and disrupted elastic fibers which usually occurs in the setting of endothelial damage [50]. This was associated with decreased expression of contractile proteins. There was also increased expression of insulin growth factor-1 (IGF1), PDGF receptors and PDGF ligands in the aortic media and VSMCs compared to wild type mice. The administration of Wnt3a ligand reversed the latter phenotype with reduction in medial thickening, VSMC proliferation, and expression levels of IGF1, PDGF receptors and PDGF ligands.
Protein expression and mRNA levels of Sp1, a ubiquitously expressed transcription factor and an established regulator of VSMC plasticity [51–53], were significantly increased in LRP6-R611C VSMCs compared to WT [50]. Unsurprisingly, LRP6-R611C VSMCs exhibited considerably lower expression of the contractile proteins, and increased expression of vimentin that is a marker for mesenchymal cells and undifferentiated VSMCs. Stimulation of LRP6 with canonical Wnt ligand (Wnt3a) caused dramatic suppression of Sp1 [50].
TCF7L2 that had been previously linked to Sp1 transcriptional regulation [32] exhibited decreased levels in VSMCs and the aortic media of LRP6-R611C homozygous mice. However, treatment with Wnt3a significantly increased TCF7L2 expression in a steady fashion peaking at 8 hours with concomitant decline in Sp1 expression that reached a nadir at 8 hours. Interestingly, Wnt3a administration had minute effects on β-catenin expression in LRP6-R611C mice. TCF7L2 suppressed Sp1 transcription by binding to T-C-A-A-A-G motif downstream from transcription initiation site, as detected with ChIP assay [50].
Reduced canonical Wnt signaling was evident in the aortic media of LRP6-R611C mice with reduction of phosphorylated LRP6 and cyclinD1 levels. Analysis of the non-canonical Wnt signaling pathway showed significantly increased activation of RhoA, JNK, and Nemo-like kinase (NLK) in LRP6-R611C mice compared to wild type. Administration of Wnt3a increased TCF7L2 and cyclin D1 expression while inhibiting the non-canonical pathway enzymes RhoA, JNK, and NLK [50].
In response to guide wire induced carotid injury, LRP6-R611C mice showed significant neointima formation compared to wild type mice. This was accompanied by reduced TCF7L2 and increased Sp1 expression. Wnt3a resulted in significant protection against neointima formation in injured mice and this correlated with increased TCF7L2 and suppressed Sp1 when compared to untreated LRP6-R611C mice [50]. Strikingly, the LRP6 R611C homozygous mice on 10 months of high-cholesterol diet developed severe coronary artery disease with extensive neointima formation and increased apoptosis in the tunica media and adventitia in contrast to mice on a chow diet. The increased apoptosis correlated with increased Sp1 expression [50]. Surprisingly, homozygous LRP6-R611C deficient mice had decreased expression of macrophage activation chemokine (MCP-1) despite increased atherosclerosis compared to LDLR deficient mice with no additional differences inflammatory plasma cytokines. Instead these mice had increased CD3+ T cells and increased eosinophil markers in atherosclerotic lesions[50]. It has been shown in LDL receptor-deficient mice fed a high-cholesterol diet that anti-CD3 antibody causes regression of atherosclerosis with via augmenting a regulatory T-cell response in mice [54]. Hypereosinophilia has been implicated in coronary artery calcification in study of subjects with a clinical suspicion of coronary heart disease, using multislice computed tomography [55]. This suggested that VSMCs produce and accumulate cholesterol and worsen the atherosclerotic burden via proliferating into the neointima [50].
2.5 LRP6, body mass, diet and glucose metabolism
While the complete LRP6 knock-out mice (LRP6−/−) were embryonically lethal, the LRP6+/− mice appeared normal but exhibited enhanced glucose tolerance and insulin sensitivity compared with their wild-type littermates [56]. When placed on a high fat diet, the LRP6+/− mice were protected from obesity and insulin resistance compared to wild type. In response to high-fat diet, the LRP6+/− mice demonstrated increased adipose tissue expression of IRS. Interestingly, insulin signaling was enhanced in the brown adipose tissue (BAT) and liver but not in skeletal muscles of LRP6+/− mice. The increased insulin sensitivity was due to reduced Wnt-dependent mammalian target of rapamycin complex 1 (mTORC1) activity and enhanced expression of brown adipose tissue PGC1-α and UCP1 in addition to increased S6K and S6 phosphorylations [56]. Of note, PGC1-x primarily regulates energy expenditure in mitochondria of the brown adipose tissue and is inhibited by Wnt-10b [57, 58].
Additionally, LRP6+/− mice on 3 weeks of high-fat diet had lower blood glucose levels compared to wild type mice. When subjected to hyperinsulinemic-euglycemic clamp studies, the endogenous hepatic glucose output was lower in LRP6+/− compared to wild type. This correlated with decreased levels of gluconeogenic enzymes (glucose-6-phosphatase and phosphoenolpyruvate carboxykinase) and decreased activity of FoxO1; a key regulator of gluconeogenesis. This implicates LRP6 as an energy-sensitive regulator of weight and glucose metabolism via the mTORC network [56].
Later studies confirmed the importance of LRP6-Wnt signaling in regulating body weight and food intake, as it is expressed in the hypothalamus and NPY neurons in the arcuate nucleus [59]. LRP6+/− mice more specifically had lower body fat content compared to wild type mice. However, this wasn’t due to decreased food intake. Although plasma leptin levels were not changed, the leptin receptor gene was significantly upregulated with corresponding phosphomodulation of downstream targets. Wnt stimulation of the hepatocytes had the opposite effect on leptin receptor expression, which means that in haploinsufficient LRP6+/−mice, the decreased inhibitory effect of canonical Wnt on leptin receptor contributes to increased insulin sensitivity [56]. Interestingly, blockade of non-canonical signaling in adipocytes with a Wnt5a antagonist (sFRP5) has been shown to improve glucose tolerance [60]. Conversely, mice lacking sFRP5 that were placed on a high-calorie diet developed evidence of metabolic syndrome with insulin resistance and NAFLD [60].
LRP6-R611C mutation carriers had significantly reduced expression insulin receptors in skeletal muscle. TCF7L2 a potent transcriptional regulator that is activated by canonical Wnt, binds a TCF/LEF binding motif within the IR promoter and promotes transcription [35]. Accordingly, overexpression of exogenous TCF7L2 in skin fibroblasts of LRP6-R611C mutation regained normal insulin receptor (IR) expression, further confirming the biological importance of this transcription factor in regulation of IR transcription and insulin signaling in humans [35]. Additional studies showed that impaired ubiquitination of IGFR by the LRP6-R611C enhances the activities of the IGF-mTORC1 contributing to insulin resistance via mTORC1/pS6K dependent serine phosphorylation of IRS-1 [35].
3.Conclusion
Our investigations uncovered an extremely important and highly conserved pathway whose dysregulation causes pleiotropic metabolic abnormalities consistent with metabolic syndrome. LRP6 mutations in humans (R611C, R473Q, R360H, and N433S) are causally linked with autosomal dominant MetS and some of these mutations reduce the canonical Wnt signaling by up to 40% [19, 61].
The LRP6 mutations interfered with LDL clearance, LDLR internalization and clathrin mediated LDL uptake as demonstrated with in vitro and in vivo experimentation in mice [27, 28]. Further contributing to the hyperlipidemia in mutation carriers, the LRP6-R611C mutation increased LDL synthesis, de novo lipogenesis, and VLDL secretion [31].
The pathogenic LRP6 mutation also caused non-alcoholic fatty liver disease due to activation of noncanonical Wnt and downstream TGF pathway thus causing liver inflammation and fibrosis [31, 36].
Patients with LRP6-R611C developed premature onset coronary artery disease which was caused by excessive proliferation of undifferentiated VSMCs. This is now attributed to increased activity of growth factors in the setting of increased non-canonical Wnt signaling and lack of TCF7L2 inhibition of Sp-1 [49, 50].
The increased body fat and insulin resistance seen with patients carrying the LRP6-R611C mutation were found to be due to decreased insulin receptor (IR) expression in the setting of increased non-canonical Wnt signaling and inhibition of TCF7L2 thus leading to activation of mTORC1 pathway and serine phosphorylation of IRS1 as demonstrated with in vitro and in vivo experimentation [35, 56].
Figure 1.
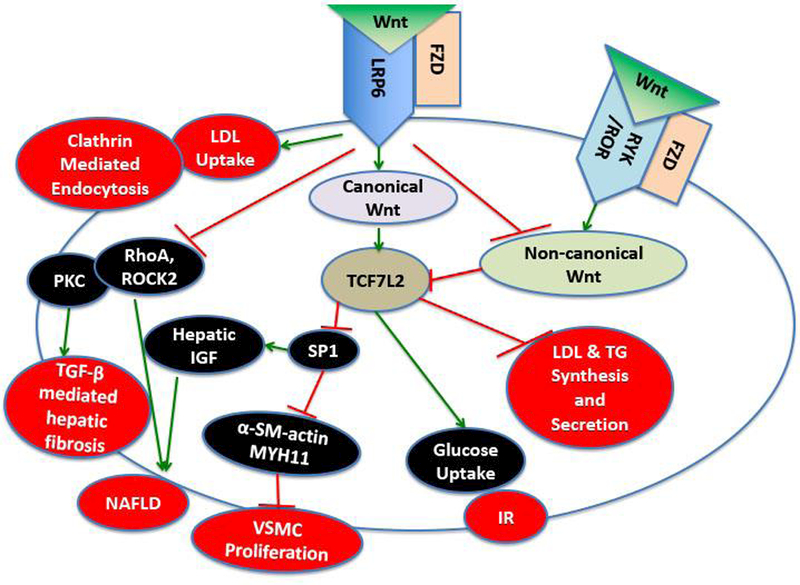
Schematic summary of the Wnt signaling derangements and the different traits of metabolic syndrome. The canonical Wnt pathway is activated when Wnt ligand binds to the Frizzeled (FZD) receptor and the LRP6 co-receptor, this in turn activates LDL uptake through Clathrin mediated endocytosis. Canonical Wnt also activates TCF7L2 transcription factor that inhibits LDL and triglyceride (TG) synthesis and excretion. In addition, TCF7L2 leads to increased glucose uptake via the insulin receptor (IR) and inhibition of vascular smooth muscle cell (VSMC) proliferation, and inhibition non-alcoholic fatty liver disease (NAFLD) via inhibiting hepatic insulin growth factor (IGF) expression. Activation of the canonical Wnt pathway reduces NAFLD and hepatic fibrosis also via inhibition of TGF-β. Interestingly the canonical and non-canonical Wnt pathways reciprocally inhibit each other. Non-canonical Wnt is activated via binding of Wnt ligand to the FZD receptor and RYK/ROR co-receptor.
Highlights:
Metabolic syndrome is a cluster of heritable metabolic traits, which centers around obesity and insulin resistance and is a major contributor to the growing prevalence of cardiovascular disease worldwide.
In genetic study of a kindred with severe manifestations of metabolic syndrome, mutations in the Wnt co-receptor LRP6 gene have been discovered that underlies the pleiotropy of these cardio-metabolic abnormalities.
Impaired canonical Wnt signaling and the activation of non-canonical Wnt signaling constitute the underlying mechanisms for these cardio-metabolic abnormalities and subsequent end-organ damage.
Acknowledgment
This manuscript was supported by R35 HL135767 grant from NIH to Arya Mani.
Abbreviation list:
- ACC1
Acetyl-CoA carboxylase 1
- ApoB
apolipoprotein B
- ApoE
apolipoprotein E
- BAT
brown adipose tissue
- BMI
body mass index
- CAD
coronary artery disease
- Cav1
Caveolin 1
- EGF
epidermal growth factor
- ERK1/2
Extra-cellular signal Regulated Protein Kinases
- FACS
Fluorescence-activated cell sorting
- FASN
Fatty acid synthase
- FoxO1
Forkhead box protein O1
- Fz
frizzled receptor
- GWAS
genome wide association studies
- HA
hemagglutinin
- HMGCR
3-Hydroxy-3-Methylglutaryl-CoA Reductase
- IGF1
insulin growth factor 1
- Insig1
Insulin Induced Gene 1
- IR
insulin receptor
- IRS1
Insulin Receptor Substrate 1
- JAK
Janus kinases
- LDL
low density lipoprotein
- LDLR
LDL receptor
- LEPR
leptin receptor
- LPL
lipoprotein lipase
- LRP6
LDL receptor-related protein 6
- LXRα
Liver X receptor alpha
- MetS
Metabolic syndrome
- mTOR
mechanistic target of rapamycin
- mTORC1
mechanistic target of rapamycin complex 1
- MTP
Microsomal triglyceride transfer protein
- NAFLD
non-alcoholic fatty liver disease
- NASH
non-alcoholic steatohepatitis
- NLK
nemo-like kinase
- NPY
Neuropeptide Y
- PDGF
platelet derived growth factor
- PDGFR
platelet derived growth factor receptor
- PGC1-α
Peroxisome proliferator-activated receptor gamma coactivator 1-alpha
- PKC
Protein kinase C
- PLC
phospholipase C
- PNPLA3
Patatin-like phospholipase domain-containing protein 3
- Rac
Ras-related C3 botulinum toxin substrate, member of the Rho family of GTPases
- RhoA
Ras homolog gene family member A
- RNA
ribonucleic acid
- ROCK2
Rho associated coiled-coil containing protein kinase 2
- ROR
receptor tyrosine kinase-like orphan receptor
- RYK
Derailed/receptor tyrosine kinase
- SCD1
Stearoyl-CoA desaturase-1
- Sp1
specificity protein 1
- SREBP1
sterol regulatory element-binding protein 1
- SREBP2
sterol regulatory element-binding protein 1
- STAT
Signal Transducer and Activator of Transcription proteins
- TCF7L2
Transcription factor 7-like 2
- TG
Triglycerides
- TGF-03B2
transforming growth factor 03B2
- UCP1
Uncoupling Protein 1
- VLDL
very low density lipoprotein
- VSMC
vascular smooth muscle cell
Footnotes
This article was part of the presentations for the 2017 Korean Nutrition Society 50th Anniversary International Conference, November 2–3, 2017 in Seoul Korea.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors have no conflicts of interest to disclose.
References:
- [1].Flegal KM, Kruszon-Moran D, Carroll MD, Fryar CD, Ogden CL. Trends in Obesity Among Adults in the United States, 2005 to 2014. JAMA 2016;315:2284–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Zaitlen N, Kraft P, Patterson N, Pasaniuc B, Bhatia G, Pollack S, et al. Using extended genealogy to estimate components of heritability for 23 quantitative and dichotomous traits. PLoS Genet 2013;9:e1003520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Visscher PM, Brown MA, McCarthy MI, Yang J. Five years of GWAS discovery. Am J Hum Genet 2012;90:7–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Abou Ziki MD, Mani A. Metabolic syndrome: genetic insights into disease pathogenesis. Curr Opin Lipidol 2016;27:162–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Daly CA, Hildebrandt P, Bertrand M, Ferrari R, Remme W, Simoons M, et al. Adverse prognosis associated with the metabolic syndrome in established coronary artery disease: data from the EUROPA trial. Heart 2007;93:1406–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Gami AS, Witt BJ, Howard DE, Erwin PJ, Gami LA, Somers VK, et al. Metabolic syndrome and risk of incident cardiovascular events and death: a systematic review and meta-analysis of longitudinal studies. J Am Coll Cardiol 2007;49:403–14. [DOI] [PubMed] [Google Scholar]
- [7].Komiya Y, Habas R. Wnt signal transduction pathways. Organogenesis 2008;4:68–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kohn AD, Moon RT. Wnt and calcium signaling: beta-catenin-independent pathways. Cell Calcium 2005;38:439–46. [DOI] [PubMed] [Google Scholar]
- [9].Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol 2004;20:781–810. [DOI] [PubMed] [Google Scholar]
- [10].Clevers H Wnt/beta-catenin signaling in development and disease. Cell 2006;127:469–80. [DOI] [PubMed] [Google Scholar]
- [11].Niemann S, Zhao C, Pascu F, Stahl U, Aulepp U, Niswander L, et al. Homozygous WNT3 mutation causes tetra-amelia in a large consanguineous family. Am J Hum Genet 2004;74:558–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Woods CG, Stricker S, Seemann P, Stern R, Cox J, Sherridan E, et al. Mutations in WNT7A cause a range of limb malformations, including Fuhrmann syndrome and Al-Awadi/Raas-Rothschild/Schinzel phocomelia syndrome. Am J Hum Genet 2006;79:402–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Chilosi M, Poletti V, Zamo A, Lestani M, Montagna L, Piccoli P, et al. Aberrant Wnt/beta-catenin pathway activation in idiopathic pulmonary fibrosis. Am J Pathol 2003;162:1495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Edeling M, Ragi G, Huang S, Pavenstadt H, Susztak K. Developmental signalling pathways in renal fibrosis: the roles of Notch, Wnt and Hedgehog. Nat Rev Nephrol 2016;12:426–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Abou Ziki MD, Mani A. Wnt signaling, a novel pathway regulating blood pressure? State of the art review. Atherosclerosis 2017;262:171–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].He X, Semenov M, Tamai K, Zeng X. LDL receptor-related proteins 5 and 6 in Wnt/beta-catenin signaling: arrows point the way. Development 2004;131:1663–77. [DOI] [PubMed] [Google Scholar]
- [17].Lu W, Yamamoto V, Ortega B, Baltimore D. Mammalian Ryk is a Wnt coreceptor required for stimulation of neurite outgrowth. Cell 2004;119:97–108. [DOI] [PubMed] [Google Scholar]
- [18].Kani S, Oishi I, Yamamoto H, Yoda A, Suzuki H, Nomachi A, et al. The receptor tyrosine kinase Ror2 associates with and is activated by casein kinase Iepsilon. J Biol Chem 2004;279:50102–9. [DOI] [PubMed] [Google Scholar]
- [19].Mani A, Radhakrishnan J, Wang H, Mani A, Mani MA, Nelson-Williams C, et al. LRP6 mutation in a family with early coronary disease and metabolic risk factors. Science 2007;315:1278–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Cheng Z, Biechele T, Wei Z, Morrone S, Moon RT, Wang L, et al. Crystal structures of the extracellular domain of LRP6 and its complex with DKK1. Nat Struct Mol Biol 2011;18:1204–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Singh R, Smith E, Fathzadeh M, Liu W, Go GW, Subrahmanyan L, et al. Rare nonconservative LRP6 mutations are associated with metabolic syndrome. Hum Mutat 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Tomaszewski M, Charchar FJ, Barnes T, Gawron-Kiszka M, Sedkowska A, Podolecka E, et al. A common variant in low-density lipoprotein receptor-related protein 6 gene (LRP6) is associated with LDL-cholesterol. Arterioscler Thromb Vasc Biol 2009;29:1316–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Yamamoto H, Komekado H, Kikuchi A. Caveolin is necessary for Wnt-3a-dependent internalization of LRP6 and accumulation of beta-catenin. Dev Cell 2006;11:213–23. [DOI] [PubMed] [Google Scholar]
- [24].Brown SD, Twells RC, Hey PJ, Cox RD, Levy ER, Soderman AR, et al. Isolation and characterization of LRP6, a novel member of the low density lipoprotein receptor gene family. Biochem Biophys Res Commun 1998;248:879–88. [DOI] [PubMed] [Google Scholar]
- [25].Hsieh JC, Lee L, Zhang L, Wefer S, Brown K, DeRossi C, et al. Mesd encodes an LRP5/6 chaperone essential for specification of mouse embryonic polarity. Cell 2003;112:355–67. [DOI] [PubMed] [Google Scholar]
- [26].Kowal RC, Herz J, Goldstein JL, Esser V, Brown MS. Low density lipoprotein receptor-related protein mediates uptake of cholesteryl esters derived from apoprotein E-enriched lipoproteins. Proc Natl Acad Sci U S A 1989;86:5810–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Liu W, Mani S, Davis NR, Sarrafzadegan N, Kavathas PB, Mani A. Mutation in EGFP domain of LDL receptor-related protein 6 impairs cellular LDL clearance. Circ Res 2008;103:1280–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Ye ZJ, Go GW, Singh R, Liu W, Keramati AR, Mani A. LRP6 protein regulates low density lipoprotein (LDL) receptor-mediated LDL uptake. J Biol Chem 2012;287:1335–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Loeffler B, Heeren J, Blaeser M, Radner H, Kayser D, Aydin B, et al. Lipoprotein lipase-facilitated uptake of LDL is mediated by the LDL receptor. J Lipid Res 2007;48:288–98. [DOI] [PubMed] [Google Scholar]
- [30].Bartz F, Kern L, Erz D, Zhu M, Gilbert D, Meinhof T, et al. Identification of cholesterol-regulating genes by targeted RNAi screening. Cell Metab 2009;10:63–75. [DOI] [PubMed] [Google Scholar]
- [31].Go GW, Srivastava R, Hernandez-Ono A, Gang G, Smith SB, Booth CJ, et al. The combined hyperlipidemia caused by impaired Wnt-LRP6 signaling is reversed by Wnt3a rescue. Cell Metab 2014;19:209–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Hatzis P, van der Flier LG, van Driel MA, Guryev V, Nielsen F, Denissov S, et al. Genome-wide pattern of TCF7L2/TCF4 chromatin occupancy in colorectal cancer cells. Mol Cell Biol 2008;28:2732–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Yecies JL, Zhang HH, Menon S, Liu S, Yecies D, Lipovsky AI, et al. Akt stimulates hepatic SREBP1c and lipogenesis through parallel mTORC1-dependent and independent pathways. Cell Metab 2011;14:21–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Hagiwara A, Cornu M, Cybulski N, Polak P, Betz C, Trapani F, et al. Hepatic mTORC2 activates glycolysis and lipogenesis through Akt, glucokinase, and SREBP1c. Cell Metab 2012;15:725–38. [DOI] [PubMed] [Google Scholar]
- [35].Singh R, De Aguiar RB, Naik S, Mani S, Ostadsharif K, Wencker D, et al. LRP6 enhances glucose metabolism by promoting TCF7L2-dependent insulin receptor expression and IGF receptor stabilization in humans. Cell Metab 2013;17:197–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wang S, Song K, Srivastava R, Dong C, Go GW, Li N, et al. Nonalcoholic fatty liver disease induced by noncanonical Wnt and its rescue by Wnt3a. FASEB J 2015;29:3436–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Rotman Y, Koh C, Zmuda JM, Kleiner DE, Liang TJ, Nash CRN. The association of genetic variability in patatin-like phospholipase domain-containing protein 3 (PNPLA3) with histological severity of nonalcoholic fatty liver disease. Hepatology 2010;52:894–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].LoGrasso PV, Feng Y. Rho kinase (ROCK) inhibitors and their application to inflammatory disorders. Curr Top Med Chem 2009;9:704–23. [DOI] [PubMed] [Google Scholar]
- [39].Lu H, Wu JY, Kudo T, Ohno T, Graham DY, Yamaoka Y. Regulation of interleukin-6 promoter activation in gastric epithelial cells infected with Helicobacter pylori. Mol Biol Cell 2005;16:4954–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ross R, Glomset JA. Atherosclerosis and the arterial smooth muscle cell: Proliferation of smooth muscle is a key event in the genesis of the lesions of atherosclerosis. Science 1973;180:1332–9. [DOI] [PubMed] [Google Scholar]
- [41].Virmani R, Kolodgie FD, Burke AP, Farb A, Schwartz SM. Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler Thromb Vasc Biol 2000;20:1262–75. [DOI] [PubMed] [Google Scholar]
- [42].Nurnberg ST, Cheng K, Raiesdana A, Kundu R, Miller CL, Kim JB, et al. Coronary Artery Disease Associated Transcription Factor TCF21 Regulates Smooth Muscle Precursor Cells that Contribute to the Fibrous Cap. Genom Data 2015;5:36–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Congrains A, Kamide K, Oguro R, Yasuda O, Miyata K, Yamamoto E, et al. Genetic variants at the 9p21 locus contribute to atherosclerosis through modulation of ANRIL and CDKN2A/B. Atherosclerosis 2012;220:449–55. [DOI] [PubMed] [Google Scholar]
- [44].Milewicz DM, Kwartler CS, Papke CL, Regalado ES, Cao J, Reid AJ. Genetic variants promoting smooth muscle cell proliferation can result in diffuse and diverse vascular diseases: evidence for a hyperplastic vasculomyopathy. Genet Med 2010;12:196–203. [DOI] [PubMed] [Google Scholar]
- [45].Visel A, Zhu Y, May D, Afzal V, Gong E, Attanasio C, et al. Targeted deletion of the 9p21 non-coding coronary artery disease risk interval in mice. Nature 2010;464:409–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Ross R, Glomset JA. The pathogenesis of atherosclerosis (second of two parts). N Engl J Med 1976;295:420–5. [DOI] [PubMed] [Google Scholar]
- [47].Sano H, Sudo T, Yokode M, Murayama T, Kataoka H, Takakura N, et al. Functional blockade of platelet-derived growth factor receptor-beta but not of receptor-alpha prevents vascular smooth muscle cell accumulation in fibrous cap lesions in apolipoprotein E-deficient mice. Circulation 2001;103:2955–60. [DOI] [PubMed] [Google Scholar]
- [48].Kozaki K, Kaminski WE, Tang J, Hollenbach S, Lindahl P, Sullivan C, et al. Blockade of platelet-derived growth factor or its receptors transiently delays but does not prevent fibrous cap formation in ApoE null mice. Am J Pathol 2002;161:1395–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Keramati AR, Singh R, Lin A, Faramarzi S, Ye ZJ, Mane S, et al. Wild-type LRP6 inhibits, whereas atherosclerosis-linked LRP6R611C increases PDGF-dependent vascular smooth muscle cell proliferation. Proc Natl Acad Sci U S A 2011;108:1914–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Srivastava R, Zhang J, Go GW, Narayanan A, Nottoli TP, Mani A. Impaired LRP6-TCF7L2 Activity Enhances Smooth Muscle Cell Plasticity and Causes Coronary Artery Disease. Cell Rep 2015;13:746–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Lin X, Wang Z, Gu L, Deuel TF. Functional analysis of the human platelet-derived growth factor A-chain promoter region. J Biol Chem 1992;267:25614–9. [PubMed] [Google Scholar]
- [52].Park GH, Plummer HK, Krystal GW 3rd. Selective Sp1 binding is critical for maximal activity of the human c-kit promoter. Blood 1998;92:4138–49. [PubMed] [Google Scholar]
- [53].Zhang X, Diab IH, Zehner ZE. ZBP-89 represses vimentin gene transcription by interacting with the transcriptional activator, Sp1. Nucleic Acids Res 2003;31:2900–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Kita T, Yamashita T, Sasaki N, Kasahara K, Sasaki Y, Yodoi K, et al. Regression of atherosclerosis with anti-CD3 antibody via augmenting a regulatory T-cell response in mice. Cardiovasc Res 2014;102:107–17. [DOI] [PubMed] [Google Scholar]
- [55].Tanaka M, Fukui M, Tomiyasu K, Akabame S, Nakano K, Yamasaki M, et al. Eosinophil count is positively correlated with coronary artery calcification. Hypertens Res 2012;35:325–8. [DOI] [PubMed] [Google Scholar]
- [56].Liu W, Singh R, Choi CS, Lee HY, Keramati AR, Samuel VT, et al. Low density lipoprotein (LDL) receptor-related protein 6 (LRP6) regulates body fat and glucose homeostasis by modulating nutrient sensing pathways and mitochondrial energy expenditure. J Biol Chem 2012;287:7213–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999;98:115–24. [DOI] [PubMed] [Google Scholar]
- [58].Barbera MJ, Schluter A, Pedraza N, Iglesias R, Villarroya F, Giralt M. Peroxisome proliferator-activated receptor alpha activates transcription of the brown fat uncoupling protein-1 gene. A link between regulation of the thermogenic and lipid oxidation pathways in the brown fat cell. J Biol Chem 2001;276:1486–93. [DOI] [PubMed] [Google Scholar]
- [59].Benzler J, Andrews ZB, Pracht C, Stohr S, Shepherd PR, Grattan DR, et al. Hypothalamic WNT signalling is impaired during obesity and reinstated by leptin treatment in male mice. Endocrinology 2013;154:4737–45. [DOI] [PubMed] [Google Scholar]
- [60].Ouchi N, Higuchi A, Ohashi K, Oshima Y, Gokce N, Shibata R, et al. Sfrp5 is an anti-inflammatory adipokine that modulates metabolic dysfunction in obesity. Science 2010;329:454–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Singh R, Smith E, Fathzadeh M, Liu W, Go GW, Subrahmanyan L, et al. Rare nonconservative LRP6 mutations are associated with metabolic syndrome. Hum Mutat 2013;34:1221–5. [DOI] [PMC free article] [PubMed] [Google Scholar]