

Abstract
Background
Atypical X-linked severe combined immunodeficiency (X-SCID) is a variant of cellular immunodeficiency due to hypomorphic mutations in the interleukin 2 receptor gamma (IL2RG) gene. Due to a leaky clinical phenotype, diagnosis and appropriate treatment are challenging in these patients.
Case presentation
We report a 16-year-old patient with a Tlow B+ NK+ cellular immunodeficiency due to a novel nonsense mutation in exon 8 (p.R328X) of the IL2RG gene. Functional impairment of the IL2RG was confirmed by IL2-Janus kinase 3-signal transducer and activator of transcription signaling pathway investigation. In addition, the characteristics of the mutations previously described in 39 patients with an atypical phenotype were reviewed and analyzed from the literature.
Conclusion
This is the first report of an atypical X-SCID phenotype due to an exon 8 mutation in the IL2RG gene. The variability in the phenotypic spectrum of classic X-SCID associated gene highlights the necessity of multi-disciplinary cooperation vigilance for a more accurate diagnostic workup.
Electronic supplementary material
The online version of this article (10.1186/s13223-018-0317-y) contains supplementary material, which is available to authorized users.
Keywords: Interleukin 2 receptor gamma, Atypical severe combined immunodeficiency, Hypomorphic mutations
Background
Interleukin 2 receptor gamma (IL2RG) is an important signaling component for IL2, IL4, IL7, IL9, IL15, and IL21 [1]. The gene encodes a common gamma chain (γC) that is essential in the ontogeny and function of immune cells, in particular T and NK cells. Mutations in the gene result in X-linked severe combined immunodeficiency (X-SCID) [2].
Approximately 200 unique mutations in the IL2RG gene have been identified to date in more than 320 patients with X-SCID. Missense and nonsense mutations comprise around 48%, while insertion/deletion and splicing mutations account for 29% and 23%, respectively according to the mutation database (Table 1). The mutations lead to the production of a nonfunctional γC or prevent the protein from being produced, resulting in an arrest in lymphocyte development.
Table 1.
Summary of IL2 RG unique mutation* and comparison with hypomorphic mutations
| Parameters | Total unique mutation | Total observed | Typical SCID | Atypical SCID | p value |
|---|---|---|---|---|---|
| Exons | |||||
| Exon 1 | 12 | 15 | 13 | 2 | 0.6642 |
| Exon 2 | 23 | 28 | 28 | 0 | 0.0574 |
| Exon 3 | 47 | 72 | 68 | 4 | 0.1377 |
| Exon 4 | 38 | 47 | 45 | 2 | 0.1998 |
| Exon 5 | 38 | 106 | 87 | 19 | 0.0046** |
| Exon 6 | 19 | 36 | 34 | 2 | 0.4018 |
| Exon 7 | 14 | 41 | 32 | 9 | 0.0253** |
| Exon 8 | 9 | 11 | 10 | 1a | 1.0000 |
| Othersb | 3 | 6 | 6 | 0 | 1.0000 |
| Type of mutation | |||||
| Missense | 61 | 138 | 108 | 30 | < 0.0001** |
| Nonsense | 36 | 73 | 70 | 3 | 0.0533 |
| Insertion | 15 | 21 | 20 | 1 | 0.7113 |
| Deletion | 43 | 52 | 50 | 2 | 0.1386 |
| Splicing | 35 | 62 | 59 | 3 | 0.1692 |
| Othersc | 13 | 16 | 16 | 0 | 0.3929 |
| Total | 202 | 362 | 323 | 39 | |
*Summary based on NCBI Clinvar database (http://www.ncbi.nlm.nih.gov/clinvar/), LOVD gene database [25] (http://www.ncbi.nlm.nih.gov/lovd/home.php?select_db=IL2RG) and OMIM database (http://www.omim.org/)
**Fisher’s exact test was used to analyze the association of exons or mutation type in the distribution of different clinical phenotypes (Typical SCID vs Atypical SCID) observed and p ≤ 0.05 was regarded as significant
aPatient in the present study
bIncludes large deletions
cIncludes complex mutations, disruption of poly-A addition, variants within the first codon
In typical X-SCID, the disease is characterized by an almost complete absence of T and NK cells, and nearly normal or high numbers of functionally deficient B cells (T–B+NK− phenotype). Infants with X-SCID are highly susceptibility to bacterial and opportunistic infections. Additional features include protracted diarrhea, rash, fever, pneumonia and sepsis. The disease is usually lethal within the 1st year of life unless reconstitution of the immune system is carried out.
Different mutations in the IL2RG gene have also been shown to be associated with less severe phenotypic variants. Several patients having hypomorphic IL2RG mutation with a milder form of combined immunodeficiency, termed “atypical X-SCID”, have been described previously [3–9]. Some patients might be less susceptible to infections, the reduction of T cells is relatively moderate and a normal lymphocyte proliferation assay may be observed. In addition, some of these patients might not be detected by newborn screening programs for SCID [10].
Due to poorly defined clinical and immunological phenotypes, the diagnosis is usually established later in childhood or even in adulthood, and the appropriate treatment is thus delayed.
Here we describe a novel nonsense mutation in the IL2RG gene consisting of a single nucleotide substitution at exon 8, in which a normal count of NK cells was found in peripheral blood.
Case presentation
The patient, a 16 years old male of Kurdish ethnicity, was admitted to the pediatric lung and allergy service of Astrid Lindgren Children’s Hospital at Karolinska University Hospital due to chronic airway hypersensitivity and recurrent sinopulmonary infections. He is the third child of consanguineous parents with a family history of several early deaths due to lung failure on the maternal side (Fig. 1). He had a normal vaccination history but a medical history of four hospitalizations due to enteroviral infection (at age 16 months presenting with skin rash and diarrhea), chronic cough and fever (at age 18 months due to Moraxella catarrhalis), otitis media, adenopathy and shingles (leading to tympanostomy at the age of 2), pneumonia and an asthmatic reaction (at the age of 6).
Fig. 1.

a Family pedigree of the patient(s). b Sanger sequencing
At the age of 8 years, a computed tomography was performed due to a progression of his pulmonary disease which revealed bronchiectasis and a right middle lobe atelectasis. Immunologic profiles were investigated as previously described [11]. Although a complete blood count and immunoglobulin levels were normal, lymphocyte populations were measured. Low CD4+ and CD8+ T cell numbers, with normal numbers of B and NK cells were detected (Fig. 1 and Table 2). The patient had low specific cell-mediated immune response in activated whole blood using mitogens and antigen, such as pokeweed mitogen (PWM), candida antigen etc. (Table 3). However, normal response to mitogens phytohemagglutinin (PHA) and concanavalin A (Con A) by CD4+ T cells, but not CD8+ T cells were detected. The observation suggested that PHA and ConA stimulations for CD4+ T cells may be different from CD8+ T cells. Despite his combined immunodeficiency, the patient was free from opportunistic infections and his condition improved with temporary substitution of subcutaneous immunoglobulin and prophylactic antibiotics.
Table 2.
Immunologic characteristics of the patient with hypomorphic/atypical X-linked severe combined immunodeficiency
| Parameters | 2004a (5 years) | 2010a, b (8 years) | 2011c (9 years) | 2012 (10 years) | Normal range |
|---|---|---|---|---|---|
| White blood cells (× 103 cells/µL) | 17↑ | 15↑ | 6.5 | 7.2 | 5.0–13.0 |
| Absolute number of lymphocytes (× 103 cells/µL) | 4.8 | 1.9 | 1.3↓ | 1.3↓ | 1.5–6.5 |
| Absolute number of neutrophils (× 103 cells/µL) | 11↑ | 12↑ | 4.4 | 4.7 | 2.0–8.4 |
| Absolute number of monocytes (cells/µL) | 760 | 900↑ | 700 | 700 | 200–800 |
| Absolute CD3+ (cells/µL) | – | 100↓ | 270↓ | 310↓ | 800–3500 |
| Absolute CD3+CD4+ (cells/µL) | – | 60↓ | 140↓ | 150↓ | 300–2000 |
| Absolute CD3+CD8+ (cells/µL) | – | 40↓ | 120↓ | 130↓ | 300–1800 |
| CD4/CD8 ratio | – | 1.5 | 1.1 | 1.1 | 0.9–2.6 |
| Absolute CD19+ (cells/µL) | – | 100↓ | 240 | 360 | 200–600 |
| Absolute CD16/56+ (cells/µL) | – | 890 | 690 | 1040 | 70–1200 |
| IgG (mg/dL) | – | 1150 | 1050 | 1130 | 610–1450 |
| IgG1 (mg/dL) | – | – | 711 | – | 350–910 |
| IgG2 (mg/dL) | – | – | 165 | – | 85–330 |
| IgG3 (mg/dL) | – | – | 146 | – | 20–104 |
| IgA (mg/dL) | – | 127 | NI | 77 | 70–365 |
| IgM (mg/dL) | – | 115 | NI | 85 | 24–210 |
NI Not investigated
aMeasurement at the time of an acute infection
bIgG were measured before receiving subcutaneous IgG replacement therapy
cIgG subclasses values were measured after 1 month withdrawal of subcutaneous IgG replacement therapy at the age of 9
Table 3.
Proliferation and specific antibody tests of the patient with hypomorphic/atypical X-linked severe combined immunodeficiency
| Parameters | Result | Year (age) | Normal range |
|---|---|---|---|
| CD4 PHA (c/µL) | 293 | 2012 (10 years) | 170–3499 |
| CD8 PHA (c/µL) | 16↓ | 2012 (10 years) | 76–3640 |
| CD4 ConA (c/µL) | 616↓ | 2012 (10 years) | 620–3800 |
| CD8 ConA (c/µL) | 44↓ | 2012 (10 years) | 180–1757 |
| CD4 PWM (c/µL) | 241 | 2012 (10 years) | 233–2189 |
| CD8 PWM (c/µL) | 15↓ | 2012 (10 years) | 50–549 |
| CD19 PWM (c/µL) | 61 | 2012 (10 years) | 42–741 |
| CD4 Staph enterotoxin (c/µL) | 1242 | 2012 (10 years) | 553–7746 |
| CD8 Staph enterotoxin (c/µL) | 275 | 2012 (10 years) | 123–2365 |
| CD4 Influenza (c/µL) | 136 | 2012 (10 years) | 19–1050 |
| CD8 Influenza (c/µL) | 0↓ | 2012 (10 years) | 5–2020 |
| CD4 Tetanus toxin (c/µL) | 0↓ | 2012 (10 years) | 5–306 |
| CD8 Tetanus toxin (c/µL) | 0↓ | 2012 (10 years) | 5–14 |
| CD4 PPD (c/µL) | 0↓ | 2012 (10 years) | 11–14 |
| CD8 PPD (c/µL) | 0↓ | 2012 (10 years) | 5–29 |
| CD4 Candida (c/µL) | 5↓ | 2012 (10 years) | 51–1014 |
| CD8 Candida (CD4, CD8) | 0↓ | 2012 (10 years) | 5–49 |
| CD4 Pneumococcus (c/µL) | 2↓ | 2012 (10 years) | 5–269 |
| CD8 Pneumococcus (c/µL) | 0↓ | 2012 (10 years) | 5–13 |
| CD4 Varicella zoster (c/µL) | 45 | 2012 (10 years) | 5–157 |
| CD8 Varicella zoster (c/µL) | 4↓ | 2012 (10 years) | 5–23 |
| Anti diphtheria (IgG, IU/mL) | 0.53 | 2010 (8 years)a | 0.1–56.2 |
| Anti tetanus (IgG, IU/mL) | 1.4 | 2010 (8 years)a | 0.09–12.87 |
| Anti tetanus (IgG1, mg/L) | 17 | 2010 (8 years)a | 0.9–228.5 |
| Anti Haemophilus influenzae B (IgG, mg/L) | 1.5 | 2010 (8 years)a | 0.15–29.5 |
| Anti PPV–23 (IgG, mg/L) | 45 | 2010 (8 years)a | 9.2–22.5 |
| Anti PPV–23 (IgG2, mg/L) | 25 | 2010 (8 years)a | 0.8–122.4 |
| Anti CMV (IgG, EIA, titer) | 21,000↑ | 2011 (9 years)a | 0–300 |
| Anti CMV (IgM, EIA, titer) | 0 | 2011 (9 years)a | 0–10 |
| Anti EBV EBNA (IgG, EIA, U/mL) | 10 | 2011 (9 years)a | 0–20 |
| Anti EBV VCA (IgM, EIA, titer) | 17 | 2011 (9 years)a | 0–25 |
| Anti HSV (IgG, EIA, titer) | 150 | 2011 (9 years)a | 0–230 |
| Anti VZV (IgG, EIA, titer) | 2600↑ | 2011 (9 years)a | 0–350 |
| Polio virus 1,2,3 neutralization test | 512,128, 2048 | 2011 (9 years)a | 300–3800 |
| Anti measles (IgG, EIA, titer) | 1500 | 2011 (9 years)a | 500–2500 |
| Anti Helicobacter pylori (IgG, EIA, titer) | 0 | 2010 (8 years)a | 0–50 |
Assays were performed in the Karolinska University Hospital according to the method described previously [26]
PHA phytohemagglutinin test, ConA concanavalin A, PWM pokeweed mitogen, PPV-23: Pneumococcal polysaccharide vaccine Pneumovax23, CMV: Cytomegalovirus, EBV: Epstein–Barr virus, EBNA: EBV nuclear antigen, VCA: viral capsid antigen, HSV: Herpes simplex virus, VZV: varicella-zoster virus, PPD: Purified protein derivative
aAll specific antibodies tested at the age of 8 was before IgG replacement and at the age of 9 after 1 month withdrawal of IgG replacement
In order to identify the molecular defect, whole exome sequencing (WES) was performed. As the patient was born in a consanguineous family and showed a family history of recurrent infections and early death on the maternal side, an autosomal recessive or X-linked inheritances pattern was expected. Analysis of all variants were performed according to a standard pipeline described previously [12]; we identified 2 homozygous (autosomal) and 5 hemizygous (X-linked) variants which were absent from dbSNP database and 1000 Genome database (Additional file 1: Table S1). Comparing with the primary immunodeficiency genes database, the only variant consistent with the patient’s immunological phenotype was a novel nonsense mutation, p.R328X (c.982C>T) in exon 8 of the IL2RG gene (Fig. 1). Based on this finding, the therapeutic plan of the patient was changed and he became a potential candidate for allogeneic hematopoietic stem cell transplantation.
Since the mutation causes a 42 amino acid truncation of the intracellular domain of the γC, including of the Janus kinase 3 (JAK3) binding site (Fig. 2), we investigated the expression of members of the IL2/JAK3 signaling pathway by western blot. Western blot (Fig. 3) demonstrated absence of IL2RG, suggesting that the mutation caused degradation of the molecule. In addition, IL2 stimulation activated JAK3 and signal transducer and activator of transcription signaling 5 (STAT5) proteins in cells from a healthy control but no activation was observed in the patient; indicating an impairment of IL-2 signaling. STAT5 expression was observed in both the control and the patient, while the main JAK3 isoform (1124 amino acids, 115kDA) was only observed in the control. However, the intensity of second isoform of JAK3 (1094 amino acids, lacking part of the kinase domain) was stronger in the patient. When the blot was reprobed with another anti-JAK3 antibody (binding to the kinase domain), expression were observed only in the control samples, indicating that the expression pattern of JAK3 is modified in the patient (Fig. 3).
Fig. 2.
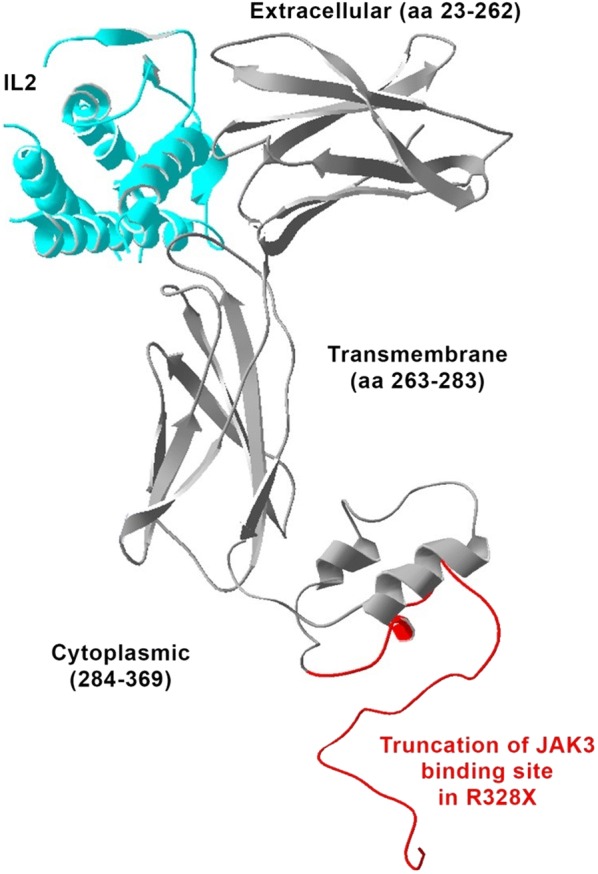
Schematic representation of the IL-2 receptor with a 42 amino-acids deletion of the intracellular domain of the γC shown in red
Fig. 3.
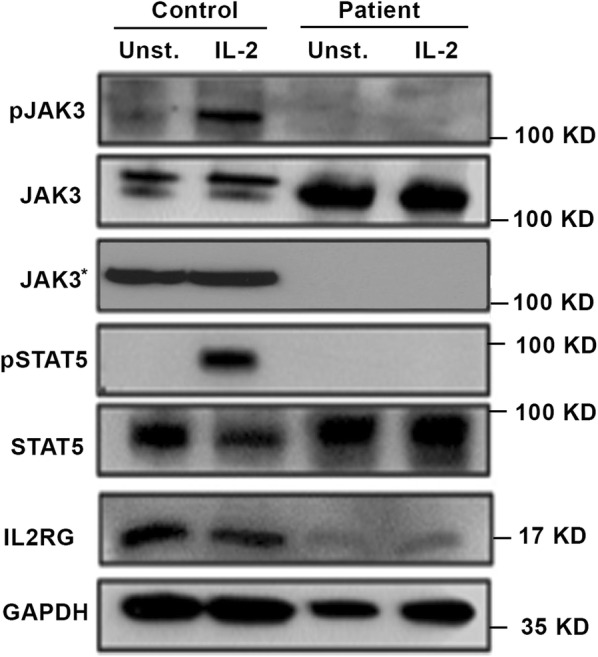
JAK3-STAT5 signaling pathway in patients with atypical X-SCID. Expression of activated JAK3 and STAT5, as well as total JAK3, JAK3* (reprobe with anti-JAK3 antibody binding to the kinase domain), STAT5, IL2R and GAPDH in unstimulated or IL2 stimulated cells, were evaluated by Western blot
Discussion and conclusions
Approximately 10% of the reported IL2RG mutations have been associated with atypical phenotypic variants (including Tlow/− B+ NK+ and Tlow/− Blow NK+/low/−, Table 1). The majority of the atypical patients present a “milder” form of immunodeficiency. Database summary shows that nearly half of the total IL2RG mutations are located in exon 5 (29.4%) and exon 3 (19.9%). Mutations in exon 5, which encodes the extracellular domain including the highly conserved WSXWS motif: a region essential for proper protein folding and thereby efficient intracellular transport as well as extracellular receptor binding, are expected to disrupt the γC configuration. Likewise, a similar clinically severe effect is predicted when the mutations occur in exon 3, which will modify the amino acids directly or close to the four conserved cysteine residues.
Mutation in exons 5 and 7, on the other hand, are significantly associated with atypical X-SCID (Fig. 4). Mutation of p.R222C in exon 5 (14 cases) is the most frequent variants among patients with NK+ phenotype [6] which leads to a differential impairment of cytokines pathways (IL21 > IL15/IL2 > IL4). Furthermore, this mutation leads to a defect in lymphocyte function rather than immune cell development since normal differentiation of lymphocytes (B and T cells) as well as a normal thymus gland has been observed [8].
Fig. 4.

IL2RG gene location of pathogenic hypomorphic mutations [27, 28] in 39 reported atypical X-SCID patients [1, 4, 6, 7, 9, 15, 17, 21, 22, 29–36] (S: Signaling sequence; C: Conserved cysteine residues, W: WSXWS box; B: box1/box2 domain)
Mutations in exon 8 are relatively rare (~ 3% including the present case). Our patient is the first atypical X-SCID with an exon 8 mutation while nine cases were reported with a mutation in exon 7 (Table 1). Exon 7 and exon 8 encode the intracellular domains of IL2RG. The exon 8 nonsense mutation in our patient results in a truncation of 42 amino acids in the cytoplasmic domain of the γC. The mutation putatively reduced the binding site of JAK3 (Fig. 2), most likely preventing the association of JAK3 with the γC. In line with this finding, Hofman et al. 2004 showed that JAK3 is dependent on γC binding for membrane localization and in the absence of IL2R, JAK3 ends up in the cytosol [13]. Moreover, Jayshree et al. [14] demonstrated that IL2 regulates the transcription of JAK3 through the concentration-dependent effects of phosphorylation-dephosphorylation of JAK3. The truncation of γC abolished the JAK3 binding site and resulted in no or low phosphorylation of JAK3. Accumulation of low or unphosphorylated JAK3 may thus modify its own transcription and hence downregulate the expression of main hematopoietic isoform JAK3. Previous reports have also described patients with atypical X-SCID with a mutation in the cytoplasmic domain of γC [3, 15]. The mutation in exon 7 (p.L293Q) impaired the association with JAK3 [16]. On the contrary, multiple cases of typical SCID phenotype have been observed with mutations in exon 8 which result in the truncation of the last 45 [17], 48 amino acids [18, 19] as well as substitution of 40 amino acids [18] and 56 amino acids [20] in the cytoplasmic domain. The discrepancies of the clinical features in patients with cytoplasmic domain mutations suggest that the level of interaction between γC and JAK3 may play an important role in defining the phenotypic manifestations.
NK+ phenotype was mainly observed in patients having mutation in exon 3 [21, 22], exon 5 [6, 18] and exon 7 [15]. Our patient is the first NK+ phenotype with an exon 8 mutation. The residual population of NK cells in selected patients with γC deficient may be due to the contribution of IL12 to IL15-independent NK cell expansion. This critical role of IL12 is evident not only by its effect on IFN-γ production by NK cells and NK cell blastogenesis during viral infections, but also in the Ilr2g−/− mouse model, where a 30-fold expansion of the absolute number of Ly49H+KLRG1+ NK cells has been noted [23]. On the other hand, increasing concentrations of dysfunctional γC due to a hypomorphic mutation could partly compensate STAT5 phosphorylation after IL15 stimulation, but not after IL7 stimulation [24]. Therefore, the presence of an NK+ phenotype in the patient suggests that the underlying IL2RG mutation results in a preferential retention of IL15 mediated signaling.
Delayed diagnostic as well as limited sample material has prevented us from investigating whether there is prolonged transplacental maternal T cell engraftment. Further investigation is required to elucidate the relationship between the mutations of the γC and clinical manifestations in patients with X-SCID to achieve a better classification of the disease. In addition, the heterogeneity of clinical presentation in primary immunodeficient patients highlights the role of a more accurate diagnostic work up with the aid of multi-omics molecular diagnostics.
Additional file
Additional file 1: Table S1. Homozygous (autosomal) and hemizygous (X-linked) variants identified by whole exome sequencing. Table S2. Summary of genetic and clinical characteristics of patients with hypomorphic/atypical X-linked severe combined immunodeficiency.
Authors’ contributions
CKL and HA interpreted data, analyzed the results, drafted and edited the manuscript. SKA performed the western blot. MS provided clinical care to the patient. LH provided guidance, supervised the findings of this work and edited the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We would like to thank the patient and family for providing the samples and their approval to share his clinical course to the healthcare and scientific community.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
The relevant data is summarized and provided within the manuscript. The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Consent for publications
Not applicable.
Ethics approval and consent to participate
Informed consent for the performed studies was obtained from the patient and his parents, in accordance with the principles of the ethics committee of the Karolinska Institutet.
Funding
This work was supported by the Swedish Research Council and funds from the Karolinska Institutet.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- X-SCID
X-linked atypical severe combined immunodeficiency
- IL2RG
interleukin 2 receptor gamma
- γC
common gamma chain
- PHA
phytohemagglutinin
- Con A
concanavalin A
- PWM
pokeweed mitogen
- WES
whole exome sequencing
- JAK3
Janus kinase 3
- STAT5
signal transducer and activator of transcription signaling 5
Contributor Information
Che Kang Lim, Email: che.kang.lim@ki.se.
Hassan Abolhassani, Email: hassan.abolhassani@ki.se.
Sofia K. Appelberg, Email: sofia.appelberg@ki.se
Mikael Sundin, Email: mikael.sundin@ki.se.
Lennart Hammarström, Phone: +46 8 5248 3586, Email: lennart.hammarstrom@ki.se.
References
- 1.Notarangelo LD, Giliani S, Mazza C, Mella P, Savoldi G, Rodriguez-Perez C, et al. Of genes and phenotypes: the immunological and molecular spectrum of combined immune deficiency. Defects of the gamma(c)-JAK3 signaling pathway as a model. Immunol Rev. 2000;178:39–48. doi: 10.1034/j.1600-065X.2000.17812.x. [DOI] [PubMed] [Google Scholar]
- 2.Noguchi M, Yi H, Rosenblatt HM, Filipovich AH, Adelstein S, Modi WS, et al. Interleukin-2 receptor gamma chain mutation results in X-linked severe combined immunodeficiency in humans. Cell. 1993;73(1):147–157. doi: 10.1016/0092-8674(93)90167-O. [DOI] [PubMed] [Google Scholar]
- 3.Brooks EG, Schmalstieg FC, Wirt DP, Rosenblatt HM, Adkins LT, Lookingbill DP, et al. A novel X-linked combined immunodeficiency disease. J Clin Investig. 1990;86(5):1623–1631. doi: 10.1172/JCI114884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.DiSanto JP, Rieux-Laucat F, Dautry-Varsat A, Fischer A, de Saint Basile G. Defective human interleukin 2 receptor gamma chain in an atypical X chromosome-linked severe combined immunodeficiency with peripheral T cells. Proc Natl Acad Sci USA. 1994;91(20):9466–9470. doi: 10.1073/pnas.91.20.9466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Felgentreff K, Perez-Becker R, Speckmann C, Schwarz K, Kalwak K, Markelj G, et al. Clinical and immunological manifestations of patients with atypical severe combined immunodeficiency. Clin Immunol. 2011;141(1):73–82. doi: 10.1016/j.clim.2011.05.007. [DOI] [PubMed] [Google Scholar]
- 6.Fuchs S, Rensing-Ehl A, Erlacher M, Vraetz T, Hartjes L, Janda A, et al. Patients with T(+)/low NK(+) IL-2 receptor gamma chain deficiency have differentially-impaired cytokine signaling resulting in severe combined immunodeficiency. Eur J Immunol. 2014;44(10):3129–3140. doi: 10.1002/eji.201444689. [DOI] [PubMed] [Google Scholar]
- 7.Mella P, Imberti L, Brugnoni D, Pirovano S, Candotti F, Mazzolari E, et al. Development of autologous T lymphocytes in two males with X-linked severe combined immune deficiency: molecular and cellular characterization. Clin Immunol. 2000;95(1 Pt 1):39–50. doi: 10.1006/clim.2000.4842. [DOI] [PubMed] [Google Scholar]
- 8.Sharfe N, Shahar M, Roifman CM. An interleukin-2 receptor gamma chain mutation with normal thymus morphology. J Clin Investig. 1997;100(12):3036–3043. doi: 10.1172/JCI119858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ursini MV, Gaetaniello L, Ambrosio R, Matrecano E, Apicella AJ, Salerno MC, et al. Atypical X-linked SCID phenotype associated with growth hormone hyporesponsiveness. Clin Exp Immunol. 2002;129(3):502–509. doi: 10.1046/j.1365-2249.2002.01823.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.King JR, Hammarstrom L. Newborn screening for primary immunodeficiency diseases: history, current and future practice. J Clin Immunol. 2018;38(1):56–66. doi: 10.1007/s10875-017-0455-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abolhassani H, Wang N, Aghamohammadi A, Rezaei N, Lee YN, Frugoni F, et al. A hypomorphic recombination-activating gene 1 (RAG1) mutation resulting in a phenotype resembling common variable immunodeficiency. J Allergy Clin Immunol. 2014;134(6):1375–1380. doi: 10.1016/j.jaci.2014.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fang M, Abolhassani H, Lim CK, Zhang J, Hammarstrom L. Next Generation sequencing data analysis in primary immunodeficiency disorders—future directions. J Clin Immunol. 2016;36(Suppl 1):68–75. doi: 10.1007/s10875-016-0260-y. [DOI] [PubMed] [Google Scholar]
- 13.Hofmann SR, Lam AQ, Frank S, Zhou YJ, Ramos HL, Kanno Y, et al. Jak3-independent trafficking of the common gamma chain receptor subunit: chaperone function of Jaks revisited. Mol Cell Biol. 2004;24(11):5039–5049. doi: 10.1128/MCB.24.11.5039-5049.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mishra J, Waters CM, Kumar N. Molecular mechanism of interleukin-2-induced mucosal homeostasis. Am J Physiol Cell Physiol. 2012;302(5):C735–C747. doi: 10.1152/ajpcell.00316.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schmalstieg FC, Leonard WJ, Noguchi M, Berg M, Rudloff HE, Denney RM, et al. Missense mutation in exon 7 of the common gamma chain gene causes a moderate form of X-linked combined immunodeficiency. J Clin Investig. 1995;95(3):1169–1173. doi: 10.1172/JCI117765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Russell SM, Johnston JA, Noguchi M, Kawamura M, Bacon CM, Friedmann M, et al. Interaction of IL-2R beta and gamma c chains with Jak1 and Jak3: implications for XSCID and XCID. Science. 1994;266(5187):1042–1045. doi: 10.1126/science.7973658. [DOI] [PubMed] [Google Scholar]
- 17.Niemela JE, Puck JM, Fischer RE, Fleisher TA, Hsu AP. Efficient detection of thirty-seven new IL2RG mutations in human X-linked severe combined immunodeficiency. Clin Immunol. 2000;95(1 Pt 1):33–38. doi: 10.1006/clim.2000.4846. [DOI] [PubMed] [Google Scholar]
- 18.Puck JM, Pepper AE, Henthorn PS, Candotti F, Isakov J, Whitwam T, et al. Mutation analysis of IL2RG in human X-linked severe combined immunodeficiency. Blood. 1997;89(6):1968–1977. [PubMed] [Google Scholar]
- 19.Ting SS, Leigh D, Lindeman R, Ziegler JB. Identification of X-linked severe combined immunodeficiency by mutation analysis of blood and hair roots. Br J Haematol. 1999;106(1):190–194. doi: 10.1046/j.1365-2141.1999.01527.x. [DOI] [PubMed] [Google Scholar]
- 20.Kumaki S, Ochs HD, Kuropatwinski KK, Konno T, Timour MS, Cosman D, et al. A novel mutant gammac chain from a patient with typical phenotype of X-linked severe combined immunodeficiency (SCID) has partial signalling function for mediating IL-2 and IL-4 receptor action. Clin Exp Immunol. 1999;115(2):356–361. doi: 10.1046/j.1365-2249.1999.00792.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jamal A, Upton JEM. IL2RG: a series of three novel mutations with clinical manifestations. LymphoSign J. 2016;3(3):8. doi: 10.14785/lymphosign-2016-0003. [DOI] [Google Scholar]
- 22.Ginn SL, Smyth C, Wong M, Bennetts B, Rowe PB, Alexander IE. A novel splice-site mutation in the common gamma chain (gammac) gene IL2RG results in X-linked severe combined immunodeficiency with an atypical NK+ phenotype. Hum Mutat. 2004;23(5):522–523. doi: 10.1002/humu.9235. [DOI] [PubMed] [Google Scholar]
- 23.Sun JC, Ma A, Lanier LL. Cutting edge: IL-15-independent NK cell response to mouse cytomegalovirus infection. J Immunol. 2009;183(5):2911–2914. doi: 10.4049/jimmunol.0901872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marcais A, Viel S, Grau M, Henry T, Marvel J, Walzer T. Regulation of mouse NK cell development and function by cytokines. Front Immunol. 2013;4:450. doi: 10.3389/fimmu.2013.00450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Puck JM. IL2RGbase: a database of gamma c-chain defects causing human X-SCID. Immunol Today. 1996;17(11):507–511. doi: 10.1016/S0167-5699(96)80903-0. [DOI] [PubMed] [Google Scholar]
- 26.Marits P, Wikstrom AC, Popadic D, Winqvist O, Thunberg S. Evaluation of T and B lymphocyte function in clinical practice using a flow cytometry based proliferation assay. Clin Immunol. 2014;153(2):332–342. doi: 10.1016/j.clim.2014.05.010. [DOI] [PubMed] [Google Scholar]
- 27.Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Itan Y, Shang L, Boisson B, Ciancanelli MJ, Markle JG, Martinez-Barricarte R, et al. The mutation significance cutoff: gene-level thresholds for variant predictions. Nat Methods. 2016;13(2):109–110. doi: 10.1038/nmeth.3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wada T, Yasui M, Toma T, Nakayama Y, Nishida M, Shimizu M, et al. Detection of T lymphocytes with a second-site mutation in skin lesions of atypical X-linked severe combined immunodeficiency mimicking Omenn syndrome. Blood. 2008;112(5):1872–1875. doi: 10.1182/blood-2008-04-149708. [DOI] [PubMed] [Google Scholar]
- 30.Stephan V, Wahn V, Le Deist F, Dirksen U, Broker B, Muller-Fleckenstein I, et al. Atypical X-linked severe combined immunodeficiency due to possible spontaneous reversion of the genetic defect in T cells. N Engl J Med. 1996;335(21):1563–1567. doi: 10.1056/NEJM199611213352104. [DOI] [PubMed] [Google Scholar]
- 31.Somech R, Roifman CM. Mutation analysis should be performed to rule out gammac deficiency in children with functional severe combined immune deficiency despite apparently normal immunologic tests. J Pediatr. 2005;147(4):555–557. doi: 10.1016/j.jpeds.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 32.Estevez OA, Ortega C, Fernandez S, Aguado R, Rumbao J, Perez-Navero J, et al. A novel IL2RG mutation presenting with atypical T(−)B(+)NK+ phenotype: rapid elucidation of NK cell origin. Pediatr Blood Cancer. 2014;61(1):178–179. doi: 10.1002/pbc.24717. [DOI] [PubMed] [Google Scholar]
- 33.Jones AM, Clark PA, Katz F, Genet S, McMahon C, Alterman L, et al. B-cell-negative severe combined immunodeficiency associated with a common gamma chain mutation. Hum Genet. 1997;99(5):677–680. doi: 10.1007/s004390050428. [DOI] [PubMed] [Google Scholar]
- 34.Jung EY, Heike T, Katamura K, Kimata H, Ohmori K, Morikawa Y, et al. X-linked severe combined immunodeficiency with gamma delta T cells. Acta Paediatrica Japonica Overseas Ed. 1997;39(4):442–447. doi: 10.1111/j.1442-200X.1997.tb03614.x. [DOI] [PubMed] [Google Scholar]
- 35.Fugmann SD, Muller S, Friedrich W, Bartram CR, Schwarz K. Mutations in the gene for the common gamma chain (gammac) in X-linked severe combined immunodeficiency. Hum Genet. 1998;103(6):730–731. doi: 10.1007/pl00008710. [DOI] [PubMed] [Google Scholar]
- 36.Morelon E, Dautry-Varsat A, Le Deist F, Hacein-Bay S, Fischer A, de Saint Basile G. T-lymphocyte differentiation and proliferation in the absence of the cytoplasmic tail of the common cytokine receptor gamma c chain in a severe combined immune deficiency X1 patient. Blood. 1996;88(5):1708–1717. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1. Homozygous (autosomal) and hemizygous (X-linked) variants identified by whole exome sequencing. Table S2. Summary of genetic and clinical characteristics of patients with hypomorphic/atypical X-linked severe combined immunodeficiency.
Data Availability Statement
The relevant data is summarized and provided within the manuscript. The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.