

Abstract
Supramolecular nanostructures formed through self-assembly can have energy landscapes, which determine their structures and functions depending on the pathways selected for their synthesis and processing, and on the conditions they are exposed to after their initial formation. We report here on the structural damage that occurs in supramolecular peptide amphiphile nanostructures, during freezing in aqueous media, and the self-repair pathways that restore their functions. We found that freezing converts long supramolecular nanofibers into shorter ones, compromising their ability to support cell adhesion, but a single heating and cooling cycle reverses the damage and rescues their bioactivity. Thermal energy in this cycle enables noncovalent interactions to reconfigure the nanostructures into the thermodynamically preferred long nanofibers, a repair process that is impeded by kinetic traps. In addition, we found that nanofibers disrupted during freeze drying also exhibit the ability to undergo thermal self-repair and recovery of their bioactivity, despite the extra disruption caused by the dehydration step. Following both freezing and freeze drying, which shorten the 1D nanostructures, their self-repair capacity through thermally driven elongation is inhibited by kinetically trapped states, which contain highly stable noncovalent interactions that are difficult to rearrange. These states decrease the extent of thermal nanostructure repair, an observation we hypothesize applies to supramolecular systems in general and is mechanistically linked to suppressed molecular exchange dynamics.
Keywords: supramolecular nanostructures, self-assembly, self-repair, biomaterials, regenerative medicine, cell-nanostructure interactions
Graphical Abstract:
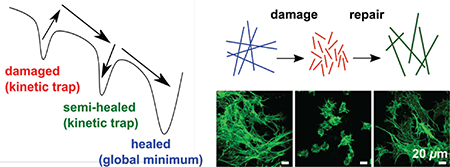
Supramolecular self-assembly directs biological molecules into quaternary protein structures, lipid membranes, and DNA double helices, and is thus essential to cell function. This natural phenomenon has inspired the design of self-assembling biomaterials for a variety of applications including regenerative medicine1–3, drug delivery,4 cancer therapies,5 and immuno-engineering.6 Although supramolecular biomaterials show great promise, a recently identified aspect of their structure and function is proper selection of their self-assembly pathway in order to optimize bioactivity.7–9 In recent work some pathways have been found to result in structures with greater cytotoxicity.8, 9 Even after the molecules have traversed an undesired self-assembly pathway, it may be possible to “repair” the “damaged” supramolecular structures, since their noncovalent nature provides inherent reversibility.10–12 The self-repair ability of supramolecular materials has been previously demonstrated in the context of mechanical defects. For example, supramolecular elastomers spontaneously self-heal when broken pieces are brought into contact, because hydrogen bonds readily reform at the fracture site.13, 14 Heat15, 16 or light17 can induce rearrangement of intermolecular interactions near a fracture site, allowing the network to fill in the defect. While these strategies can mend mechanical defects, they cannot repair defective supramolecular nanostructures that have undergone incorrect self-assembly pathways and reached non-functional positions in their energy landscapes. For example, misfolded protein configurations are considered non-functional positions in their energy landscape.18 The repair of misfolded proteins has been reported but the processes are extremely cumbersome, requiring finely-tuned mixtures of small molecules and chaperones,19–21 because the energy landscapes of protein folding are extremely complex.
Our laboratory has developed a broad platform of self-assembling molecules known as peptide amphiphiles (PAs), which in canonical form contain a hydrophobic tail, a β-sheet forming peptide domain to trigger self-assembly into highly one-dimensional fiber-like nanostructures, and charged amino acid headgroups to promote solubility in water.1, 22 In some cases, the peptide sequence terminates in a bioactive domain designed to interact directly with cell receptors or bind specific signaling proteins such as growth factors.2, 23–25 We have demonstrated their functionality in a variety of in vivo pre-clinical models for the regeneration of bone25, cartilage23, muscle,26 blood vessels,24 as well as spinal cord27 and peripheral nerves.28 For regenerative medicine applications, our group has extensively studied the use of PAs with the peptide sequence V3A3E3 (Figure S1). This PA self-assembles into high-aspect ratio nanofibers that can mimic the architecture of mammalian extracellular matrices, and also forms self-supporting gels when the negatively charged glutamic acids are ionically crosslinked with divalent counterions (usually Ca2+).29–32 Since PA nanostructures in solution are dynamic, we typically freeze dry PA solutions and store the dried powders until they are to be used in biological applications. Knowing that self-assembly of PA molecules into nanostructures is sensitive to the preparation pathway,8, 9 we were motivated to study how the freeze drying process affects the PA nanoscale filaments. Amyloid fibrils, another self-assembling peptide system, can fragment into shorter pieces33 or undergo liquid crystalline phase transformations34 when exposed to freeze-thaw cycles. Other organic nanostructures such as proteins,35 nanoparticles,36 and liposomes,37 as well as cells38 are often preserved by freezing or freeze drying, and thus sometimes are structurally damaged by ice nucleation, the use of extreme temperatures, and dehydration.35–38 The objective of this work has been to characterize freezing and freeze drying damage to PA nanostructures and explore mechanisms and strategies to repair their structure and function.
Before freezing and freeze drying V3A3E3 PA solutions, we equilibrated the nanostructures at their thermodynamic minimum by applying a previously discovered, thermally activated self-assembly pathway (annealing at 80°C for 30 minutes followed by slow cooling overnight).9, 31 These solutions contained 1 wt% (8.66 mM) PA dissolved in 15 mM NaOH, conditions that lead to a low charge density on the nanostructures, which in turn favor β-sheet formation and thus formation of long nanofibers.9 Therefore, the annealing procedure results in formation of highly viscous solutions. When this solution is flash frozen and allowed to thaw at room temperature, its viscosity decreases drastically (Figure 1a-b, Figure S2–4), suggesting disruption of the nanofiber structure. Using nanoparticle tracking analysis (NTA) to visualize light scattered by nanofibers (Figure S5), we estimated their lengths in annealed and flash frozen solutions, and found that annealed PA nanofibers tend to be well over 10 μm long, while flash frozen nanofibers are generally less than 5 μm (Figure 1c). Viewed under cross-polarizers, annealed solutions of PA nanostructures contain large birefringent monodomains (Figure 1d), while flash frozen PA contains smaller ones (Figure 1e), indicating shorter fibers forming domains with less alignment. These observations are consistent with the lyotropic liquid crystalline nature of aqueous solutions formed by the nanoscale filaments.31 We examined individual nanofibers by cryogenic transmission electron microscopy (cryoTEM) from both annealed (Figure 1f) and flash frozen solutions (see Figure 1g), confirming the previous observations. Taken together, viscosity, light scattering, and microscopy data demonstrate that flash freezing long nanofibers in solution fractures them into significantly shorter ones. Although long nanofibers should be thermodynamically favored, we infer the short freeze-damaged nanofibers are kinetically trapped in a local energy minima and do not spontaneously grow back to their original lengths during our time of observation.
Figure 1.

(a) Schematic representation summarizing the effects of flash freezing annealed PA solutions. (b) Viscosity of annealed and flash frozen PA solutions (p < 0.0001, two-tailed paired t-test; error bars represent standard error of mean). (c) Box-and-whisker plot of apparent nanofiber lengths in annealed and flash frozen PA using nanoparticle tracking analysis (NTA) video frames (p < 0.0001, two-tailed unpaired t-test) (d, e) Cross-polarized light micrograph of annealed and flash frozen PA, respectively. (f, g) CryoTEM of annealed and flash frozen PA, respectively. (h, i) SEM micrographs of annealed and flash frozen PA gels, respectively. (j, k) Confocal micrographs of MC3T3 mouse preosteoblast cells encapsulated inside annealed and flash frozen PA gels, respectively (insets are TEM micrographs of cell cross-sections; scale bar for inset: 1 μm). (l) Fluorescence recovery after photobleaching (FRAP) experiments on FITC-conjugated BSA within annealed and flash frozen PA solutions and gels (error bars are 95% confidence intervals).
Knowing that flash freezing shortens nanofibers in PA solutions, we studied the bioactivity implications for the PA gels made up of these one-dimensional nanostructures. The nanostructures can localize and deliver growth factors and provide a scaffold to support cell infiltration.23, 25, 39 We prepared PA gels by introducing CaCl2 to annealed and flash frozen PA solutions, which crosslinks negatively charged glutamic acids with positively charged Ca2+ ions, and then examined the resultant structure and rheological properties (Figure S6–8) of the gels, as well as cell and protein interactions with the gels. Scanning electron micrographs (SEM) reveal that annealed PA gels contain long nanofibers (Figure 1h) while flash frozen PA gels contain shorter nanofibers (Figure 1i), which is consistent with our observations in solution. Since cells are sensitive to nanoscale topographical cues,40–42 we studied whether this difference would affect cellular response to the nanofibers within the PA gels. We encapsulated MC3T3 mouse preosteoblasts in annealed and flash frozen PA gels, allowed the cells to equilibrate for 24 hours, and then imaged the cell morphology using actin staining. Confocal microscopy images show that cells within annealed PA gels spread and elongate (Figure 1j), but those within flash frozen PA gels remain more rounded (Figure 1k). For a closer view of the interface between cells and PA, we cut thin sections of cells inside PA gels and observed them with transmission electron microscopy (TEM). These thin sections revealed that cells inside the annealed PA gels have a few small protrusions along otherwise smooth plasma membranes (Figure 1j, inset), while cells inside the flash frozen PA gels show intense membrane blebbing (Figure 1k, inset). Previous work has shown that cell membrane blebbing occurs in the earliest stages of cell spreading, with blebs maturing to adherent lamellipodia in later stages.43–45 Therefore, we hypothesize that the cell membrane continues blebbing after unsuccessful attempts to attach to shorter, freeze-damaged fibers. These results are consistent with previous work from our laboratory, which showed that chemically identical PA surfaces support cell adhesion better when fibers are longer.9 Taken together, these results indicate that freezing damage is detrimental for either encapsulation of cells within PA gels or the recruitment of endogenous cells when they need to infiltrate implanted PA gels.
Next we investigated protein interactions with annealed and flash frozen PA gels, since PA gels are known to be effective vehicles for growth factor delivery.23, 25 We examined the effect of flash freezing on the ability of PA nanofibers to bind proteins using bovine serum albumin (BSA). Although BSA has an overall negative charge, it contains both negatively and positively charged residues, and is known to attach to either positively or negatively charged surfaces.46 We mixed FITC (fluorescein isothiocyanate)-conjugated BSA into annealed and flash frozen PA solutions, induced gelation with CaCl2, and imaged the gels with confocal microscopy. We performed fluorescence recovery after photobleaching (FRAP) experiments by bleaching 10 μm diameter circles in the gels and monitoring fluorescence recovery. Since photobleaching is irreversible, fluorescence recovery only occurs if unbleached FITC-BSA diffuses into the bleached area. Less or slower recovery indicates less protein mobility, signifying stronger association with nanofibers. FRAP data shows that annealed and flash frozen PA are equally capable of immobilizing BSA within their gels, with only 10–20% of the initial fluorescence recovered in both cases (Figure 1l). This suggests that protein retention by nanofibers derives from surface properties, such as electrostatic forces and is independent of fiber length. Furthermore, BSA diffuses freely through PA solutions and recovers approximately 90% of initial fluorescence (Figure 1l), demonstrating that gelation of PA or immobilization of nanofibers into a solid structure is responsible for immobilization of proteins. For applications where PA gels are intended only as growth factor delivery vehicles, freezing damage to the nanostructures does not seem to be harmful. Although interactions with cells are sensitive to nanofiber length, our data indicate that their ability to retain proteins is controlled by their surface properties and therefore the chemical structure of PA molecules.
Considering the sensitivity of cells to nanofiber length, we proceeded to determine if this effect was reversible. In light of recent work on the energy landscapes of peptide amphiphile self-assembly,9 we hypothesized that we could restore the thermodynamically preferred long nanofibers by overcoming the activation barrier to reach this energy minimum. To test this hypothesis, we re-annealed the flash frozen PA (Figure 2a, left pathway), giving it the same amount of thermal energy used to create the original annealed PA. The viscosity of flash frozen PA was found to increase upon re-annealing (Figure 2b), suggesting the formation of long nanofibers again. Although viscosity does not reach that of the original annealed PA solutions (Figure 2b), several techniques show the reappearance of long nanofibers. Cross-polarized light microscopy shows large birefringent monodomains (Figure 2c) and cryoTEM reveals long nanofibers (Figure 2d), similar to the original annealed PA (Figure 1). Most importantly, cells spread and elongate within re-annealed PA gels (Figure 2e), demonstrating that flash frozen and re-annealed nanofibers regain the ability to promote cell adhesion. Using FRAP, we confirmed that flash frozen and re-annealed PA gels still have the ability to immobilize BSA protein (Figure S9). These data demonstrate that, given thermal energy to rearrange supramolecular interactions, freeze-damaged PA nanostructures successfully self-repair.
Figure 2.

(a) Two flash freezing pathways for PA solutions, with (left) and without (right) annealing prior to freezing. (b) Viscosity of annealed, flash frozen, and re-annealed PA (*p = 0.0002 **p = 0.0006, two-tailed paired t-tests; error bars represent standard error of mean). (c) Cross-polarized micrograph of re-annealed PA. (d) CryoTEM of flash frozen and re-annealed PA. (e) Confocal micrograph of MC3T3 mouse preosteoblast cells encapsulated within flash frozen and re-annealed PA gel and visualized by actin staining (green). (f) Viscosity of PA solutions after annealing freshly dissolved material, and after annealing freshly dissolved material that was flash frozen prior to annealing (p = 0.0341, two-tailed paired t-test; error bars represent standard error of mean). (g) CryoTEM of freshly dissolved PA solution. (h) CryoTEM of freshly dissolved PA solution that has been flash frozen (right pathway).
Although re-annealing successfully repairs structural and functional damage, we wanted to understand why the solution viscosity does not return to its original value. We hypothesized that when PA solutions are flash frozen, forces exerted by ice crystals mechanically break the supramolecular nanofibers. However, since they were formed under conditions of high charge screening, the long annealed nanofibers likely contain highly cohesive β-sheets,9 which become kinetically trapped in the short fibers. We omitted the first annealing step before flash freezing, and instead flash froze freshly dissolved PA and immediately annealed it (Figure 2a, right pathway). We found that this flash freezing pathway did not result in a reduction of the post-annealing viscosity. In fact, this pathway resulted in a small but statistically significantly increase in viscosity after annealing (Figure 2f). CryoTEM images of freshly dissolved PA (Figure 2g) and flash frozen freshly dissolved PA (Figure 2h) show no obvious differences between the two conditions, in contrast to the drastic change observed after flash freezing annealed PA (Figure 1). Flash freezing annealed PA activates a pathway that results in a metastable state of short fibers with cohesive β-sheets, an energy landscape position that is not reached when flash freezing non-equilibrium nascent nanostructures in freshly dissolved PA. Indeed, circular dichroism (CD) experiments show that flash frozen PA solutions retain a characteristic β-sheet signature (Figure S10), in contrast to thermodynamically stable short nanofibers that exhibit a random coil CD signature, reported in previous work from our laboratory.9 Using super-resolution microscopy, our laboratory previously showed that molecular exchange occurs between different nanofibers in solution,47 and we presume that this exchange is necessary for elongation of nanofibers. We hypothesize that annealed PA nanofibers have slower exchange rates than freshly dissolved metastable assemblies, possibly due to a higher coherence length of β-sheets along the nanofiber. This β-sheet configuration and slower exchange rate persists into the short freeze-damaged nanofibers, making them more resistant to change during re-annealing. In contrast the un-annealed assemblies prior to freezing are in non-equilibrium states which must have shorter β-sheet coherence lengths, and are thus more likely to exchange molecules and molecular clusters to build stable nanofibers. Furthermore, we speculate that nanofiber endcaps in metastable assemblies contain “defects” in the β-sheet structure, and are thus highly dynamic areas where PA molecules can more easily join the nanofiber or break off to join a longer nanofiber. When long annealed nanofibers are physically fractured, perhaps the newly created endcaps retain order in their hydrogen bonds and are thus less dynamic. We also note that small angle x-ray scattering (SAXS) of PA nanofibers show very minor changes during flash freezing and subsequent re-annealing (Figure S11–13), demonstrating that the fibrous geometry is maintained, even as hydrogen bonds are rearranging and nanofiber length is changing.
To further explore how freezing affects PA assemblies, we performed experiments where we slowly froze annealed PA nanofibers, as opposed to flash freezing them in liquid nitrogen. A slower freezing process should result in larger ice crystals, as well as prolong the time that nanostructures in solution are exposed to cold temperatures and ice nucleation. Instead of flash freezing the PA solutions with liquid nitrogen, the solutions were allowed to equilibrate in a −20°C freezer. Slowly freezing PA solutions decreases the viscosity (Figure 3a), similar to flash freezing PA (Figure 1b, Figure S14–15). However, we found that re-annealing slowly frozen PA solutions allows the solutions to recover, on average, their original viscosity (Figure 3a). To determine if exposure to cold temperature alone disrupted nanostructures, we equilibrated PA solutions in a 4°C refrigerator overnight. This refrigeration, after which no solid ice was observed, does not decrease the PA solution’s viscosity (Figure 3b, Figure S14–15). Furthermore, cross-polarized microscopy shows smaller monodomains in slowly frozen PA solutions (Figure 3c), similar to flash frozen PA solutions (Figure 1e), and larger monodomains in refrigerated PA solutions (Figure 3d), similar to the original annealed PA (Figure 1d). In both slowly frozen (Figure 3e) and refrigerated (Figure 3f) samples, cryoTEM images show long nanofibers extending beyond the field of view, which must not be sufficiently large to capture fiber length differences at this scale. Interestingly, cryoTEM was able to capture the shorter length of flash frozen PA nanofibers (Figure 1g), which showed an approximate 75% decrease in viscosity (Figure 1b) relative to annealed PA, while slowly frozen PA nanofibers showed an approximate 53% decrease (Figure 3a). These data suggest that slowly frozen nanofibers are longer than their flash frozen counterparts, possibly because slower ice nucleation reduces the rate of nanofiber rupture, thus requiring less repair. Furthermore, slowly frozen nanofibers may have less cohesive β-sheets than flash frozen nanofibers, which allows them to rearrange more easily during subsequent annealing. While flash frozen nanofibers solidify long before their hydrogen bonds can be destabilized, PA nanofibers in the liquid portion of a partially frozen solution may adsorb to ice crystals, which may destabilize their hydrogen bonds. For proteins, infrared spectroscopy has shown that, in partially frozen systems, adsorption to ice crystals can result in loss of secondary structure.48
Figure 3.

(a) Viscosity of annealed, slowly frozen, and re-annealed PA (*p = 0.0062 **p = 0.0053, two-tailed paired t-tests (error bars represent standard error of mean). (b) Viscosity of annealed PA and refrigerated PA (two-tailed paired t-test). (c, d) Cross-polarized micrographs of slowly frozen PA and refrigerated PA, respectively. (e, f) CryoTEM of slowly frozen PA and refrigerated PA, respectively.
After characterizing the self-repair of PA nanofibers from freezing, both rapidly with liquid nitrogen and slowly within a freezer, we were motivated to explore the consequences on bioactivity after freeze drying. From a functional point of view in clinical settings, the nanostructures should be stored dried rather than frozen in the hydrated state before coming into contact with living tissues. Since flash freezing breaks long nanofibers into shorter ones (Figure 1), we expected that freeze drying would have a similar effect. Also, since the volume of PA nanofibers contains water molecules that contribute to their self-assembly,32 we expected that the added dehydration step would disrupt the nanofibers even further than freezing alone. Since the presence of water triggers self-assembly and directs hydrogen bonding of PA molecules as a result of hydrophobic collapse, we hypothesized that nanofibers would easily “re-assemble” after freeze drying as the nanostructured material is reconstituted as aqueous solutions. We freeze dried (flash freeze with liquid nitrogen followed by sublimation of water under vacuum) and reconstituted annealed PA solutions (Figure 4a, left pathway) and found that the viscosity decreases, similar to what we found in the case of the flash freezing pathway (Figure 4b). Re-annealing this reconstituted PA (Figure 4a, left pathway) increases the viscosity, but not to the original value (Figure 4b). Despite the lower viscosity, PA nanofibers “re-created” after freeze drying form large aligned monodomains (Figure 4c), appear long by cryoTEM (Figure 4d), and the gels they form are capable of supporting cell spreading and adhesion (Figure 4e) as well as retaining BSA protein (Figure S9). Thus these nanostructures remain bioactive, similar to those exposed to flash freezing and re-annealed.
Figure 4.

(a) Two freeze drying pathways for PA, with (left) and without (right) annealing prior to freeze drying. (b) Viscosity of annealed, freeze dried, and re-annealed PA (*p = 0.0007 **p = 0.0007 ***p = 0.0102, two-tailed paired t-tests, error bars represent standard error of mean). (c) Cross-polarized light micrograph of freeze dried and re-annealed PA. (d) CryoTEM of freeze dried and re-annealed PA. (e) Confocal micrograph of MC3T3 mouse preosteoblast cells encapsulated inside freeze dried and re-annealed PA gel, visualized by actin staining. (f) Viscosity of PA after annealing freshly dissolved solution, and annealing freshly dissolved solution that has been freeze dried (not significant, two-tailed paired t-test, error bars represent standard error of mean). (g) CryoTEM of PA immediately after reconstitution of powder, freeze dried from an annealed PA solution (left pathway). (h) CryoTEM of PA immediately after reconstitution of powder, freeze dried from a freshly dissolved solution (right pathway).
We then investigated whether omitting the annealing step before freeze drying would prevent the decrease in viscosity (Figure 4a, right pathway), similar to our experiment with flash freezing freshly dissolved PA before annealing (Figure 2a, right pathway). Bypassing annealing before freeze drying will generally prevent a decrease in viscosity (Figure 4f), similar to bypassing annealing before flash freezing (Figure 2f). However, this trend is not as consistent when freeze drying PA, with some experiments showing a decrease in viscosity and others showing an increase, and thus on average a change in viscosity is not observed (Figure S4). CryoTEM images of PA immediately after reconstitution, following freeze drying from annealed PA solution (Figure 4g) and freshly dissolved solution (Figure 4h), show no obvious differences, and both also appear similar to the original freshly dissolved PA (Figure 2g). While the cryoTEM micrographs appear very similar, we speculate that freezing annealed PA preserves the cohesive β-sheets of long nanofibers, which persist into the melted (flash frozen) or dried (freeze dried) states. Although nanostructures are disrupted during freeze drying, the preserved β-sheets diminish the system’s ability to rearrange during subsequent annealing. These preserved β-sheets can be avoided if PA is not annealed before freeze drying, however, in contrast to the case of just freezing this processing pathway will not always prevent a decrease in viscosity (Figure S4). We therefore suspected that the dehydration process was causing other changes to PA self-assembly in addition to those introduced by freezing alone, which do not appear to occur in a reproducible manner.
To investigate how freeze drying affects self-assembly of the PAs investigated, we varied freeze drying conditions. We hypothesized that freeze drying could increase the electrostatic charge on headgroups of PA molecules by converting the glutamic acids into their conjugate salt form. The added negative charges would add repulsive forces among PA molecules, thus decreasing their propensity to self-assemble and changing their energy landscape.9 Previous work on freeze drying of proteins has shown that their ionization states in solution persist into the dried state, leading to a phenomenon called “pH memory.”49, 50 Because the solution pH determines the ionization states, freeze dried proteins “remember” the pH of the aqueous solution from which they were dried.49, 50 This “remembered” ionization state persists if the proteins are reconstituted into organic solvents, but is “erased” if proteins are reconstituted into aqueous media with a new pH value.49 In the case of freeze drying PAs, the three headgroups are “remembered” as either protonated glutamic acids or deprotonated glutamate salts. Although these ionization states can change upon reconstitution with aqueous media, the relative amount of glutamic acids versus glutamate salts in the PA powder will be determined by the previous freeze drying step. If this powder is reconstituted with pure water, which has no buffering capacity, the amount of conjugate acid versus conjugate base that was present can continue to exert an effect. During self-assembly, the three glutamic acid side chains interact with each other and with neighboring molecules, which can affect their ionization state. Under our specific stoichiometric conditions (15 mM NaOH to 8.66 mM PA, which contain 26 mM glutamic acids), we expect a wide range of possible ionization state configurations within the assembled nanostructures. When the PA is freshly dissolved and metastable, these configurations are likely to be highly dynamic, and perhaps highly variable from sample to sample. Upon drying in the presence of a non-volatile base like NaOH, we speculate that this variable configuration of ionization states gets kinetically trapped, with some Na+ ions bound to the glutamate residues, maintaining them as glutamate salts, and others existing as phase separated NaOH powder. Thus, the large variation in possible ionization state configurations and NaOH powder amounts after freeze drying can lead to variations in energy landscapes upon reconstitution of PA.
To better control the ionization states of the PA’s glutamic acids after freeze drying, we freeze dried PA from solutions containing a volatile base (NH4OH instead of NaOH). Previous work has shown that the pH memory effect in proteins is suppressed when they are dried from solutions containing volatile buffers.50 The drying process removes the volatile buffers, leaving behind uncharged residues, effectively removing any “memory” of the ionization state in buffered solution.50 Unlike NaOH, NH4OH cannot remain in the freeze dried powder as a solid, and NH4+ ions are not likely to remain bound to glutamate residues, as the removal of NH3 gas under vacuum would deplete the NH4+ ions by Le Chatelier’s principle (NH4+COO- → NH3 + COOH; ionic reactions during freeze drying are further discussed in the Supporting Information, Section S15). Thus, our strategy was to add excess volatile NH4OH (1% volume/volume) to deprotonate PA as much as possible before freeze drying, and to add a specific amount of NaCl (non-volatile salt) to control the number of glutamic acid residues that remain ionized after freeze drying. While PA powder freeze dried from NaOH dissolves easily when reconstituted in pure water, PA powder freeze dried from NH4OH is not soluble (Figure 5a), suggesting that the glutamic acids are left non-ionized and the base has also been removed. However, when we add 15 mM NaCl to the solution before freeze drying, the resulting powder was found to be water soluble (Figure 5a). In contrast, the PA powder is also not soluble if the NaCl is added later (by freeze drying without salt and reconstituting in 15 mM NaCl). This suggests that the positive Na+ ions form sodium glutamate salts with the PA, increasing its solubility above that of PA with glutamic acids. We measured UV-visible absorbance of the supernatant in these samples to estimate the concentration of soluble peptide (Figure 5b), which confirmed our visual observations on the solubility.
Figure 5.

(a) Photographs of PA solutions after freeze drying and reconstitution, using solvents indicated in chart. (b) UV-visible absorbance spectra of freeze dried PA solutions from panel a. (c) UV-visible absorbance spectra of freeze dried PA solutions immediately after reconstitution, with indicated concentrations of NaCl present during freeze drying. (d) Approximate pH values of freeze dried PA solutions after reconstitution and subsequent annealing, with indicated concentrations of NaCl present during freeze drying. (e) Viscosity of freeze dried PA solutions after annealing, with indicated concentrations of NaCl present during freeze drying (15mM NaCl sample is significantly different from all other samples, p < 0.05, two-tailed paired t-test; error bars represent standard error of mean).
We then varied the concentration of NaCl in solution before freeze drying to see if we could control the extent of ionization in the final dried PA. We dissolved 8.66 mM (1 wt%) PA with a fixed amount of NH4OH (1% volume/volume), and varied the concentration of NaCl in the solution (4, 8.5, 15, 26, 50, 150 mM). These samples, containing a total of 26 mM ionizable carboxylic acids (8.66 mM PA, three glutamic acids per V3A3E3 PA), were freeze dried and reconstituted with pure water. UV-visible spectroscopy shows that as low as 4 mM NaCl increases the PA solubility, and that PA solubility improves up to a maximum at 15 mM NaCl (Figure 5c). To further explore the idea that the NaCl controls the ionization state of the PA’s glutamic acids, we then annealed the fully soluble PA solutions (those containing 15 – 150 mM NaCl) and measured the approximate pH. Since PA immediately after reconstitution is metastable and highly dynamic, we compared the annealed solutions, which should contain more stable nanostructures. The PA solution freeze dried from 15 mM NaCl is more acidic than PA freeze dried from higher NaCl concentrations (Figure 5d), suggesting that it contains fewer carboxylic acids in the deprotonated, ionized state than samples with more added NaCl. All samples had been exposed to the same amount of NH4OH base and NaCl alone does not affect the pH of water (Figure S16). At 15 mM NaCl, there are enough Na+ ions to maintain approximately two out of three carboxylate anions per PA molecule, while at least 26 mM of Na+ ions is sufficient to stabilize all three carboxylate groups. Taken together, the solubility and pH data suggest that the presence of NaCl during freeze drying determines how many glutamate salt residues on the PA’s headgroups will be maintained into the dried state, and thus the amount of negative charges on the PA nanostructures after reconstitution in pure water. In this specific case, PA freeze dried with 15 mM NaCl contains less negative charge than PA freeze dried with higher amounts of NaCl since this sample has less Na+ ions to maintain negatively charged carboxylate groups during freeze drying.
Since increased charges on the PA are known to promote repulsive forces and change the energy landscape,9 we expected that the change in the PA’s charged state, induced by certain freeze drying conditions, would affect PA self-assembly. Indeed, PA freeze dried from 15 mM NaCl is significantly more viscous than PA samples with higher NaCl concentrations (Figure 5e, Figure S17), which is likely due to the lower negative charge on the 15 mM NaCl sample, suggested by the solution’s more acidic pH (Figure 5d). Previous work has shown that increased pH can increase the electrostatic charge on acidic PA molecules, which disrupts their self-assembly,51 and our experiments show that simply adding more NaOH base to a PA solution will result in lower viscosity after annealing (Figure S19). When approximately all three glutamic acids are deprotonated instead of two, the added charge frustrates self-assembly by introducing repulsion between PA molecules, shifting the thermodynamic minimum to relatively shorter, less cohesive nanofibers. The self-repair capacity of PA nanostructures relies on the “functional” long nanofibers existing at an energy minimum that can be accessed with equilibration through the use of thermal energy. The NH4OH and NaCl experiments suggest that certain processing steps, particularly freeze drying, can shift this energy landscape minimum and thus complicate self-repair. When freeze drying freshly dissolved PA in NaOH, these energy landscape shifts varied from sample to sample, due to the wide range of possible ionization states within highly dynamic nanostructures and the ability of Na+ ions to exist in either phase-separated NaOH powder or sodium glutamate conjugates on the PA. Using NH4OH and NaCl, we were able to better control the PA’s ionization states and shed light on the possible mechanism through which freeze drying causes shifts in energy landscape (further discussion of PA ionization states in these two buffers can be found in Supporting Information, Section S15). Taken together, the solubility, pH, and viscosity experiments (Figure 5) suggest that counterions present during freeze drying can increase the net charge on the PA by maintaining conjugate salt groups, which can change the energy minimum in the PA’s energy landscape. In this specific case, Na+ ions present during freeze drying of V3A3E3 PA maintain the glutamic acid residues as negatively charged glutamate salts, which introduces charge repulsion and disrupts self-assembly, thus shifting the energy minimum to shorter nanofibers that produce a less viscous solution. When the source of these Na+ ions is NaOH, these energy landscape shifts happen in a highly variable way because the amount of glutamate salts after freeze drying is not easily controlled. This variation is reduced when NaCl is the source of Na+ ions, and this way the amount of glutamate salts after freeze drying can be controlled.
We have described a thermally activated self-repair pathway for PA nanostructures, which can restore long nanofibers after they break into shorter ones. Long nanofibers are superior to shorter ones in supporting cell adhesion, and the repair pathway was able to recover this biological function. This self-repair pathway works by overcoming the activation barrier to return the system to the thermodynamically preferred long nanofibers. Some thermal histories will result in kinetic traps that impede this thermal repair process, specifically annealing prior to freezing or freeze drying will result in short nanofibers containing highly cohesive internal structures that suppress the ability of nanostructures to re-elongate. Since this self-repair pathway succeeds when thermal energy reconfigures the system towards an energy minimum containing the long nanofibers necessary for bioactivity, any pathway that shifts this minimum will complicate the repair process. This was demonstrated by our freeze drying experiments, where the dehydration process can increase the electrostatic charges on PA nanostructures. In contrast, freezing alone should not change the PA’s net charge and shift the energy minimum, so “incomplete” self-repair may be attributed purely to kinetic traps. In principle, longer heating times or higher temperatures should overcome the energy barrier to achieve “complete” self-heating, but could lead to solvent evaporation or precipitation, thus altering solution concentrations (Supporting Information, Section S17). Such kinetic traps should therefore be avoided through the design of processing pathways. The self-repair capacity of supramolecular nanostructures is highly possible, but must be optimized by careful selection of processing pathways, which no doubt are necessary in the actual use of the systems for biological applications. We hypothesize that the self-repair capacity of supramolecular nanostructures when their environment disrupts them should be broadly applicable to many chemical structures.
Supplementary Material
ACKNOWLEDGMENTS
Synthesis of PAs and study of the self-assembly landscapes were supported by the Center for Bio-Inspired Energy Sciences (CBES), an Energy Frontiers Research Center (EFRC) funded by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under award number DE-SC0000989. Biological studies were funded by the NIH/NIDCR (grant no. 2R01DE015920–11) and the Northwestern University Center for Regenerative Nanomedicine Catalyst Award. C.H.C. has received support from a National Defense Science and Engineering Graduate Fellowship and a NIH T32 HD07418. The following research facilities at Northwestern University were used in this work: Peptide Synthesis and Analytical BioNano Technology Equipment Cores of the Simpson Querrey Institute (U.S. Army Research Office, U.S. Army Medical Research and Materiel Command, Northwestern University, Soft and Hybrid Nanotechnology Experimental [SHyNE] Resource [NSF NNCI-1542205]), Optical Microscopy and Metallography Facility (MRSEC Program [DMR-1121262]), Keck Biophysics Facility (NCI CCSG P30 CA060553 grant awarded to the Robert H Lurie Comprehensive Cancer Center), Biological Imaging Facility (Northwestern University Office for Research), EPIC Facility at the NUANCE Center (SHyNE Resource [NSF NNCI-1542205], MRSEC Program [NSF DMR-1121262]; International Institute for Nanotechnology [IIN], Keck Foundation, the State of Illinois through the IIN), Center for Advanced Microscopy (NCI CCSG P30 CA060553 awarded to the Robert H Lurie Comprehensive Cancer Center). This work also made use of the DuPont-Northwestern-Dow Collaborative Access Team (Northwestern University, E.I. DuPont de Nemours & Co., and The Dow Chemical Company) located at Sector 5 of the Advanced Photon Source (U.S. Department of Energy [DOE] Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02–06CH11357). We are grateful to Theint Aung (Keck Biophysics Facility) for performing all NTA experiments and Charlene Wilke (Biological Imaging Facility) for preparing thin sections for TEM imaging.
ABBREVIATIONS
- PA
peptide amphiphile
- NTA
nanoparticle tracking analysis
- cryoTEM
cryogenic electron transmission microscopy
- SEM
scanning electron microscopy
- TEM
transmission electron microscopy
- FRAP
fluorescence recovery after photobleaching
Footnotes
Supporting Information
Details on material synthesis and purification, flash freezing and freeze drying experiments, and materials characterization are in the Supporting Information.
References
- 1.Hartgerink JD; Beniash E; Stupp SI Science 2001, 294, (5547), 1684–1688. [DOI] [PubMed] [Google Scholar]
- 2.Silva GA; Czeisler C; Niece KL; Beniash E; Harrington DA; Kessler JA; Stupp SI Science 2004, 303, (5662), 1352–1355. [DOI] [PubMed] [Google Scholar]
- 3.Hendricks MP; Sato K; Palmer LC; Stupp SI Acc. Chem. Res 2017, 50, (10), 2440–2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang P; Cheetham AG; Lin Y.-a.; Cui H ACS Nano 2013, 7, (7), 5965–5977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kuang Y; Du X; Zhou J; Xu B Adv. Healthc. Mater 2014, 3, (8), 1217–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rudra JS; Tian YF; Jung JP; Collier JH Proc. Natl. Acad. Sci. U.S.A 2010, 107, (2), 622–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Korevaar PA; George SJ; Markvoort AJ; Smulders MM; Hilbers PA; Schenning AP; De Greef TF; Meijer E Nature 2012, 481, (7382), 492–496. [DOI] [PubMed] [Google Scholar]
- 8.Korevaar PA; Newcomb CJ; Meijer E; Stupp SI J. Am. Chem. Soc 2014, 136, (24), 8540–8543. [DOI] [PubMed] [Google Scholar]
- 9.Tantakitti F; Boekhoven J; Wang X; Kazantsev RV; Yu T; Li J; Zhuang E; Zandi R; Ortony JH; Newcomb CJ; Palmer LC; Shekhawat GS; Olvera de la Cruz M; Schatz GC; Stupp SI Nat. Mater 2016, 15, (4), 469–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sijbesma RP; Beijer FH; Brunsveld L; Folmer BJ; Hirschberg JK; Lange RF; Lowe JK; Meijer E Science 1997, 278, (5343), 1601–1604. [DOI] [PubMed] [Google Scholar]
- 11.Kaitz JA; Possanza CM; Song Y; Diesendruck CE; Spiering AJH; Meijer E; Moore JS Polym. Chem 2014, 5, (12), 3788–3794. [Google Scholar]
- 12.Yu Z; Tantakitti F; Yu T; Palmer LC; Schatz GC; Stupp SI Science 2016, 351, (6272), 497–502. [DOI] [PubMed] [Google Scholar]
- 13.Cordier P; Tournilhac F; Soulié-Ziakovic C; Leibler L Nature 2008, 451, (7181), 977–980. [DOI] [PubMed] [Google Scholar]
- 14.Chen Y; Kushner AM; Williams GA; Guan Z Nat. Chem 2012, 4, (6), 467–472. [DOI] [PubMed] [Google Scholar]
- 15.Burattini S; Colquhoun HM; Fox JD; Friedmann D; Greenland BW; Harris PJ; Hayes W; Mackay ME; Rowan SJ Chem. Commun 2009, (44), 6717–6719. [DOI] [PubMed] [Google Scholar]
- 16.Burattini S; Greenland BW; Merino DH; Weng W; Seppala J; Colquhoun HM; Hayes W; Mackay ME; Hamley IW; Rowan SJ J. Am. Chem. Soc 2010, 132, (34), 12051–12058. [DOI] [PubMed] [Google Scholar]
- 17.Burnworth M; Tang L; Kumpfer JR; Duncan AJ; Beyer FL; Fiore GL; Rowan SJ; Weder C Nature 2011, 472, (7343), 334–337. [DOI] [PubMed] [Google Scholar]
- 18.Noé F; Schütte C; Vanden-Eijnden E; Reich L; Weikl TR Proc. Natl. Acad. Sci. U.S.A 2009, 106, (45), 19011–19016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Janovick JA; Stewart MD; Jacob D; Martin LD; Deng JM; Stewart CA; Wang Y; Cornea A; Chavali L; Lopez S Proc. Natl. Acad. Sci. U.S.A 2013, 110, (52), 21030–21035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Simola M; Hänninen AL; Stranius SM; Makarow M Mol. Microbiol 2000, 37, (1), 42–53. [DOI] [PubMed] [Google Scholar]
- 21.Marques C; Guo W; Pereira P; Taylor A; Patterson C; Evans PC; Shang F FASEB J 2006, 20, (6), 741–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hartgerink JD; Beniash E; Stupp SI Proc. Natl. Acad. Sci. U.S.A 2002, 99, (8), 5133–5138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shah RN; Shah NA; Lim MMDR; Hsieh C; Nuber G; Stupp SI Proc. Natl. Acad. Sci. U.S.A 2010, 107, (8), 3293–3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Webber MJ; Tongers J; Newcomb CJ; Marquardt K-T; Bauersachs J; Losordo DW; Stupp SI Proc. Natl. Acad. Sci. U.S.A 2012, 109, (23), 9220–9220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee SS; Hsu EL; Mendoza M; Ghodasra J; Nickoli MS; Ashtekar A; Polavarapu M; Babu J; Riaz RM; Nicolas JD Adv. Healthc. Mater 2015, 4, (1), 131–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sleep E; Cosgrove BD; McClendon MT; Preslar AT; Chen CH; Sangji MH; Pérez CMR; Haynes RD; Meade TJ; Blau HM Proc. Natl. Acad. Sci. U.S.A 2017, 114, (38), E7919–E7928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pan L; North HA; Sahni V; Jeong SJ; Mcguire TL; Berns EJ; Stupp SI; Kessler JA PLoS One 2014, 9, (8), e104335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li A; Hokugo A; Yalom A; Berns EJ; Stephanopoulos N; McClendon MT; Segovia LA; Spigelman I; Stupp SI; Jarrahy R Biomaterials 2014, 35, (31), 8780–8790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Greenfield MA; Hoffman JR; Olvera de la Cruz M; Stupp SI Langmuir 2009, 26, (5), 3641–3647. [DOI] [PubMed] [Google Scholar]
- 30.Pashuck ET; Cui H; Stupp SI J. Am. Chem. Soc 2010, 132, (17), 6041–6046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang S; Greenfield MA; Mata A; Palmer LC; Bitton R; Mantei JR; Aparicio C; Olvera de la Cruz M; Stupp SI Nat. Mater 2010, 9, (7), 594–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ortony JH; Qiao B; Newcomb CJ; Keller TJ; Palmer LC; Deiss-Yehiely E; Olvera de la Cruz M; Stupp SI J. Am. Chem. Soc 2017, 139, (26), 8915–8921. [DOI] [PubMed] [Google Scholar]
- 33.Domigan LJ; Healy JP; Meade SJ; Blaikie RJ; Gerrard JA Biopolymers 2012, 97, (2), 123–133. [DOI] [PubMed] [Google Scholar]
- 34.Zhao J; Bolisetty S; Adamcik J; Han J; Fernández-Ronco MP; Mezzenga R Langmuir 2016, 32, (10), 2492–2499. [DOI] [PubMed] [Google Scholar]
- 35.Bhatnagar BS; Bogner RH; Pikal MJ Pharm. Dev. Technol 2007, 12, (5), 505–523. [DOI] [PubMed] [Google Scholar]
- 36.Fonte P; Soares S; Costa A; Andrade JC; Seabra V; Reis S; Sarmento B Biomatter 2012, 2, (4), 329–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van Winden EC; Zhang W; Crommelin DJ Pharm. Res 1997, 14, (9), 1151–1160. [DOI] [PubMed] [Google Scholar]
- 38.Mazur P Science 1970, 168, (3934), 939–949. [DOI] [PubMed] [Google Scholar]
- 39.Berns EJ; Sur S; Pan L; Goldberger JE; Suresh S; Zhang S; Kessler JA; Stupp SI Biomaterials 2014, 35, (1), 185–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McMurray RJ; Gadegaard N; Tsimbouri PM; Burgess KV; McNamara LE; Tare R; Murawski K; Kingham E; Oreffo RO; Dalby MJ Nat. Mater 2011, 10, (8), 637–644. [DOI] [PubMed] [Google Scholar]
- 41.Abagnale G; Steger M; Nguyen VH; Hersch N; Sechi A; Joussen S; Denecke B; Merkel R; Hoffmann B; Dreser A Biomaterials 2015, 61, 316–326. [DOI] [PubMed] [Google Scholar]
- 42.Park J; Kim D-H; Kim H-N; Wang CJ; Kwak MK; Hur E; Suh K-Y; An SS; Levchenko A Nat. Mater 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Erickson C; Trinkaus J Exp. Cell Res 1976, 99, (2), 375–384. [DOI] [PubMed] [Google Scholar]
- 44.Bereiter-Hahn J; Luck M; Miebach T; Stelzer H; Voth M J. Cell Sci 1990, 96, (1), 171–188. [DOI] [PubMed] [Google Scholar]
- 45.Charras G; Paluch E Nat. Rev. Mol. Cell Biol 2008, 9, (9), 730–736. [DOI] [PubMed] [Google Scholar]
- 46.Phan HT; Bartelt-Hunt S; Rodenhausen KB; Schubert M; Bartz JC PloS one 2015, 10, (10), e0141282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.da Silva RM; van der Zwaag D; Albertazzi L; Lee SS; Meijer E; Stupp SI Nat. Commun 2016, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schwegman JJ; Carpenter JF; Nail SL J. Pharm. Sci 2009, 98, (9), 3239–3246. [DOI] [PubMed] [Google Scholar]
- 49.Costantino HR; Griebenow K; Langer R; Klibanov AM Biotechnol. Bioeng 1997, 53, (3), 345–348. [DOI] [PubMed] [Google Scholar]
- 50.Zacharis E; Halling PJ; Rees DG Proc. Natl. Acad. Sci. U.S.A 1999, 96, (4), 1201–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cote Y; Fu IW; Dobson ET; Goldberger JE; Nguyen HD; Shen JK J. Phys. Chem. C 2014, 118, (29), 16272–16278. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.