

Abstract
Central to CD8+ Tcell-mediated immunity is the recognition of peptide-major histocompatibility complex class I (p-MHC I) proteins displayed by antigen-presenting cells. Chaperone-mediated loading of high-affinity peptides onto MHC I is a key step in the MHC I antigen presentation pathway. However, the structure of MHC I with a chaperone that facilitates peptide loading has not been determined.We report the crystal structure of MHC I in complex with the peptide editor TAPBPR (TAP-binding protein-related), a tapasin homolog. TAPBPR remodels the peptide-binding groove of MHC I, resulting in the release of low-affinity peptide. Changes include groove relaxation, modifications of key binding pockets, and domain adjustments. This structure captures a peptide-receptive state of MHC I and provides insights into the mechanism of peptide editing by TAPBPR and, by analogy, tapasin.
Cell surface peptide-major histocompatibility complex class I (p-MHC I) complexes play a crucial role in adaptive and innate immunity by functioning as ligands for immuno-surveillance by CD8+ T cells and natural killer cells (1,2). Key to the intracellular assembly of p-MHC I complexes is the selective loading of high-affinity peptides, a process termed “peptide editing.” A dynamic series of steps involving peptide binding and exchange is orchestrated in the endoplasmic reticulum (ER) by tapasin or the closely related TAPBPR (TAP-binding protein-related) (3) to ensure that peptides of optimal affinity and length are assembled with MHC I for display on the cell surface. Although the x-ray structure of tapasin in complex with the peptide-loading complex component ERp57 has been determined (4), structural data addressing the mechanism of peptide editing have been limited, likely because of the difficulty in generating crystallizable peptide-free (PF) or peptide-receptive (PR) MHC I.
TAPBPR interacts directly with PF or PR MHC I molecules, such as HLA-A*02:01 [HLA-A2 (HLA, human leukocyte antigen)] and H2-Dd (Dd), generated by photolysis of coassembled photosensitive peptides (5, 6). Attempts to crystallize purified TAPBPR-MHC I complexes generated in this manner (fig. S1) were unsuccessful. Thus, we developed an alternative strategy to prepare partially peptide-filled yet stable p-MHC I molecules. Previous studies of MHC I interactions with the related molecule, tapasin (7–11), as well as molecular dynamics (MD) simulations (12–17) suggest that the flexibility of the MHC I F pocket, which accommodates the C-terminal amino acid of the peptide (18), is modulated by tapasin. Thus, we reasoned that p-MHC I molecules assembled with peptides truncated from the C terminus, leaving the F pocket unoccupied, would serve as effective ligands for TAPBPR. However, C-terminal-truncated peptides failed to efficiently refold many MHC I molecules, so we adapted an approach developed for the HLA-DM-HLA-DR1 complex (19,20) and examined both human and mouse p-MHC I structures to identify those that would be amenable to disulfide linkage at central peptide positions (see materials and methods). By exploiting the stabilizing effect of a GL dipeptide (see materials and methods), we successfully generated Dd73C molecules disulfide-linked to each of three peptides (table S1): full-length pI10C5 (RGPGCAFVTI) and two truncated versions [pA6C5 (RGPGCA) and pR5C5 (RGPGC)].
We next examined the binding of peptide- Dd73C complexes to human TAPBPR by surface plasmon resonance (SPR) (Fig. 1). The Dd73C-10mer (10mer, 10-amino acid oligomer) bound to TAPBPR with an equilibrium constant, KD, of 0.19 μM and a dissociation rate constant, kd, of 0.024 s−1 (Fig. 1A). By contrast, the Dd73C-6mer (Fig. 1B) and the Dd73C-5mer (Fig. 1C) bound with ~10-fold greater affinity (KD of 0.0094 and 0.0091 μM, respectively), primarily owing to smaller kd values of 0.0013 and 0.0011 s−1, respectively. Statistical evaluation of the binding parameters is shown in Fig. 1, D and E. As shown previously for TAPBPR-MHC I complexes prepared with HLA-A2, H2-Db,H2-Ld, and H2-Dd (6), high-affinity peptide led to the dissociation of TAPBPR from the Dd73C-5mer (fig. S2). The rate of dissociation by peptide was related to the affinity of the peptide for the MHC I molecule (fig. S2) (5).
Fig. 1. Kinetics and affinity of the interaction between TAPBPR and Dd73C containing disulfide-linked peptide.

(A to C) SPR analysis of the interaction between TAPBPR and the Dd73C-10mer (A), the Dd73C-6mer (B), and the Dd73C-5mer (C). Data are shown in red, and fits of the data to a 1:1 binding model are overlaid in black. (D and E) Results of three independent experiments (mean ± SD) are shown as scatter plots. ***P< 0.05 [one-way analysis of variance (ANOVA)]; ns, not significant. KD, equilibrium constant; ka, association rate constant; kd, dissociation rate constant; RU, resonance units.
We crystallized the chromatographically purified TAPBPR-Dd73C-5mer complex, merged the four best x-ray data sets, and solved a structure consisting of the TAPBPR lumenal domains, Dd73C, and hβ2m. Crystals were of the P31 space group, with four complexes in the asymmetric unit (table S2). The refined structure, at 3.4-Å resolution, had Rwork and Rfree values of 24.2 and 26.9%, respectively, and satisfied standard validation criteria for proteins solved at this resolution. We focus our discussion on the first of four complexes in the asymmetric unit and note differences where relevant. In addition, crystal structures of the unliganded Dd73C-10mer, Dd73C-6mer, and Dd73C-5mer were determined as summarized in table S2 and fig. S3.
The overall structure (Fig. 2) of the TAPBPR- Dd73C-5mer complex revealed a heterotrimer of TAPBPR-Dd73C heavy chain-β2m, consistent with previous analytical ultracentrifugation and SPR data (6). The Dd73C-β2m heterodimer was nestled in a glove-like basket consisting of the N- and immunoglobulin V (IgV)-like domains of TAPBPR that clasp the α2–1 helix of Dd73C and extend a loop [residues 210 to 213, connecting strands β13 and β14 (fig. S4)] beneath the peptide-binding platform. This loop interacted with β2m and supported Dd73C β strands 6 to 8 on the floor of the binding groove (Fig. 2, A to C). The composite TAPBPR N/IgV domain, which extends from the N terminus to residue 281, consisted of 19 β strands with one short 310-helix at residues 254 to 256. These β strands were structurally similar to those of tapasin in the tapasin-ERp57 complex and reflect the sequence conservation among different TAPBPR species (fig. S4). The C-terminal domain (residues 282 to 377) revealed a typical IgC structure that notably forms part of an Ig domain trimer coordinating with the Ig-like β2m and Dd73C α3 domains (Fig. 2D). The TAPBPR- Dd73C interface (Fig. 2, E and F) buried an area of 1884 Å2, with 1310 Å2 contributed by Dd73C and 574 Å2 by β2m. The interface was dominated by 17 hydrogen bonds between TAPBPR and the Dd73C heavy chain, three salt bridges, and a total of 22 TAPBPR residue contacts (table S3). Interactions with β2m were less extensive, with five hydrogen bonds and a total of nine TAPBPR residue contacts. This TAPBPR-Dd73C structure was consistent with extensive mutagenesis data reported previously (21) (tables S4 and S5).
Fig. 2. Overall structure of the TAPBPR-Dd73C-hβ2m-5mer complex.
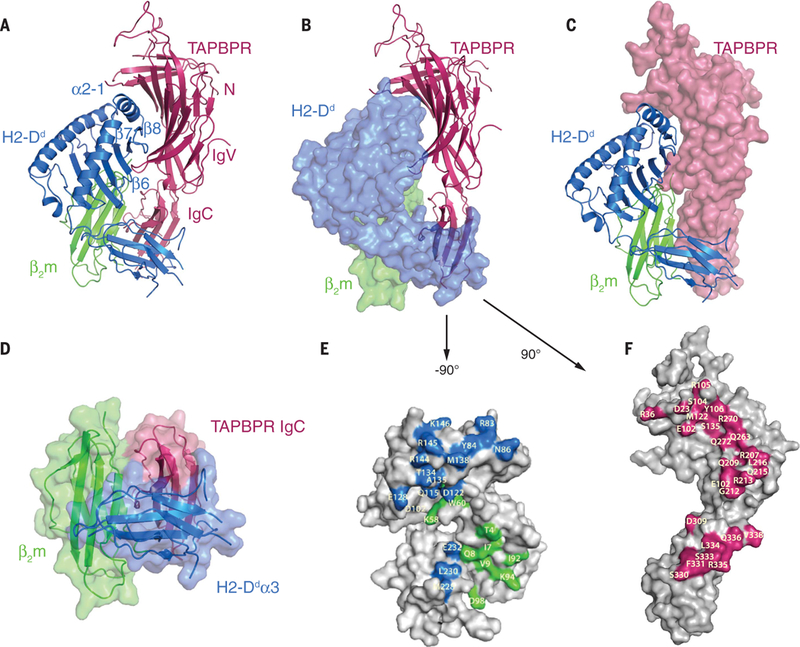
(A) Ribbon representation of TAPBPR (pink), Dd73C heavy chain (H2-Dd, blue), and β2m (green), with individual domains of TAPBPR and structural elements of Dd73C labeled. (B) Similar to (A) but with Dd73C and β2m in surface representation. (C) Similar to (A) but with TAPBPR in surface representation. (D) Ribbon and surface representation of the Ig domain trimer composed of the TAPBPR IgC domain, Dd73C α3, and β2m. (E) Surface representation of Dd73C and β2m with interface residues highlighted in blue and green, respectively. Dd73C-β2m was rotated to the left by ~90° (F) Surface representation of TAPBPR with interface residues highlighted in pink (rightward rotation of 90°). Single-letter abbreviations for the amino acid residues are as follows: A, Ala; C, Cys; D, Asp; E, Glu; F, Phe; G, Gly; H, His; I, Ile; K, Lys; L, Leu; M, Met; N, Asn; P, Pro; Q, Gln; R, Arg; S, Ser; T, Thr; V, Val; W, Trp; and Y, Tyr.
A comparison of the free and TAPBPR-bound forms of the Dd73C-5mer revealed a distortion of the platform domain in the TAPBPR-complexed state (Fig. 3, A and B). In the region of direct contact between the Dd73C-5mer and TAPBPR, the α2–1 helix was pulled away by as much as 3 Å [by interaction between residues 138 to 150 (particularly R144, R145, and E148 of Dd73C) and residues of the palm of the “glove” of the TAPBPR N/IgV domain (Fig. 3, A and C, and table S3)]. In addition, β strand 8 of Dd73C was drawn down ~1.8 Å (Fig. 3B and movie S1), and β strand 5 was pulled by an interdigitating loop at the heel of the TAPBPR glove by as much as 1.4 Å (Fig. 3D). Other intermolecular interactions are listed in table S3. Interaction with TAPBPR distorted the peptide-binding groove. Notably, the side chain of Y84, which canonically coordinates both the C terminus of bound peptide and K146 of α2–1, flipped away from the groove to interact with E102 of TAPBPR (Fig. 3, E and F). The changes in this region deformed the F pocket, which normally accommodates the side chain of the C-terminal amino acid of the bound peptide. TAPBPR not only distorted the localized region of contact but also exerted distal effects on the organization of A and B pocket residues that coordinate the N terminus of the bound peptide. In two of the four molecules in the asymmetric unit, the Dd73C R66 side chain extended across the binding groove to hydrogen bond with Y159, effectively closing off this section (Fig. 3, G and H). As a consequence of these dynamic structural changes in the binding groove, electron density for the covalently bound 5mer peptide, the GL dipeptide (used in refolding), and the accompanying peptide-stabilizing hydrogen bonding network, clearly seen in the unliganded Dd73C-5mer (fig. S3, C, F, and G), was no longer visualized in the TAPBPR complex. This lack of electron density of the covalently bound peptide, taken together with the widening of the groove at the F pocket region and rearrangements of key peptide-binding residues of Dd73C, is consistent with the view that the covalently bound 5mer is mobile in the complex and is no longer tethered to the binding groove.
Fig. 3. TAPBPR remodels the Dd73C binding groove.

(A to C) Superposition of the platform domain of Dd73C in the unliganded (gray) and TAPBPR-bound (blue) forms.TAPBPR is shown in pink. Groove widening at the α2–1 helix is indicated by arrows in (B) and (C). (D) Comparison of the floor of the peptide- binding groove of Dd73C in the unliganded (gray) and TAPBPR-bound (blue) forms. Movement of the β strands is depicted the arrow. (E) Contact between Y84 in the a1 helix of Dd73C (blue) and E102 of TAPBPR (pink). (F) Y84 of the Dd73C-5mer (gray) contacts a GL dipeptide (light blue) in the F pocket in the absence of TAPBPR. (G) Steric blocking of the A and B pockets of Dd73C in the TAPBPR-bound form by side-chain interactions between R66 and Y159. Electron density for these residues is depicted in dark blue. (H) Disposition of R66 and Y159 in the unliganded Dd73C-5mer when the groove is occupied by a peptide.
A comparison of the free and TAPBPR-bound forms of the Dd73C-5mer also revealed extensive movements of the MHC I α3 and β2m domains, which shifted 6.4 and 4.4 A, respectively, to engage TAPBPR (fig. S5 and movie S2). The repositioning of the Dd73C α3 and β2m domains in the TAPBPR complex illustrates the coordinated and dynamic structural changes that occur when Dd73C interchanges between PR and peptide-loaded conformations.
An unanticipated result was the identification of important contacts of TAPBPR with β2m. In the complex, several regions of TAPBPR-β2m contact were noted, including residues of the β1 strand at the N terminus of β2m; the loop at residues 58 to 60, which abuts the 210-to-213 loop of TAPBPR; and C-terminal residues 92 to 94 of strand β7 that bind residues 330 to 332 of the IgC domain of TAPBPR (Fig. 4, A to C). The importance of the β2m58-to-60 loop is evidenced by reduced TAPBPR binding to the Dd73C-5mer assembled with a β2m D59A (D59→A59) mutant (Fig. 4, E and G to I). Ala substitutions of β2m residues I7 or I92/K94 do not affect TAPBPR binding as severely as D59A (Fig. 4, D and F to I). Functional and binding studies have suggested the mutual contribution of β2m and bound peptide to MHC I stability (22–25). Additionally, nuclear magnetic resonance studies of MHC I reveal peptide-influenced changes in chemical shifts of the β2m 58-to-60 loop, which abuts the floor of the peptide-binding platform (26). Thus, β2m 58-to-60 loop interactions with TAPBPR may not only stabilize PR MHC I but also communicate the occupancy of the peptide- binding groove—whether empty, partially occupied, or occupied by a low- or high-affinity peptide.
Fig. 4. Importance of β2m-TAPBPR contacts and mechanism of peptide editing as viewed through the structure of the TAPBPR-MHC I complex.

(A to C) Close-up view of contact residues between hβ2m and TAPBPR: (A) β2m I7 to TAPBPR L334; (B) β2m D59 to TAPBPR K211; and (C) β2m I92/K94 to TAPBPR S330/F331. (D to F) Summary of binding of Dd73C-5mer complexes assembled with the indicated mutant hβ2m chains. (G to I) Summary of binding parameters as compared with those of the wild type (WT) (mean ± SD). Asterisks indicate the degree of statistical significance of means of multiple determinations: ****P< 0.001; ***P< 0.006; **P< 0.03 (one-way ANOVA). ns, not significant. (J) Model for TAPBPR function in peptide presentation. (1) Various peptides, ranging from low affinity (LA) to high affinity (HA), are provided to the ER after cytoplasmic proteolysis and TAP-mediated transport. (2) TAPBPR binds to MHC I, bearing a low-affinity peptide (MHC I/LA) or a peptide of suboptimal length, and catalyzes peptide dissociation. (3) TAPBPR binding remodels the peptide groove, causing release of the low-affinity peptide and stabilization of PF MHC I (TAPBPR-MHC I—PR). (4) Upon binding a high-affinity peptide, MHC I—PR changes conformation to MHC I—HA (high affinity), dissociates from TAPBPR (fig. S2), and is transported to the cell surface. This model addresses only the effects of interactions of TAPBPR with MHC I. Further experiments are needed to structurally clarify the recently described recycling pathway that employs UDP-glucose: glycoprotein glucosyltransferase 1 (UGT1) (30)
The crystal structure of the TAPBPR-Dd73C- 5mer complex provides an x-ray structure of TAPBPR and permits direct comparison with the related tapasin. As implied by previous low- resolution small-angle x-ray scattering structures (6) and anticipated by mutational and MD studies (21, 27), TAPBPR and tapasin, despite only 22% protein sequence identity, reveal marked structural similarity (Fig. 2G). Mutational analyses (4,6,8,9,21,28,29)and MD simulations(12,15,17), mapped the general region of the tapasin-MHC I interaction to the MHC I α2–1 helix and the α3 domain, sites that overlap with the TAPBPR binding site (21). Residues that interact with MHC I are broadly conserved between TAPBPR and tapasin (fig. S4). Finally, the TAPBPR-Dd73C complex structure provides a basis for building a homology model of tapasin-HLA-B*44:02 (fig. S6) to guide further tests of the mechanism underlying the catalytic role of tapasin.
A comparison of the role of TAPBPR with that of HLA-DM, which similarly stabilizes a peptide-receptive conformation of the MHC II molecule, revealed that these two classes of chaperone function by destabilizing diametrically opposite regions of the binding groove: TAPBPR affects the F pocket (C-terminal portion) of the MHC I groove directly, whereas HLA-DM distorts the P1 pocket (N-terminal portion) of MHC II. Although large structural changes in HLA-DR on HLA-DM binding in the P9 region have not been observed, a clear-cut effect of P9-substituted peptides on the stability of HLA-DM-HLA-DR complexes has been noted (19).
TAPBPR and tapasin function as both chaperones and peptide editors. As viewed through the x-ray structure of the peptide-dissociable TAPBPR- Dd73C-5mer complex described here, in which the 5mer represents a low-affinity peptide, chaperone and editor functions are the result of the same structural effects: stabilization of an MHC I conformation showing global differences in the α2–1 helix; the accessibility of the full peptide- binding groove, including the A and F pockets; and the relative disposition of the α3 and β2m domains. We propose a model of TAPBPR- mediated peptide editing in which TAPBPR catalyzes dissociation of low-affinity peptides, stabilizes PR (empty) MHC I, and is released by high-affinity peptide (Fig. 4J). By analogy, tapasin follows the same general structural principles.
This structural description of the interaction between MHC I and TAPBPR provides insights into the molecular mechanism of peptide editing, an evolutionarily conserved and critical step in generating stable cell surface p-MHC I complexes for immunological surveillance. Additionally, this work provides a basis for further structural and computational approaches modeling additional components of the TAPBPR- and tapasin-mediated peptide presentation pathways.
Supplementary Material
ACKNOWLEDGMENTS
We thank P. Sun and P. Roche for discussions and comments on the manuscript and J. Skinner for guidance on statistics. This research was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases, NIH. Data were collected at SER-CAT (22-ID) and GM/CA-CAT (23-ID) beamlines at the Advanced Photon Source (APS), Argonne National Laboratory. Use of the APS was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under contract no. W-31–109-Eng-38. GM/CA @ APS has been funded in whole or in part with federal funds from the National Cancer Institute (ACB-12002) and the National Institute of General Medical Sciences (AGM-12006). X-ray structure factors and refined coordinates have been deposited in the Protein Data Bank (www.wwpdb.org) under accession numbers 5WER, 5WES, 5WET, and 5WEU for TAPBPR-Dd73C-5mer, Dd73C-5mer, Dd73C- 6mer, and Dd73C-10mer, respectively. J.J., K.N., L.F.B., G.I.M., M.G.M., and D.H.M. planned experiments, generated constructs, expressed and purified proteins, and performed and analyzed binding data. J.J. and K.N. performed crystallization screens, collected and scaled diffraction data, solved the structures by molecular replacement, and performed crystallographic refinement. J.J. performed data merging, scaling, extensive refinement, and model building. J.J., K.N., L.F.B., M.G.M., and D.H.M. wrote the paper. We dedicate this paper to the memory of our friend and colleague, William E. Paul.
Footnotes
SUPPLEMENTARY MATERIALS
REFERENCES AND NOTES
- 1.Blum JS, Wearsch PA, Cresswell P, Annu. Rev. Immunol. 31, 443–473 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rock KL, Reits E, Neefjes J, Trends Immunol. 37, 724–737 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boyle LH et al. , Proc. Natl. Acad. Sci. U.S.A. 110, 3465–3470 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dong G, Wearsch PA, Peaper DR, Cresswell P, Reinisch KM, Immunity 30, 21–32 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hermann C et al. , eLife 4, e09617 (2015).26439010 [Google Scholar]
- 6.Morozov GI et al. , Proc. Natl. Acad. Sci. U.S.A. 113, E1006–E1015 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen M, Bouvier M, EMBO J. 26, 1681–1690 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peace-Brewer AL et al. , Immunity 4, 505–514 (1996). [DOI] [PubMed] [Google Scholar]
- 9.Williams AP, Peh CA, Purcell AW, McCluskey J, Elliott T, Immunity 16, 509–520 (2002). [DOI] [PubMed] [Google Scholar]
- 10.Turnquist HR et al. , Immunol. Res. 25, 261–269 (2002). [DOI] [PubMed] [Google Scholar]
- 11.Turnquist HR et al. , J. Immunol. 167, 4443–4449 (2001). [DOI] [PubMed] [Google Scholar]
- 12.Abualrous ET et al. , Eur. J. Immunol. 45, 1248–1257 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Sieker F, Straatsma TP, Springer S, Zacharias M, Mol. Immunol. 45, 3714–3722 (2008). [DOI] [PubMed] [Google Scholar]
- 14.Sieker F, Springer S, Zacharias M, Protein Sci. 16, 299–308 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ostermeir K, Springer S, Zacharias M, Mol. Immunol. 63, 312–319 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Garstka MA et al. , FASEB J. 25, 3989–3998 (2011). [DOI] [PubMed] [Google Scholar]
- 17.Fisette O, Wingbermühle S, Tampe R, Schäfer LV, Sci. Rep. 6,19085 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saper MA, Bjorkman PJ, Wiley DC, J. Mol. Biol. 219, 277–319 (1991). [DOI] [PubMed] [Google Scholar]
- 19.Anders AK et al. , Nat. Immunol. 12, 54–61 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pos W et al. , Cell 151, 1557–1568 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hermann C, Strittmatter LM, Deane JE, Boyle LH, J. Immunol. 191, 5743–5750 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kozlowski S et al. , Nature 349, 74–77 (1991). [DOI] [PubMed] [Google Scholar]
- 23.Cook JR, Myers NB, Hansen TH, Mol. Immunol. 35, 929–934 (1998). [DOI] [PubMed] [Google Scholar]
- 24.Smith RA, Myers NB, Robinson M, Hansen TH, Lee DR, J. Immunol. 169, 3105–3111 (2002). [DOI] [PubMed] [Google Scholar]
- 25.Shields MJ, Hodgson W, Ribaudo RK, Mol. Immunol. 36, 561–573 (1999). [DOI] [PubMed] [Google Scholar]
- 26.Beerbaum M et al. , J. Biomol. NMR 57, 167–178 (2013). [DOI] [PubMed] [Google Scholar]
- 27.Thomas C, Tampe R, Front. Immunol. 8, 65 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Simone LC, Wang X, Tuli A, Solheim JC, Immune Cell Biol. 88, 57–62 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu YY et al. , J. Immunol. 163, 4427–4433 (1999). [PubMed] [Google Scholar]
- 30.Neerincx A et al. , eLife 6, e23049 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.