

Abstract
Breast cancer patients with BRCA1/2‐driven tumors may benefit from targeted therapy. It is not clear whether current BRCA screening guidelines are effective at identifying these patients. The purpose of our study was to evaluate the prevalence of inherited BRCA1/2 pathogenic variants in a large, clinically representative breast cancer cohort and to estimate the proportion of BRCA1/2 carriers not detected by selectively screening individuals with the highest probability of being carriers according to current clinical guidelines. The study included 5,122 unselected Swedish breast cancer patients diagnosed from 2001 to 2008. Target sequence enrichment (48.48 Fluidigm Access Arrays) and sequencing were performed (Illumina Hi‐Seq 2,500 instrument, v4 chemistry). Differences in patient and tumor characteristics of BRCA1/2 carriers who were already identified as part of clinical BRCA1/2 testing routines and additional BRCA1/2 carriers found by sequencing the entire study population were compared using logistic regression models. Ninety‐two of 5,099 patients with valid variant calls were identified as BRCA1/2 carriers by screening all study participants (1.8%). Only 416 study participants (8.2%) were screened as part of clinical practice, but this identified 35 out of 92 carriers (38.0%). Clinically identified carriers were younger, less likely postmenopausal and more likely to be associated with familiar ovarian cancer compared to the additional carriers identified by screening all patients. More BRCA2 (34/42, 81.0%) than BRCA1 carriers (23/50, 46%) were missed by clinical screening. In conclusion, BRCA1/2 mutation prevalence in unselected breast cancer patients was 1.8%. Six in ten BRCA carriers were not detected by selective clinical screening of individuals.
Keywords: BRCA1, BRCA2, clinical testing, next‐generation sequencing, screening criteria, prediction, breast cancer
Short abstract
What's new?
In order to provide personalized therapy to patients with pathogenic BRCA1/2 variants, it's necessary to find them. Currently, patients are screened based on risk factors, such as family history. How many BRCA1/2 carriers are missed? What if everyone were screened? Here, the authors sequenced DNA from more than 5,000 Swedish breast cancer patients looking for pathogenic BRCA1/2 variants, and they found 92 carriers. Of these, only 35 had been identified by clinical screening. 60% of cancer‐causing BRCA variants had not been detected. This is one of the largest population‐based studies to date examining BRCA1/2 prevalence.
Introduction
Estimates of the prevalence of BRCA1 or BRCA2 germline pathogenic variants vary considerably depending on the technology used for mutation screening, population size and to what extent the genes are tested.1 Although BRCA1/2 pathogenic variants are major determinants of hereditary breast cancers, women diagnosed with BRCA1/2‐associated breast cancer do not necessarily exhibit worse survival patterns than breast cancer patients without such pathogenic variants.2 On the contrary, patients diagnosed with BRCA1/2‐associated breast cancers have advantages in terms of treatment options when compared to patients with BRCA1/2 wild‐type breast cancer (reviewed in Ref. 3). Evidence from clinical trials showed significantly greater sensitivity and higher response rate of BRCA1/2‐associated cancers to neoadjuvant and standard adjuvant chemotherapy than their wild‐type BRCA1/2 counterparts.3 Treatment options for BRCA1/2 breast cancers are also broadened with the introduction of new therapeutic agents, such as poly (ADP‐ribose) polymerase (PARP) inhibitors, which selectively target BRCA1/2‐deficient cancer cells.4, 5, 6, 7
Recommendation for counseling and genetic screening for BRCA1/2 pathogenic variants is mainly based on personal and family history of breast and/or ovarian cancer, young age at disease onset, male breast cancer and multiple tumors (bilateral breast cancer or breast and ovarian cancer in the same patient).8 However, BRCA testing guidelines vary by region and country.9, 10 In Sweden, the Swedish Breast Cancer Group BRCA1 and BRCA2 screening criteria are used.8 A report by Nilsson et al. estimated that the Swedish BRCA testing criteria has an effectiveness of only 18% and concluded that clinical genetic testing criteria for BRCA1 and BRCA2 should be critically revised.8 As the effective identification of BRCA1/2 germline pathogenic variants has potential to influence treatment decision and has implications for the family of the patients,3, 4, 5, 6, 11, 12 the pros and cons of testing all women diagnosed with breast cancer for such pathogenic variants need to be examined. In a large, clinically representative breast cancer cohort, we examined the prevalence and characteristics of BRCA1/2 germline mutation carriers and compared our results with BRCA mutation carriers already identified through a national clinical BRCA screening program.
Methods
Study participants
All women under the age of 80 and diagnosed with breast cancer from 2001 to 2008 in Stockholm, Sweden were identified through the Stockholm‐Gotland Regional Breast Cancer quality register.13, 14 Women were invited to participate in the LIBRO1 study in 2009. In all, 5,715 women of the LIBRO1 study gave informed consent to the retrieval of data from medical records and national registers, answered a detailed questionnaire on background and lifestyle risk factors, and provided a blood specimen for genetic analysis.13, 14 Of these women, 5,125 were successfully genotyped in a large‐scale genotyping study on breast cancer risk (see eTable 1 in Data Supplement 1 for exclusion criteria, Supporting Information).15 Of these women, 5,122 had enough DNA remaining for targeted sequencing. The final analytical dataset comprised 5,099 samples which passed quality control. our study was approved by the Regional Ethical Review Board in Stockholm, Sweden (Karolinska Institutet, DNR2009/254–31/4).
Patient characteristics
Self‐reported information on education level, age at menarche, body mass index (BMI), number of children, oral contraceptive use, hormone replacement therapy and details of family history of breast and ovarian cancer were obtained from the questionnaire. Patients were asked if their biological mothers and sisters have been diagnosed with breast or ovarian cancer, and if so, at what age. Mammograms were retrieved from radiology departments. Percent mammographic density was measured using an automated method described in Ref. 16. Information on whether the patients have an ovarian cancer or any nonbreast malignancy was retrieved via linkage to the Swedish Cancer Register using unique personal identity numbers of study participants (personnummer, 10 or 12 digit number used in Sweden to identify individuals).17
Tumor characteristics
Tumor characteristics were retrieved from the Stockholm‐Gotland Regional Breast Cancer Quality Register18, 19 using unique personal identity numbers.17 Tumor size was measured in millimeters. Lymph node involvement was dichotomized into positive or negative. Estrogen receptor (ER) status was recorded as negative or positive in the registers, determined by radioimmunoassay or immunohistochemistry with cutoff values of more than 10% positive cells for IHC and more than 0 fmol/μg DNA for radioimmunoassay assays. The completeness of the registry data was 98% for tumor size and lymph node status and 80% for ER status. Information on grade (Nottingham histologic grade for invasive cancer and nuclear grade for cancer in situ) was available from 2004, with 93% completeness.19
Data on molecular markers were retrieved in 2015–2016 from medical and pathology records at treating hospitals (previously described in Ref. 20). HER2 status was dichotomized (positive/negative) in accordance with the Swedish Society of Pathology's guidelines: negative if protein expression showed 0 or 1+, or was higher with no confirmed gene amplification by FISH, and positive if FISH showed gene amplification.20 Proliferation marker Ki67 was measured according to contemporary guidelines and reported as percent staining (low if <20% and high otherwise).20 HER2 and Ki67 markers were not assessed, and thus not available in medical records, prior to 2005. Breast cancer subtype was assigned using a random forest algorithm (caret R package, v. 6.0.58) described in Ref. 20. The algorithm was trained to predict subtype based on a subset of individuals with PAM50 subtype derived from gene expression data (n = 237). Breast cancer subtype was then assigned to the remaining cases based on age at diagnosis, ER, PR, HER2 and Ki67 status.
Targeted sequencing and data processing
Target‐enriched sequencing libraries of germline DNA from 5,122 breast cancer patients were prepared at the Centre for Cancer Genetic Epidemiology (University of Cambridge), as part of a larger effort that included samples from other cohorts. Briefly, target sequence enrichment was performed using 48.48 Fluidigm Access Arrays according to the manufacturer's protocol (Fluidigm, South San Francisco, CA). Fluidigm D3 assay design software was used to select primer pairs, which were multiplexed into pools selected for GC content and avoidance of off‐target primer‐primer and primer‐product complementarity (eTable 2 in Data Supplement 2, Supporting Information). Target sequences were amplified with Illumina sequencing adaptors and one of 1,536 unique sample barcodes (supplied by Fluidigm, South San Francisco, CA). Robotic liquid handling and barcode plate identification were used in all steps of the library preparation process. The amplicon library was quantified with the KAPA Library Quantification Kit (KapaBiosystems, Boston, MA) and then sequenced on the Illumina Hi‐Seq 2,500 instrument using v4 chemistry, according to the manufacturer's protocol (Illumina, San Diego, CA). Each library was sequenced 2–3 times to provide sufficient coverage. Details on sequence data processing and quality control are shown in eMethods in Data Supplement 1, Supporting Information. A total of 5,099 samples had valid variant calls. The mean read depth across the coding sequences of BRCA1 and BRCA2 was 792.2 (standard deviation: 587.4) and 631 (standard deviation: 516), respectively. More than 90% of targeted bases had more than 15x coverage (94.8 [15.9] and 92.5 [20.4] for BRCA1 and BRCA2, respectively).
Definition of pathogenic variants
As described previously in Borg et al.,21 sequence variants were categorized based on their predicted effect on the mRNA and amino acid level and defined as pathogenic if they were (1) frameshift and nonsense variants with the exception of the BRCA2 c.9976A > T (BIC: K3326X) and other variants located 3′ thereof (n = 105) and (2) all consensus splice acceptor or donor sequence sites, except those predicted to lead to naturally occurring in‐frame RNA isoforms that may rescue gene function.22 Public data on pathogenic BRCA variants (includes frameshift insertion/deletions, nonsense, splice sites and missense variants conclusively demonstrated to be pathogenic) that have been curated and classified by an international expert panel, the ENIGMA consortium, were also downloaded from http://brcaexchange.org/ (access date: Feb 22, 2017) for the annotation of the sequence data.
Identification of women who have undergone BRCA testing in Sweden
Mutation screening for all oncogenetic clinics in Sweden (Lund, Stockholm, Uppsala, Göteborg, Linköping and Umeå) were conducted at the Department of Oncology, Lund University as part of a national BRCA testing program (eMethods in Data Supplement 1, Supporting Information). We cross‐referenced the personal identity numbers of all study participants in LIBRO1 with the BRCA testing unit at Lund University to identify women who have been tested for BRCA1/2 pathogenic variants previously. The SweBRCA criteria are the only BRCA1/2 testing criteria used in Sweden (eTable 3 in Data Supplement 1, Supporting Information).8 Clinicians do not have any obligation to comply with the guidelines.8
Statistical analysis
Predictor variables which include patient and tumor characteristics were described by the counts of each category and corresponding proportions. Binary logistic regression models were fitted for the dichotomous outcome (BRCA1 [reference] and BRCA2) and multinomial logistic regression models were fitted for the three‐category outcome (BRCA1, BRCA2 and non‐BRCA [reference category]), adjusting for age and year of diagnosis. Logistic regression models were also used to compare estimates (odds ratios [OR] and corresponding 95% confidence intervals [CI]) of patient and tumor characteristics between BRCA1/2 carriers already identified among a subset of 416 patients screened as part of clinical BRCA testing routines and additional BRCA1/2 carriers found by sequencing the entire study population (i.e., those not tested by the Swedish BRCA testing program).
Results
The median time from date of diagnosis to study entry is 4.8 years (range: 1.3–9.2). The median age of breast cancer diagnosis of the study cohort was 59.6 years (range: 25.1–79.9). Nine of ten breast cancers were invasive (89.4%).
Spectrum of BRCA1 and BRCA2 pathogenic variants
Of the 5,099 breast cancer patients, 92 (1.8%) were identified as BRCA1/2 carriers (50 BRCA1 carriers and 42 BRCA2 carriers) and 5,007 were non‐BRCA.
Among the 50 BRCA1 carriers, there were 28 unique germline BRCA1 pathogenic variants (11 frameshift deletions, 2 frameshift insertions, 8 truncating, 4 splice sites and 3 missense) (Fig. 1 and eTable 4 in Data Supplement 1, Supporting Information). Frameshift insertions and deletions made up 26/50 (52%) of the BRCA1 pathogenic variants. Exon 11 harbored 33/50 (66%) of the BRCA1 pathogenic variants. The most common pathogenic variant was c.3048_3052dupTGAGA (n = 8), which is a founder mutation originating from the West coast of Sweden.23 Three other Swedish founder pathogenic variants were also identified (c.1082_1092del [n = 5], c.2475delC [n = 2]) and c.3626delT [n = 3]).23, 24, 25, 26
Figure 1.
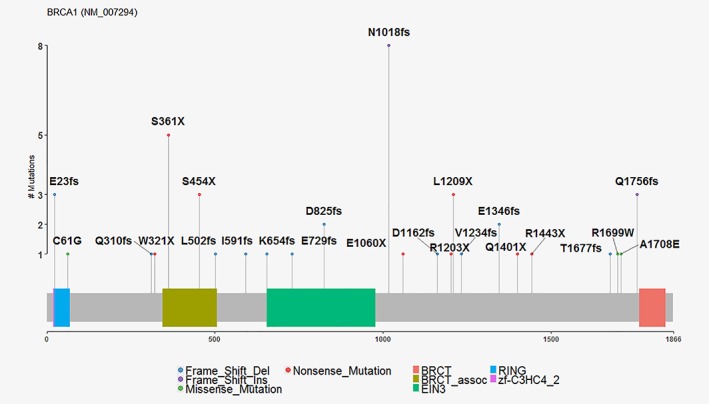
Mutation plot of BRCA1. Four and three splice variants for BRCA1 (NM_007294.3) are not shown.
Among the 42 BRCA2 carriers, there were 33 unique BRCA2 pathogenic variants (18 frameshift deletions, 3 frameshift insertions, 9 truncating and 3 splice sites) (Fig. 2 and eTable 5 in Data Supplement 1, Supporting Information). Over half of all BRCA2 carriers (24/42, 57.1%) had a pathogenic variant on exon 11.
Figure 2.
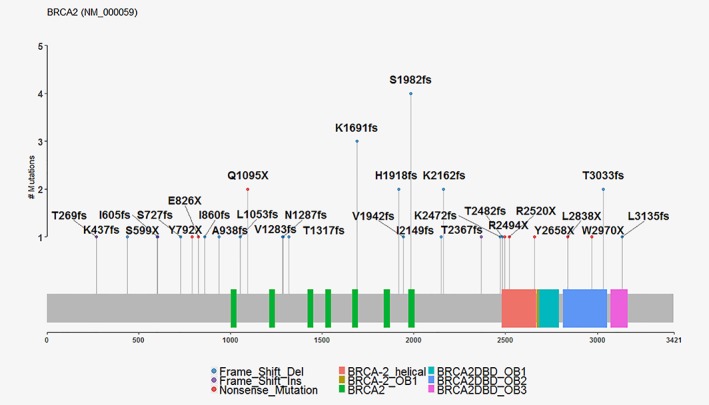
Mutation plot of BRCA2. Three splice variants for BRCA2 (NM_000059.3) are not shown.
Patient characteristics of non‐BRCA, BRCA1 and BRCA2 carriers
Half of the non‐BRCA women were at least 60 years old, compared to 26.0 and 33.3% for women with BRCA1 and BRCA2 pathogenic variants, respectively (eTable 6 in Data Supplement 1, Supporting Information). In the crude analyses controlling for age and year of diagnosis, BRCA1 and BRCA2 carriers were more likely than non‐BRCA women to report family history of both breast (ORBRCA1 vs non‐BRCA: 4.00 [2.27–7.05] and ORBRCA2 vs non‐BRCA: 2.23 [1.17–4.26]) and family history of ovarian cancer (ORBRCA1 vs non‐BRCA: 7.53 [3.82–14.82] and ORBRCA2 vs non‐BRCA: 3.62 [1.50–8.71]) (eTable 6 in Data Supplement 1, Supporting Information). BRCA1 carriers, in particular, were also more likely to be also diagnosed with an ovarian cancer themselves (ORBRCA1 vs non‐BRCA: 28.02 [10.72–73.29] and ORBRCA2 vs non‐BRCA: 8.11 [1.87–35.24]) than non‐BRCA patients (eTable 6 in Data Supplement 1, Supporting Information). BRCA1 carriers were more likely to have a personal history of another malignant cancer in addition to their breast cancer than patients with non‐BRCA patients (ORBRCA1 vs non‐BRCA: 2.93 [1.37–6.27]). This association was driven by ovarian cancers (ORBRCA1 vs non‐BRCA for all non‐breast and non‐ovarian malignancies: 0.83 [0.25–2.73]). BRCA2 carriers were significantly less likely to be ever users of hormone replacement therapy (HRT) than non‐BRCA breast cancer patients (26.2% vs 53.8%) (eTable 6 in Data Supplement 1, Supporting Information). In multivariable models shown in Table 1, all variables remained significantly associated, with the exception of personal history of any non‐breast malignancy.
Table 1.
Odds ratio (OR) and corresponding 95% confidence intervals (CI) of predictors according to BRCA status
| BRCA1 vs non‐BRCA OR (95% CI) | BRCA2 vs non‐BRCA OR (95% CI) | BRCA2 vs BRCA1 OR (95% CI) | |
|---|---|---|---|
| Model 1: Patient characteristics | |||
| Age at diagnosis: 50–59 | 0.21 (0.10–0.45) | 0.78 (0.36–1.69) | 3.55 (1.05–11.97) |
| Age at diagnosis: ≥60 | 0.14 (0.06–0.31) | 0.55 (0.24–1.23) | 3.91 (1.11–13.84) |
| Year of diagnosis: 2005–2008 | 1.68 (0.91–3.08) | 1.03 (0.55–1.92) | 0.90 (0.33–2.48) |
| HRT ever: Yes | 1.08 (0.56–2.10 | 0.36 (0.17–0.76) | 0.31 (0.10–0.93) |
| Familiy history of breast cancer: Yes | 3.57 (1.99–6.41) | 2.08 (1.08–3.99) | 0.60 (0.24–1.55) |
| Familiy history of ovarian cancer: Yes | 6.99 (3.43–14.24) | 3.57 (1.47–8.68) | 0.38 (0.11–1.35) |
| Personal history of ovarian cancer: Yes | 19.21 (5.89–62.72) | 8.01 (1.61–39.94) | 0.49 (0.04–6.74) |
| Personal history of any malignant cancer (not breast): Yes | 1.35 (0.52–3.54) | 0.81 (0.26–2.56) | 0.49 (0.07–3.59) |
| Model 2: Tumor characteristics, adjusted for age and year of diagnosis | |||
| Detection mode: Interval | 1.34 (0.38–4.79) | 1.16 (0.45–3.03) | 0.44 (0.05–3.50) |
| Detection mode: Clinical cancer in women without previous mammograms | 2.61 (0.81–8.37) | 0.66 (0.20–2.12) | 0.35 (0.05–2.38) |
| Detection mode: Clinical cancer in women who had previous mammograms (i.e., interval > 24 months) | 3.54 (1.15–10.89) | 1.57 (0.63–3.94) | 0.34 (0.06–2.02) |
| ER status: Negative | 5.19 (2.68–10.06) | 1.17 (0.48–2.87) | 0.22 (0.07–0.77) |
| Grade: Intermediate‐differentiated | 1.97 (0.24–16.23) | 1.82 (0.52–6.34) | 1.32 (0.10–18.26) |
| Grade: Poorly differentiated | 7.11 (0.91–55.30) | 1.55 (0.39–6.22) | 0.36 (0.03–4.92) |
| Tumor size: ≥20 | 0.87 (0.48–1.59) | 1.26 (0.67–2.39) | 1.17 (0.37–3.76) |
| Nodal involvement: Yes | 1.60 (0.79–3.27) | 2.54 (1.20–5.37) | 1.67 (0.43–6.51) |
| Model 3: Breast cancer subtype, adjusted for age and year of diagnosis | |||
| Subtype: Luminal B | 2.83 (0.54–14.77) | 0.49 (0.06–3.73) | 0.19 (0.01–2.60) |
| Subtype: HER2‐enriched | 0.93 (0.11–8.07) | 0.33 (0.04–2.52) | 0.38 (0.02–8.07) |
| Subtype: Basal‐like | 40.07 (14.26–112.59) | 0.84 (0.11–6.43) | 0.02 (0.00–0.17) |
Tumor characteristics of non‐BRCA, BRCA1 and BRCA2 carriers
In the crude analyses controlling for age and year of diagnosis, BRCA2 carriers were in general not significantly different from non‐BRCA women in terms of tumor characteristics, with the exception of nodal involvement (ORBRCA2 vs non‐BRCA: 2.71 [1.31–5.62], eTable 7 in Data Supplement 1, Supporting Information). On the contrary, tumors of BRCA1 carriers were more aggressive than those of non‐BRCA breast cancer patients for all tumor characteristics examined (ER and PR status, grade, tumor size, nodal involvement and breast cancer subtype) except for the proportion of invasive tumors (eTable 7 in Data Supplement 1, Supporting Information).
In multivariable multinomial models including all tumor characteristics that were significantly different between non‐BRCA and BRCA1‐positive breast cancer patients, only ER‐negativity remained significant (ORBRCA1 vs non‐BRCA: 5.19 [2.68–10.06]) (Table 1). ER status was also the only independent tumor characteristic that distinguished between BRCA1 and BRCA2 carriers (ORBRCA2 vs BRCA1: 0.22 [0.07–0.77]). This observation was mirrored in a separate multinomial model considering breast cancer subtypes, where BRCA1 tumors were found to be 40 times more likely to be of the basal‐like subtype (ORBRCA1 vs non‐BRCA: 40.07 [14.26 to 112.59]). Only nodal involvement remained significant in the comparison between BRCA2 and non‐BRCA breast cancer cases in the multivariable model (ORBRCA2 vs non‐BRCA: 2.54 [1.20–5.37) (Table 1).
Comparison of BRCA1/2 carriers identified versus not identified through clinical screening
Linkage with the Swedish BRCA register found 416 patients (8.2%) that were screened for pathogenic variants as part of routine clinical practice. Among these 416 women, clinical screening identified 39 carriers in the study cohort, of which our study confirmed 35 (Fig. 3). Four pathogenic variants were missed (BRCA1: c.4186‐1785_4,358‐1667dup and c.4358‐1729_4986 + 736dup; BRCA2: c.7805 + 1538_8331 + 560del and c.9097_9098insT) (Fig. 3). Three of these were large exonic deletions or duplications that the Fluidigm Access Array system is not suitable for detecting. This gives the Fluidigm Access Array method an estimated sensitivity of about 90%, or 97% when excluding large exonic variants.
Figure 3.
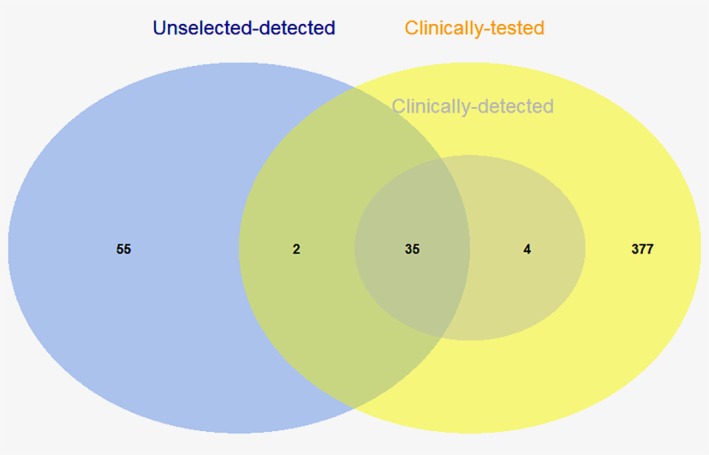
Overlap between women attending BRCA screening (clinically tested), BRCA carriers identified through selective clinical testing routine (clinically detected carriers) and BRCA carriers identified through screening all unselected LIBRO1 breast cancer patients (unselected‐detected). Of the 416 women who were clinically tested, 39 were found to be BRCA1/2 carriers (39/416, 9.3%). Our study confirmed 35 of these pathogenic variants. Four pathogenic variants were missed (BRCA1: c.4186‐1785_4,358‐1667dup and c.4358‐1729_4986 + 736dup; BRCA2: c.7805 + 1538_8331 + 560del and c.9097_9098insT). By sequencing the entire Swedish study, we found 55 more carriers who were not screened as part of clinical routine. [Color figure can be viewed at wileyonlinelibrary.com]
Overall, 57/92 carriers (62.0%) were not already clinically identified: Two additional carriers were detected by the Fluidigm Access Array method among clinically screened patients (BRCA2: c.2578delA [confirmed by Sanger sequencing to be a false positive] and c.7443delT [missed carrier, screened with DHPLC and MLPA in 2008]); the remaining 55 out of 92 carriers (59.8%) identified by the Fluidigm Access Array method in the complete study cohort were never screened as part of clinical routine (Fig. 3).
More BRCA2 (34/42, 80%) than BRCA1 pathogenic variants (23/50, 46%) were missed by selectively testing only high‐risk individuals who were recommended for genetic testing and counseling (Table 2). Controlling for only year of diagnosis, BRCA carriers identified by clinical routine screening were younger (37.2% aged 50 years and above, compared to 73.7%), less likely to have experienced menopause (ORidentified versus not identified: 0.17 [0.07–0.44]) and more likely to be associated with a family history of ovarian cancer (ORidentified versus not identified: 3.11 [1.06–9.09]) (Table 2). Further adjustment for gene revealed a significant association with age at menarche (ORidentified versus not identified: 2.99 [1.00–8.94]). There was also a trend between the likelihood of being identified as a carrier by selective testing and more children (Table 2). Tumors of BRCA1/2 carriers identified by selective testing were more often detected clinically (ORidentified versus not identified: 5.52 [1.38–22.18]), higher grade (ORidentified versus not identified: 0.28 [0.08–0.92]), larger size (ORidentified versus not identified: 2.48 [1.00–6.16]) and of a basal subtype (ORidentified versus not identified: 6.07 [1.49–24.76]) (eTable 8 in Data Supplement 1, Supporting Information). The differences observed for all tumor characteristics and selective testing detection did not remain significant after adjusting for gene.
Table 2.
Frequency, odds ratio (OR) and corresponding 95% confidence intervals (CI) of patient characteristics among BRCA carriers identified versus not identified through selective clinical screening
| Patient characteristic | Not identified by selective testing (n = 57) | Identified by selective testing (n = 35) | OR (95% CI)1 | OR (95% CI)2 | OR (95% CI)3 |
|---|---|---|---|---|---|
| n (%) | n (%) | ||||
| Gene, 1unadjusted | |||||
| BRCA1 | 23 (40.4) | 27 (77.1) | 1.00 (Reference) | ||
| BRCA2 | 34 (59.6) | 8 (22.9) | 0.20 (0.08–0.52) | ||
| Age at diagnosis, 1unadjusted | |||||
| <50 | 15 (26.3) | 22 (62.9) | 1.00 (Reference) | ||
| 50–59 | 20 (35.1) | 8 (22.9) | 0.27 (0.10–0.78) | ||
| ≥60 | 22 (38.6) | 5 (14.3) | 0.15 (0.05–0.50) | ||
| Year of diagnosis, 1unadjusted | |||||
| 2001–2004 | 26 (45.6) | 12 (34.3) | 1.00 (Reference) | ||
| 2005–2008 | 31 (54.4) | 23 (65.7) | 1.61 (0.67–3.84) | ||
| Education | |||||
| University | 29 (50.9) | 21 (60.0) | 1.00 (Reference) | 1.00 (Reference) | 1.00 (Reference) |
| Intermediate | 12 (21.1) | 9 (25.7) | 1.06 (0.37–2.98) | 1.40 (0.45–4.39) | 2.08 (0.59–7.40) |
| Elementary | 7 (12.3) | 0 (0.0) | ‐ | ‐ | ‐ |
| Other | 9 (15.8) | 5 (14.3) | 0.78 (0.23–2.68) | 0.65 (0.17–2.46) | 1.63 (0.35–7.66) |
| Age at menarche in years | |||||
| <13 | 21 (36.8) | 7 (20.0) | 1.00 (Reference) | 1.00 (Reference) | 1.00 (Reference) |
| ≥13 | 36 (63.2) | 28 (80.0) | 2.17 (0.79–5.94) | 2.99 (1.00–8.94) | 4.12 (1.19–14.26) |
| Menopause status before breast cancer diagnosis | |||||
| Premenopause | 14 (24.6) | 23 (65.7) | 1.00 (Reference) | 1.00 (Reference) | 1.00 (Reference) |
| Postmenopause | 43 (75.4) | 12 (34.3) | 0.17 (0.07–0.44) | 0.17 (0.06–0.45) | 0.18 (0.03–1.25) |
| BMI in kg/m2 | |||||
| <25 | 29 (50.9) | 24 (68.6) | 1.00 (Reference) | 1.00 (Reference) | 1.00 (Reference) |
| ≥25 | 27 (47.4) | 11 (31.4) | 0.52 (0.21–1.27) | 0.42 (0.16–1.12) | 0.32 (0.11–0.94) |
| Missing | 1 (1.8) | 0 (0.0) | |||
| Percentage mammographic density | |||||
| <25 | 22 (38.6) | 10 (28.6) | 1.00 (Reference) | 1.00 (Reference) | 1.00 (Reference) |
| ≥25 | 16 (28.1) | 14 (40.0) | 1.97 (0.69–5.62) | 1.54 (0.51–4.69) | 0.93 (0.27–3.21) |
| Missing | 19 (33.3) | 11 (31.4) | |||
| Number of children | |||||
| 0 | 12 (21.1) | 3 (8.6) | 1.00 (Reference) | 1.00 (Reference) | 1.00 (Reference) |
| 1 | 13 (22.8) | 7 (20.0) | 2.39 (0.49–11.65) | 2.64 (0.50–13.83) | 5.34 (0.84–33.79) |
| 2 | 22 (38.6) | 14 (40.0) | 2.91 (0.68–12.53) | 3.12 (0.68–14.24) | 4.76 (0.89–25.43) |
| ≥3 | 10 (17.5) | 11 (31.4) | 4.64 (1.00–21.66) | 4.69 (0.93–23.60) | 10.55 (1.62–68.68) |
| HRT ever | |||||
| No | 34 (59.6) | 25 (71.4) | 1.00 (Reference) | 1.00 (Reference) | 1.00 (Reference) |
| Yes | 21 (36.8) | 10 (28.6) | 0.61 (0.24–1.54) | 0.45 (0.16–1.24) | 0.84 (0.26–2.70) |
| Missing | 2 (3.5) | 0 (0.0) | |||
| Oral contraceptives ever | |||||
| No | 19 (33.3) | 5 (14.3) | 1.00 (Reference) | 1.00 (Reference) | 1.00 (Reference) |
| Yes | 37 (64.9) | 30 (85.7) | 3.04 (1.01–9.15) | 2.90 (0.91–9.24) | 2.36 (0.71–7.85) |
| Missing | 1 (1.8) | 0 (0.0) | |||
| Family history of breast cancer | |||||
| No | 37 (64.9) | 18 (51.4) | 1.00 (Reference) | 1.00 (Reference) | 1.00 (Reference) |
| Yes | 20 (35.1) | 17 (48.6) | 1.84 (0.77–4.39) | 1.58 (0.63–3.99) | 1.46 (0.54–3.90) |
| Family history of ovarian cancer | |||||
| No | 50 (87.7) | 24 (68.6) | 1.00 (Reference) | 1.00 (Reference) | 1.00 (Reference) |
| Yes | 7 (12.3) | 11 (31.4) | 3.11 (1.06–9.09) | 2.87 (0.91–9.11) | 3.41 (0.99–11.73) |
| Ovarian cancer | |||||
| No | 51 (89.5) | 33 (94.3) | 1.00 (Reference) | 1.00 (Reference) | 1.00 (Reference) |
| Yes | 6 (10.5) | 2 (5.7) | 0.60 (0.11–3.26) | 0.37 (0.06–2.17) | 0.46 (0.07–3.01) |
| Other malignant cancer | |||||
| No | 48 (84.2) | 31 (88.6) | 1.00 (Reference) | 1.00 (Reference) | 1.00 (Reference) |
| Yes | 9 (15.8) | 4 (11.4) | 0.76 (0.21–2.75) | 0.54 (0.14–2.12) | 0.66 (0.15–2.96) |
Adjusted for year of diagnosis (2001–2004, 2005–2008).
Adjusted for year of diagnosis and gene (BRCA1, BRCA2).
Adjust for year of diagnosis, gene and age at diagnosis (<50, 50–59, ≥60).
Discussion
BRCA1/2 pathogenic variants were found in 1.8% of unselected breast cancer patients. In contrast to studies reporting BRCA1/2 prevalence for a subset of high risk women,27, 28 the present sample reflects the general breast cancer population. None of the breast cancer risk factors examined differed between BRCA1 and BRCA2 carriers. However, BRCA1 and BRCA2 breast cancers differed in the proportions of patients with ER‐negative disease and basal‐like subtype. Six out of ten BRCA1/2 carriers were not identified through genetic testing in the clinic.
BRCA1 and BRCA2 mutation frequencies in breast and ovarian cancer patients unselected for family history or age at onset are generally low (<1–7% for BRCA1 and 1–3% for BRCA2).29 The combined BRCA1/2 mutation frequency in a Swedish population of unselected breast cancer cases recruited from 1998 through 2000 in Stockholm has been previously estimated to be not more than 1% in the work by Margolin et al.1 In that study, screening for BRCA1 pathogenic variants was limited to exon 11, which covers over half the coding region of BRCA1. 30 More than 70% of diagnosed pathogenic variants including four founder pathogenic variants in the Swedish population are known to be located on this exon.31, 32, 33 Prevalence of BRCA2 pathogenic variants in the Swedish population was deemed by Margolin et al. to be negligible among unselected breast cancer patients due to the low frequency of such pathogenic variants even in high‐risk groups in the region.1 On the contrary, only 33 of 50 BRCA1 pathogenic variants were identified on exon 11 in our study, thus suggesting that 34% of BRCA1 carriers would have been missed if exon 11 alone were screened. Through testing the entire sequences of BRCA1/2 genes with improved methodology and techniques, we estimate the combined prevalence of BRCA1/2 pathogenic variants among unselected breast cancer patients in Sweden to be closer to 2%.
There are close to 2,000 known BRCA1 germline pathogenic variants, many of which are loss‐of‐function frameshift pathogenic variants.34 Nine of 28 (32%) unique BRCA1 and 6 of 33 (18%) unique BRCA2 pathogenic variants were found to be recurrent in Swedish breast cancer patients (i.e., pathogenic variants that were found to occur in at least two unrelated individuals). The relatively low recurrent mutation frequency, including that of Swedish founder pathogenic variants, would mean that screening of selected pathogenic variants alone may not be a sensitive approach in this population as majority of BRCA1 and BRCA2 carriers will have been missed. While BRCA1 pathogenic variants confer a more aggressive tumor phenotype, BRCA2 pathogenic variants typically resemble sporadic breast cancer.35 There is good agreement between our observed results regarding the tumor characteristic differences between BRCA1/2 and non‐BRCA breast cancer cases and what has been previously reported in literature. It has been observed by others that tumors in BRCA1 carriers more frequently exhibited high mitotic count, high grade, ER and PR negativity.36, 37, 38 A large proportion of BRCA1 mutation cases (~80%) have also been documented to be triple negative and basal‐like breast cancers.36, 37, 38 In a Swedish study where 54 female breast cancer patients from 22 families with BRCA2 germ line pathogenic variants from Sweden and Denmark were compared to 214 age‐ and date of diagnosis‐matched controls identified among breast cancer patients from South Sweden, BRCA2‐associated cases were more often node‐positive than non‐BRCA cases.39 Other than nodal involvement, BRCA2‐associated breast carcinomas were generally associated with less aggressive tumor characteristics than BRCA1 cancers, and were more likely to be hormone‐related.37, 38
Thirty‐eight percent of BRCA1/2 carriers were identified through selective clinical testing of 8.2% of breast cancer patients. Grindedal et al. evaluated the results of BRCA1/2 testing in South‐Eastern Norway and found that 65% of the BRCA1/2 carriers would have been missed if using age of onset below 40 or triple negative breast cancer as criteria for testing.40 It is also conceivable that, due to an emphasis on disease family history in current guidelines, a smaller family size may compromise the identification of high risk individuals who would otherwise benefit from genetic testing.41 In a Swedish retrospective study by Nilsson et al. where all breast cancer patients were tested, it was found that while 65% of the BRCA1/2 carriers fulfilled Swedish criteria for testing, only 18% had been identified in regular clinical routine.8 Other factors such as varying compliance with guidelines for the recommendation of BRCA testing by clinicians will lead to even more BRCA1/2 carriers being missed. It may thus be of benefit to test all newly diagnosed breast cancers in light of available targeted therapy options.
To our knowledge, this is the largest population‐based breast cancer testing study for BRCA1/2 published outside of founder populations. Despite the richness of the data which encompasses patient and tumor, some risk groups were too small to be examined with adequate statistical power (e.g., benign breast disease). The Swedish health care system is mainly government‐funded and decentralized, making it possible to identify all women who went for clinical BRCA testing. Nonetheless, private health care also exists, and some BRCA1/2 carriers may have been identified by commercial testing outside the public sector. However, the number of patients tested outside of the national BRCA testing program is likely negligible during the period 2001–2008.8 It should be also noted that the Fluidigm Access Array method used cannot detect large rearrangements and has a sensitivity of ~90%, hence further analytical validity studies are needed. More sensitive methods and the universal BRCA testing of newly breast cancer patients will help to increase the number of women getting the best treatment for their disease.
In summary, BRCA1/2 pathogenic variants were found in 1.8% of an unselected Swedish breast cancer cohort. Six out of ten BRCA carriers were not identified through selective clinical testing routines. Our results give fruitful information for further decisions of BRCA testing for all breast cancer patients at time of diagnosis. The presented data can be a starting point for further studies dealing with issues such as cost effectiveness of screening patients with different tumor characteristics and patient health attitudes.
Supporting information
Data Supplement 1
Data Supplement 2
Acknowledgements
We thank Don Conroy, Caroline Baynes, Patricia Harrington, Martine Dumont and Stéphane Dubois for assistance with the sequencing experiments. We also thank Hanis Mariyah Mohd Ishak and Chek Mei Bok for careful reading of the study. This work was supported by AstraZeneca, the Swedish Research Council (grant no: 2014‐2271 to KC), Swedish Cancer Society (grant no: CAN 2016/684 to KC), FORTE (grant no: 2016‐00081 to KC) and ALF Medicine (grant no: 20170088 to KC). This study was also supported by the Cancer Risk Prediction Center (CRisP; www.crispcenter.org), a Linnaeus Centre [grant no: 70867902] financed by the Swedish Research Council. Targeted sequencing was supported by Cancer Research UK grants C1287/A16563 to DFE and C8197/A16565 to AMD, and the PERSPECTIVE project, funded from the Government of Canada through Genome Canada and the Canadian Institutes of Health Research, the Ministère de l’Économie, de la Science et de l'Innovation du Québec through Genome Québec, and the Quebec Breast Cancer Foundation. BD was supported by the Intramural Research Program of the National Human Genome Research Institute. JL is a recipient of a National Research Foundation Fellowship Singapore (NRF‐NRFF2017‐02) and an award from the Alex and Eva Wallström Foundation.
Conflict of interest: Helene Nordahl Christensen and Astrid Torstensson are employed by AstraZeneca. No other author declared competing financial interests.
References
- 1. Margolin S, Werelius B, Fornander T, et al. BRCA1 mutations in a population‐based study of breast cancer in Stockholm County. Genet Test 2004;8:127–32. [DOI] [PubMed] [Google Scholar]
- 2. Foulkes WD. BRCA1 and BRCA2: chemosensitivity, treatment outcomes and prognosis. Fam Cancer 2006;5:135–42. [DOI] [PubMed] [Google Scholar]
- 3. Niravath P, Cakar B, Ellis M. The role of genetic testing in the selection of therapy for breast cancer: a review. JAMA Oncol 2017;3(2):262–68. [DOI] [PubMed] [Google Scholar]
- 4.Olaparib Keeps Hereditary Breast Tumors in Check. Cancer Discov 2017;7: OF10. [DOI] [PubMed] [Google Scholar]
- 5. Robson M, Im S‐A, Senkus E, et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N Engl J Med 2017;377:523–33. [DOI] [PubMed] [Google Scholar]
- 6. Tutt A, Robson M, Garber JE, et al. Oral poly(ADP‐ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof‐of‐concept trial. Lancet 2010;376:235–44. [DOI] [PubMed] [Google Scholar]
- 7. Tutt A, Ellis P, Kilburn L, et al. Abstract S3‐01: the TNT trial: a randomized phase III trial of carboplatin (C) compared with docetaxel (D) for patients with metastatic or recurrent locally advanced triple negative orBRCA1/2breast cancer (CRUK/07/012). Cancer Res 2015;75: S3‐01. [Google Scholar]
- 8. Nilsson MP, Winter C, Kristoffersson U, et al. Efficacy versus effectiveness of clinical genetic testing criteria for BRCA1 and BRCA2 hereditary mutations in incident breast cancer. Fam Cancer 2017;16:187–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gadzicki D, Evans DG, Harris H, et al. Genetic testing for familial/hereditary breast cancer—comparison of guidelines and recommendations from the UK, France, The Netherlands and Germany. J Community Genet 2011;2:53–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Valencia OM, Samuel SE, Viscusi RK, et al. The role of genetic testing in patients with breast cancer: a review. JAMA Surg 2017;152:589–94. [DOI] [PubMed] [Google Scholar]
- 11. Rosenberg SM, Ruddy KJ, Tamimi RM, et al. BRCA1 and BRCA2 mutation testing in young women with breast cancer. JAMA Oncol 2016;2:730–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Desmond A, Kurian AW, Gabree M, et al. Clinical actionability of multigene panel testing for hereditary breast and ovarian cancer risk assessment. JAMA Oncol 2015;1:943–51. [DOI] [PubMed] [Google Scholar]
- 13. Wendt C, Lindblom A, Arver B, et al. Tumour spectrum in non‐BRCA hereditary breast cancer families in Sweden. Hered Cancer Clin Pract 2015;13:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Holm J, Humphreys K, Li J, et al. Risk factors and tumor characteristics of interval cancers by mammographic density. J Clin Oncol 2015;33:1030–7. [DOI] [PubMed] [Google Scholar]
- 15. Michailidou K, Hall P, Gonzalez‐Neira A, et al. Large‐scale genotyping identifies 41 new loci associated with breast cancer risk. Nat Genet 2013;45:353–61. 61e1‐2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Eriksson M, Li J, Leifland K, et al. A comprehensive tool for measuring mammographic density changes over time. Breast Cancer Res Treat 2018;169:371–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ludvigsson JF, Otterblad‐Olausson P, Pettersson BU, et al. The Swedish personal identity number: possibilities and pitfalls in healthcare and medical research. Eur J Epidemiol 2009;24:659–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Emilsson L, Lindahl B, Koster M, et al. Review of 103 Swedish healthcare quality registries. J Intern Med 2015;277:94–136. [DOI] [PubMed] [Google Scholar]
- 19. Holm J, Li J, Darabi H, et al. Associations of breast cancer risk prediction tools with tumor characteristics and metastasis. J Clin Oncol 2016;34:251–8. [DOI] [PubMed] [Google Scholar]
- 20. Holm J, Eriksson L, Ploner A, et al. Assessment of breast cancer risk factors reveals subtype heterogeneity. Cancer Res 2017;77:3708–17. [DOI] [PubMed] [Google Scholar]
- 21. Borg A, Haile RW, Malone KE, et al. Characterization of BRCA1 and BRCA2 deleterious mutations and variants of unknown clinical significance in unilateral and bilateral breast cancer: the WECARE study. Hum Mutat 2010;31:E1200–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. ENIGMA Consortium . ENIGMA BRCA1/2 Gene Variant Classification Criteria https://enigmaconsortiumorg/wp‐content/uploads/2017/12/ENIGMA_Rules_2017–06‐29pdf 2017: Version 2.5 (29 June) (See Table 6).
- 23. Bergman A, Einbeigi Z, Olofsson U, et al. The western Swedish BRCA1 founder mutation 3171ins5; a 3.7 cM conserved haplotype of today is a reminiscence of a 1500‐year‐old mutation. Eur J Hum Genet 2001;9:787–93. [DOI] [PubMed] [Google Scholar]
- 24. Johannsson O, Ostermeyer EA, Hakansson S, et al. Founding BRCA1 mutations in hereditary breast and ovarian cancer in southern Sweden. Am J Hum Genet 1996;58:441–50. [PMC free article] [PubMed] [Google Scholar]
- 25. Janavicius R. Founder BRCA1/2 mutations in the Europe: implications for hereditary breast‐ovarian cancer prevention and control. EPMA J 2010;1:397–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Loman N, Johannsson O, Kristoffersson U, et al. Family history of breast and ovarian cancers and BRCA1 and BRCA2 mutations in a population‐based series of early‐onset breast cancer. J Natl Cancer Inst 2001;93:1215–23. [DOI] [PubMed] [Google Scholar]
- 27. Winter C, Nilsson MP, Olsson E, et al. Targeted sequencing of BRCA1 and BRCA2 across a large unselected breast cancer cohort suggests that one‐third of mutations are somatic. Ann Oncol 2016;27:1532–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. de Sanjose S, Leone M, Berez V, et al. Prevalence of BRCA1 and BRCA2 germline mutations in young breast cancer patients: a population‐based study. Int J Cancer 2003;106:588–93. [DOI] [PubMed] [Google Scholar]
- 29. Balmana J, Diez O, Rubio IT, et al. BRCA in breast cancer: ESMO clinical practice guidelines. Ann Oncol 2011;22:vi31–vi4. [DOI] [PubMed] [Google Scholar]
- 30. Miki Y, Swensen J, Shattuck‐Eidens D, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994;266:66–71. [DOI] [PubMed] [Google Scholar]
- 31. Zelada‐Hedman M, Wasteson Arver B, Claro A, et al. A screening for BRCA1 mutations in breast and breast‐ovarian cancer families from the Stockholm region. Cancer Res 1997;57:2474–7. [PubMed] [Google Scholar]
- 32. Arver B, Claro A, Langerod A, et al. BRCA1 screening in patients with a family history of breast or ovarian cancer. Genet Test 1999;3:223–6. [DOI] [PubMed] [Google Scholar]
- 33. Arver B, Borg A, Lindblom A. First BRCA1 and BRCA2 gene testing implemented in the health care system of Stockholm. Genet Test 2001;5:1–8. [DOI] [PubMed] [Google Scholar]
- 34. Petrucelli N, Daly MB, Feldman GL. Hereditary breast and ovarian cancer due to mutations in BRCA1 and BRCA2. Genet Med 2010;12:245–59. [DOI] [PubMed] [Google Scholar]
- 35. Atchley DP, Albarracin CT, Lopez A, et al. Clinical and pathologic characteristics of patients with BRCA‐positive and BRCA‐negative breast cancer. J Clin Oncol 2008;26:4282–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Peshkin BN, Alabek ML, Isaacs C, et al. BRCA1/2 mutations and triple negative breast cancers. Breast Dis 2011;32:25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lakhani SR, Reis‐Filho JS, Fulford L, et al. Prediction of BRCA1 status in patients with breast cancer using estrogen receptor and basal phenotype. Clin Cancer Res 2005;11:5175–80. [DOI] [PubMed] [Google Scholar]
- 38. Lakhani SR, Jacquemier J, Sloane JP, et al. Multifactorial analysis of differences between sporadic breast cancers and cancers involving BRCA1 and BRCA2 mutations. J Natl Cancer Inst 1998;90:1138–45. [DOI] [PubMed] [Google Scholar]
- 39. Loman N, Johannsson O, Bendahl P, et al. Prognosis and clinical presentation of BRCA2‐associated breast cancer. Eur J Cancer 2000;36:1365–73. [DOI] [PubMed] [Google Scholar]
- 40. Grindedal EM, Heramb C, Karsrud I, et al. Current guidelines for BRCA testing of breast cancer patients are insufficient to detect all mutation carriers. BMC Cancer 2017;17:438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sibert A, Goldgar DE. The effect of disease penetrance, family size, and age of onset on family history with application to setting eligibility criteria for genetic testing. Fam Cancer 2003;2:35–42. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Supplement 1
Data Supplement 2