

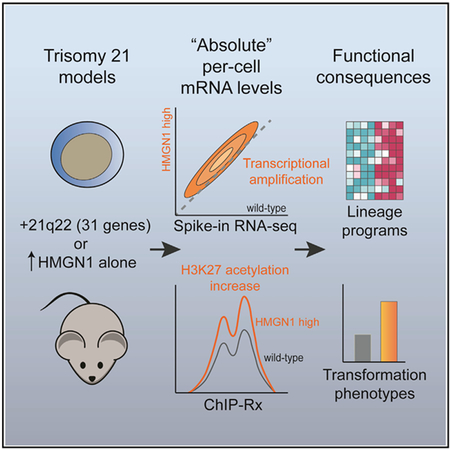
SUMMARY
Down syndrome (DS, trisomy 21) is associated with developmental abnormalities and increased leukemia risk. To reconcile chromatin alterations with transcriptome changes, we performed paired exogenous spike-in normalized RNA and chromatin immunoprecipitation sequencing in DS models. Absolute normalization unmasks global amplification of gene expression associated with trisomy 21. Overexpression of the nucleosome binding protein HMGN1 (encoded on chr21q22) recapitulates transcriptional changes seen with triplication of a Down syndrome critical region on distal chromosome 21, and HMGN1 is necessary for B cell phenotypes in DS models. Absolute exogenous-normalized chromatin immunoprecipitation sequencing (ChIP-Rx) also reveals a global increase in histone H3K27 acetylation caused by HMGN1. Transcriptional amplification downstream of HMGN1 is enriched for stage-specific programs of B cells and B cell acute lymphoblastic leukemia, dependent on the developmental cellular context. These data offer a mechanistic explanation for DS transcriptional patterns and suggest that further study of HMGN1 and RNA amplification in diverse DS phenotypes is warranted.
Graphical Abstract
In Brief
How trisomy 21 contributes to Down syndrome phenotypes, including increased leukemia risk, is not well understood. Mowery et al. use per-cell normalization approaches to reveal global transcriptional amplification in Down syndrome models. HMGN1 overexpression is sufficient to induce these alterations and promotes lineage-associated transcriptional programs, signaling, and B cell progenitor phenotypes.
INTRODUCTION
Down syndrome, or constitutional trisomy of chromosome 21 (+21), causes numerous developmental and phenotypic changes at the level of the whole organism. Cell biological studies of Down syndrome compared to euploid cells have reported diverse alterations associated with +21, but the molecular basis for most of these is not clear. Two general theories, which are not mutually exclusive, attempt to explain Down syndrome phenotypes as either related to aneuploidy itself (i.e., simply having an additional copy of genetic material) or due to dosage increases of specific genes on chromosome 21 (Beach et al., 2017; Bonney et al., 2015; Roper and Reeves, 2006).
Trisomy 21 is highly associated with acute leukemia. Individuals with Down syndrome have at least a 20-fold increased risk of developing B cell acute lymphoblastic leukemia (B-ALL) compared to non-Down syndrome individuals (Berger, 1997). Chromosome 21 is also the most common somatically gained whole chromosome in the leukemia cells of individuals without Down syndrome (Heerema et al., 2007). Additionally, interstitial amplification of a portion of the long arm of chromosome 21 (iAMP21) is seen in a specific subtype of B-ALL and is associated with a poor prognosis (Harrison et al., 2014; Li et al., 2014). Furthermore, some individuals with Down syndrome developmental phenotypes have triplication of only focal segments of chromosome 21 (Korenberg et al., 1994). This can involve one of the so-called Down syndrome critical regions (DSCRs) on chr21q22, which overlaps with the iAMP21 region in B-ALL and a similar region of recurrent somatic amplification in acute myeloid leukemia (AML) (Moorman et al., 2010; Mrózek et al., 2002; Rand et al., 2011). Together, these data suggest that genes in the DSCR might be responsible for at least some Down syndrome developmental and cancer phenotypes.
Many studies indicate that Down syndrome cells have genome-wide epigenomic alterations, not confined to chromosome 21, when compared to euploid cells. These include changes in gene expression (Costa et al., 2011; Letourneau et al., 2014; Lockstone et al., 2007), RNA content (Hamurcu et al., 2006), histone modifications (Lane et al., 2014; Letourneau et al., 2014), nucleosome spacing (Kahmann and Rake, 1993), and DNA methylation (Lu et al., 2016; Mendioroz et al., 2015). Yet, linking transcriptional and epigenomic changes directly to chromosome 21 or specific triplicated genes has been challenging because of other genetic and phenotypic heterogeneity within cohorts of Down syndrome individuals. A recent study analyzed otherwise isogenic cells from a pair of identical twins who were discordant only for trisomy 21 (Letourneau et al., 2014). Those experiments revealed alterations in gene expression and histone modifications across all chromosomes in Down syndrome cells, in a pattern that suggested trisomy 21 modulates global gene regulation in discrete domains. The authors coined the term “gene expression dysregulation domains” (GEDDs) as a possible unifying characteristic of +21 cells, and they also detected similar expression patterns in an animal model of Down syndrome that triplicates 65 mouse chromosome 21 orthologs. The culprit gene(s) were not identified but they and others postulated that future studies should attempt to identify chromosome 21 gene products that could globally modify the epigenome (Pope and Gilbert, 2014).
We previously showed that B cells from Down syndrome mouse models and B-ALL leukemias from patients with Down syndrome have epigenomic changes compared to euploid cells (Lane et al., 2014). We found that among the 31 genes triplicated in the Ts1Rhr Down syndrome mouse model, the most critical gene for maintaining aberrant B cell phenotypes associated with +21 was Hmgn1, which encodes a nucleosome binding protein that modulates chromatin compaction and gene expression. We therefore hypothesized that HMGN1 overexpression could mediate trisomy 21-associated epigenomic and chromatin regulatory alterations.
RESULTS
Genome-wide Transcriptome Amplification in +21 Models Revealed by Absolute Normalization
Progenitor B cells from Ts1Rhr mice, which model Down syndrome by triplicating 31 genes on mouse chr16 orthologous to a DSCR on human chr21q22, have genome-wide differences in histone modifications compared to wild-type cells (Lane et al., 2014). Despite these widespread epigenomic changes, transcriptome analysis in the same B cells revealed only a small subset of genes significantly increased or decreased in expression. This is consistent with studies in Down syndrome cells and other +21 models that show a modest number of genes with differential expression compared to diploid cells, despite global changes in epigenomic modifications. To reconcile this conundrum, we performed a series of experiments to evaluate the transcriptome and epigenome of cells that model trisomy 21.
To improve whole transcriptome profiling, we performed RNA-sequencing (RNA-seq) in the presence of “spiked-in” nonmammalian synthetic ERCC (External RNA Controls Consortium) RNA controls for normalization of RNA content per cell (Lin et al., 2012; Lovén et al., 2012). The advantage of normalizing to an exogenous RNA control is that it allows comparison of samples based on absolute RNA per cell, rather than normalizing to the median measured RNA in each sample. To our surprise, we found that pro-B cells from the Ts1Rhr mouse produced more RNA transcripts per gene per cell than wild-type pro-B cells (p < 0.0001 by t test for difference from no change) (Figures 1A and 1B). The increase in RNA output was evident in genes located on all chromosomes, but the magnitude of transcriptional change was not the same across all mRNAs. Genes that were not expressed in wild-type cells remained silenced, and the highest expressed genes were less strongly affected in Ts1Rhr pro-B cells. The per-cell increase in RNA expression in cells from the Ts1Rhr model was most evident among the genes expressed the low- to mid-range (Figure 1C).
Figure 1. Triplication of a Down Syndrome Critical Region or HMGN1 Overexpression Alone Results in Increased RNA per Transcript per Cell.

(A) Log2 fold change per transcript from RNA-sequencing of progenitor B cell colonies from Ts1Rhr compared to wild-type (top) and from HMGN1-OE transgenic versus wild-type (bottom) bone marrow. n = 3 biological replicates per genotype. Plots on the left are median read count normalized between samples (“Relative”), and plots on right are ERCC spike-in per cell-normalized (“Absolute”). Distribution compared to the null hypothesis of no difference between genotypes using single sample t test.
(B) The data from (A) are plotted with each dot representing a single gene’s expression quantitated in the indicated genotypes. The dotted line represents the unity line of no difference between genotypes. The contour lines and legend represent high (green) to low (blue) relative bin density. Red and blue numbers represent the number of genes that increase or decrease, respectively, in Ts1Rhr or HMGN1 versus wild-type (fold-change >1.5, p < 0.05). FPKM, fragments per kilobase of transcript per million mapped reads; nFPKM, ERCC-normalized FPKM.
(C)Fold change in expressed transcripts (FPKM ≥0.5) quantitated by relative or absolute spike-in normalization in progenitor B cells of the indicated genotypes were binned into 100 groups ranked by expression level in wild-type cells. Each point is the mean log2 fold change in the bin, error bars are 95% confidence intervals. The red line represents non-linear curve fit of the binned data points.
(D) Venn diagrams showing (top) the overlap of the top 200 most upregulated genes in Ts1Rhr B cell progenitors compared to wild-type, or (bottom) the significantly enriched gene sets (FDR <0.05) in gene set enrichment analysis (GSEA) in the 3,402 gene sets in “C2 CGP” collection in MSigDB (Broad Institute) using data from either relative or absolute normalized expression analysis.
Spike-in normalization did not interfere with detection of the most differentially expressed genes or resulting gene expression signatures in these B cell progenitors. The 200 most increased genes in Ts1Rhr as compared to wild-type cells were highly overlapping in the absolute- and relative-normalized datasets (p < 0.0001 by Fisher’s exact test, Figure 1D). Gene set enrichment analysis (GSEA) (Subramanian et al., 2005) applied to all expressed genes in both datasets identified many of the same signatures (Figure 1D; Data S1). Therefore, per-cell normalization added additional insights to traditional gene expression profiling by revealing a low-level global increase in RNA per-gene in Ts1Rhr cells. At the same time, absolute normalization preserved identification of the highly differentially expressed genes as seen with relative normalization techniques.
To validate the association between polysomy of human chromosome 21 and increased RNA, we analyzed human retinal pigment epithelium (RPE) cells that were engineered to carry extra copies of specific chromosomes using microcell-mediated chromosome transfer (MMCT) (Lane et al., 2014). Consistent with our findings from murine Ts1Rhr pro-B cells, RPE cells with 1 or 2 extra copies of human chromosome 21 also contained more RNA per cell than euploid RPE cells (Figure S1A).
HMGN1 Overexpression Recapitulates RNA Amplification Seen in the Larger DSCR Triplication
We previously found that Hmgn1 was the chr21q22 orthologous gene most essential to promote progenitor B cell self-renewal when amplified in the Ts1Rhr mouse model (Lane et al., 2014). Given that HMGN1 promotes chromatin accessibility, modifications in post-translational histone marks, and expression changes in genes regulated by nucleosome configuration (Catez et al., 2002; Lim et al., 2005; Rochman et al., 2009), we hypothesized that it might be responsible for the transcriptional amplification associated with Down syndrome and the Ts1Rhr model. Therefore, we analyzed cells from a transgenic model that overexpresses only HMGN1 (HMGN1-OE), at ~2-fold protein levels compared to wild-type cells (Bustin et al., 1995). When we performed RNA-seq with spike-in controls, we saw global amplification of expressed genes in HMGN1-OE cells that was not evident when using median normalization, similar to what we observed in Ts1Rhr cells (Figures 1A and 1B). There were no significant differences in cell-cycle status between HMGN1-OE and wild-type cells that might contribute to changes in RNA content (Figure S1B).
We next measured median-normalized RNA expression in low, moderate, and high FPKM (fragments per kilobase of transcript per million mapped reads) bins. We observed a small relative increase in “low-expressed” genes and decrease in “high-expressed” genes in Ts1Rhr when compared to wild-type cells using median normalization (Figure 2A). This was similar to what was reported in the description of GEDDs in human +21 cells and mouse cells from the Ts65Dn Down syndrome model (Letourneau et al., 2014). However, when we used spike-in normalization, we found that all gene groups were increased in expression in Ts1Rhr and HMGN1-overexpressing cells (Figure 2A). Thus, the dominant expression change in Ts1Rhr B cells may be global transcriptional amplification, possibly via the activity of HMGN1.
Figure 2. Transcriptome Changes in Ts1Rhr and HMGN1-Overexpressing B Cells Are Widely Distributed but Enriched at Genes Associated with Certain Histone Modifications.

(A) Distribution of fold change in individual gene expression between Ts1Rhr (top) or HMGN1-OE (bottom) and wild-type B cell progenitors from relative (orange) or cell count-normalized (“Absolute,” red) RNA-seq analysis. Positive fold change = higher in Ts1Rhr or HMGN1 compared to WT. Plots are all expressed genes (FPKM >0.1) divided into low, mid, and high terciles of equal size by gene expression in wild-type cells. Distributions compared by t test.
(B) Median H3K4m3 and H3K27me3 histone marks in wild-type B cell progenitors, plotted as median mark intensity in each of 50 bins of genes ranked by expression in wild-type cells, as in Figure 1C.
(C) Left: gene set enrichment analysis of all expressed genes in HMGN1 overexpressing compared to wild-type B cell progenitors of polycomb repressor complex 2 and H3K27-related datasets in the C2 CGP collection of MSigDB (Broad Institute) shows significant enrichment of EZH2 target genes in hematopoietic stem cells (KAMMINGA_EZH2_TARGETS) and genes marked by H3K27me3 in reprogrammed iPS cells (MIKKELSEN_MCV6_HCP_WITH_H3K27ME3). ES, enrichment score; FDR, false discovery rate (adjusted p value). Right: heatmap of expression level of the “leading edge” genes in the Kamminga gene set from GSEA analysis in biological triplicate samples of wild-type, Ts1Rhr, and HMGN1-OE B cell progenitors (blue, low; red, high expression).
We previously found that the genes whose expression was most affected in Ts1Rhr pro-B cells and in patients with Down syndrome-associated leukemias overlapped with polycomb repressor complex 2 (PRC2) “targets” and genes associated with H3K27 trimethylation (the histone mark catalyzed by PRC2 activity) (Lane et al., 2014). These were particularly enriched in so-called “bivalent” genes in wild-type cells marked simultaneously by both active (H3K4me3) and inactive (H3K27me3) histone marks. Here, we found that although absolute normalization revealed a larger number of genes whose expression was amplified in the setting of HMGN1 overexpression compared to median normalization, the amplified genes in the “low to mid” expression level at baseline in wild-type cells were those genes marked with both active and repressive histone marks (Figure 2B) and enriched for known targets of EZH2 (the catalytic subunit of PRC2) in hematopoietic stem cells (Figure 2C). These findings are consistent with the established function of HMGN1 to relax chromatin compaction and modify, but not on its own initiate, transcription. Genes that are active but “poised” for higher expression, as indicated by bivalent chromatin marks at baseline, may be most likely to increase in expression when additional HMGN1 is present.
RNA Amplification by HMGN1 Is Rapid and Requires Its Nucleosome Binding Residues
To determine if increased RNA output in the setting of HMGN1 overexpression is direct, we generated human B cell lines (euploid for HMGN1 copy number at baseline) with a doxycycline-inducible HMGN1 cassette (Figure 3A). As a control, we used an HMGN1 cDNA encoding a protein with two serine residues in the nucleosome binding domain mutated to glutamate (HMGN1-SE), which abrogates its ability to bind to nucleosomes (Prymakowska-Bosak et al., 2001). Overexpression of HMGN1 increased the total RNA content per cell within hours after the addition of doxycycline (Figure 3B). The increase in RNA output required the ability of HMGN1 to bind to the nucleosome because induction of the HMGN1-SE mutant did not increase RNA content compared to cells containing an empty expression cassette. To rule out that the increased RNA per cell in HMGN1-overexpressing cells was due to a change in the rate of RNA turnover, we performed a pulse-chase experiment before and after induction of HMGN1. The rate of labeled RNA decay in HMGN1 overexpressing cells was similar to that in control HMGN1-SE overexpressing cells (Figure S2A).
Figure 3. HMGN1 Increases RNA Content after Short-Term Induction, Requires Residues Necessary for Nucleosome Binding, and Induces Greater Nascent Transcription.

(A) Western blotting of lysates from Nalm6 B-ALL cells stably infected with a lentivirus that contains a doxycycline-inducible human HMGN1 cDNA (N1), an HMGN1 mutant that lacks the ability to bind to the nucleosome, HMGN1-S20,24E (SE), or empty vector (V) at baseline (0 hr), 3 hr, and 6 hr after the addition of doxycycline to the culture medium.
(B) Total RNA content measured by fluorometric assay in biological triplicates of the indicated cells relative to empty vector over a time course after addition of doxycycline to the culture medium. n = 3 biological replicates. Samples compared by t test.
(C) Left: expression ratios across all expressed genes from RNA-sequencing of Nalm6 cells 6 hr after addition of doxycycline to overexpress HMGN1 or HMGN1-SE, calculated using relative or absolute spike-in normalization (n = 4 biological replicates per condition). Right: dot plot of individual gene expression comparing HMGN1 to HMGN1-SE expressing Nalm6 cells after doxycycline induction using relative or absolute normalization. The dotted line represents the unity line of no difference between conditions. The contour lines and legend represent high (green) to low (blue) relative bin density. Red and blue numbers represent the number of genes that increase or decrease, respectively, in HMGN1-WT versus HMGN1-SE (fold-change >1.5, p < 0.05). FPKM, fragments per kilobase of transcript per million mapped reads; nFPKM, ERCC-normalized FPKM.
(D) Distribution of fold change in individual gene expression between HMGN1 and HMGN1-SE overexpressing cells from relative (orange) or absolute normalized (red) RNA-seq analysis. Positive fold change = higher in HMGN1 compared to HMGN1-SE. Plots are divided into low, mid, and high terciles by gene expression in HMGN1-SE cells, similarly to Figure 2A. Distributions compared by t test.
(E) Cumulative distribution function plot of SLAM-seq data for active genes showing the log2 fold change in nascent reads during the labeling period in induced compared to uninduced cells. The red line represents cells after induction of HMGN1 and the gray line represents cells after induction of the HMGN1-SE mutant (n = 3 biological replicates). The rightward shift of the curves suggests that per gene there are more transcripts generated after HMGN1 induction compared to uninduced cells or to cells expressing the HMGN1-SE mutant. Changes in distributions are significant at p < 1e–9 by two-tailed t test.
(F) Binned plot of active genes ranked by nascent transcript fraction in uninduced cells (x axis) versus the mean log2 fold change in the conversion ratio (number of nascent transcripts generated in the labeling period relative to pre-existing transcripts) for genes in a bin, in induced or uninduced cells (y axis). Error bars represent 95% confidence intervals of the mean.
(G) Conversion fraction (nascent transcripts per total transcripts) of an illustrative gene, PRDM1, in uninduced cells, cells after induction of HMGN1, or cells after induction of the HMGN1-SE mutant. Error bars represent SEM.
In some contexts, the amount of RNA per cell correlates with cell-cycle status, cell size, and growth rate (Darzynkiewicz et al., 1979; Padovan-Merhar et al., 2015). To test for this possibility at the single-cell level, we analyzed collections of cells in a time course after induction of HMGN1 using several methods, including a suspended microchannel resonator (SMR) device to measure the buoyant mass of a cell with femtogram precision (Cermak et al., 2016), a Coulter counter to measure single-cell volume, and a microfluidic hydrodynamic trap array to directly observe single-cell proliferation kinetics (Kimmerling et al., 2016). We saw no difference in buoyant mass, cell volume, or single-cell interdivisionary time between HMGN1 overexpressing cells and controls within the first 6 hr after doxycycline induction (Figures S2B–S2D). These data suggest that HMGN1-overexpressing cells are not cycling or generating mass from cell-extrinsic sources more rapidly than controls.
We next performed RNA-seq with exogenous controls in B cell lines 6 hr after induction of wild-type HMGN1 or the nucleosome binding incompetent HMGN1-SE mutant. Similar to what we observed in the primary progenitor B cells, absolute normalization revealed a global increase in per transcript RNA content measured per cell, averaging ~1.3-fold per gene across the genome (Figures 3C, S3A, and S3B). When we compared gene expression in HMGN1-overexpressing cells to HMGN1-SE overexpressing cells, ranked by expression level in the HMGN1-SE control, we observed transcript amplification in most genes with baseline expression of >0.5 FPKM (Figures 3C and 3D). Standard relative read count normalization between samples did not detect the same degree of global change in gene expression.
To ask if the RNA increase in cells overexpressing HMGN1 is directly related to greater nascent transcription per gene, we performed thiol(SH)-linked alkylation for the metabolic labeling of RNA (SLAM-seq) (Herzog et al., 2017). SLAM-seq utilizes 4-thiouridine incorporation to quantitate newly synthesized transcripts as a fraction of the total transcripts of a given gene. The data can be expressed as the “conversion fraction,” referring to the iodoacetamide-induced alkylation that converts labeled residues during the reverse transcription step prior to next generation sequencing. Cells induced to overexpress HMGN1 generated more nascent transcripts per gene during the 5-hr labeling period compared to cells overexpressing the HMGN1-SE mutant or uninduced controls (p < 1e–9, Figure 3E). An increase in conversion fraction was evident globally (Figures 3F and S3C) and at the single gene level (Figure 3G) across measurable genes. Consistent with the increased RNA per cell being secondary to increased transcription, we also found that pre-mRNA levels, measured using nascent intron expression from SLAM-seq and validated by exon and intron real-time qPCR, increased after HMGN1 induction (Figures S3D and S3E). While several mechanistic functions, some indirect, attributed to HMGN1 at the chromatin could be contributing to expression changes (Catez et al., 2002; Deng et al., 2015; Rattner et al., 2009), these data support the assertion that HMGN1 overexpression results in an increase in nascent transcription, and this phenotype is dependent on HMGN1 binding to the nucleosome.
Spike-In Chromatin Immunoprecipitation and Sequencing Unmasks Global H3K27 Hyperacetylation by HMGN1 Overexpression
Next, we wanted to quantitate the immediate consequences of HMGN1 induction on histone marks. Quantitation of locus-specific histone mark changes in the setting of perturbations that cause global alterations in the epigenome is challenging. Local changes associated with HMGN1 overexpression could be obscured using traditional chromatin immunoprecipitation and sequencing (ChIP-seq) bioinformatics, which also relies on normalization to median read counts between samples and might hide a widespread effect (Chen et al., 2015).
Therefore, we performed ChIP-seq for histone marks in cells with inducible HMGN1 expression normalized to spiked-in exogenous nuclei from Drosophila S2 cells (“ChIP-Rx,” or chromatin immunoprecipitation with reference exogenous genome) (Orlando et al., 2014). Six hours after induction of HMGN1, we observed a global increase in H3K27 acetylation compared to baseline (p < 0.0001, Figures 4A and 4B). This finding is consistent with more globally active chromatin at promoters and/or enhancers, and with the increase in transcriptional output per cell we detected using per cell-normalized RNA-seq. We observed a similar genome-wide increase in promoter-associated H3K4me3 after induction of HMGN1, also consistent with the observed increased transcription. Notably, ChIP-seq analysis using traditional median read count normalization did not detect the global increase in H3K27ac or H3K4me3 (Figures 4A and 4B).
Figure 4. Absolute per Cell-Normalized Chromatin Immunoprecipitation Reveals Global Effects of HMGN1 Overexpression on Histone Marks.

(A) Metagene plots and read quantitation of H3K27ac and H3K4me3 ChIP-seq in Nalm6 cells stably expressing a doxycycline-inducible HMGN1 cDNA 6 hours after addition of doxycycline or vehicle to the culture medium. Plots on the left are relative median read count-normalized “traditional” ChIP-seq, and on the right, absolute per cell-normalized to sequencing reads from ChIP-seq of exogenous spiked-in Drosophila S2 cell nuclei. RPM, reads per million; nRPM, normalized reads per million; x axis of metagene plots shows the transcriptional start site (TSS) and 5 kb upstream and downstream. Boxplots represent the distribution of ChIP-seq peak height at each detected gene (compared by t test).
(B) Gene tracks showing a representative locus on chr17 where an increase in H3K27 acetylation after addition ofdoxycycline is only evident after applying per cell absolute spike-in normalization.
(C) Relative read counts of histone marks at peaks within 1 kb of the TSS of genes that significantly (fold-change >1.5, p < 0.05) increased (UP, red) or decreased (DOWN, blue) in expression after induction of HMGN1 overexpression. Distributions compared by t test.
Next, we performed an integrated analysis of ChIP-seq and RNA-seq in B cells after induction of HMGN1 or the HMGN1-SE mutant. The normalized ChIP-seq peak heights for H3K27ac were significantly higher within 1 kb of the promoters of genes that were most increased in expression in the setting of HMGN1 induction (Figure 4C). These data show that gene expression changes within 6 hr following induction of HMGN1 are associated with specific histone marks and suggest that measurement of histone marks “classically” associated with active expression (e.g., H3K27ac) are preserved when measured by per cell-normalized ChIP-seq.
In addition to its association with promoters and near transcriptional start sites, H3K27ac is also associated with active enhancers. Therefore, we tested the hypothesis that even in the setting of global expression changes, the genes with the greatest increase in expression after HMGN1 induction might be enriched for those associated with broad H3K27ac deposition in regulatory regions of developmental stage and lineage-defining genes, the so-called “super enhancers” (Whyte et al., 2013). Indeed, among genes expressed at baseline (FPKM >1) in Nalm6 cells, those near super enhancers were more likely to have a significant increase in gene expression (fold-change >1.5, adjusted p value [adjP] <0.05) than those without an associated super enhancer (16.7% versus 10.2%, p = 0.003 by Chi square test with Yates correction). These data suggested that HMGN1 overexpression might augment a cell’s pre-existing transcriptional programs, rather than induce a wholesale change in cell state. In support of this hypothesis, we observed no significant differences in the pattern of chromatin accessibility after short-term HMGN1 induction, as measured by an Assay for Transposase-Accessible Chromatin sequencing (ATAC-seq), including at loci with increased expression and H3K27 acetylation (Figure S4A). Together, these findings suggest that HMGN1 supports increased expression of pre-existing transcriptional programs.
HMGN1 Promotes Upregulation of Developmental Stage-Specific Pathways in B Cells
“Transcriptional addiction” may be a hallmark of cancer (Franco and Kraus, 2015). Based on our data, HMGN1 overexpression and trisomy 21 could contribute to leukemia by enhancing transcription. However, it remains unclear specifically how transcriptional amplification promotes tumor growth and whether this process differs by cellular context. One possibility is that increased transcription allows a cell to overcome growth-limiting conditions by increasing the activity of survival pathways. Therefore, we tested for effects of HMGN1 overexpression or chr21q22 triplication on developmental stage-specific and leukemia-associated signaling pathways. Developing B cells and B-ALLs can be classified as pre-B, with predominant growth signaling via B cell receptor (BCR) pathways to SRC family kinase activation and targets such as BCL6; or pro-B, with signaling via cytokine receptor pathways to JAK/STAT activation and targets such as the D type cyclins (Geng et al., 2015). The model systems we tested represent these two B cell subtypes based on developmental stage, cell surface markers, and gene expression profile (human Nalm6 is a pre-B cell; mouse B cells colonies grown in the presence of interleukin-7 are pro-B cells). Therefore, we hypothesized that upon HMGN1 overexpression, Nalm6 cells might preferentially increase expression of BCR signaling and pre-B ALL pathways, and pro-B cells from Ts1Rhr or HMGN1-OE bone marrow may increase JAK/STAT pathways.
We observed transcriptome changes supporting each of these hypotheses. RNA-seq of Nalm6 cells after induction of wild-type HMGN1 compared to the nucleosome binding-incompetent HMGN1-SE demonstrated specific enrichment of B cell receptor (BCR) signaling pathway-associated genes and genes that distinguish pre-B ALL from other subtypes of B-ALL (Figure 5A; Data S2) (Geng et al., 2015). At the chromatin, HMGN1 induction resulted in increased expression, H3K27 acetylation, and HMGN1 binding to promoters of genes important in B cells, such as PRDM1 and IRF4 (Figures S4A and S4B). Conversely, BCL6, a zinc-finger transcription factor with repressive activity at many BCR pathway genes in pre-B cells (Geng et al., 2015) had decreased binding at the same promoters (Figure S4B), consistent with the observed increased transcription. These changes were associated with modest increases in protein level of some BCR pathway members and enhanced phosphorylation of AKT, a downstream target of tonic BCR signaling in Nalm6 cells (Figures S4C–S4E). Histone acetylation driven by transcriptional co-activators such as the histone acetyltransferases (HATs) CBP and p300 are known to act in opposition to BCL6 and its associated co-repressor complexes in B cells (Hatzi et al., 2013). Furthermore, HMGN1 promotes HAT activity, possibly via local steric changes that allow histone modifying enzymes greater access to individual nucleosomes (Lim et al., 2005). These data support the hypothesis that HMGN1 modulates gene expression and histone mark alterations at “tunable” loci, such as at poised promoters and enhancers, possibly in association with relative changes in activity of transcriptional proteins.
Figure 5. Lineage- and Leukemia-Specific Pathway Expression Is Enriched in Cells with Overexpression of HMGN1, and Hmgn1 Triplication Is Necessary for Pro-B Cell Phenotypes Associated with the Ts1Rhr Model.

(A) GSEA of RNA-sequencing data from Nalm6 preB-ALL cells after induction of HMGN1 or HMGN1-SE showing enrichment in B cell receptor signaling pathway genes (BIOCARTA_BCR_SIGNALING) and in genes that distinguish preB-ALL from other types of ALL (PreB-ALL). Distribution plots shown on left (ES, enrichment score; FDR, false discovery rate [adjusted p value]), and heatmaps of gene expression (blue, low; red, high) genes in the “core enrichment” from those GSEA analyses are shown on the right.
(B) GSEA of RNA-sequencing data from primary B cell progenitors cultured from Ts1Rhr or HMGN1-OE transgenic bone marrow showing enrichment of STAT5 target genes and genes bound by STAT5 and B cell transcription factors in Ts1Rhr and HMGN1-OE B cells compared to wild-type (ES, enrichment score; FDR, false discovery rate [adjusted p value]).
(C) Heatmaps showing expression of the top 50 genes in the “leading edge” of the STAT5 target gene signature from GSEA in wild-type (WT), Ts1Rhr, and HMGN1-OE B cell progenitors.
(D) Mouse genotypes tested in panels E-G and their associated copy number profiles for Hmgn1 and 30 other genes in the Ts1Rhr model. The Ts1Rhr/Hmgn1+/− intercross has three copies of the chromosome 21q22 orthologs, except for only two copies of Hmgn1.
(E) Progenitor B cell colony numbers in 1st and 2nd passage derived from bone marrow of mice of the indicated genotypes (for E–G, n = 3 biological replicates, genotypes compared by t test, *p < 0.05).
(F) Relative H3K27 acetylation measured by intracellular flow cytometry in progenitor B cells of the indicated genotypes harvested after passage 1.
(G) Relative RNA expression by real-time qPCR for Fgf13, Kit, and Hmgn1 in progenitor B cells of the indicated genotypes harvested after passage 1.
Consistent with lineage-specific activity, in pro-B cells, triplication of 21q22 orthologs or overexpression of HMGN1 alone was associated with enrichment of B-ALL-associated STAT5 target genes and genes in chromatin regions marked by binding of STAT5 and B cell lineage transcription factors (Figure 5B; Data S2) (Katerndahl et al., 2017). Ts1Rhr and HMGN1-OE pro-B cells were remarkably similar in their gene expression changes compared to wild-type cells, especially in the subset of known leukemia-associated STAT5 and B cell transcription factor-bound genes (Figure 5C). The observed associations with HMGN1 overexpression were cell-type-specific and not only due to global increased transcription, because STAT5/pro-B cell genes were not specifically enriched in Nalm6 pre-B cells after HMGN1 induction, and BCR/preB-ALL signatures were not enriched in pro-B cell colonies from Ts1Rhr or HMGN1-OE mice. Together, these data suggest that in addition to modest global transcriptional amplification, within a given cell type, HMGN1 overexpression most significantly affects genes associated with lineage and maturation stage-specific transcriptional programs.
Hmgn1 Triplication Is Necessary for Progenitor B Cell Phenotypes Associated with the Ts1Rhr Down Syndrome Model
To test if three copies of Hmgn1 were necessary for the increased colony-forming activity (Lane et al., 2014) and transcriptional phenotypes associated with triplication of 31 chromosome 21q22 orthologs, we crossed Ts1Rhr mice with mice engineered to lack one copy of Hmgn1 (Birger et al., 2003). The resulting progeny included wild-type (two copies of Hmgn1 and all other genes), Ts1Rhr (three copies of the 31 genes, including Hmgn1), and Ts1Rhr/Hmgn1+/− (two copies of Hmgn1, but three copies of 30 other genes; black, red, and gray, respectively, in Figures 5D–5G). Reversion of Hmgn1 to two copies abrogated the increase in progenitor B cell colony-forming activity (Figure 5E), increase in global H3K27 acetylation (Figure 5F), and gene-specific increases in RNA expression (Figure 5G) associated with Ts1Rhr B cells. These data suggest that triplication of Hmgn1 is required for at least several progenitor B cell phenotypes associated with 21q22 DSCR triplication.
DISCUSSION
We have attempted to reconcile seemingly conflicting observations comparing epigenomic modifications and transcriptional profiles in Down syndrome and euploid cells. Several studies have noted global, genome-wide alterations in the epigenome of +21 cells, including in CpG methylation (Lu et al., 2016; Mendioroz et al., 2015), global and locus-specific changes in the histone marks H3K4me3, H3K27me3, and H3K27ac (Lane et al., 2014; Letourneau et al., 2014), and even changes in the length of spacing between sequential nucleosomes (Kahmann and Rake, 1993). However, gene expression profiling has detected only modest changes in small numbers of genes that are often not consistent across cell types or between studies. There is even controversy as to whether the genes on chromosome 21 itself have a higher expression level in Down syndrome cells by virtue of trisomy (Antonarakis, 2017).
Using cell count-normalized RNA-seq and ChIP-seq, we found that otherwise isogenic models of Down syndrome have increased RNA per cell compared to euploid cells. This transcriptional amplification was evident across thousands of genes and was further enriched at genes associated with certain chromatin marks. Overexpression of HMGN1, a nucleosome binding protein encoded on chr21q22 that is known to relax chromatin compaction and globally modulate epigenomic control, was sufficient to recapitulate these transcriptional phenomena and directly caused an increase in nascent transcription. These data may provide a mechanistic explanation for a prior observation of increased RNA in peripheral blood mononuclear cells of Down syndrome compared to non-Down syndrome individuals (Hamurcu et al., 2006).
We propose that trisomy 21/HMGN1-mediated transcriptional amplification could also offer a mechanism contributing to other genome-wide changes observed in Down syndrome cells. While the previously described specific domains of gene expression dysregulation, “GEDDs” (Letourneau et al., 2014), are controversial (Do et al., 2015), many studies have nonetheless observed genome-wide epigenomic alterations in Down syndrome, not confined to chromosome 21. HMGN1 is one possible culprit chromosome 21 gene to cause global transcriptional changes, given its ability to modulate chromatin (Pope and Gilbert, 2014), but it was unclear how HMGN1 might cause specific expression patterns. Trisomy 21/HMGN1 transcriptional amplification may offer one model, which could be relevant to many transcriptional measurements in Down syndrome, not only for interpreting GEDDs. By fitting to an equivalent median signal between Down syndrome and euploid cells, genes with low expression in euploid cells might appear relatively higher in Down syndrome, and those with high expression might appear relatively lower. Thus, the observed “flattening” of transcription attributed to trisomy 21 (Pope and Gilbert, 2014) could be a product of median normalization on an amplified transcriptome (Figure 6).
Figure 6. Model for Gene Expression Changes Associated with Trisomy 21 or HMGN1 Overexpression.

Absolute per cell normalization of transcriptome data may provide a mechanistic explanation for some previous, incompletely explained observations in trisomy 21 cells, including and global RNA and epigenomic changes. If the actual expression pattern in trisomy 21 (or with HMGN1 overexpression) is amplification that differentially affects areas of baseline “high” or “low” expression (left), then forced normalization to an equivalent median signal between samples (blue arrow and dotted line) would instead be interpreted as decreased expression in baseline “high” expressed genes and increased expression in “low” expressed genes (right).
While the transcriptional changes we observed were “global,” in that they affected many genes throughout the genome and in aggregate were associated with increased expression, the effect size was not identical at all loci, and not every gene was affected. Transcriptome reshaping by HMGN1 was associated with chromatin signatures at promoters, and in B cells, lineage-defining genes were enriched for having susceptible baseline chromatin and expression states. This led to an overall effect where, although many genes were upregulated, there was more pronounced activation of specific sets of B-lineage genes. This may have contributed to the functional consequences in B cell growth and signaling we observed. Knockout of HMGNs changes the DNase hypersensitivity landscape and gene expression in a tissue-specific manner (Deng et al., 2015), which fits logically with what we found in the setting of HMGN1 overexpression. Furthermore, the concept that pre-existing chromatin states shape the most affected genes in the setting of “global” transcriptional amplification is similar to what has been reported for MYC-dependent changes (Nie et al., 2012; Walz et al., 2014; Zeid et al., 2018), suggesting that this may be a recurrent mechanism of oncogenic gene expression alteration.
In patients with B-ALL, somatic gain of chromosome 21 frequently co-occurs with the t(9;22) BCR-ABL1 tyrosine kinase rearrangement (Wetzler et al., 2004). Similarly, Down syndrome-associated ALL is enriched for acquired mutations in cytokine receptor and/or kinase signaling genes, such as CRLF2 and JAK2 (Lee et al., 2016). One mechanistic hypothesis for this epidemiologic association is that transcriptional amplification via HMGN1 cooperates by facilitating increased output of activated signaling pathways in transformed B cells at the chromatin. Alternatively, transcriptional amplification could be more generally oncogenic by enhancing multiple growth and survival phenotypes, as some studies have shown that increased RNA output is a common feature of transformed cells (Lin et al., 2012; White, 2005). In either case, RNA increases mediated by HMGN1 might offer improved relative fitness to a leukemia cell. However, additional chromosome 21 genes are almost certainly involved in Down syndrome phenotypes. For example, enhanced interferon signaling was observed in several +21 cell types, including B lymphoblastoid lines, attributed to increased expression of interferon receptors located on chromosome 21 (Sullivan et al., 2016). We did not observe enrichment of interferon pathways in our models, but neither the Ts1Rhr triplicated region nor the minimally amplified chromosome 21 segment in iAMP21 B-ALL (Rand et al., 2011) includes interferon receptor gene loci. This underscores the complexity of directly relating specific chromosome 21 gene dosage to widespread Down syndrome phenotypes across cell types.
The HMGN family of proteins may have a more general role in cancer. The closely related HMGN2 enhances STAT5 function and chromatin accessibility downstream of the prolactin receptor in breast cancer cells (Schauwecker et al., 2017), similar to what we hypothesized for HMGN1 in proB-ALL cells. Also, of potential therapeutic relevance, overexpression of HMGN family members is associated with relative chemoresistance whereas knockdown enhances chemosensitivity (He et al., 2015; Yang et al., 2014). However, neither Ts1Rhr nor HMGN1-OE mice develop spontaneous leukemia (Lane et al., 2014; Malinge et al., 2012), which suggests, like in children with Down syndrome, cooperating events may be required to achieve fully malignant transformation. Additional studies will be required to understand the specific mechanisms by which RNA amplification contributes to transformation and cancer cell survival, and whether HMGN1 overexpression creates unique dependencies that could be exploited for treatment.
It remains unclear how transcriptional amplification might contribute to the developmental characteristics of Down syndrome. While Down syndrome phenotypes are complex and likely polygenic (i.e., not simply caused by three copies of a single gene), HMGN1 could shape the Down syndrome transcriptome across lineages via its ability to decompact chromatin and enhance transcription in tissue-specific contexts. In fact, some Down syndrome-associated epigenomic changes might even be secondary to transcription induced by HMGN1, because chromatin density and transcription can modulate epigenomic enzyme activity in some scenarios, rather than the reverse (Yuan et al., 2012). These hypotheses should be tested using existing models, keeping in mind that Down syndrome abnormalities manifest in a variety of tissues, often with distinct clinical and cellular features. We know that HMGNs can affect transcription differently based on cell or tissue type (Kugler et al., 2013). Therefore, the contribution of HMGN1 and transcriptional amplification to diverse Down syndrome phenotypes may need to be studied separately in each developmental context.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Andrew Lane (andrew_lane@dfci.harvard.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal studies
All animal experiments were performed with approval of the DFCI Institutional Animal Care and Use Committee (IACUC) and according to Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) standards. All mice used were as previously described (Lane et al., 2014), except for Hmgn1 knockout mice were as described in Birger et al. (2003)). All transgenic strains were crossed > 10 generations to a C57BL/6J background. Ts1Rhr/Hmgn1+/− mice were generated by crossing Hmgn1−/− with Ts1Rhr (Jackson Laboratory, 005383) animals. Bone marrow harvested for progenitor B cell analysis was from 6-8-week-old randomly selected male and female littermates, co-housed separately by sex in specific pathogen-free environments.
Cell lines
The human pre-B leukemia cell line Nalm6 was from DSMZ, cell identity verified by STR profiling, and the mouse pro-B cell line FL5.12 was obtained from the laboratory of Anthony Letai at DFCI. Retinal pigment epithelial (RPE) cells harboring extra copies of chromosomes generated by microcell-mediated chromosome transfer were as previously described (Lane et al., 2014). Unless otherwise noted, all cells were cultured at 37°C with 5% CO2 in RPMI 1640 supplemented with 10% fetal bovine serum (GIBCO, 10438026), 1% penicillin/streptomycin (GIBCO, 15140122), and 1% GlutaMAX (GIBCO, 35050061). FL5.12 cells were cultured in complete RPMI media supplemented with 10 ng/ml mouse rIL-3 (Gold Biotechnology 1310-03-2).
METHOD DETAILS
Primary progenitor B cell colonies
Whole bone marrow was harvested from 6-8-week-old mice and red blood cells were lysed. Cells were plated in B cell methylcel-lulose media containing interleukin-7 (Methocult M3630, Stem Cell Technologies) in 35 mm dishes at 2×105 cells/ml. Colonies were collected by pooling the cultures after 7 days of growth.
Inducible expression of HMGN1
Human or mouse HMGN1 cDNA (wild-type or encoding the nucleosome binding-deficient HMGN1 S20,24E mutant (Prymakowska-Bosak et al., 2001)) was cloned into the pCW57.1 (pLIX_401) lentiviral tet-ON vector (Addgene). Virus was produced in 293T cells using standard procedures with Lipofectamine 2000 (Invitrogen, 11668027). Virus was harvested at 48 and 72 hours post-transfection and subsequently concentrated by ultracentrifugation at 23,000 g for 2 hours at 4°C. Actively dividing Nalm6 and FL5.12 cells were then spinfected in the presence of virus and polybrene at 2000 g for 2 hours at 32°C, and then were selected in puromycin after 48 hours. To induce overexpression of HMGN1 in stably infected cells, 100 ng/ml doxycycline was added to the cultures for the indicated times. Cell concentration was standardized across conditions. RNA was quantitated using either a Nanodrop spectrophotometer, Qubit fluorometric quantitation (ThermoFisher), or QuantiFluor RNA System (Promega) following manufacturers’ protocols.
Antibodies
Antibodies for western blotting and ChIP were HMGN1 (Bethyl, 302-363A), BCL6 (Cell Signaling Technologies, 14895S), beta-actin (Sigma, SAB5500001), CD79A (Santa Cruz, sc-25604), LYN (Santa Cruz, sc-15), SYK (Santa Cruz, sc12-40), H3K27me3 (Cell Signaling Technologies, 9733), H3K4me3 (Abcam, ab8580), H3K27ac (Abcam, ab4729), or control IgG (Cell Signaling Technologies, 2729S).
Pulse-chase measurements of RNA decay
RNA labeling and detection using bromouridine (BrU) was performed as previously described (Larsen et al., 2001). Briefly, cells were cultured in the presence of doxycycline for 5 hours to induce HMGN1 or HMGN1-SE expression. 1 mM BrU was added to the media and incubated for one hour. Cells were washed and media with doxycycline was replaced, and samples were taken for analysis at the indicated time points. Harvested cells were fixed, permeabilized, stained with an anti-BrU antibody in PBS, and analyzed by flow cytometry. Controls included unlabeled cells and BrU-labeled cells treated with RNase.
Single-cell buoyant mass and interdivisionary time
Suspended microchannel resonator (SMR) measurements of cell mass and microfluidic trap-based time-lapse measurements of single-cell inderdivisionary time were performed as previously described (Kimmerling et al., 2016; Son et al., 2012). Briefly, as in previous SMR analyses, the device was placed on a copper heat sink/source connected to a heated water bath, maintained at 37°C for the duration of the experiment. The sample was loaded into the device from vials pressurized under air with 5% CO2. To measure a sample of cells, we first flushed one bypass channel of the device with the cell sample. We next equalized the pressures upstream and downstream of this bypass and induced a pressure drop across the SMR to begin flowing cells across the sensor. Each cell transiting the sensor led to a resonant frequency shift directly proportional to the buoyant mass of the cell. To calculate this proportionality constant and determine the final buoyant mass values, the SMR was calibrated by measuring a population of polystyrene beads with a known buoyant mass (ThermoFisher, 4207a).
Briefly, for single-cell interdivisionary time measurements, cells were loaded into a microfluidic device fabricated from a silicon-on-insulator wafer with 17-μm deep flow channels. Fluidic connections were established by securing the devices to a Teflon manifold with PEEK tubing maintained at 37°C with a recirculating water bath. Pressure-driven flow in the device was controlled with electronic pressure regulators; all fluids were pressurized with 5% CO2. Single cells were manually loaded into the device by introducing a cell sample at a concentration of 2 × 105 cells per ml and flowing it into the trap lanes. For long-term growth and kinetics measurements, a single cell was loaded in each of the 20 lanes of the device. Once a single cell was loaded in each lane, the bypass channels were flushed to remove any remaining untrapped cells. For continued nutrient repletion, cell growth media was perfused through the bypass channels at a flow rate of 100 μl h−1 with a pressure drop applied along the bypass channels. A slight pressure drop was concurrently introduced across the traps to ensure that the cells remained trapped and their progeny flowed downstream to unoccupied traps. Time-since-division measurements were determined by manually tracking division events and subsequent trap locations for single cells throughout time-lapse image stacks. Single-cell volume measurements from bulk cultures were collected on an automated cell counter (Beckman Coulter).
RNA sequencing
For RNA sequencing experiments, cells were stimulated in complete media with or without doxycycline or in methylcellulose media as indicated. Each condition was processed in biological triplicate or quadruplicate, as indicated in the text (n = 3 or 4 biological replicates per viral transduction per time point). After the indicated time, one million cells were counted, and RNA was extracted using Trizol reagent (Invitrogen, 15596018) with attention paid to ensuring equal and consistent volume transfers. For per cell normalization, 1 μl of Mix #1 ERCC exogenous spike-in RNA (Ambion, 4456740, diluted 1:1000) was added to each RNA sample. Quality control of total RNA was performed using the RNA Qubit Assay (Invitrogen) and the Bioanalyzer RNA Nano 6000 Chip Kit (Agilent). At least 100 ng of total RNA and a Bioanalyzer RNA Integrity Number of > 7.0 were required. Libraries were prepared using TruSeq RNA Library Preparation Kit v2, Set A (Illumina, RS-122-2001). Sequencing was performed at the DFCI Molecular Biology Core Facilities (MBCF). Bioinformatic processing of RNA-seq data, including with spike-in controls was as previously described (Lin et al., 2012; Lovén et al., 2012). Briefly, sequences were aligned using Bowtie (version 0.12.2) to a human genome build to which sequences of the ERCC synthetic spike-in RNAs (https://tools.thermofisher.com/content/sfs/manuals/cms_095047.txt) had been added. The RPKM (Reads Per Kilobase of exon per Million) was then computed for each gene and synthetic spike-in RNA. We used a loess regression to re-normalize the RPKM values, using only the spike-in values to fit the loess. The affy package in R provides a function, loess.normalize, which performs loess regression on a matrix of values (defined using the parameter mat) and allows for the user to specify which subset of data to use when fitting the loess (defined using the parameter subset, see the affy package documentation for further details). For this application, the parameters mat and subset were set as a matrix of all RPKM values and the row-indices of the ERCC spike-ins, respectively. The default settings for all other parameters were used. The result of this was a matrix of RPKM values normalized to the control ERCC spike-ins.
Gene set enrichment analysis (GSEA)
GSEA (https://www.broadlnstitute.org/gsea/) was performed using the Molecular Signatures Database (MSigDB) version 6.0 (Subramanian et al., 2005). Genes in each gene set are available at MSigDB or are in publications referenced in the text. Leading edge analysis was performed in GSEA and visualized as heatmaps using Morpheus (https://software.broadinstitute.org/morpheus/).
SLAM-seq
For cell processing and sequencing, thiol(SH)-linked alkylation for the metabolic sequencing of RNA (SLAM-seq) was performed as described (Herzog et al., 2017). Briefly, Nalm6 cells harboring tet-on HMGN1 or HMGN1-SE mutant constructs were induced with vehicle or 100 ng/ml doxycycline for 6 hours. During the last 5 hours of induction, 4-thiouridine was added to the culture medium at 100 μM final concentration. After the 6-hour induction period, cells were harvested in Trizol, and RNA processing and thiol modification using 4 μg input RNA were performed as previously described (Herzog et al., 2017). For sequencing, libraries were prepared using Illumina’s TruSeq Stranded mRNA kit according to the manufacturer’s instructions and sequencing was performed on a Next-Seq 500 using a NextSeq 500/550 Mid Output v2 kit (150 cycles).
For data processing and analysis, forward and reverse reads from fastq files were merged and SlamDunk v0.2.4 (https://github.com/t-neumann/slamdunk) was run using “slamdunk all” with default parameters (trimming 12bp from the 5′ end, reporting up to 100 alignments for multi-mappers and activating the multi-mapper retention strategy, filtering for variants with a variant fraction of 0.2, and filtering for base-quality cutoff of ≥ 27). Whole gene and 3′ UTR annotations were obtained in BED format from the UCSC table browser (https://genome.ucsc.edu/cgi-bin/hgTables, GRCh37/hg19, RefSeq Curated). Reads were filtered for having ≥ 2 T > C conversions. For SLAM-seq analysis, we sought to examine the effects of HMGN1 or HMGN1-SE overexpression on nascent transcription. First, we limited analysis to genes with a peak expression in the top 25% of overall mRNA expression in at least one sample. We also filtered for genes for which read conversion was detectable across all samples (suggesting at least a baseline level of nascent transcription). These filtered steps resulted in 5,275 active genes utilized in all analyses. Fold changes in nascent transcription upon HMGN1 or HMGN1-SE induction were calculated relative to the uninduced cells. The statistical significance of the difference between distributions was assessed using a two-tailed t test (Figure 3E). To examine the relationship between basal nascent transcription and the effect of HMGN1 induction, active genes were ranked by initial nascent transcription fraction and binned into 50 evenly distributed bins (~105 genes per bin). For each bin, the mean log2 fold change in nascent transcription was plotted. Error bars represent 95% confidence Intervals of the mean (Figure 3F). To visualize changes in nascent transcription at individual genes, the mean nascent transcription fraction and standard error of the mean were plotted (Figure 3G).
To quantify changes In nascent pre-mRNA levels, we utilized the aligned read file (.bam file) of nascent (a.k.a. “converted”) reads generated by the SlamDunk pipeline. We filtered for genes with at least 10 total concordant (i.e., positive strand read upstream of minus strand read) read pairs. Read pairs were considered emanating for pre-mRNA If their sequences overlapped Introns by at least 5 bases. Pre-mRNA levels were quantified In units of fragments (read pairs) per million mapped reads (fpm) In the original dataset (which Included both converted and unconverted reads). To quantify only genes with sufficient coverage, a cut-off of 1 fpm averaged in at least one condition was applied. After this threshold was applied, 7,402 genes remained. The log2 fold change of HMGN1-WT overexpression or HMGN1-SE overexpression was computed versus the no doxycycline control (Figure S3D). The statistical significance of the difference In distributions was assessed by a two-tailed Welch’s t test.
Spike-in normalized ChIP-seq (ChIP-Rx)
For each immunoprecipitation, ~8 million cells were cross-linked with 1% formaldehyde In RPMI medium for 10 min at 37°C, washed In cold phosphate-buffered saline (PBS), resuspended In lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris-HCl, pH 8.1, and complete protease Inhibitors (Roche)). Cross-linked Drosophila S2 cells were spiked Into experimental cells In lysis buffer at one S2 cell per five experimental cells and then sonicated to obtain chromatin fragments between 200 bp and 1200 bp, as previously (Orlando et al., 2014). Sonicated chromatin was resuspended In IP buffer (1% triton, 2 mM EDTA, 150 mM NaCl, 20 mM Tris-HCl, pH 8.1) and Incubated overnight at 4°C with protein A/G magnetic beads (Dynal) conjugated to 10 μg of one of the following antibodies: H3K27me3 (Cell Signaling Technologies, 9733), H3K4me3 (Abcam, ab8580), H3K27ac (Abcam, ab4729), or control IgG (Cell Signaling Technologies, 2729S). The IP was washed 6 times with RIPA buffer (50 mM HEPES, pH 7.6, 1 mM EDTA, 0.7% Na deoxycholate, 1% NP-40, 0.5 M LICl), the DNA recovered by reversing the cross-links In 1% SDS, 0.1 M NaHCO3 for 8 h at 65°C, and then purified using a QIAquick PCR purification kit (QIAGEN, #28104). Bioinformatic processing of ChIP-seq data with and without S2 chromatin normalization (ChIP-Rx) were as previously described (Orlando et al., 2014). Briefly, libraries were sequenced using Illumina NextSeq500 with single-end reads. Sequences were demultiplexed and aligned using Bowtie against a “genome” that combines the human hg19 genome and the Drosophila dm3 genome. A complete description of the basis and derivation of the ChIP-Rx normalization factor Is detailed In Orlando et al. (2014). Briefly, we derived a normalization constant such that after normalization the signal per reference cell Is the same across all samples. The total ChIP-seq signal derived from the reference cells Is the count of reads aligning to the Drosophila genome, which was used to normalize the read counts In the experimental cells. Super-enhancers were Identified using ROSE (Whyte et al., 2013). ChIP-PCR validation was performed using the SimpleChIP enzymatic chromatin IP kit (Cell Signaling Technology, #9005) per the manufacturer’s Instructions.
H3K27ac and phospho-Akt flow cytometry
Cells were harvested and washed twice with PBS. For H3K27ac measurement, cells were then treated with 4% paraformaldehyde in PBS for 15 minutes on ice, followed by centrifugation for 5 minutes at 300 g. Next, cells were resuspended in cold 70% EtOH added drop by drop while vortexing gently and then incubated overnight at −20°C. Then, cells were washed twice with PBS and once with PBS/1% FBS. Cells were permeabilized for 20 minutes with 0.25% Triton X-100 in PBS/1% FBS followed by incubation with primary H3K27ac (Cell Signaling Technology 8173S) or phospho-Akt (Cell Signaling Technologies, 4060S) antibodies at 1:500 dilution for 30 minutes, and then secondary antibody (1:1000) (Life Technologies Goat anti-rabbit IgG cross-adsorbed antibody Alexa Fluor 555, A21428) for 2 hours at 4°C in the dark, prior to flow cytometry analysis.
ATAC-seq
An Assay for Transposase-Accessible Chromatin using sequencing (ATAC-seq) was performed using the Omni-ATAC protocol as previously described (Corces et al., 2017). Briefly, 105 cells were resuspended in 1 mL of cold ATAC-seq resuspension buffer (RSB; 10 mM Tris-HCl pH 7.4, 10 mM NaCl, and 3 mM MgCl2 in water). Cells were centrifuged at 500 g for 5 min in a pre-chilled (4°C) fixed-angle centrifuge, and the supernatant was carefully aspirated. Cell pellets were then resuspended in 50 μL RSB containing 0.1% NP40, 0.1% Tween-20, and 0.01% digitonin by pipetting up and down three times. This cell lysis reaction was incubated on ice for 3 min. After lysis, 1 mL RSB containing 0.1% Tween-20 (without NP40 or digitonin) was added, and the tubes were inverted to mix. Nuclei were then centrifuged for 10 min at 500 g in a pre-chilled fixed-angle centrifuge. Supernatant was removed and nuclei were resuspended in 50 μL of transposition mix (Corces et al., 2017) (2.5 μL transposase26 (100 nM final), 16.5 μL PBS, 0.5 μL 1% digitonin, 0.5 μL 10% Tween-20, and 5 μL water) by pipetting up and down six times. Transposition reactions were incubated at 37°C for 30 min in a thermomixer with shaking at 1000 rpm. Reactions were cleaned up with QIAquick PCR spin columns. Library quantitation and number of amplification cycles was determined as described (Buenrostro et al., 2015). After sequencing on a NextSeq 500 per the manufacturer’s instructions, we used ChiLin pipeline 2.0.0 for QC and preprocessing (Qin et al., 2016), Burrows-Wheeler Aligner (BWA) for read mapping (Li and Durbin, 2009), Model-based Analysis of ChIP-Seq (MACS) as a peak caller (Zhang et al., 2008), and DESeq2 for differential peak analysis (Love et al., 2014).
Quantitative RT-PCR
RNA was prepared using the Trizol reagent (Invitrogen, 15596018), DNA was removed from the final RNA product using the DNA-free DNA Removal Kit (Invitrogen, AM1906), cDNA was prepared using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, 4368814), and quantitative PCR was performed using the Power SYBR Green PCR Master Mix (Applied Biosystems, 4367659), all per the manufacturers’ instructions.
Primers
Oligonucleotide primer sequences for qRT-PCR and ChIP-PCR are in Table S1.
QUANTIFICATION AND STATISTICAL ANALYSIS
Except as where otherwise stated (e.g., specific R packages named in the Method Details section), Prism software (GraphPad) was used for calculating statistical significance. Statistical tests used, number of biological and technical replicates, dispersion and precision measures for each experiment are detailed in the figure legends. Unless otherwise stated, for boxplots comparing per-gene expression by genotype, boxes represent the interquartile range and the whiskers extend to 1.5x of the range in either direction. The horizontal line within the box represents the median. For testing statistical significance between distributions (including RNA expression and ChIP-seq peak read counts), a two-tailed t test was used. For binned analyses, genes were ranked by increasing expression, binned (number of bins in each experiment detailed in figure and legend), and mean was plotted with error bars representing the 95% confidence interval of the mean as determined by empirical resampling with replacement (1000 iterations). Best fit line in binned analyses was added using a locally weighted regression (loess). For bar charts of colony numbers, H3K27 acetylation, RNA expression by quantitative RT-PCR, and ChIP-PCR, unless otherwise specified in figure or legend, bars represent the mean of 3-4 replicates and error bars are standard error of the mean. Statistical significance was compared by unpaired two-tailed t test, with P value < 0.05 considered significant. Gene set enrichment analysis (GSEA) was performed using 1000 permutations by gene set, max size set 500, min size set 15, weighted enrichment statistic, genes ranked by Signal2Noise. The GSEA enrichment score reflects the degree to which a gene set is overrepresented at the upper or lower ends of a ranked list of genes. A running-sum statistic is generated from the ranked list of genes, with the magnitude of the increment depending on the correlation of the gene with the phenotype. The enrichment score is the maximum deviation from zero. The false discovery rate (FDR) in GSEA is the estimated probability that a gene set with a given enrichment score represents a false positive, and is calculated as a ratio of two distributions: (1) the actual enrichment score versus the enrichment score for all gene sets against all permutations of the dataset, and (2) the actual enrichment score versus the enrichment score for all gene sets against the actual dataset.
DATA AND SOFTWARE AVAILABILITY
The accession number for the sequencing data reported in this paper is GEO: GSE121071.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-human HMGN1 dilution 1:1000 | Bethyl | A302-363A |
| Rabbit anti-human BCL6 dilution 1:1000 | Cell Signaling Technology | 14895S |
| Rabbit anti-human beta-Actin dilution 1:1000 | Sigma | SAB5500001 |
| Rabbit anti-human CD79A dilution 1:1000 | Santa Cruz | sc-25604 |
| Rabbit anti-human Lyn dilution 1:1000 | Santa Cruz | sc-15 |
| Mouse anti-human Syk dilution 1:1000 | Santa Cruz | sc12-40 |
| Rabbit anti-human H3K27me3 (ChIP) | Cell Signaling Technology | 9733 |
| Rabbit anti-human H3K4me3 (ChIP) | Abcam | ab8580 |
| Rabbit anti-human H3K27acetyl (ChIP) | Abcam | ab4729 |
| Rabbit IgG | Cell Signaling Technology | 2729 |
| Rabbit anti-human H3K27acetyl dilution 1:500 (Flow) | Abcam | ab4729 |
| Rabbit anti-human phospho-Akt (Ser473) dilution 1:500 (Flow) | Cell Signaling Technology | 4060 |
| Goat anti-Rabbit IgG Alexa Fluor 555 dilution 1:1000 | Invitrogen | A21428 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant mouse IL3 | Gold Biotechnology | 1310-03-2 |
| B cell methylcellulose with IL7 | Stem Cell Technologies | M3630 |
| Doxycycline hydrochloride | Fisher Scientific | BP26531 |
| 5-Bromouridine | Sigma | B7166 |
| Trizol | Life Technologies | 15596018 |
| ERCC Spike-in | Thermo Fisher/Ambion | 4456740 |
| 4-Thiouridine | Sigma | T4509 |
| Puromycin dihydrochloride | Gold Biotechnology | P-600 |
| Polybrene | Santa Cruz | sc-134220 |
| Lipofectamine 2000 | Life Technologies | 11668500 |
| RNase | Sigma | R6513 |
| Critical Commercial Assays | ||
| Qubit RNA BR assay kit | Life Technologies | Q10210 |
| Quantifluor RNA System | VWR | PAE3310 |
| Agilent RNA 6000 Nano Kit | Agilent | 5067-1511 |
| SimpleChIP enzymatic chromatin IP kit | Cell Signaling Technology | 9005 |
| DNA-free DNA removal kit | Invitrogen | AM1906 |
| High-Capacity cDNA Reverse Transcription Kit | Applied Biosystems | 4368814 |
| Power SYBR Green PCR master mix | Applied Biosystems | 4367659 |
| Deposited Data | ||
| Raw and analyzed RNA-seq and ChIP-seq data | This paper | GEO: GSE121071 https://www.ncbi.nlm.nih.gov/geo/ |
| Experimental Models: Cell Lines | ||
| Human: Nalm6 | DSMZ | ACC 128 |
| Mouse: FL5.12 | Laboratory of Anthony Letai | N/A |
| RPE with MMCTof chr21 | This paper and Lane et al.,2014 | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: Ts1Rhr B6.129S6-Dp(16Cbr1-Fam3b)1Rhr/J, backcrossed to C57BL/6J > 10 generations | Jackson Laboratory | 005383 |
| Mouse: HMGN1-OE, backcrossed to C57BL/6J > 10 generations | Laboratory of Michael Bustin | N/A |
| Mouse: C57BL/6J wild-type | Jackson Laboratory | 0000664 |
| Oligonucleotides | ||
| Primers for RT-PCR and ChIP-PCR, see Table S1 | This paper | N/A |
| Recombinant DNA | ||
| Plasmid: pCW57.1 (TRE-gateway; PGK-rtTA-2A-puro) | This paper | Addgene 41393 |
| Software and Algorithms | ||
| GraphPad Prism 7 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| Morpheus | N/A | https://software.broadinstitute.org/morpheus/ |
| Gene Set Enrichment Analysis | Subramanian et al., 2005 | https://www.broadinstitute.org/gsea/ |
| SlamDunk v0.2.4 | Herzog et al., 2017 | https://github.com/t-neumann/slamdunk |
| ROSE | Whyte et al., 2013 | https://bitbucket.org/young_computation/rose |
Highlights.
Transcriptional amplification in trisomy 21 models is revealed by spike-in controls
Overexpression of HMGN1 (on 21q22) is necessary and sufficient for RNA effect
HMGN1 overexpression causes global H3K27 hyperacetylation, detected by ChIP-Rx
Transcriptome amplification enriches cell-type-specific programs of B cells/B-ALL
ACKNOWLEDGMENTS
The authors thank Zach Herbert and the DFCI Molecular Biology Core Facility for assistance with next generation sequencing. This work was supported by the National Cancer Institute (CA181340-01 and CA225191-01 to A.A.L., CA215452-01 to C.Y.L., and CA215226-01 to T.F.W.), Alex’s Lemonade Stand Foundation (to A.A.L.), Curing Kids Cancer (to A.A.L.), the Anna Fuller Fund (to A.A.L.), the Cancer Prevention Research Institute of Texas (RR150093 to C.Y.L.), the Center for Cancer Research of the Intramural Program of the National Cancer Institute (to M.B.), and the McNair Medical Institute (to J.H.W. and T.F.W). C.Y.L. is a Pew-Stewart Scholar for Cancer Research (Alexander and Margaret Stewart Trust).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes, four figures, one table, and two data files and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.10.061.
DECLARATION OF INTERESTS
C.Y.L. is a consultant for Jnana Therapeutics and is a shareholder and inventor of intellectual property licensed to Syros Pharmaceuticals. A.A.L. receives research support from Stemline Therapeutics and is a consultant for N-of-One.
REFERENCES
- Antonarakis SE (2017). Down syndrome and the complexity of genome dosage imbalance. Nat. Rev. Genet 18, 147–163. [DOI] [PubMed] [Google Scholar]
- Beach RR, Ricci-Tam C, Brennan CM, Moomau CA, Hsu PH, Hua B, Silberman RE, Springer M, and Amon A (2017). Aneuploidy causes non-genetic individuality. Cell 169, 229–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger R (1997). Acute lymphoblastic leukemia and chromosome 21. Cancer Genet. Cytogenet. 94, 8–12. [DOI] [PubMed] [Google Scholar]
- Birger Y, West KL, Postnikov YV, Lim JH, Furusawa T, Wagner JP, Laufer CS, Kraemer KH, and Bustin M (2003). Chromosomal protein HMGN1 enhances the rate of DNA repair in chromatin. EMBO J. 22, 1665–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonney ME, Moriya H, and Amon A (2015). Aneuploid proliferation defects in yeast are not driven by copy number changes of a few dosage-sensitive genes. Genes Dev. 29, 898–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Chang HY, and Greenleaf WJ (2015). ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol. 109, 21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustin M, Alfonso PJ, Pash JM, Ward JM, Gearhart JD, and Reeves RH (1995). Characterization of transgenic mice with an increased content of chromosomal protein HMG-14 in their chromatin. DNA Cell Biol. 14, 997–1005. [DOI] [PubMed] [Google Scholar]
- Catez F, Brown DT, Misteli T, and Bustin M (2002). Competition between histone H1 and HMGN proteins for chromatin binding sites. EMBO Rep. 3, 760–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cermak N, Olcum S, Delgado FF, Wasserman SC, Payer KR, A Murakami M, Knudsen SM, Kimmerling RJ, Stevens MM, Kikuchi Y, et al. (2016). High-throughput measurement of single-cell growth rates using serial microfluidic mass sensor arrays. Nat. Biotechnol. 34, 1052–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Hu Z, Xia Z, Zhao D, Li W, and Tyler JK (2015). The overlooked fact: fundamental need for spike-in control for virtually all genome-wide analyses. Mol. Cell. Biol. 36, 662–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Arm-strong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, Wu B, et al. (2017). An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa V, Angelini C, D’Apice L, Mutarelli M, Casamassimi A, Sommese L, Gallo MA, Aprile M, Esposito R, Leone L, et al. (2011). Massive-scale RNA-seq analysis of non ribosomal transcriptome in human trisomy 21. PLoS ONE 6, e18493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darzynkiewicz Z, Evenson D, Staiano-Coico L, Sharpless T, and Melamed MR (1979). Relationship between RNA content and progression of lymphocytes through S phase of cell cycle. Proc. Natl. Acad. Sci. USA 76, 358–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng T, Zhu ZI, Zhang S, Postnikov Y, Huang D, Horsch M, Furusawa T, Beckers J, Rozman J, Klingenspor M, et al. (2015). Functional compensation among HMGN variants modulates the DNase I hypersensitive sites at enhancers. Genome Res. 25, 1295–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do LH, Mobley WC, and Singhal N (2015). Questioned validity of gene expression dysregulated domains in Down’s syndrome. F1000Res. 4, 269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco HL, and Kraus WL (2015). No driver behind the wheel? Targeting transcription in cancer. Cell 163, 28–30. [DOI] [PubMed] [Google Scholar]
- Geng H, Hurtz C, Lenz KB, Chen Z, Baumjohann D, Thompson S, Goloviznina NA, Chen WY, Huan J, LaTocha D, et al. (2015). Self-enforcing feedback activation between BCL6 and pre-B cell receptor signaling defines a distinct subtype of acute lymphoblastic leukemia. Cancer Cell 27, 409–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamurcu Z, Demirtas H, and Kumandas S (2006). Flow cytometric comparison of RNA content in peripheral blood mononuclear cells of Down syndrome patients and control individuals. Cytometry B Clin. Cytom. 70, 24–28. [DOI] [PubMed] [Google Scholar]
- Harrison CJ, Moorman AV, Schwab C, Carroll AJ, Raetz EA, Devidas M, Strehl S, Nebral K, Harbott J, Teigler-Schlegel A, et al. ; Ponte di Legno International Workshop in Childhood Acute Lymphoblastic Leukemia (2014). An international study of intrachromosomal amplification of chromosome 21 (iAMP21): cytogenetic characterization and outcome. Leukemia 28, 1015–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzi K, Jiang Y, Huang C, Garrett-Bakelman F, Gearhart MD, Giannopoulou EG, Zumbo P, Kirouac K, Bhaskara S, Polo JM, et al. (2013). A hybrid mechanism of action for BCL6 in B cells defined by formation of functionally distinct complexes at enhancers and promoters. Cell Rep. 4, 578–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J, Liu C, Wang B, Li N, Zuo G, and Gao D (2015). HMGN5 blockade by siRNA enhances apoptosis, suppresses invasion and increases chemosensitivity to temozolomide in meningiomas. Int. J. Oncol. 47, 1503–1511. [DOI] [PubMed] [Google Scholar]
- Heerema NA, Raimondi SC, Anderson JR, Biegel J, Camitta BM, Cooley LD, Gaynon PS, Hirsch B, Magenis RE, McGavran L, et al. (2007). Specific extra chromosomes occur in a modal number dependent pattern in pediatric acute lymphoblastic leukemia. Genes Chromosomes Cancer 46, 684–693. [DOI] [PubMed] [Google Scholar]
- Herzog VA, Reichholf B, Neumann T, Rescheneder P, Bhat P, Burkard TR, Wlotzka W, von Haeseler A, Zuber J, and Ameres SL (2017). Thiol-linked alkylation of RNA to assess expression dynamics. Nat. Methods 14, 1198–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahmann NH, and Rake AV (1993). Altered nucleosome spacing associated with Down syndrome. Biochem. Genet. 31, 207–214. [DOI] [PubMed] [Google Scholar]
- Katerndahl CDS, Heltemes-Harris LM, Willette MJL, Henzler CM, Frietze S, Yang R, Schjerven H, Silverstein KAT, Ramsey LB, Hubbard G, et al. (2017). Antagonism of B cell enhancer networks by STAT5 drives leukemia and poor patient survival. Nat. Immunol. 18, 694–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmerling RJ, Lee Szeto G, Li JW, Genshaft AS, Kazer SW, Payer KR, de Riba Borrajo J, Blainey PC, Irvine DJ, Shalek AK, and Manalis SR (2016). A microfluidic platform enabling single-cell RNA-seq of multigenerational lineages. Nat. Commun. 7, 10220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korenberg JR, Chen XN, Schipper R, Sun Z, Gonsky R, Gerwehr S, Carpenter N, Daumer C, Dignan P, Disteche C, et al. (1994). Down syndrome phenotypes: the consequences of chromosomal imbalance. Proc. Natl. Acad. Sci. USA 91, 4997–5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kugler JE, Horsch M, Huang D, Furusawa T, Rochman M, Garrett L, Becker L, Bohla A, Hölter SM, Prehn C, et al. (2013). High mobility group N proteins modulate the fidelity of the cellular transcriptional profile in a tissue- and variant-specific manner. J. Biol. Chem 288, 16690–16703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane AA, Chapuy B, Lin CY, Tivey T, Li H, Townsend EC, van Bodegom D, Day TA, Wu SC, Liu H, et al. (2014).Triplication of a 21q22 region contributes to B cell transformation through HMGN1 overexpression and loss of histone H3 Lys27 trimethylation. Nat. Genet. 46, 618–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen JK, Jensen PØ, and Larsen J (2001). Flow cytometric analysis of RNA synthesis by detection of bromouridine incorporation. Curr. Protoc. Cytom. Chapter 7, Unit 7.12. [DOI] [PubMed] [Google Scholar]
- Lee P, Bhansali R, Izraeli S, Hijiya N, and Crispino JD (2016). The biology, pathogenesis and clinical aspects of acute lymphoblastic leukemia in children with Down syndrome. Leukemia 30, 1816–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letourneau A, Santoni FA, Bonilla X, Sailani MR, Gonzalez D, Kind J, Chevalier C, Thurman R, Sandstrom RS, Hibaoui Y, et al. (2014). Domains of genome-wide gene expression dysregulation in Down’s syndrome. Nature 508, 345–350. [DOI] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Schwab C, Ryan S, Papaemmanuil E, Robinson HM, Jacobs P, Moorman AV, Dyer S, Borrow J, Griffiths M, et al. (2014). Constitutional and somatic rearrangement of chromosome 21 in acute lymphoblastic leukaemia. Nature 508, 98–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim JH, West KL, Rubinstein Y, Bergel M, Postnikov YV, and Bustin M (2005). Chromosomal protein HMGN1 enhances the acetylation of lysine 14 in histone H3. EMBO J. 24, 3038–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CY, Lovén J, Rahl PB, Paranal RM, Burge CB, Bradner JE, Lee TI, and Young RA (2012). Transcriptional amplification in tumor cells with elevated c-Myc. Cell 151, 56–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockstone HE, Harris LW, Swatton JE, Wayland MT, Holland AJ, and Bahn S (2007). Gene expression profiling in the adult Down syndrome brain. Genomics 90, 647–660. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovén J, Orlando DA, Sigova AA, Lin CY, Rahl PB, Burge CB, Levens DL, Lee TI, and Young RA (2012). Revisiting global gene expression analysis. Cell 151, 476–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Mccarter M, Lian G, Esposito G, Capoccia E, Delli-Bovi LC, Hecht J, and Sheen V (2016). Global hypermethylation in fetal cortex of Down syndrome due to DNMT3L overexpression. Hum. Mol. Genet. 25, 1714–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinge S, Bliss-Moreau M, Kirsammer G, Diebold L, Chlon T, Gurbuxani S, and Crispino JD (2012). Increased dosage of the chromosome 21 ortholog Dyrk1a promotes megakaryoblastic leukemia in a murine model of Down syndrome. J. Clin. Invest. 122, 948–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendioroz M, Do C, Jiang X, Liu C, Darbary HK, Lang CF, Lin J, Thomas A, Abu-Amero S, Stanier P, et al. (2015). Trans effects of chromosome aneuploidies on DNA methylation patterns in human Down syndrome and mouse models. Genome Biol. 16, 263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorman AV, Ensor HM, Richards SM, Chilton L, Schwab C, Kinsey SE, Vora A, Mitchell CD, and Harrison CJ (2010). Prognostic effect of chromosomal abnormalities in childhood B-cell precursor acute lymphoblastic leukaemia: results from the UK Medical Research Council ALL97/99 randomised trial. Lancet Oncol. 11, 429–438. [DOI] [PubMed] [Google Scholar]
- Mrózek K, Heinonen K, Theil KS, and Bloomfield CD (2002). Spectral karyotyping in patients with acute myeloid leukemia and a complex karyotype shows hidden aberrations, including recurrent overrepresentation of 21q, 11q, and 22q. Genes Chromosomes Cancer 34, 137–153. [DOI] [PubMed] [Google Scholar]
- Nie Z, Hu G, Wei G, Cui K, Yamane A, Resch W, Wang R, Green DR, Tessarollo L, Casellas R, et al. (2012). c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell 151, 68–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlando DA, Chen MW, Brown VE, Solanki S, Choi YJ, Olson ER, Fritz CC, Bradner JE, and Guenther MG (2014). Quantitative ChIP-seq normalization reveals global modulation of the epigenome. Cell Rep. 9, 1163–1170. [DOI] [PubMed] [Google Scholar]
- Padovan-Merhar O, Nair GP, Biaesch AG, Mayer A, Scarfone S, Foley SW, Wu AR, Churchman LS, Singh A, and Raj A (2015). Single mammalian cells compensate for differences in cellular volume and DNA copy number through independent global transcriptional mechanisms. Mol. Cell 58, 339–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope BD, and Gilbert DM (2014). Genetics: up and down in Down’s syndrome. Nature 508, 323–324. [DOI] [PubMed] [Google Scholar]
- Prymakowska-Bosak M, Misteli T, Herrera JE, Shirakawa H, Birger Y, Garfield S, and Bustin M (2001). Mitotic phosphorylation prevents the binding of HMGN proteins to chromatin. Mol. Cell. Biol. 21, 5169–5178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Q, Mei S, Wu Q, Sun H, Li L, Taing L, Chen S, Li F, Liu T, Zang C, et al. (2016). ChiLin: a comprehensive ChIP-seq and DNase-seq quality control and analysis pipeline. BMC Bioinformatics 17, 404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand V, Parker H, Russell LJ, Schwab C, Ensor H, Irving J, Jones L, Masic D, Minto L, Morrison H, et al. (2011). Genomic characterization implicates iAMP21 as a likely primary genetic event in childhood B-cell precursor acute lymphoblastic leukemia. Blood 117, 6848–6855. [DOI] [PubMed] [Google Scholar]
- Rattner BP, Yusufzai T, and Kadonaga JT (2009). HMGN proteins act in opposition to ATP-dependent chromatin remodeling factors to restrict nucleosome mobility. Mol. Cell 34, 620–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochman M, Postnikov Y, Correll S, Malicet C, Wincovitch S, Karpova TS, McNally JG, Wu X, Bubunenko NA, Grigoryev S, and Bustin M (2009). The interaction of NSBP1/HMGN5 with nucleosomes in euchromatin counteracts linker histone-mediated chromatin compaction and modulates transcription. Mol. Cell 35, 642–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roper RJ, and Reeves RH (2006). Understanding the basis for Down syndrome phenotypes. PLoS Genet. 2, e50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauwecker SM, Kim JJ, Licht JD, and Clevenger CV (2017). Histone H1 and chromosomal protein HMGN2 regulate prolactin-induced STAT5 transcription factor recruitment and function in breast cancer cells. J. Biol. Chem. 292, 2237–2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son S, Tzur A, Weng Y, Jorgensen P, Kim J, Kirschner MW, and Manalis SR (2012). Direct observation of mammalian cell growth and size regulation. Nat. Methods 9, 910–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan KD, Lewis HC, Hill AA, Pandey A, Jackson LP, Cabral JM, Smith KP, Liggett LA, Gomez EB, Galbraith MD, et al. (2016). Trisomy 21 consistently activates the interferon response. eLife 5, e16220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walz S, Lorenzin F, Morton J, Wiese KE, von Eyss B, Herold S, Rycak L, Dumay-Odelot H, Karim S, Bartkuhn M, et al. (2014). Activation and repression by oncogenic MYC shape tumour-specific gene expression profiles. Nature 511, 483–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetzler M, Dodge RK, Mrózek K, Stewart CC, Carroll AJ, Tantravahi R, Vardiman JW, Larson RA, and Bloomfield CD (2004). Additional cytogenetic abnormalities in adults with Philadelphia chromosome-positive acute lymphoblastic leukaemia: a study of the Cancer and Leukaemia Group B. Br. J. Haematol. 124, 275–288. [DOI] [PubMed] [Google Scholar]
- White RJ (2005). RNA polymerases I and III, growth control and cancer. Nat. Rev. Mol. Cell Biol. 6, 69–78. [DOI] [PubMed] [Google Scholar]
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, and Young RA (2013). Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 753, 307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Gao R, Wang J, Yuan W, Wang C, and Zhou X (2014). High-mobility group nucleosome-binding domain 5 increases drug resistance in osteosarcoma through upregulating autophagy. Tumour Biol. 35, 6357–6363. [DOI] [PubMed] [Google Scholar]
- Yuan W, Wu T, Fu H, Dai C, Wu H, Liu N, Li X, Xu M, Zhang Z, Niu T, et al. (2012). Dense chromatin activates Polycomb repressive complex 2 to regulate H3 lysine 27 methylation. Science 337, 971–975. [DOI] [PubMed] [Google Scholar]
- Zeid R, Lawlor MA, Poon E, Reyes JM, Fulciniti M, Lopez MA, Scott TG, Nabet B, Erb MA, Winter GE, et al. (2018). Enhancer invasion shapes MYCN-dependent transcriptional amplification in neuroblastoma. Nat. Genet. 50, 515–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.