

SUMMARY
Our recent ERK1/2 inhibitor analyses in pancreatic ductal adenocarcinoma (PDAC) indicated ERK1/2-independent mechanisms maintaining MYC protein stability. To identify these mechanisms, we determined the signaling networks by which mutant KRAS regulates MYC. Acute KRAS suppression caused rapid proteasome-dependent loss of MYC protein, through both ERK1/2-dependent and -independent mechanisms. Surprisingly, MYC degradation was independent of PI3K-AKT-GSK3β signaling and the E3 ligase FBWX7. We then established and applied a high-throughput screen for MYC protein degradation and performed a kinome-wide proteomics screen. We identified an ERK1/2-inhibition-induced feed-forward mechanism dependent on EGFR and SRC, leading to ERK5 activation and phosphorylation of MYC at S62, preventing degradation. Concurrent inhibition of ERK1/2 and ERK5 disrupted this mechanism, synergistically causing loss of MYC and suppressing PDAC growth.
Keywords: EGFR, ERK, ERK5, FBXW7, KRAS, PI3K, MYC, pancreatic cancer, SRC
In Brief
Vaseva et al. find that mutant KRAS regulates MYC via ERK1/2-dependent and independent mechanisms in pancreatic cancer (PDAC). ERK1/2 blockade activates a compensatory EGFR-SRC-ERK5 cascade that stabilizes MYC, and combined ERK1/2 and ERK5 inhibition promotes synergistic loss of MYC and suppresses PDAC growth.
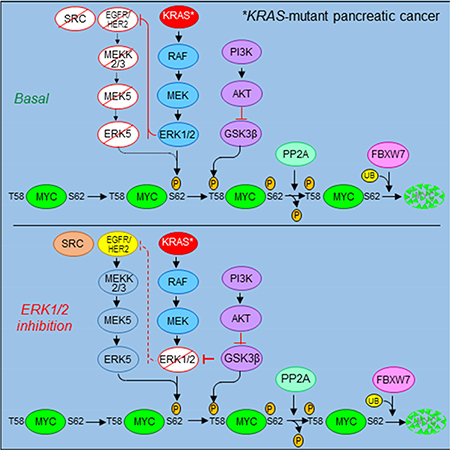
Graphical Abstract
INTRODUCTION
The KRAS oncogene is mutated in ~95% of pancreatic ductal adenocarcinoma (PDAC) (Waters and Der, 2017). Cell culture and mouse model studies show that ablation of KRAS function has a significant impact on PDAC progression and growth. Thus, the development of KRAS targeted therapies is one of four key initiatives for pancreatic cancer research. Yet, after more than three decades, a clinically effective anti-KRAS therapy remains to be developed (Papke and Der, 2017).
Growth transformation of rodent fibroblasts by mutant HRAS was shown long ago to require cooperating MYC overexpression, thereby providing the first demonstration that MYC can facilitate RAS-mediated oncogenesis (Land et al., 1983). Subsequent studies in mouse models demonstrated the critical requirement for MYC in Kras-driven oncogenesis (Soucek et al., 2008; Soucek et al., 2013). Several studies then showed that genetic ablation of Myc impaired mutant Kras-driven PDAC development and growth (Saborowski et al., 2014; Walz et al., 2014). Conversely, Myc overexpression alone was sufficient to cause formation of metastatic PDAC (Lin et al., 2013). These findings established MYC as a critical mediator of KRAS function and support the idea that targeting MYC could be a viable therapeutic strategy for targeting KRAS-driven PDAC.
Like RAS, MYC has been widely considered to be “undruggable” (Dang et al., 2017). Unlike recent early-stage progress in developing direct inhibitors of RAS (Ostrem and Shokat, 2016), MYC inhibitor development has focused on indirect strategies including inhibition of MYC gene expression (e.g., bromodomain inhibitors), MYC-MAX dimerization and DNA binding, and MYC target function (Dang, 2012). However, while RAS-driven mechanisms that regulate MYC protein stability have been described (Farrell and Sears, 2014), surprisingly limited effort has been made to exploit these mechanisms as a therapeutic strategy for targeting RAS (Farrell et al., 2014).
In normal cells, MYC protein levels are tightly regulated by both transcriptional and posttranslational mechanisms, and the half-lives of both MYC mRNA (~30 min) and MYC protein (~20 min) are very short (Dang, 2012). In cancer, MYC protein overexpression can be facilitated by gene amplification, increased transcription, and/or increased protein stability. Immunohistochemical (IHC) analyses of a limited number of PDAC cases revealed MYC protein overexpression in 44% of primary tumors and 32% of metastases, but overexpression did not correlate with MYC gene amplification (Schleger et al., 2002). Furthermore, MYC amplification was limited to several copies and therefore could not account for the high levels of MYC protein. A second study found that normal pancreatic tissue was negative for MYC staining, whereas 38% of PDAC exhibited positive staining (Lin et al., 2013).
Early studies of mutant RAS-transformed rodent fibroblasts showed that RAS activation of the RAF-MEK1/2-ERK1/2 mitogen-activated protein kinase (MAPK) cascade resulted in ERK1/2 phosphorylation of MYC at S62 (pS62) and in increased MYC protein stability (Farrell et al., 2014). Phosphorylation at S62 also allowed subsequent phosphorylation at MYC residue T58 (pT58) by GSK3β {Sears, 2000 #17}. Upon PP2A-catalyzed loss of pS62, pT58 then comprised a phospho-degron signal for FBXW7 E3 ligase-mediated MYC protein degradation. Indirect pharmacologic activation of PP2A can decrease pS62, increasing MYC degradation {Farrell, 2014 #32}. Since RAS activation of the PI3K-AKT effector pathway can cause AKT phosphorylation and inactivation of GSK3β, there are at least two distinct effector cascades, RAF-MEK-ERK and PI3K-AKT-GSK3β, by which RAS could promote MYC protein stability.
Recently, we evaluated ERK1/2 inhibitors as a therapeutic strategy for KRAS-mutant PDAC and identified both ERK inhibitor-sensitive and -resistant PDAC cell lines (Hayes et al., 2016). We determined that ERK inhibitor-mediated loss of MYC protein correlated with ERK inhibitor sensitivity in vitro and in vivo. These findings suggested that ERK inhibitor-resistant PDAC must possess ERK-independent mechanisms that maintain MYC protein stability. This prompted our evaluation of KRAS effector signaling that regulates MYC protein levels in KRAS-mutant PDAC.
RESULTS
MYC Inhibition Impairs the Growth of KRAS-Mutant Pancreatic Cancer
Mouse model studies demonstrated that Myc is required for mutant Kras-driven tumor initiation and progression (Saborowski et al., 2014; Walz et al., 2014). Demonstrating a similar requirement for MYC in maintaining human PDAC growth, we determined that siRNA-mediated MYC suppression strongly reduced anchorage-dependent clonogenic growth (>90% reduction) and anchorage-independent soft agar colony formation (Figures 1A-D) of KRAS-mutant human PDAC cell lines. Overall, we found that MYC was important for the growth of all 15 KRAS-mutant lines tested (Figures 1A-D, S1A-G).
Figure 1. MYC Inhibition Impairs the Growth of KRAS-mutant PDAC growth.

(A) Indicated KRAS-mutant PDAC cell lines were transfected with control nonspecific (NS) or siRNA oligos targeting two distinct MYC sequences. Suppression of MYC expression (24 hr) was assessed by immunoblotting.
(B) Representative 6-well plates from panel A were stained with crystal violet to visualize colonies of proliferating cells, ~10 days after plating.
(C) Quantitation of data in panel B. Colonies were counted for each cell line transfected with MYC siRNA and counts were normalized to those of NS. Data are presented as the mean of three biological replicates, with error bars representing the standard error of the mean (SEM).
(D) Soft agar colonies at 15 days after plating. Scale bar = 1 mm.
(E) Tet (doxycycline)-driven SMART vector for inducible expression of Myc shRNA.
(F) INK4.1syn_Luc mouse pancreatic tumor cells stably expressing the SMART Myc shRNA vector were treated with 250 ng/ml doxycycline (DOX). Myc knockdown was confirmed by immunoblotting (left) and its effect on proliferation was visualized by crystal violet staining (right). Images from 6-well plates are shown.
(G) INK4.1syn_Luc mouse pancreatic tumor cells stably expressing the SMART Myc shRNA vector and luciferase were injected orthotopically into the head of the pancreas of syngeneic FVB/n mice. Ten days later, tumors were detected by IVIS imaging and Myc silencing was initiated by feeding DOX-containing chow
(H) Kaplan-Meier survival plot of control (n=10) and Myc shRNA (n=10) mice fed on DOX chow.
See also Figure S1.
We next evaluated if MYC was also essential for maintenance of the tumorigenic growth of established tumors, by employing a syngeneic orthotopic PDAC mouse model (Collisson et al., 2012). We stably infected the mouse pancreatic tumor-derived cell line INK4.1syn_Luc, established from the KrasG12D/Ink4a−/− mouse model, using a lentivirus shRNA vector encoding a murine Myc shRNA driven by a doxycycline-inducible promoter (Figure 1E). After verifying that doxycycline induced loss of MYC protein and suppression of the growth of this line in vitro (Figure 1F), we injected it into the pancreas of syngeneic mice. Upon silencing Myc, we observed significant impairment of tumor growth (Figure 1G) and increased survival (Figure 1H). The median survival increase was 11 days (control: 46 days vs. Myc shRNA: 57 days, p=0.005), where 50% of the mice lived at least 60+ days and one of them was cured. At the time of mouse sacrifice (~40 day), MYC protein expression was incompletely reduced in harvested tumors (Figure S1H), likely accounting for the incomplete suppression of tumor growth. Consistent with our cell culture analyses, we conclude that suppression of MYC impairs PDAC growth.
KRAS Suppression Causes Polyubiquitination and Proteasomal Degradation of MYC Protein
To examine the role of KRAS in regulating MYC protein levels, we transiently (24 hr) suppressed KRAS expression using siRNA. This suppression decreased MYC protein in KRAS-mutant but not wild-type PDAC cell lines (Figures 2A and S2A). Conversely, stable ectopic expression of mutant KRAS in two immortalized human pancreatic cell lines was associated with KRAS-dependent elevated MYC protein levels (Figure 2B, compare basal (NS) MYC levels in HPDE versus HPDE-K and in HPNE versus HPNEK). Thus, mutant KRAS is both necessary and sufficient to support increases in MYC protein.
Figure 2. KRAS Suppression Causes Proteasome-dependent Loss of MYC Protein.

(A) PDAC cell lines were transfected with NS or two independent KRAS siRNAs. Immunoblotting was done after 24 hr to monitor changes in MYC and KRAS4B protein levels.
(B) Control and KRASG12V-transformed human pancreatic ductal epithelial cells (HPDE and HPDE-K, respectively) or human pancreatic nestin-expressing cells (HPNE and HPNE-K) were transfected as described in panel A and immunoblotted as indicated.
(C) PDAC cells were transfected with NS or KRAS siRNAs for 24 hr and RNA was extracted to measure MYC expression. Real-time qPCR was performed using two sets of MYC-specific primers (MYC A and MYC B). MYC mRNA levels were normalized to GAPDH mRNA.
(D) To assess proteasome-dependent MYC downregulation, PDAC cells were transfected with NS or KRAS siRNAs for 20 hr and treated with 5 μM proteasome inhibitor MG132 for an additional 6 hr. Cell lysates were immunoblotted as indicated.
(E) To detect polyubiquitinated (Poly-Ub) MYC protein, MIA PaCa-2 cells were transfected for 20 hr with NS or KRAS siRNA and then treated with 5 μM MG132 for an additional 6 hr. Cell lysates were immunoblotted as indicated. Short (s) and long (l) exposures are shown for MYC protein.
(F) To measure MYC protein half-life, protein translation was blocked by treatment with cycloheximide (CHX), and MYC levels were monitored by immunoblotting every 15 min for a total of 90 min. Right, MYC half-life in minutes (t1/2 (min)) was calculated for each cell line using GraphPad Prism.
See also Figure S2.
To determine the basis for these mutant KRAS-dependent increases in MYC protein, we first monitored mRNA levels by RT-PCR. We found no significant reduction in MYC gene transcription after 24 hr of KRAS suppression (Figure 2C). Thus, because MYC mRNA was reduced only well after loss of MYC protein (Figure S2B), we concluded that KRAS regulates MYC largely through a translational or post-translational mechanism. Accordingly, we found that the proteasome inhibitor MG132 blocked KRAS suppression-induced loss of MYC protein (Figure 2D), and this was associated with accumulation of polyubiquitinated MYC (Figure 2E). Consistent with KRAS promotion of MYC stability, we found that MYC half-life was significantly prolonged in KRAS-transformed HPNE (t1/2 = 37 min) and KRAS-mutant PDAC lines (t1/2 = 43–50 min) compared with untransformed HPNE cells (t1/2 = 22 min) (Figures 2F and S2C). Finally, since KRAS suppression did not alter the cell cycle after 24 hr (Figures S2D and S2E), MYC protein reduction was not a secondary consequence of perturbation in cell cycle progression. We conclude that KRAS suppression-induced loss of MYC protein involves a proteasome-dependent post-translational mechanism.
ERK but not PI3K-AKT-mTOR Effector Signaling Contributes to KRAS Modulation of MYC Protein Stability
We next addressed the role of KRAS effector signaling in regulating MYC protein steady-state levels. We first determined if ERK or PI3K-AKT signaling was reduced upon acute KRAS suppression (24 and 48 hr) (Figures 3A, S3A and S3B). Extending our previous findings (Figure 2A), we observed that acute KRAS suppression caused MYC protein loss in 9 of 10 cell lines. Consistent with a role for KRAS-ERK signaling in promoting MYC protein stability, KRAS suppression reduced phosphorylation of the ERK substrate RSK in 8 of 10 cell lines. In contrast, it did not reduce pAKT, pGSK3β or pS6, indicating that MYC protein loss did not result from impaired PI3K-AKTpGSK3β/mTOR effector signaling. Also consistent with a critical role for ERK signaling in MYC protein stability, we determined that pharmacologic inhibition of MEK or ERK decreased MYC protein levels (Figure 3B). In contrast, pharmacologic inhibition of PI3K or AKT, despite decreasing pGSK3β as expected, did not alter total or phosphorylated (T58) MYC levels (Figure 3C).
Figure 3. KRAS Regulation of MYC Expression is Dependent on MEK-ERK1/2 Signaling, but Not PI3K-AKT-GSK3β.

(A) PDAC cell lines were transfected with NS or KRAS siRNA (24 hr). Immunoblotting of cell lysates was done to determine total KRAS and MYC, phosphorylated ERK1/2 (pERK) and its substrate p90RSK (pRSK), and phosphorylated AKT (pAKT) and its substrate GSK3β (pGSK3β).
(B) PDAC cell lines were treated with the ERK1/2 inhibitor SCH772984 (ERKi) or the MEK1/2 inhibitor selumetinib (MEKi) for 24 hr and cell lysates were immunoblotted as indicated.
(C) PDAC cell lines were treated with the PI3K inhibitor AZD8186 (PI3Ki) or the AKT inhibitor AZD5363 (AKTi) for 24 hr and cell lysates were immunoblotted as indicated.
(D) IHC staining of PDAC patient samples was performed for total MYC, pERK1/2 and pAKT. Representative sections are shown. Scale bar = 100 μm.
(E) Kaplan-Meier survival curves for patients with high (IHC scores of 2+ or higher) or low (1+ or lower) levels of total MYC (left panel) or phosphorylated ERK1/2 (right panel). N=72 patients per group.
The KRAS WT PDAC cell line BxPC-3 harbors a deletion-activated BRAF mutant that acts downstream of KRAS (Chen et al., 2016; Foster et al., 2016). Thus, KRAS suppression would not be expected to reduce MYC levels in BxPC-3 cells, and did not do so (Figure 2A). However, BxPC-3 growth was impaired by pharmacologic inhibition of RAF (Chen et al., 2016; Foster et al., 2016), and MYC levels were decreased in BxPC-3 cells upon inhibition of MEK or ERK (Figure S3C), but not PI3K or AKT (Figure S3D). Although treatment with the AKT inhibitor AZD5363 caused increased AKT phosphorylation, likely due to feedback loss and upstream activation, efficient inhibition of AKT activity was achieved as demonstrated by loss of GSK3β phosphorylation. Direct suppression of MYC also impaired BxPC-3 growth (Figures S3E-G). Since BxPC-3 cells are wild-type for PTEN and PIK3CA, we suggest that activation of the RAF-ERK effector pathway alone is sufficient to drive MYC overexpression.
We then extended our analyses to patient tumor samples. Immunohistochemical analyses of 120 formalin-fixed and paraffin-embedded (FFPE) PDAC cases showed a very high positive correlation (p<0.0001, rs=0.485) between pERK and MYC (Figure 3D and Table S1), but not between pAKT and MYC (p=0.193). Taken together with our cell line analyses, we conclude that ERK but not AKT effector signaling contributes to KRAS-mediated MYC protein stabilization. Interestingly, among the 72 cases with available patient survival data, we also found that high levels of MYC protein, but not pERK alone, correlated significantly with poor overall survival (log rank, p=0.003) (Figure 3E).
KRAS Regulates MYC Stability Through an ERK/T58/FBXW7-Independent Mechanism
Our finding that inhibition of PI3K-AKT effector signaling is not associated with loss of MYC protein suggested that MYC phosphorylation at residue T58 and subsequent FBXW7-dependent degradation alone cannot account for KRAS regulation of MYC protein stability. To address this directly, we transiently suppressed FBXW7 expression and determined that loss of this E3 ligase prevented the loss of MYC upon transient suppression of ERK1 and ERK2 but not of KRAS (Figures 4A-D). Thus, KRAS suppression involves the loss of both ERK-dependent and ERK-independent effector signaling mechanisms that are critical to promote MYC protein stability.
Figure 4. KRAS Regulates MYC Protein Stability In Part Independently of FBXW7 and MYC Residues T58 and S62.

(A) MIA PaCa-2 cells were transfected with NS or two distinct FBXW7 siRNA for 24 hr, followed by transfection with NS or two ERK1/ERK2 siRNA for an additional 24 hr. “E 1+ F 1” and “E 2+ F 2” indicate samples transfected with both ERK1/ERK2 and FBW7 siRNAs. FBXW7 silencing was assessed by real-time qPCR using FBXW7-specific primers. FBXW7 mRNA levels were normalized to GAPDH mRNA levels.
(B) MIA PaCa-2 cells were transfected as described in (A) and cell lysates were immunoblotted as indicated.
(C) MIA PaCa-2 cells were transfected with NS or FBXW7 siRNA for 24 hr, followed by transfection with NS or KRAS siRNA for an additional 24 hr. FBXW7 silencing was confirmed as in (A). ‘K 1+ F 1’ and ‘K 2+ F 2’ indicate samples transfected with both KRAS and FBW7 siRNAs.
(D) MIA PaCa-2 cells were transfected as in (C) and cell lysates were immunoblotted as indicated.
(E) MIA PaCa-2 cells stably expressing exogenous FLAG epitope-tagged WT or phospho-deficient mutants of MYC (T58A and S62A) were transiently transfected with NS or siRNA oligos targeting KRAS or ERK1//ERK2. After 24 hr, cell lysates were immunoblotted as indicated.
(F) PDAC cell lines were transiently transfected with NS or KRAS siRNA. MG132 was added to block degradation of endogenous MYC. After 24 hr, MYC phosphorylation at T58 and S62 was detected by immunoblotting with phospho-specific MYC antibodies.
See also Figure S4.
We next determined the role of MYC phosphorylation at T58 and S62 by evaluating exogenous phospho-deficient MYC proteins in MIA PaCa-2 cells (Figure 4E). As with endogenous MYC, exogenous WT MYC was decreased upon KRAS or ERK suppression. However, phosphodeficient T58A, S62A and T58/S62 MYC was resistant to ERK inhibition but not KRAS suppression-mediated loss (Figures 4E, S4A and S4B). Finally, in the presence of proteasome inhibitor, transient KRAS knockdown significantly reduced phosphorylation of endogenous MYC at S62 in AsPC-1 (~75%) and to a lesser degree in MIA PaCa-2 cells. In contrast, pMYC T58 levels were not substantially reduced (Figure 4F). We conclude that KRAS regulates MYC protein stability through an ERK-, FBXW7- and MYC T58/S62 phosphorylation-dependent mechanism and additionally ERK-independent mechanism(s) less dependent on FBXW7 and MYC T58/S62 phosphorylation.
Determination of Additional KRAS-dependent Mechanisms that Regulate MYC Protein Stability
We applied three different strategies to identify additional KRAS-regulated ERK-independent mechanisms that control MYC protein stability. First, we performed a kinome-wide chemical proteomics screen using Multiplexed kinase Inhibitor Beads and mass spectrometry (MIB/MS) (Duncan et al., 2012) to determine changes in protein kinase expression/activity caused by transient siRNA suppression of KRAS (Figures S5A and S5B). Multiple up- and down-regulated changes were identified (Figure 5A). In particular, one upregulated protein kinase identified in three PDAC lines was MEK5, a MAPKK that phosphorylates and activates the nonclassical MAPK ERK5 (Hoang et al., 2017; Simoes et al., 2016) (Figure 5B). We verified that acute KRAS suppression was associated with increased MEK5 activity, as determined by increased pERK5 levels (Figure 5C).
Figure 5. Kinase Activation and MYC Protein Degradation Assays Identify a Role for MEK5-ERK5 Signaling in KRAS Regulation of MYC Protein Degradation.

(A) MIB/MS whole-kinome profiling upon transient siRNA KRAS knockdown in PDAC cells. Cell lysates were prepared from Panc 10.05 cells transfected with NS or KRAS 1 siRNA 1 for 24 hr, then subjected to affinity capture of endogenous kinases, followed by quantitative mass spectrometry. The kinase abundance ratios (KRAS siRNA/NS), as determined by label-free quantitative (LFQ) analysis, were averaged from three independent experiments. A ratio ≤ 0.7 denotes decreased MIB binding while a ratio ≥ 1.4 denotes increased MIB binding.
(B) Boxplots of MIB/MS analysis data (log2 LFQ) for MEK5 dynamics upon KRAS knockdown. The upper line of the box is the 75th percentile of MEK5 log2 LFQ, the middle line is the 50th percentile, and the bottom line is the 25th percentile. Upper and lower lines outside of the box represent the distance to the point closest to but not greater than 1.5 times the interquartile range (75th – 25th percentile difference) in each direction. PDAC cells were treated as described in panel 5A. Data shown for each condition represents three biological replicates.
(C) PDAC cell lines were transfected with NS or two independent KRAS siRNAs (24 hr). Cell lysates were blotted as indicated.
(D) A schematic illustration of the pGPS-LP lentivirus vector encoding MYC (GPSMYC). GPS-MYC contains a single CMV promoter and an IRES that permits the cotranslation of DsRed and chimeric EGFP-MYC from a single RNA transcript at a fixed ratio.
(E) Immunofluorescent images of MIA PaCa-2 cells expressing GPS-MYC (left) or GPSKRAS4B (right). Scale bar = 10 μm.
(F) MIA PaCa-2 cells stably expressing the GPS-MYC reporter were transfected with NS or FBXW7 siRNA for 24 hr, followed by transfection with NS or KRAS siRNA for an additional 24 hr. Immunoblotting was done as indicated. The EGFP-MYC fusion was detected with MYC antibody.
(G) Histogram of EGFP/DsRed ratio measured by flow-cytometry. MIA PaCa-2 cells stably expressing the GPS-MYC reporter were treated as in (F), then EGFP and DsRed fluorescence were measured by FACS. Data were plotted using Summit 5.2.
See also Figure S5.
Second, we developed a fluorescence-based sensor to monitor MYC protein stability (Figure 5D). We introduced human MYC cDNA sequences into the pGPS-LP lentivirus expression vector (Emanuele et al., 2011), whereby DsRed fluorescent protein and EGFP-MYC fusion protein are co-expressed under control of the same promoter (designated GPS-MYC) leading to transcription of one common mRNA, while IRES ensures translation of two separate proteins. Thus, the EGFP/DsRed expression ratio reflects the stability of the EGFP-MYC fusion protein. We stably infected MIA PaCa-2 cells with GPS-MYC and verified that exogenously expressed EGFP-MYC shared the same properties as endogenous MYC (Figure S5C): nuclear localization (Figure 5E), short half-life (Figure S5D), and reduced levels upon acute KRAS suppression (Figure S5E). Parallel analyses by FACS showed a reduced EGFP/DsRed fluorescence ratio upon treating with cycloheximide (CHX) to block protein translation (Figure S5F) or upon KRAS suppression (Figure S5G). As observed with endogenous MYC (Figures 4C and 4D), the FBXW7 suppression-mediated increase in EGFP-MYC levels and the EGFP/DsRed ratio were still reduced by KRAS suppression (Figures 5F and 5G). We conclude that the exogenously expressed EGFP-MYC fusion protein can accurately model KRAS regulation of endogenous MYC protein stability.
We first applied the EGFP-MYC reporter in a gain-of-function screen using the Cancer Toolkit, a lentivirus expression vector-based library encoding 36 activated components representing 17 different cancer signaling pathways (Martz et al., 2014). As expected, activated RAS and components of the RAF (MEK1) or PI3K (PI3Kα, AKT and GSK3β) effector pathways enhanced EGFP-MYC protein levels (Figure S6A). In contrast, no significant enhancement was seen upon activation of the mTORC1, Notch, β-catenin or JNK MAPK pathways. Interestingly, we also observed increased EGFP/DsRed ratios upon expression of two different activated variants of MEK5 (Figure 6A).
Figure 6. Identification of the MEK5-ERK5 Pathway as a Regulator of MYC Protein Stability in PDAC Cells.

(A) Histograms of EGFP/DsRed ratio in MIA PaCa-2 cells stably expressing the GPSMYC reporter and infected with control or two lentivirus expression vectors encoding constitutively-activated variants of MEK5 (Myr-FLAG-MEK5 or MEK5S311/T315D/DD). In a well-by-well format, MIA PaCa-2 cells expressing the GPS-MYC reporter were infected with individual lentiviral vectors encoding activated signaling components from the Cancer Toolkit). Forty-eight hr after infection, EGFP and DsRed fluorescence were measured by FACS and EGFP/DsRed ratio histograms were generated using Summit 5.2.
(B) Chemical structure of the compound UNC10225170 (GW284543), a 4-anilinoquin(az)oline chemotype, from the PKIS2 library.
(C) MIA PaCa-2 cells were treated with the indicated doses of UNC10225170 for 6 hr. Cell lysates were immunoblotted as indicated.
(D) MIA PaCa-2 cells were transfected with expression vectors encoding ubiquitin and HA epitope-tagged WT or kinase-deficient ERK5 (K84R) for 48 hr. Following 6 hr MG132 treatment, MYC protein was isolated by immunoprecipitation and immunoblotted for ubiquitination using anti-ubiquitin antibody. MYC protein precipitates and heavy chain as loading control are also shown. Immunoblotting of cell lysates was also done to determine total MYC and HA-ERK5 input protein levels.
(E) 293T cells were transiently transfected with expression vectors encoding GFP (control), or HA epitope-tagged ERK5. After 48 hr, the cultures were treated with CHX for the indicated times. Cell lysates were then immunoblotted with the indicated antibodies (left panel). Following quantitation of blot data to the left, MYC half-life was calculated using Prism (right panel).
(F) PDAC cell lines were transfected with siRNA oligos targeting ERK5 for 42 hr, then treated with 5 μM MG132 for an additional 6 hr. Cell lysates were immunoblotted as indicated.
(G) PDAC cells were transfected with NS or ERK5 siRNAs for 48 hr and RNA was extracted to measure gene expression. Following cDNA synthesis, real-time qPCR was performed using MYC or ERK5 specific primers. MYC or ERK5 mRNA expression levels were normalized to GAPDH mRNA levels. Data are presented as the mean of three replicates with error bars representing SEM.
(H) PDAC cell lines were treated with the indicated doses of ERK5-selective inhibitor XMD8–92 (ERK5i) or MEK5-selective inhibitor BIX02189 (MEK5i) for 8 hr. Cell lysates were immunoblotted as indicated.
See also Figure S6.
We next utilized GPS-MYC to establish a flow cytometry-based 384-well high-throughput screen to identify small molecules that regulate MYC protein stability. We screened the Published Kinase Inhibitor Set (PKIS) and PKIS2 libraries of ATP-competitive protein kinase inhibitors (Drewry et al., 2017; Elkins et al., 2016), identifying over 30 compounds that altered the EGFP/DsRed ratio (Figure S6B). Since we had identified MEK5 in both the MIB/MS and Cancer Toolkit analyses as a positive regulator of MYC stability, we further focused on a MEK5-selective inhibitor, UNC10225170 (Figures 6B and S6C). UNC10225170 treatment dose-dependently inhibited MEK5, as determined by reductions in pERK5, and decreased endogenous MYC protein (Figure 6C). Thus, in three different unbiased screens, we identified MEK5 as a regulator of MYC protein stability.
MEK5-ERK5 Signaling Promotes MYC Protein Stability
We next characterized the mechanism whereby MEK5 positively regulates MYC protein stability. We found that ectopic expression of WT but not kinase-dead (K84R) ERK5 in MIA PaCa-2 cells reduced polyubiquitination of endogenous MYC protein (Figure 6D) and increased its half-life from 35 to 48 min (Figure 6E). Conversely, siRNA suppression of ERK5 caused proteasome-dependent loss of MYC protein but not of MYC mRNA (Figures 6F and 6G). We also determined that pharmacologic inhibitors of either MEK5 (BIX02188; MEK5i) or ERK5 (XMD8–92; ERK5i) reduced MYC protein in a dose-dependent manner (Figure 6H).
ERK1/2 Inhibition Activates a MEK5-ERK5 Compensatory Mechanism to Promote MYC Stability
To determine if ERK5 can directly phosphorylate MYC, we first performed coimmunoprecipitation experiments and found that KRAS knockdown or ERK1/2 inhibitor treatment stimulated endogenous ERK5 association with endogenous MYC (Figures 7A and S7A). This association was dependent on ERK5 activation, since it was blocked by concurrent treatment with MEK5 or ERK5 inhibitors (Figure S7A).
Figure 7. Concurrent ERK5 and ERK1/2 Inhibition Synergistically Destabilizes MYC, Reduces MYC S62 Phosphorylation and Inhibits PDAC Cell Growth.

(A) MIA PaCa-2 cells were transfected with NS or KRAS siRNA for 20 hr and treated with MG132 for an additional 6 hr. Cell lysates (Input) were used for immunoprecipitation (IP) of ERK5 and the amount of co-precipitated MYC was detected by immunoblotting (WB).
(B) MIA PaCa-2 cells were treated with the ERK1/2i SCH772984 for the indicated times. Cell lysates were immunoblotted for total or phospho-specific antibodies (pEGFR, Y1068; pHER2 Y1248; pSRC, Y416).
(C) MIA PaCa-2 cells were treated for 12 hr with 200 nM ERK1/2i SCH772984 in the presence or absence of 500 nM inhibitors: EGFR inhibitors poziotinib and erlotinib, SRC inhibitor saracatinib or pan-FGFR inhibitor LY2874455. Immunoblotting of cell lysates was done as indicated.
(D) MIA PaCa-2 cells were transiently transfected with NS or ERK5 siRNA for 30 hr, then treated with vehicle (DMSO), SCH772984 (100 nM) or trametinib (2 nM) for an additional 8 hr. Where indicated, MG132 treatment was for 6 hr. Cell lysates were immunoblotted with the indicated antibodies. Short (s) and long (l) exposures are shown for phosphorylated and total MYC proteins.
(E) The indicated cell lines were treated with increasing doses of SCH772984 in combination with increasing doses of either ERK5i XMD8–92 or MEK5i BIX02189 for 96 hr, and proliferation was monitored by the CellTiter-Blue Cell Viability Assay. Bliss scores were calculated using Combenefit and heatmaps were generated using Graphpad Prism 7.
(F) Immunocompromised (NSG) mice with the implanted KRAS-mutant EMC1222 PDX tumors were treated with SCH772984 (ERK1/2i) alone or together with XMD8–92 (ERK5i) for the indicated days. The relative tumor volume and standard error of the mean were graphed and the p-value was determined by ANOVA analysis from combination treatment relative to the control or to each of the single agent treatments. EMC1222 PDX tumors have the following mutations: KRAS G12D, KMT2C R4541*, SMAD4 INDEL and CDKN2A p.D74G.
(G) Hematoxylin and eosin (H&E) and cleaved caspase-3 immunohistochemical staining of tumor tissue isolated from EMC1222 PDX tumors at maximum size ( day 12 for ERK5i group) or end of the treatment ( day 18). Scale bar = 100 μm.
(H) Images of representative EMC1222 PDX tumors from Figure 7F at day 12 for ERK5i group and day 18 for the rest. Scale bar = 1 cm.
(I) Tumor tissue was harvested from EMC1222 PDX tumors when tumors either reached maximum size (days 12–18) or at the end of the treatment (day 18), then immunoblotted as indicated.
See Figure S7.
We next assessed the importance of T58 and S62 phosphorylation in ERK5 promotion of MYC stability. Phospho-deficient (T58A and/or S62A) MYC proteins were protected from degradation upon siRNA suppression of ERK5 (Figure S7B), similar to our observation with siRNA suppression of ERK1/2 (Figures 4E and S4A). Taken together, our results support a model wherein, similar to ERK1/2, activated ERK5 promotes MYC protein stability through direct association with MYC and regulation of S62 phosphorylation.
We next determined the relationship between MEK5-ERK5 activity and ERK1/2 signaling. Whereas immunoblotting showed that pERK1/2 levels were efficiently suppressed after 2 hr treatment with MEKi or ERKi, robust increases in pERK5 were seen only after 4 hr, and this coincided with a rebound in MYC protein levels (Figures 7B and S7C). This result suggests that the increased MEK5-ERK5 signaling may be a secondary, compensatory response to ERK1/2 inhibition. Consistent with this, our MIB/MS analyses also demonstrated that ERK inhibitor treatment as short as 1 hr caused strong reductions in ERK1 (MAPK3) and ERK2 (MAPK1) activity without a concurrent increase in MEK5-ERK5 activity at that early time point (Figures S7D and S7E).
ERK1/2 activation can initiate negative feedback mechanisms, whereby ERK1/2 phosphorylates upstream signaling components to prevent excessive ERK activity. Conversely, inhibition of ERK1/2 can lead to loss of this negative feedback and result in a feed-forward activation of growth factor receptor and other tyrosine kinase signaling, for example through EGFR (Duncan et al., 2012; Lito et al., 2012). The MEK5-ERK5 cascade is activated by many of the same pathways, including EGF in a SRC-dependent manner (Sun et al., 2003) and HER2 (Drew et al., 2012; Esparis-Ogando et al., 2002). We tested whether ERK1/2 inhibition-induced activation of MEK5-ERK5 is mediated through known upstream activators of MEK5. We found that ERK1/2 inhibition caused a delayed increase in pEGFR, as well as in pHER2 and pSRC (Figures 7B and S7C). Accordingly, the ERK1/2 inhibition-induced increase in pERK5 was blocked by concurrent treatment with inhibitors of EGFR family members (poziotinib, erlotinib) or SRC (saracatinib), but not FGFR (LY2874455) (Figures 7C and S7F). Interestingly, we also identified EGFR, HER2 and SRC inhibitors in an unbiased 525-drug screen to define combinations with the ERKi SCH772984 that caused synergistic growth suppression (Figure S7G). We propose a model whereby ERK1/2 inhibition induces EGFR/HER2/SRC-dependent feed-forward activation of MEK5-ERK5, leading to MYC phosphorylation at S62, preventing MYC degradation.
Concurrent ERK1/2 and ERK5 Inhibition Causes Synergistic Loss of MYC and Suppression of PDAC Growth
Based on our model that MEK5-ERK5 activation is a compensatory response that prevents ERK1/2 inhibition-induced MYC degradation, we speculated that concurrent pharmacologic or genetic inhibition of ERK1/2 and ERK5 would cause synergistic loss of MYC protein. Indeed, whereas ERK5 silencing alone did not cause a significant reduction in MYC, it synergistically enhanced proteasome-dependent MYC loss caused by pharmacologic inhibition of ERK1/2 or MEK1/2, and this was associated with synergistic reduction of MYC S62 phosphorylation (Figures 7D and S7H). Flow cytometry analyses also demonstrated that concurrent short-term (8 hr) pharmacological inhibition of ERK1/2 and ERK5 caused more significant reduction in EGFP-MYC than seen with ERK1/2i or ERK5i alone (Figure S7I). Finally, concurrent inhibition of ERK1/2 and MEK5 or ERK5 resulted in proteasome-dependent synergistic loss of endogenous MYC protein (Figure S7J).
Based on our findings that MYC is essential for PDAC growth, and that concurrent inhibition of ERK1/2 and ERK5 caused loss of MYC, it follows that ERK5 inhibition would also enhance ERK1/2i growth inhibitory activity. We found that concurrent inhibition of ERK1/2 and MEK5 or ERK5 resulted in highly synergistic growth inhibition of MIA PaCa-2 and AsPC-1 cells in vitro (Figures 7E and S7K). Either genetic suppression of ERK5 via siRNA (Figure S7L) or pharmacologic inhibition of ERK5 kinase activity (Figure S7M) sensitized four different PDAC cell lines to ERK1/2imediated growth suppression. Finally, supporting the contribution of MYC in the activity of this combination, we determined that ectopic expression of phospho-deficient (S62A and/or T58) MYC proteins in MIA PaCa-2 and AsPC-1 cells decreased sensitivity to combination inhibition of ERK1/2 and ERK5 that corresponded to retention of MYC protein (Figures S7N-P).
To extend these findings to in vivo tumor growth, we evaluated the combination of ERK1/2i and ERK5i in KRAS-mutant pancreatic patient-derived xenograft (PDX) tumors that model the stroma-containing patient tumors (Knudsen et al., 2018). Although PDX EMC1222 was completely resistant to treatment either ERK1/2i or ERK5i alone, the combination potently and completely blocked tumor progression and induced cell death (Figures 7F-H, S7Q). We conclude that concurrent inhibition of ERK1/2 and ERK5 suppresses PDAC growth more effectively than ERK1/2 inhibition alone.
We found more heterogeneous responses in other PDX models. PDX AZ97 was highly sensitive to ERKi alone, which caused almost complete shutdown of tumor growth. Adding ERK5i neither increased nor antagonized ERKi anti-tumor activity (Figure S7R). Impressively, in PDX 1013, whereas ERKi alone caused tumor growth delay, adding ERK5i resulted in robust tumor regression (Figure S7S). The combination treatment was well tolerated, with no weight loss observed (Figure S7T).
To determine whether the tumor growth suppression was related to loss of MYC, we evaluated MYC protein levels in inhibitor-treated tumors. As we observed in cell culture, treatment of PDX EMC1222 with ERKi alone was associated with increased pERK5 and pMYC S62 levels in vivo (Figure 7I), and concurrent treatment with both ERKi and ERK5i was associated with loss of phospho-MYC S62 and total MYC protein. Finally, supporting the contribution of MYC loss in the activity of this combination in vivo, we determined that ectopic expression of phospho-deficient MYC S62A in AsPC-1 cells decreased sensitivity to combination inhibition of ERK1/2 and ERK5 (Figures S7U,V). Thus, concurrent inhibition of ERK1/2 and ERK5 effectively caused loss of MYC protein and impaired growth of KRAS-driven PDAC both in vitro and in vivo.
DISCUSSION
Our recent analyses of ERK inhibitors in PDAC identified loss of MYC protein as a marker of inhibitor sensitivity (Hayes et al., 2016). In contrast, de novo resistant cell lines exhibited ERK-independent mechanisms that maintained MYC protein expression. Since PDAC is a KRAS-driven cancer (Waters and Der, 2017), these findings prompted our evaluation of the mechanisms by which mutant KRAS regulates MYC expression in PDAC. We first determined that acute KRAS suppression caused rapid proteasome-dependent MYC degradation. Consistent with our previous observations (Hayes et al., 2016), we found that ERK1/2 MAPKs are critical to support KRAS-dependent MYC protein stability, yet surprisingly, we eliminated a role for PI3K-AKT-GSK3β. While we did find that activated PI3K and AKT could increase MYC protein stability, this was independent of KRAS signaling, consistent with our previous observation that PI3K-AKT signaling was not associated with mutant KRAS signaling or growth regulation in PDAC cells (Hayes et al., 2016). We utilized kinome-wide profiling and a MYC degradation screen in our search for other ERK-independent mechanisms responsible for KRAS-dependent MYC regulation and for ERKi resistance in PDAC. Our MIB/MS kinome analyses identified a spectrum of kinases whose activity increased or decreased upon KRAS suppression, and our GPS-MYC degradation screen identified protein kinase inhibitors that promote MYC degradation or stability. Together, these two datasets will guide future identification of additional KRAS-regulated protein kinases involved in regulation of MYC protein stability. We suspect that the kinase networks regulating MYC protein stability are considerably more complex than is currently understood.
Both our kinome-wide profiling and our GPS-MYC degradation screen identified an ERK1/2 inhibition-induced compensatory mechanism that caused activation of the MEK5-ERK5 cascade and prevented MYC degradation. Accordingly, we observed that concurrent inhibition of ERK5 blocked this compensation and synergistically enhanced ERK1/2 inhibitor anti-tumor activity in KRAS-mutant PDAC. A similar compensatory mechanism involving ERK5 (also known as BMK1) drove resistance to combined treatment with MEK1/2 and BRAF inhibitors in BRAF-mutant melanoma (Song et al., 2017). Likewise, genetic depletion of ERK1 and ERK2 or pharmacological inhibition of MEK1/2 induced ERK5 activity in KRAS-mutant colorectal cancer (CRC) cell lines, and concurrent inhibition of MEK1/2 and ERK5 enhanced CRC growth inhibition in vitro (de Jong et al., 2016). In contrast, another study showed no enhancement of growth inhibition in either BRAF- or KRAS-mutant CRC cell lines upon concurrent inhibition of MEK1/2 and ERK5 (Lochhead et al., 2016). Thus, whether combined inhibition of ERK1/2 and ERK5 will be effective in KRAS-mutant cancers other than PDAC remains to be determined. Additionally, whether this combination effectively causes loss of MYC protein in non-PDAC cancers remains to be addressed.
While there is increasing interest in ERK5 as a therapeutic target for cancer treatment (Hoang et al., 2017; Simoes et al., 2016), progress in developing ERK5 inhibitors has been limited and none are currently under clinical evaluation. In contrast to the considerable effort and progress made in targeting the RAF-MEK-ERK1/2 pathway for cancer treatment (Ryan et al., 2015), there is no clear cancer type or genetic driver to define the patient populations that will benefit from ERK5 inhibition. Furthermore, as is the case with targeted therapies in general, ERK5 inhibitor treatment as a monotherapy will likely not be effective in RAS-mutant cancers. Instead, inhibitor cocktails that target multiple MYC-regulatory protein kinases will likely be needed for long-term suppression of MYC protein and an effective therapeutic response. In addition to ERK1/2, CDK2 was identified as another protein kinase that can phosphorylate MYC at S62 (Hydbring et al., 2010) and more will likely be discovered.
Like ERK1/2, ERK5 phosphorylates proline-directed X-S/T-P-X motifs. Unlike ERK1/2, for which numerous phosphoproteomic studies have identified hundreds of direct substrates (Unal et al., 2017), there has been no detailed characterization of ERK5 substrates. The best characterized ERK5 substrates are the transcription factors MEF2A/C/D, SAP1 and PML (Hoang et al., 2017; Simoes et al., 2016), the latter of which was reported to play a critical role in responses to the ERK5i XMD8–92 (Yang et al., 2010). Additionally, a chimeric ERK2-ERK5 protein was shown to phosphorylate a MYC fragment peptide in vitro, but whether this was a physiologically relevant interaction and whether MYC is a direct ERK5 substrate in vivo was not determined (English et al., 1998). Our studies support MYC as another key ERK5 substrate and ERK5 as yet another protein kinase that phosphorylates MYC at S62. Our ongoing studies utilize ERK1/2-selective inhibitors to define ERK1/2 substrates that are physiologically relevant in KRAS-mutant PDAC. Parallel studies with ERK5-selective inhibitors to identify ERK5 substrates in the same setting will help to further elucidate how ERK5 reactivation reverses the consequences of ERK1/2 inhibition.
The long-established finding that RAS effector signaling can regulate MYC protein stability (Farrell and Sears, 2014) has been neither fully understood nor exploited. Targeting MYC degradation as a means of targeting KRAS has not been pursued by the field. Our finding that Myc suppression alone impaired mouse PDAC tumorigenic growth supports the critical role of MYC in KRAS-driven PDAC growth and is consistent with observations in other mouse models (Saborowski et al., 2014; Walz et al., 2014). However, MYC itself is considered even more undruggable than KRAS, and even the most promising and intensively pursued anti-MYC approaches, such as bromodomain inhibitors (e.g., JQ1), are highly nonselective for MYC (Dang, 2012). Our studies support an alternate anti-MYC strategy to target KRAS-driven PDAC growth: targeting KRAS-regulated protein kinases that promote MYC protein stability. Further, our discovery of compensatory MEK5-ERK5 activation identifies a mechanism by which PDAC cells evolve resistance to inhibitors of the ERK MAPK cascade and supports the need for more concerted efforts to develop ERK5 inhibitors for cancer treatment.
STAR★METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-MYC | Cell Signaling Tech. | Cat# 5605 |
| Mouse anti-MYC | Invitrogen | Cat# MA1–980 |
| Mouse anti-β-actin | Sigma-Aldrich | Cat# A5441 |
| Mouse anti-KRAS4B | Millipore Sigma | Cat# OP24 |
| Rabbit anti-phospho-ERK1/2 T202/Y204 | Cell Signaling Tech. | Cat# 4370 |
| Rabbit anti-ERK1/2 | Cell Signaling Tech. | Cat# 9102 |
| Rabbit anti-phospho-AKT S473 | Cell Signaling Tech. | Cat# 9271 |
| Rabbit anti-AKT | Cell Signaling Tech. | Cat# 9272 |
| Rabbit anti-phospho-p90RSK T359/S363 | Cell Signaling Tech. | Cat# 9344 |
| Rabbit anti-p90RSK | Cell Signaling Tech. | Cat# 9355 |
| Rabbit anti-phospho-GSK3β S9 | Cell Signaling Tech. | Cat# 5558 |
| Rabbit anti-GSK3β | Cell Signaling Tech. | Cat# 12456 |
| Species anti-phospho-MYC T58 | Abcam | Cat# ab28842 |
| Species anti-phospho-MYC S62 | Abcam | Cat# ab106952 |
| Rabbit anti-ubiquitin | Cell Signaling Tech. | Cat# 3933 |
| Rabbit anti-phospho-ERK5 T218/T220 | Cell Signaling Tech. | Cat# 3371 |
| Rabbit anti-ERK5 | Cell Signaling Tech. | Cat# 3372 |
| Mouse anti-ERK5 | Santa Cruz | Cat# sc-398015 |
| Rabbit anti-phospho EGF receptor Y1068 | Cell Signaling Tech. | Cat# 3777 |
| Rabbit anti-EGF receptor | Cell Signaling Tech. | Cat# 4267 |
| Rabbit anti-phospho-HER2/ErbB2 Y1248 | Cell Signaling Tech. | Cat# 2247 |
| Rabbit anti-HER2/Erbb2 | Cell Signaling Tech. | Cat# 4290 |
| Rabbit anti-phospho-Src Family Y416 | Cell Signaling Tech. | Cat# 6943 |
| Rabbit anti-Src | Cell Signaling Tech. | Cat# 2109 |
| Rabbit anti-MEKK2 | Cell Signaling Tech. | Cat# 19607 |
| Rabbit anti-phospho-S6 Ribosomal Protein (Ser235/236) | Cell Signaling Tech. | Cat# 2211 |
| Rabbit anti-cleaved Caspase-3 (Asp175) | Cell Signaling Tech. | Cat# 9661 |
| Mouse anti-IgG | Santa Cruz | Cat# sc-2025 |
| Rabbit anti-IgG | Cell Signaling Tech. | Cat# 2729 |
| Species anti-MYC (IHC) | Abcam | Cat# ab32072 |
| Species anti-phospho-ERK1/2 (IHC) | Cell Signaling Tech. | Cat# 4376 |
| Species anti-phospho-AKT T308 (IHC) | Abcam | Cat# ab38449 |
| Bacterial and Virus Strains | ||
| DH5α | Thermo Fisher | Cat# 18258012 |
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Doxycycline | Sigma-Aldrich | Cat# D3072 |
| MG132 | Sigma-Aldrich | Cat# M7449 |
| Dimethyl sulfoxide | Fisher Scientific | Cat# BP231 |
| Cycloheximide | Sigma-Aldrich | Cat# C4859 |
| SCH772984 | Merck | N/A |
| AZD6244 | Astra-Zeneca | N/A |
| AZD5363 | Astra-Zeneca | N/A |
| AZD8186 | Astra-Zeneca | N/A |
| UNC10225170A | (Matossian et al., 2017) | N/A |
| XMD8–92 in vivo | MedChem Express | Cat# HY-14443 |
| XMD8–92 | Selleckchem | Cat# S7525 |
| BIX02189 | Selleckchem | Cat# S1531 |
| Trametinib | Selleckchem | Cat# S2673 |
| Poziotinib | Selleckchem | Cat# S7358 |
| Erlotinib | Selleckchem | Cat# S1023 |
| Saracatinib | Selleckchem | Cat# S1006 |
| LY2874455 | Selleckchem | Cat# S7057 |
| Critical Commercial Assays | ||
| CellTiter-Blue Cell Viability Assay | Promega | Cat# G8081 |
| XenoLight D-Luciferin - K+ | Perkin Elmer | Cat# 122799 |
| Doxycycline Diet (2020, 625 Dox, R) | Tekland | Cat# TD.130141 |
| Deposited Data | ||
| N/A | N/A | N/A |
| Experimental Models: Cell Lines | ||
| INK4.1syn_Luc | (Collisson et al., 2012) | N/A |
| MIA PaCa-2 | ATCC | Cat# CRL-1420 |
| PANC-1 | ATCC | Cat# CRL-1469 |
| AsPC-1 | ATCC | Cat# CRL-1682 |
| Panc 10.05 | ATCC | Cat# CRL-2547 |
| HPAF-II | ATCC | Cat# CRL-1997 |
| SW1990 | ATCC | Cat# CRL-2172 |
| Capan-2 | ATCC | Cat# HTB-80 |
| BxPC-3 | ATCC | Cat# CRL-1687 |
| Pa01C | (Jones et al., 2008) | N/A |
| Pa02C | (Jones et al., 2008) | N/A |
| Pa03C | (Jones et al., 2008) | N/A |
| Pa14C | (Jones et al., 2008) | N/A |
| Pa16C | (Jones et al., 2008) | N/A |
| Pa18C | (Jones et al., 2008) | N/A |
| HPAC | ATCC | Cat# CRL-2119 |
| HPDE | (Radulovich et al., 2008) | N/A |
| HPDE-KRAS | (Radulovich et al., 2008) | N/A |
| HPNE | (Campbell et al., 2007) | N/A |
| HPNE-KRAS | (Campbell et al., 2007) | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: FVB/n | The Jackson Laboratory | Cat# 001800 |
| Mouse: NSG | The Jackson Laboratory | Cat# 005557 |
| Mouse : NOD scid | The Jackson Laboratory | Cat# 001303 |
| Oligonucleotides – See Table S2 | ||
| Recombinant DNA | ||
| SMART | Dharmacon | V3SH7675 |
| pMSCV MYC-FLAG | (Hayes et al., 2016) | N/A |
| pMSCV MYCT58A-FLAG | (Hayes et al., 2016) | N/A |
| pMSCV MYCS62A-FLAG | (Hayes et al., 2016) | N/A |
| pMSCV MYCT58/S62A-FLAG | (Hayes et al., 2016) | N/A |
| pGPS-LP | (Emanuele et al., 2011) | N/A |
| GPS-MYC | This paper | N/A |
| Kras (G12V)-pcw107 | (Martz et al., 2014) | N/A |
| Hras (G12V)-pcw107 | (Martz et al., 2014) | N/A |
| MEK1 (S218/222D)-pcw107 | (Martz et al., 2014) | N/A |
| myr-FLAG-PIK3CA-pcw107 | (Martz et al., 2014) | N/A |
| myr-FLAG-AKT1-pcw107 | (Martz et al., 2014) | N/A |
| FLAG-Rheb1 (Q64L)-pcw107 | (Martz et al., 2014) | N/A |
| beta-catenin (S33A/S37A/T41A)-pcw107 | (Martz et al., 2014) | N/A |
| beta-catenin (S33Y)-pcw107 | (Martz et al., 2014) | N/A |
| GSK3beta (K85A)-pcw107 | (Martz et al., 2014) | N/A |
| JNK2 WT O/E (MAPK7)-pcw107-V5 | (Martz et al., 2014) | N/A |
| FLAG-Mkk7-JNK2 fusion-pcw107 | (Martz et al., 2014) | N/A |
| p38 WT O/E (MAPK14)-pcw107-V5 | (Martz et al., 2014) | N/A |
| FLAG-MKK6 (S207/T211E)-pcw107 | (Martz et al., 2014) | N/A |
| Notch1 intracellular domain-pcw107-V5 | (Martz et al., 2014) | N/A |
| Notch3 intracellular domain-pcw107-V5 | (Martz et al., 2014) | N/A |
| Myr-FLAG-MEK5 | (Martz et al., 2014) | N/A |
| MEK5 DD(S311D/T315D)-pcw107-V5 | (Martz et al., 2014) | N/A |
| myr-FLAG-MEK5-pcw107 | (Martz et al., 2014) | N/A |
| HA-ERK5K84R | (Brady et al., 2014) | Addgene Cat# 53171 |
| HA-ERK5 WT | (Brady et al., 2014) | Addgene Cat# 53175 |
| HA-ERK5 WT | (Buschbeck et al., 2002) | Addgene Cat# 65244 |
| HA-Ubiquitin | (Kamitani et al., 1997) | Addgene Cat# 18712 |
| Software and Algorithms | ||
| GraphPad Prism 5 Software | GraphPad Software | N/A |
| SPSS Statistics | IBM | N/A |
| Summit 5.2 | Beckman Coulter | N/A |
| Other | ||
| Multiplexed Kinase Inhibitor Beads (MIB) | ||
| A/G agarose beads | Santa Cruz Biotech. | Cat# sc-2003 |
CONTACT FOR REAGENT AND RESOURCE SHARING
Requests for further information and for reagents may be directed to, and will be fulfilled by, the Lead Contact, Channing J. Der (cjder@med.unc.edu)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Culture
Human pancreatic cancer cell lines were obtained from ATCC or were gift from Dr. Anirban Miatra (MD Anderson Cancer Center) and were maintained in Dulbecco’s Modified Eagle Medium supplemented with 10% fetal bovine serum (FBS) (HPAC, PANC-1, and MIA PaCa-2), or in RPMI-1640 supplemented with 10% FBS (AsPC-1, CFPAC-1, HPAF-II, HuPT3, Panc 10.05, SW-1990, Pa01C, Pa02C, Pa04C, Pa14C, Pa16C and Pa18C) or with 15% FBS (Capan1 and Capan-2). Short Tandem Repeat (STR) analysis was done to verify identity of all PDAC cell lines and all cell lines were tested for mycoplasma within three months of use.
Patient-derived xenograft tumors
The PDX models were derived from resected pancreatic cancers that were engrafted into NSG mice as described previously (Knudsen et al., 2018). All tissues were obtained on IRB approved protocols at the University Texas Southwestern or University of Arizona.
Mice
For the syngeneic orthotopic PDAC mouse model, adult FVB/N mice were purchased from The Jackson Laboratory. These animals were cared for according to guidelines set forth by the American Association for Accreditation of Laboratory Animal Care and the U.S. Public Health Service policy on Human Care and Use of Laboratory Animals. All mouse studies were approved and supervised by the Institutional Animal Care and Use Committees of the University of North Carolina at Chapel Hill and the University of Arizona.
For PDAC PDX mouse models, NSG mice were purchased from The Jackson Laboratory and experiments were performed at the University of Arizona. For subcutaneous PDAC mouse analyses, NOD scid mice were purchased from The Jackson Laboratory and experiments were performed at the University of Texas Health Science Center at San Antonio. All mice were maintained in a specific pathogen-free facility and all animal experiments were performed in accordance with protocols approved by the Institutional Animal Care and Use Committees of the University of Arizona, the University of North Carolina at Chapel Hill and at the University of Texas Health Science Center at San Antonio.
METHOD DETAILS
Immunoblotting
For lysis, cell pellets were resuspended in 1% Triton Buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% Triton) supplemented with phosphatase (Sigma) and protease (Roche) inhibitors, and incubated on ice for 30 min, with brief vortexing every 5–10 min. Lysates were then cleared by centrifugation at 15,000×g for 10 min at 4oC, and protein concentration was determined using the Bradford Assay (Bio-Rad). Standard immunoblotting procedures were followed. Membranes were blocked in 5% nonfat dried milk (diluted in TBS 0.05%/Tween 20 [TBST]). To determine the levels of activated proteins, cell lysates were immunoblotted with phospho-specific antibodies to ERK1/2, pan-AKT, MYC, GSK3β, and ERK5. Antibodies recognizing total MYC, ERK1/2, AKT, MEK5, ERK5, and β-actin controlled for total protein levels.
Immunoprecipitation
To detect endogenous MYC and ERK5 complexes following transfection or inhibitor treatments, cells were lysed in 1% Triton Buffer with 2% glycerol, supplemented with a protease/phosphatase inhibitor cocktail (Roche). 500 μg of total protein at a concentration of 1 mg/ml were incubated with 20 μl protein A/G agarose beads and 5 μg anti-ERK5 antibody (Cell Signaling Tech, #3371) or 5 μg normal rabbit IgG (Cell Signaling Tech, #2729), overnight at 4oC with continuous inversion. The next day, the beads were pelleted by centrifugation at 2000 rpm for 2 min and washed 3 times with 1% Triton Buffer and 3 times with 1× PBS. Immunocomplexes were eluted and denatured by boiling for 5 min in 2× Laemmli Buffer, then analyzed by immunoblotting. 15 μg cell lysate were used to determine relative protein input levels.
For ubiquitination assays, ubiquitin and ERK5 WT or K84R were expressed in MIA PaCa-2 cells for 48 h, which were then treated with 5 μM MG132 for 6 h. Cells were lysed in 1% Triton Buffer and endogenous MYC protein was precipitated from 500 μg protein lysates at a concentration of 1 mg/ml with 10 μg anti-MYC antibody (Invitrogen, MA1–980) and 30 μl protein A/G agarose beads at 4 degrees overnight, with gentle rotation. The next day, the beads were pelleted by centrifugation at 2000 rpm for 2 min and washed 3 times with 1% Triton Buffer and 3 times with 1× PBS. Protein complexes were eluted with 2× Laemmli buffer and analyzed by immunoblotting. Fifteen μg of cell lysate were used to determine relative protein input levels.
siRNA and Plasmid Transfections
All siRNA transfection experiments were performed using Lipofectamine RNAiMAX (Life Technologies) following manufacturer instructions. The siRNA concentration used was 10 nM. The sequences of all siRNA oligos are listed in the Resource table. Plasmid transfections were done using Fugene 6 (Roche) or Lipofectamine 3000 (Life Technologies), following manufacturer instructions.
Expression Vectors
SMART lentiviral shRNA vectors for doxycycline-inducible suppression of mouse Myc gene expression in vivo were purchased from Dharmacon as viral particles. Three vectors containing different shRNA sequences targeting mouse Myc were tested for Myc suppression in INK4.1syn-Luc cells. The sequence with most potent suppression in vitro was selected for in vivo studies.
To generate GPS-MYC, the human MYC cDNA sequence was amplified by PCR using Platinum Pfx polymerase (Invitrogen) and the following gateway attB primers:
attB1:5’GGGGACAAGTTTGTACAAAAAAGCAGGCTTAGAAGGAGATAGAACCATGCCCCTCA ACGTTAGCTTCAC 3’
attB2: 5’GGGGACCACTTTGTACAAGAAAGCTGGGTCCTACGCACAAGAGTTCCGTAGC-3’ PCR product was purified and cloned into pDONR223 (Addgene) via BP reaction (Invitrogen). MYC was then transferred into the pGPS-LP vector via LR reaction (Invitrogen).
The pMSCV puro retrovirus vector and pMSCV puro encoding FLAG epitope-tagged MYC (T58A) were provided by Juan Belmonte (Addgene plasmid 20076) (Aasen et al., 2008). pMSCV puro vectors encoding FLAG epitope-tagged MYC S62A and S58/62A were generated as we described previously (Hayes et al., 2016).
Viral Expression Vector Infections
Retroviral vectors were transiently transfected into 293T cells with a pCL-10A1 packaging system (Novus Biologicals) using the Fugene 6 transfection reagent. Viral supernatant was collected at 48–72 hr post-transfection, filtered through 0.2 μm PES syringe filters (Nalgene) and used immediately or stored in aliquots at −80oC. Virus infection was done in the presence of polybrene (8 μg/ml) and spinnoculation at 2200 × g for 1 hr at 37oC.
Lentiviral vectors were produced and used as described above for retroviruses with the use of pMD2.G and the pSPAX2 packaging system (Addgene).
Cycloheximide (CHX) Chase Assay
Cells were treated with 10 μg/ml cycloheximide (CHX) and harvested at the indicated time points. Protein was extracted from the cell lysates and subjected to immunoblotting. Protein levels were quantitated by densitometric intensity.
Cell Cycle Distribution Assay
Cells were washed with PBS and fixed with 70% ethanol for 24 hr at −20oC. Then cells were centrifuged for 5 min at 200 × g, washed with PBS, and incubated with propidium iodide (PI)/Triton X-100 staining solution with RNase A (Sigma) for 2–4 hr at room temperature. DNA content was measured using a Beckman Coulter Cyan flow cytometer, and Summit 4.4 software was used to acquire and analyze the data.
Mouse Studies
To evaluate the requirement for Myc in Kras-driven pancreatic cancer growth in vivo, INK4.1syn-Luc (KrasG12D/Ink4a−/−) mouse pancreatic tumor-derived cells (Collisson et al., 2012), kindly provided by Dr. Eric Collisson (University of California, San Francisco) were stably infected with the SMART lentivirus empty vector (control) or encoding shRNA targeting mouse Myc. INK4.1syn-Luc cells were trypsinized, washed, and resuspended in Hanks’ balanced salt solution (HBSS; Gibco), and injected into the head of the pancreas of 10 syngeneic FVB/n mice per group. Cells were injected into each mouse at 103 cells in 50 μl 1:1 mixture of HBSS and BD Matrigel. Mice were between 6–8 weeks of age. Mice were anesthetized prior to surgery with ketamine (80 mg/kg) + xylazine (8 mg/kg) + acepromazine (1 mg/kg). Following fur removal and sterile skin preparation, an incision was made in the abdomen to visualize the pancreas underneath the spleen. A 1 ml tuberculin syringe with a 30-g needle was used to inject the cell suspension directly into the head of the pancreas. After injection, the skin incision was closed using surgical clips and the mice were observed until fully recovered. Ten days after injection, upon tumor detection, mice were fed doxycycline-containing chow, and luciferase-labeled tumor progression was monitored using an IVIS Lumina optical imaging system and D-Luciferin substrate (Perkin Elmer) per manufacturer instructions. For the survival study, each mouse was monitored daily until it became moribund and was then sacrificed.
Three PDX models were used to evaluate the anti-tumor activity of ERK1/2i SCH772984 or ERK5i XMD8–92 alone, or in combination. PDX tumors were passaged in NSG mice to develop treatment cohorts. Mice were randomized to treatment arms when tumors reached 100200 mm3. SCH772984 was delivered at 50 mg/kg 6 days per week by oral gavage. XMD8–92 was delivered at 50 mg/kg 6 days per week via IP injection. Tumor volumes were measured by digital calipers every 48 hr. The average tumor volume and standard error of the mean were graphed and statistical significance was determined by ANOVA. Mice were sacrificed when tumors reached >1000 cm or at the end of 21 days of treatment.
To evaluate the contribution of MYC in the anti-tumor activity of combination treatment with ERK1/2i and ERK5i, AsPC1 cells stably infected with the pBabe empty vector or encoding MYC S62A were injected subcutaneously into the right flank of NOD scid mice. Cells were injected into each mouse at 5×106 cells in 100 μl at a 1:1 mixture of HBSS and BD Matrigel. Mice were randomized to treatment arms when tumors reached 100–200 mm3. SCH772984 was delivered at 65 mg/kg 6 days per week via IP injection. XMD8–92 was delivered at 35 mg/kg 6 days per week via IP injection. Tumor volumes were measured by digital calipers once a week. Mice were sacrificed when tumors reached >2000 cm or at the end of 21 days of treatment.
Immunohistochemistry
The human PDAC tissue microarrays (TMAs) used in this study were the same as those described previously (Whatcott et al., 2015). All tissues were archived samples that were provided to this study without any Protected Health Information (PHI). The PDAC diagnosis and histologic subtype information was obtained from de-identified pathology reports and histomorphologically confirmed by a board-certified pathologist (GH). The IHC staining procedures for the TMA slides were also similar to those described previously (Whatcott et al., 2015). Briefly, the TMA blocks were sectioned at 5 μm thickness, deparaffinized with xylene, rehydrated, and antigen retrieved on-line using the BOND-MAX™ autostainer (Leica Microsystems). The slides were incubated with the primary antibodies using the following conditions: anti-Myc (Abcam, Cat# ab32072, 1:75 for 60 min); anti-pERK1/2 (Cell Signaling Technology, Cat# 4376, 1:175 for 60 min); and anti-pan-AKT (phospho T308) (Abcam, Cat# ab38449, 1:50 for 60 min) with subsequent signal detection by the Mach 3 Rabbit HRP Polymer Detection kit (Biocare Medical). Slides were scanned using an Aperio Scanscope (Leica Microsystems) and visualized using the Aperio Image scope software (Leica Microsystems). The images were assessed and reviewed by a board-certified pathologist (GH). The staining intensity for each of the antibodies in each case was scored using a 0–3+ scoring system, where 0 is negative, 1+ is weak staining, 2+ is moderate staining, and 3+ is strong staining. For Kaplan-Meier survival analysis, cases with a score of 0 or 1+ were combined to form the “low” group and cases with a score of 2+ or 3+ were combined to form the “high” group.
For the PDX studies, immunohistochemistry to detect Ki67 and cleaved caspase 3 was performed on a Leica Bond auto-stainer. The cleaved caspase 3 antibody was from Cell Signaling (#9661) and used at a dilution of 1:200. The Ki67 was ready to use from Leica Biosystems (PA0230).
Growth Assays
To measure 2D growth, cells were plated at low density in 6 well plates. Within 7 to 10 days, colonies were fixed with 4% paraformaldehyde and stained with 1% crystal violet. For siRNA experiments, cells were plated 24 hr after transfection. For drug treatment studies, cells were plated and treated the next day.
To monitor anchorage-independent cell growth, a soft agar colony formation assay was done. At 24 hr after siRNA transfection, 2×103 to 104 cells were suspended between layers of 0.6% (bottom) and 0.3% (top) agar in each of three wells of a 6-well dish. Cells were supplemented with growth medium twice a week and allowed to form colonies for 7–20 days. Colonies were visualized by staining with MTT (2 mg/ml). Colonies were imaged and quantitated using ImageJ (National Institutes of Health).
To measure cell viability upon inhibitor treatments, cells were plated in 96 well plates at a density of 103 cells/well and treated the next day. At 5 days after treatment, cell viability was measured using the CellTiter-Blue Cell Viability Assay (Promega), following manufacturer instructions.
MIB/MS
To monitor changes in protein kinase expression/activity, PDAC cell lines were treated with either KRAS siRNA for 24 hr or with 1 μM SCH772984 for 1 hr prior to being subjected to MIB/MS analysis. Lysates were prepared and MIB/MS was performed as described previously (Duncan et al., 2012). Briefly, cells were lysed on ice in MIB lysis buffer [50 mM HEPES (pH 7.5), 0.5% Triton X-100, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 10 mM sodium fluoride, 2.5 mM sodium orthovanadate, 1× protease inhibitor cocktail (Roche), 1% phosphatase inhibitor cocktail 2 (Sigma-Aldrich), and 1% of phosphatase inhibitor cocktail 3 (Sigma-Aldrich)]. Cell lysates were sonicated for 3×10 seconds on ice and centrifuged at 10,000×g for 10 min at 4°C. The supernatant was collected and syringe-filtered through a 0.2 μm SFCA membrane. The filtered lysate (approximately 5 mg of protein per experiment) was brought to 1 M NaCl and passed through a column of mixed inhibitor-conjugated beads (MIBs) consisting of Sepharoseconjugated, UNC2147A, CTx-0294885, UNC8088A. Purvalanol B, PP58 and VI16832. The MIBs were washed with 5 mL of high-salt buffer and 5 mL of low-salt buffer [50 mM HEPES (pH 7.5), 0.5% Triton X-100, 1 mM EDTA, 1 mM EGTA, and 10 mM sodium fluoride, and 1 M NaCl or 150 mM NaCl, respectively]. The columns were washed a final time with 1 ml 0.1% SDS before elution in 1 ml of elution buffer [0.5% SDS, 1% 2 mercaptoethanol, and 0.1 M Tris (pH 6.8)] (100°C, 5 min). Eluted kinases were reduced ( dithiothreitol) and alkylated (iodoacetamide) prior to being concentrated with Amicon Ultra centrifugal filters (Millipore), and detergent was removed from the concentrated eluate by chloroform/methanol extraction. Protein pellets were resuspended in 50 mM HEPES (pH 8.0) and were digested for overnight at 37ºC 24 hr with sequencing grade modified trypsin (Promega). Digested peptides were cleaned using PepClean C18 spin columns (Thermo Scientific).
LC/MS/MS analysis
The peptide samples (n=3) were analyzed by LC/MS/MS using an Easy nLC 1000 coupled to a QExactive HF mass spectrometer (Thermo Scientific). Samples were injected onto an Easy Spray PepMap C18 column (75 μm id × 25 cm, 2 μm particle size; Thermo Scientific) and separated over a 2 hr method. The gradient for separation consisted of 5–32% mobile phase B at a 250 nl/min flow rate, where mobile phase A was 0.1% formic acid in water and mobile phase B consisted of 0.1% formic acid in ACN. The QExactive HF was operated in datadependent mode where the 15 most intense precursors were selected for subsequent fragmentation. Resolution for the precursor scan (m/z 400–1600) was set to 120,000 with a target value of 3 × 106 ions. MS/MS scans resolution was set to 15,000 with a target value of 2 × 104 ions. The normalized collision energy was set to 27% for HCD. Peptide match was set to preferred, and precursors with unknown charge or a charge state of 1 and ≥ 8 were excluded.
MYC Protein Degradation Screen
To establish a high-throughput screen to identify protein kinases regulating MYC protein stability, we first generated a large scale supply of frozen MIA PaCa-2 cells stably expressing the GPS-MYC reporter, to minimize variability during the screening campaign. First, cells were plated at 20,000 cells/well into a 384-well tissue culture plate using an automated liquid dispenser (Multidrop Combi Reagent Dispenser; Thermo Scientific). The following day, a portion of the growth medium was removed with a robotic liquid handler (Biomek FX; Beckman Coulter) and growth medium supplemented with test compound or DMSO was added to a final concentration of 20 μM or 0.1% respectively. After 4 or 6 hr incubation, the cells were washed with buffer then gently dissociated with Accumax cell dissociation solution. Plates were then processed using a 20 sec sip time on an Intellicyte iQue high throughput flow cytometer. The Published Kinase Inhibitor Sets (PKIS) 1 and 2, comprised of 843 ATP-competitive protein inhibitors, were used (Drewry et al., 2017; Elkins et al., 2016). Each plate was run in duplicate at two time points (4 and 6 hr). Each plate contained 32 control wells treated with DMSO, 16 wells treated with 10 μg/ml CXH and 16 wells treated with 5 μM MG132. Raw data was uploaded into the CICBDD database (ScreenAble Solutions) and test compounds were normalized to control wells. Normalized data was exported to Spotfire for visualization and further analysis. Both the EGFP/DsRed ratio as well as the absolute DsRed value were used to evaluate the screening data. Compounds that reduced DsRed by more than 50% alone were filtered from the hits. Compounds that reduced the EGFP/DsRed ratio more than 30% were selected for follow-up studies.
Cancer Toolkit Analyses
Cancer Toolkit lentivirus expression vectors were provided by Kris Wood (Duke) (Martz et al., 2014). Open reading frame (ORF)-expressing lentiviruses were produced and MIA PaCa-2 cells stably expressing MYC-GPS were infected at an MOI of 2, in 6 well plates. Virus infection was done in the presence of polybrene (8 μg/ml) and spinnoculation at 2200 × g for 1 hr at 37oC. Forty-eight hr later, cells were trypsinized and EGFP/DsRed fluorescence was measured by FACS using BD FACSCalibur and FlowJo acquisition software.
Drug Sensitivity and Resistance Testing (DSRT) Chemical Library Screen
To identify inhibitors that enhance the growth suppressing activity of SCH772984, DSRT was performed on pancreatic cancer cell lines essentially as described previously (Pemovska et al., 2013). The DSRT library is comprised of 525 conventional chemotherapeutics and a broad range of targeted oncology compounds. The compounds were dissolved in 100% dimethyl sulfoxide (DMSO) and dispensed into tissue culture-treated 384-well plates (Corning) using an acoustic liquid handling device, Echo 550 (Labcyte Inc.). The compounds were plated at five different concentrations in 10-fold dilutions covering a 10,000-fold concentration range relevant for each drug (e.g., 1–10,000 nmol/L). The predrugged plates were kept in pressurized StoragePods (Roylan Developments Ltd.) under inert nitrogen gas until needed. The compounds were dissolved in 5 μl of culture medium while shaking for 30 min. Twenty μl of a single-cell suspension (1,000 cells) was transferred to each well using a MultiDrop Combi peristaltic dispenser (Thermo Scientific). The plates were incubated in a humidified environment at 37oC and 5% CO2, and cell viability was measured after 72 hr using the CellTiter-Glo Luminescent Assay (Promega) with a Molecular Devices Paradigm plate reader, according to manufacturer instructions. The data were normalized to negative control (DMSO only) and positive control wells (containing 100 μmol/L benzethonium chloride, effectively killing all cells). Curve fitting and calculations of drug sensitivity scores were performed as previously described (Yadav et al., 2014).
QUANTITATION AND STATISTICAL ANALYSIS
IHC Statistical Analysis
Correlations between marker expression (immunohistochemical staining intensity) and patient survival were analyzed using Kaplan-Meier survival curves. The statistical significance of the median survival times vs. marker expression (high and low expression groups) was assessed by Log-rank test using GraphPad Prism 5 software. The association between the staining intensity of different protein markers (MYC, pERK, and pAKT) was analyzed using the nonparametric Spearman’s rho correlation coefficient (rs). The significance level was set at p value ≤ 0.01 with a two-tailed test. Data were analyzed using standard statistical software (IBM SPSS Statistics).
MIB/MS Data Analysis
Raw data files were processed using MaxQuant version 1.5.3.17 and searched against the reviewed Uniport human database (containing 20,203 entries), using Andromeda within MaxQuant. Enzyme specificity was set to trypsin, up to two missed cleavage sites were allowed, carbamidomethylation of C was set as a fixed modification and oxidation of M was set as a variable modification. A 1% FDR was used to filter all data. Match between runs was enabled (2 min match time window, 20 min alignment window), and a minimum of two peptides was required for label-free quantitation using the LFQ intensities. Further analyses were performed in Perseus version 1.6.0.2 and R.
Supplementary Material
Acute suppression of KRAS causes proteasome-dependent degradation of MYC protein
KRAS regulates MYC protein stability by ERK but not by PI3K-AKT effector signaling
ERK1/2-inhibition-induced feed-forward activation of EGFR-MEK5-ERK5 blocks MYC loss
Inhibiting ERK1/2 and ERK5 synergistically causes MYC loss and inhibits PDAC growth
Significance.
Functional interplay between RAS and MYC oncogenes in cancer is well established. Despite the critical involvement of MYC in RAS-driven cancer growth, targeting MYC has not been widely pursued as an anti-RAS therapeutic strategy. Here we identify ERK1/2-dependent and -independent mechanisms by which mutant KRAS stabilizes MYC in KRAS-mutant pancreatic cancer. We further dissected an EGFR/HER2dependent feedback mechanism that activates the MEK5-ERK5 pathway to prevent ERK1/2 inhibition-mediated loss of MYC. Concurrent inhibition of ERK1/2 and ERK5 synergistically promotes MYC degradation and suppression of pancreatic cancer growth. Taken together, our findings establish targeting MYC degradation as an effective anti-KRAS strategy and identify concurrent ERK5 and ERK1/2 inhibition as a potential MYC-destabilizing and growth inhibitory therapeutic approach for PDAC.
ACKNOWLEDGMENTS
We thank Eric Collisson (University of California, San Francisco) for the INK4.1syn_Luc cell line, Kris Wood (Duke) for the Cancer Toolkit expression vectors, Michael Emanuele (University of North Carolina at Chapel Hill) for the pGPS-LP plasmid, and Edward Favours and Abhik Bandyopadhyay for technical assistance (The Greehey Children’s Cancer Research Institute, The University of Texas Health Science Center at San Antonio). Support was provided by grants from the National Cancer Institute (NCI) (CA199235 and CA203657) to C.J.D. and A.D.C., and from the NCI (CA42978, CA196510), the Pancreatic Cancer Action Network/AACR, Department of Defense, and the Lustgarten Pancreatic Cancer Foundation to C.J.D. A.V.V. was supported by fellowships from the NCI (T32CA009156) and American Cancer Society (128014-PF15–059-01-TBE). D.R.B. was supported by NCI fellowships (T32CA071341 and F31CA216965), I.O.D. by the Slomo and Cindy Silvian Foundation and K.L.B. was supported by T32CA009156 and a grant from the Pancreatic Cancer Action Network/AACR. This research is based in part upon work conducted using the UNC Proteomics Core Facility, which is supported in part by the P30 CA016086 Cancer Center Core Support Grant to UNC.
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Aasen T, Raya A, Barrero MJ, Garreta E, Consiglio A, Gonzalez F, Vassena R, Bilic J, Pekarik V, Tiscornia G, et al. (2008). Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat Biotechnol 26, 1276–1284. [DOI] [PubMed] [Google Scholar]
- Brady DC, Crowe MS, Turski ML, Hobbs GA, Yao X, Chaikuad A, Knapp S, Xiao K, Campbell SL, Thiele DJ, et al. (2014). Copper is required for oncogenic BRAF signalling and tumorigenesis. Nature 509, 492–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buschbeck M, Eickhoff J, Sommer MN, and Ullrich A (2002). Phosphotyrosinespecific phosphatase PTP-SL regulates the ERK5 signaling pathway. J Biol Chem 277, 29503–29509. [DOI] [PubMed] [Google Scholar]
- Campbell PM, Groehler AL, Lee KM, Ouellette MM, Khazak V, and Der CJ (2007). K-Ras promotes growth transformation and invasion of immortalized human pancreatic cells by Raf and phosphatidylinositol 3-kinase signaling. Cancer Res 67, 2098–2106. [DOI] [PubMed] [Google Scholar]
- Chen SH, Zhang Y, Van Horn RD, Yin T, Buchanan S, Yadav V, Mochalkin I, Wong SS, Yue YG, Huber L, et al. (2016). Oncogenic BRAF deletions that function as homodimers and are sensitive to inhibition by RAF dimer inhibitor LY3009120. Cancer Discov 6, 300–315. [DOI] [PubMed] [Google Scholar]
- Collisson EA, Trejo CL, Silva JM, Gu S, Korkola JE, Heiser LM, Charles RP, Rabinovich BA, Hann B, Dankort D, et al. (2012). A central role for RAF->MEK-->ERK signaling in the genesis of pancreatic ductal adenocarcinoma. Cancer Discov 2, 685–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV (2012). MYC on the path to cancer. Cell 149, 22–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV, Reddy EP, Shokat KM, and Soucek L (2017). Drugging the ‘undruggable’ cancer targets. Nat Rev Cancer 17, 502–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong PR, Taniguchi K, Harris AR, Bertin S, Takahashi N, Duong J, Campos AD, Powis G, Corr M, Karin M, et al. (2016). ERK5 signalling rescues intestinal epithelial turnover and tumour cell proliferation upon ERK1/2 abrogation. Nat Commun 7, 11551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew BA, Burow ME, and Beckman BS (2012). MEK5/ERK5 pathway: the first fifteen years. Biochim Biophys Acta 1825, 37–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drewry DH, Wells CI, Andrews DM, Angell R, Al-Ali H, Axtman AD, Capuzzi SJ, Elkins JM, Ettmayer P, Frederiksen M, et al. (2017). Progress towards a public chemogenomic set for protein kinases and a call for contributions. PLoS One 12, e0181585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan JS, Whittle MC, Nakamura K, Abell AN, Midland AA, Zawistowski JS, Johnson NL, Granger DA, Jordan NV, Darr DB, et al. (2012). Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell 149, 307–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkins JM, Fedele V, Szklarz M, Abdul Azeez KR, Salah E, Mikolajczyk J, Romanov S, Sepetov N, Huang XP, Roth BL, et al. (2016). Comprehensive characterization of the Published Kinase Inhibitor Set. Nat Biotechnol 34, 95–103. [DOI] [PubMed] [Google Scholar]
- Emanuele MJ, Elia AE, Xu Q, Thoma CR, Izhar L, Leng Y, Guo A, Chen YN, Rush J, Hsu PW, et al. (2011). Global identification of modular cullin-RING ligase substrates. Cell 147, 459–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English JM, Pearson G, Baer R, and Cobb MH (1998). Identification of substrates and regulators of the mitogen-activated protein kinase ERK5 using chimeric protein kinases. J Biol Chem 273, 3854–3860. [DOI] [PubMed] [Google Scholar]
- Esparis-Ogando A, Diaz-Rodriguez E, Montero JC, Yuste L, Crespo P, and Pandiella A (2002). Erk5 participates in neuregulin signal transduction and is constitutively active in breast cancer cells overexpressing ErbB2. Mol Cell Biol 22, 270285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell AS, Allen-Petersen B, Daniel CJ, Wang X, Wang Z, Rodriguez S, Impey S, Oddo J, Vitek MP, Lopez C, et al. (2014). Targeting inhibitors of the tumor suppressor PP2A for the treatment of pancreatic cancer. Mol Cancer Res 12, 924–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell AS, and Sears RC (2014). MYC degradation. Cold Spring Harb Perspect Med 4, a014365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster SA, Whalen DM, Ozen A, Wongchenko MJ, Yin J, Yen I, Schaefer G, Mayfield JD, Chmielecki J, Stephens PJ, et al. (2016). Activation mechanism of oncogenic deletion mutations in BRAF, EGFR, and HER2. Cancer Cell 29, 477–493. [DOI] [PubMed] [Google Scholar]
- Hayes TK, Neel NF, Hu C, Gautam P, Chenard M, Long B, Aziz M, Kassner M, Bryant KL, Pierobon M, et al. (2016). Long-term ERK inhibition in KRAS-mutant pancreatic cancer Is associated with MYC degradation and senescence-like growth suppression. Cancer Cell 29, 75–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang VT, Yan TJ, Cavanaugh JE, Flaherty PT, Beckman BS, and Burow ME (2017). Oncogenic signaling of MEK5-ERK5. Cancer Lett 392, 51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hydbring P, Bahram F, Su Y, Tronnersjo S, Hogstrand K, von der Lehr N, Sharifi HR, Lilischkis R, Hein N, Wu S, et al. (2010). Phosphorylation by Cdk2 is required for Myc to repress Ras-induced senescence in cotransformation. Proc Natl Acad Sci U S A 107, 58–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, et al. (2008). Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 321, 1801–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamitani T, Kito K, Nguyen HP, and Yeh ET (1997). Characterization of NEDD8, a developmentally down-regulated ubiquitin-like protein. J Biol Chem 272, 2855728562. [DOI] [PubMed] [Google Scholar]
- Knudsen ES, Balaji U, Mannakee B, Vail P, Eslinger C, Moxom C, Mansour J, and Witkiewicz AK (2018). Pancreatic cancer cell lines as patient-derived avatars: genetic characterisation and functional utility. Gut 67, 508–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Land H, Parada LF, and Weinberg RA (1983). Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature 304, 596–602. [DOI] [PubMed] [Google Scholar]
- Lin WC, Rajbhandari N, Liu C, Sakamoto K, Zhang Q, Triplett AA, Batra SK, Opavsky R, Felsher DW, DiMaio DJ, et al. (2013). Dormant cancer cells contribute to residual disease in a model of reversible pancreatic cancer. Cancer Res 73, 18211830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lito P, Pratilas CA, Joseph EW, Tadi M, Halilovic E, Zubrowski M, Huang A, Wong WL, Callahan MK, Merghoub T, et al. (2012). Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas. Cancer Cell 22, 668–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochhead PA, Clark J, Wang LZ, Gilmour L, Squires M, Gilley R, Foxton C, Newell DR, Wedge SR, and Cook SJ (2016). Tumor cells with KRAS or BRAF mutations or ERK5/MAPK7 amplification are not addicted to ERK5 activity for cell proliferation. Cell Cycle 15, 506–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martz CA, Ottina KA, Singleton KR, Jasper JS, Wardell SE, Peraza-Penton A, Anderson GR, Winter PS, Wang T, Alley HM, et al. (2014). Systematic identification of signaling pathways with potential to confer anticancer drug resistance. Sci Signal 7, ra121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matossian MD, Elliott S, Hoang VT, Burks HE, Phamduy TB, Chrisey DB, Zuercher WJ, Drewry DH, Wells C, Collins-Burow B, et al. (2017). Novel application of the published kinase inhibitor set to identify therapeutic targets and pathways in triple negative breast cancer subtypes. PLoS One 12, e0177802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrem JM, and Shokat KM (2016). Direct small-molecule inhibitors of KRAS: from structural insights to mechanism-based design. Nat Rev Drug Discov 15, 771–785. [DOI] [PubMed] [Google Scholar]
- Papke B, and Der CJ (2017). Drugging RAS: Know the enemy. Science 355, 11581163. [DOI] [PubMed] [Google Scholar]
- Pemovska T, Kontro M, Yadav B, Edgren H, Eldfors S, Szwajda A, Almusa H, Bespalov MM, Ellonen P, Elonen E, et al. (2013). Individualized systems medicine strategy to tailor treatments for patients with chemorefractory acute myeloid leukemia. Cancer Discov 3, 1416–1429. [DOI] [PubMed] [Google Scholar]
- Radulovich N, Qian JY, and Tsao MS (2008). Human pancreatic duct epithelial cell model for KRAS transformation. Methods Enzymol 439, 1–13. [DOI] [PubMed] [Google Scholar]
- Ryan MB, Der CJ, Wang-Gillam A, and Cox AD (2015). Targeting RAS-mutant cancers: is ERK the key? Trends Cancer 1, 183–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saborowski M, Saborowski A, Morris J.P.t., Bosbach B, Dow LE, Pelletier J, Klimstra DS, and Lowe SW (2014). A modular and flexible ESC-based mouse model of pancreatic cancer. Genes Dev 28, 85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleger C, Verbeke C, Hildenbrand R, Zentgraf H, and Bleyl U (2002). c-MYC activation in primary and metastatic ductal adenocarcinoma of the pancreas: incidence, mechanisms, and clinical significance. Mod Pathol 15, 462–469. [DOI] [PubMed] [Google Scholar]
- Simoes AE, Rodrigues CM, and Borralho PM (2016). The MEK5/ERK5 signalling pathway in cancer: a promising novel therapeutic target. Drug Discov Today 21, 16541663. [DOI] [PubMed] [Google Scholar]
- Song C, Wang L, Xu Q, Wang K, Xie D, Yu Z, Jiang K, Liao L, Yates JR, Lee JD, et al. (2017). Targeting BMK1 impairs the drug resistance to combined inhibition of BRAF and MEK1/2 in melanoma. Sci Rep 7, 46244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soucek L, Whitfield J, Martins CP, Finch AJ, Murphy DJ, Sodir NM, Karnezis AN, Swigart LB, Nasi S, and Evan GI (2008). Modelling Myc inhibition as a cancer therapy. Nature 455, 679–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soucek L, Whitfield JR, Sodir NM, Masso-Valles D, Serrano E, Karnezis AN, Swigart LB, and Evan GI (2013). Inhibition of Myc family proteins eradicates KRasdriven lung cancer in mice. Genes Dev 27, 504–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, Wei X, Kesavan K, Garrington TP, Fan R, Mei J, Anderson SM, Gelfand EW, and Johnson GL (2003). MEK kinase 2 and the adaptor protein Lad regulate extracellular signal-regulated kinase 5 activation by epidermal growth factor via Src. Mol Cell Biol 23, 2298–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unal EB, Uhlitz F, and Bluthgen N (2017). A compendium of ERK targets. FEBS Lett 591, 2607–2615. [DOI] [PubMed] [Google Scholar]
- Walz S, Lorenzin F, Morton J, Wiese KE, von Eyss B, Herold S, Rycak L, Dumay-Odelot H, Karim S, Bartkuhn M, et al. (2014). Activation and repression by oncogenic MYC shape tumour-specific gene expression profiles. Nature 511, 483–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters AM, and Der CJ (2018). KRAS: the critical driver and therapeutic target for pancreatic cancer. Cold Spring Harb Perspect Med 8, a031435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whatcott CJ, Diep CH, Jiang P, Watanabe A, LoBello J, Sima C, Hostetter G, Shepard HM, Von Hoff DD, and Han H (2015). Desmoplasia in primary tumors and metastatic lesions of pancreatic cancer. Clin Cancer Res 21, 3561–3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav B, Pemovska T, Szwajda A, Kulesskiy E, Kontro M, Karjalainen R, Majumder MM, Malani D, Murumagi A, Knowles J, et al. (2014). Quantitative scoring of differential drug sensitivity for individually optimized anticancer therapies. Sci Rep 4, 5193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q, Deng X, Lu B, Cameron M, Fearns C, Patricelli MP, Yates JR 3rd, Gray NS, and Lee JD (2010). Pharmacological inhibition of BMK1 suppresses tumor growth through promyelocytic leukemia protein. Cancer Cell 18, 258–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.