

Increasing evidence supports the critical role of active stromal adipocytes in breast cancer development and spread. However, the mediators and the mechanisms of action are still elusive.
KEYWORDS: IL-8, STAT3, adipocytes, breast cancer, reparixin
ABSTRACT
Increasing evidence supports the critical role of active stromal adipocytes in breast cancer development and spread. However, the mediators and the mechanisms of action are still elusive. We show here that cancer-associated adipocytes (CAAs) isolated from 10 invasive breast carcinomas are proinflammatory and exhibit active phenotypes, including higher proliferative, invasive, and migratory capacities compared to their adjacent tumor-counterpart adipocytes (TCAs). Furthermore, all CAAs secreted higher level of interleukin-8 (IL-8), which is critical in mediating the paracrine procarcinogenic effects of these cells. Importantly, ectopic expression of IL-8 in TCA cells activated them and enhanced their procarcinogenic effects both in vitro, in a STAT3-dependent manner, and in vivo. In contrast, inhibition of the IL-8 signaling using specific short hairpin RNA, anti-IL-8 antibody, or reparixin suppressed the active features of CAAs, including their non-cell-autonomous tumor-promoting activities both on breast luminal cells and in orthotopic tumor xenografts in mice. IL-8 played also an important role in enhancing the proangiogenic effects of breast adipocytes. These results provide clear indication that IL-8 plays key roles in the activation of breast CAAs and acts as a major mediator for their paracrine protumorigenic effects. Thus, targeting CAAs by inhibiting the IL-8 pathway could have great therapeutic value.
INTRODUCTION
Obesity is correlated with highly aggressive tumors and higher mortality rates for the majority of malignancies, including breast cancer. Indeed, increased inflammation, angiogenesis, hypoxia, and changes in endocrine factors are very frequent in obese females. Furthermore, several studies have shown that adipocytes adjacent to cancer cells, or cancer-associated adipocytes (CAAs), exhibit different molecular and cellular features and can increase the proliferation and invasion capacities of cancer cells via conferring metabolic advantages (1, 2). In addition, most cancer cells express receptors for adipocyte-secreted adipokines, which play major roles in tumor onset and spread in a paracrine fashion (3).
Leptin (LEP product) is a major oncogenic adipokine, which activates several downstream cancer-promoting pathways, including JAK2/STAT3, mitogent-activated protein kinase (MAPK), and phosphatidylinositol 3-kinase (PI3K)/AKT (4). Based on its autocrine, endocrine, and paracrine effects, leptin acts at different levels of breast tumorigenesis, from initiation and primary tumor growth to metastatic progression. Leptin can also affect breast tumor microenvironment through promoting angiogenesis and recruiting immune cells such as macrophages and monocytes (5, 6).
Interleukin-8 (IL-8), another key inflammatory adipokine, is a member of the CXC chemokine family, which plays important roles in breast tumorigenesis, and has important prognostic and/or predictive values. The expression of IL-8, which is under the control of nuclear factor κB (7), correlates well with angiogenesis and metastatic potential of several solid tumors in various animal models (8–11). In breast cancer, IL-8 is most commonly overexpressed in estrogen receptor-negative tumors, and the serum levels of IL-8 correlate well with advanced disease (12). IL-8 secreted by tumor cells potentiates tumor progression through inducing the epithelial-to-mesenchymal transition (EMT) process (13). The IL-8 function is mainly mediated through its association with specific cell surface G protein-coupled receptors CXCR-1 and CXCR-2 (14). Considering the presence of both IL-8 receptors in cancer cells, IL-8 could act as an autocrine motility and growth factor, providing tumors with additional growth and progression advantages. Furthermore, stress- and drug-induced IL-8 signaling enhances the resistance of cancer cells to various chemotherapeutic agents (11). Thereby, targeting the IL-8 signaling could constitute a powerful therapeutic approach.
Based on the well-known link between obesity/adipocytes, IL-8, and breast cancer, we sought in the present report to investigate the role of IL-8 in activating breast adipocytes and in promoting their paracrine procarcinogenic effects. We have shown that CAAs from breast cancer are active and express higher levels of IL-8 than their adjacent tumor-counterpart adipocytes (TCAs). Interestingly, whereas ectopic expression of IL-8 transactivated TCAs to active adipocytes, IL-8 downregulation in CAAs “normalized” these adipocytes and repressed their procarcinogenic effects.
RESULTS
Cancer-associated adipocytes display features of active adipocytes.
We started the present study by delineating various molecular and cellular features of CAAs relative to their TCAs. To this end, adipose tissues were collected from 10 human invasive ductal carcinomas of different subtypes, which were obtained from patients with various body mass index (BMI) values (Table 1). Adipose tissues were dissociated, and preadipocytes were obtained and cultured (CAAs). We also isolated the corresponding preadipocyte populations taken from histologically normal regions of the same breasts at least 2 cm away from the outer tumor margins (TCAs). CAAs and TCAs were both differentiated into mature adipocytes. Whole-cell extracts were prepared, and specific antileptin antibody, as a marker of active adipocytes (15), was utilized for immunoblotting with anti-glyceraldehyde-3-phosphate dehydrogenase (anti-GAPDH) antibody as an internal control. Figure 1A, upper panel, shows that the level of leptin was higher in 8 of 10 CAAs (80%) than in their corresponding TCAs. However, the leptin levels were similar in the two CAA/TCA pairs in patient 2 (P2) and P3. In addition, a great interindividual variation in the leptin expression was observed between the various CAAs and also TCAs (Fig. 1A, upper panel). Similarly, the levels of IL-1β, IL-8, and NF-κB (p65) were also higher in all CAAs (100%) compared to their corresponding TCAs, with interindividual variations (Fig. 1A, upper panel). Figure 1A, lower panel, shows a clear difference in the expression of both IL-8 and NF-κB (p65) in TCA cells compared to their counterpart CAAs. This shows the proinflammatory status of CAA cells compared to TCA cells.
TABLE 1.
Patient BMI and pathological characteristics
| Patient | BMI | ER | PR | Her2neu |
|---|---|---|---|---|
| P1 | 37 | + | + | – |
| P2 | 33.9 | – | – | + |
| P3 | 22.1 | + | + | – |
| P4 | 25.9 | + | + | – |
| P5 | 30.1 | + | + | – |
| P6 | 24.1 | + | – | – |
| P7 | 42.4 | – | – | + |
| P8 | 22.7 | + | + | – |
| P9 | 35.1 | + | – | – |
| P10 | 31.4 | + | + | – |
FIG 1.


Mature breast cancer-associated adipocytes are active. (A) Whole-cell lysates were prepared from the indicated mature adipocytes and utilized for immunoblotting with antibodies against the indicated proteins. (Lower panel, left) Quantification of leptin expression levels. (Right) Box plot showing IL-8 and NF-κB (p65) expression levels in the indicated types of breast adipocytes. (B and C) Total RNA was prepared from the indicated adipocytes and utilized to assess the levels of the CXCL8 and RELA mRNAs by qRT-PCR using GAPDH as a reference gene. Error bars represent means ± the SD, and values reflect three independent experiments. Upper panels show the difference in the expression of individual transcripts between CAA and TCA cells isolated from the same patients, as indicated. Lower panels show the difference between all the CAAs compared to their corresponding TCAs. (D) ChIP assay. Chromatin was purified from the indicated adipocytes and then immunoprecipitated with the indicated antibodies. Subsequently, the CXCL8 promoter bearing the NF-κB-p65 binding site was amplified by qPCR using specific primers. The GAPDH promoter was used as an unlinked locus control, and the abundance of the promoter was plotted relative to GAPDH after normalization of each sample against its own input. These experiments were performed three times, and error bars represent means ± the SD. *, P ≤ 4.1 × 10−5. (E) The indicated CAAs and TCAs were seeded in SFM in the upper wells of the CIM plates, and the migration/invasion abilities were assessed by the RTCA-xCELLigence system. For proliferation, adipocytes were seeded in complete medium in the E-plate, and the proliferation rate was assessed by the RTCA-xCELLigence system. (F) SFCM from four subtypes of CAAs and their corresponding TCA cells were utilized to assess the secreted levels of IL-8 by ELISA. Error bars represent means ± the SD; values were determined from three different experiments. *, P ≤ 0.04.
Next, the levels of the CXCL8 and RELA mRNAs were assessed in the same cells by quantitative RT-PCR (qRT-PCR). Figure 1B and C show a clear increase in the CXCL8 and RELA mRNA levels in all CAAs (100%) compared to their adjacent TCAs. This shows that the expression of the CXCL8 and RELA mRNA reflects the corresponding proteins in all TCA/CAA pairs (Fig. 1B and C), indicating that IL-8 and NF-κB upregulation is due, at least in part, to an increase in the level of their corresponding transcripts.
The expression of IL-8 is primarily regulated by the NF-κB-mediated transcriptional activity (7). Therefore, to elucidate the molecular mechanism that underlies the IL-8 mRNA upregulation in CAAs, we assessed the binding of NF-κB to the CXCL8 promoter and the effect of this binding in vivo by the chromatin immunoprecipitation (ChIP) assay. To this end, the TCA/CAA1, -2, and -5 pairs were utilized to prepare chromatin, and the NF-κB–DNA and the H3-DNA complexes were pulled down using anti-NF-κB (p65) antibody. Anti-IgG was used as a negative control. The CXCL8 promoter was then amplified by qPCR. The GAPDH promoter served as an unlinked locus control. Although the CXCL8 promoter was robustly amplified when ChIP assayed with anti-NF-κB and anti-H3 antibodies in CAA cells, this amplification was decreased in TCA cells (Fig. 1F). However, whereas no amplification product was found with anti-IgG, the GAPDH promoter was amplified only when chromatin was immunoprecipitated with anti-H3 antibody (Fig. 1D). Thus, the elevated level of the CXCL8 mRNA in CAA cells is due to the binding of the NF-κB protein to its promoter, which enhanced its expression compared to the TCA counterparts.
Next, we assessed the migration/invasion and proliferation abilities of four subtypes of CAAs—CAA1, -2, -3, and -5—and their corresponding TCAs using the xCelligence system. Figure 1E shows that all four CAAs migrate/invade and proliferate more efficiently than TCAs. Interestingly, the levels of both basal and active forms of the AKT and STAT3 proteins, the downstream effectors of the IL-8 signaling (16), were higher in all (100%) CAAs than in TCAs (Fig. 1A, upper panel). These findings suggested the possible active status of CAAs.
Furthermore, serum-free conditioned media (SFCM) were collected from CAA1, -2, -3, and -5 and their corresponding TCAs, and the level of the secreted IL-8 protein was assessed by enzyme-linked immunosorbent assay (ELISA). Figure 1F shows that CAAs secrete higher levels of IL-8 than their corresponding TCA cells.
CAA cells enhance EMT in normal breast luminal cells in an IL-8-dependent fashion.
Next, we examined the potential paracrine procarcinogenic influence of CAAs on normal breast luminal cells (LeaL-10). Therefore, the four CAA and TCA pairs were incubated for 48 h, and the resulting SFCM were applied onto LeaL-10 cells for 24 h (serum-free medium [SFM] was used as a negative control). Figure 2A shows that CAA1- and CAA2-SFCM enhanced the proliferation and the migration/invasion abilities of LeaL-10 cells relative to the slight effect of TCA1- and TCA2-SFCM compared to SFM. In addition, while the expression level of total AKT was only slightly affected, SFCM from four CAA cells activated this protein kinase compared to SFM (Fig. 2B). However, SFCM from four TCA cells did not activate AKT (Fig. 2B). This suggests that CAA cells can enhance the proliferation, migration, and invasion capacities of normal breast luminal cells. Similar results were obtained for the breast cancer MCF-7 cells (Fig. 2C).
FIG 2.


CAAs promote EMT in normal breast luminal cells in an IL-8-dependent manner. LeaL-10 cells were exposed to SFM or SFCM from four CAAs and their four corresponding TCA cells for 24 h. (A and C) Migration/invasion and proliferation capabilities were assessed for the indicated pairs. The graphs are representative of different experiments. (B and D) Cell lysates were prepared from the indicated cells and used for immunoblotting. (E) LeaL-10 cells were grown in 3D cultures and then either exposed to two CAAs and their two corresponding TCAs or SFM. Total RNA was prepared and used to assess the level of the indicated transcripts by qRT-PCR. Error bars represent means ± the SD, and values were drawn from three different experiments. (F) LeaL-10 cells were treated as for panel E, cytospun, and used for immunofluorescence. Scale bars, 50 µm. (G) LeaL-10 cells were exposed to SFM or SFCM from CAA1 containing either anti-IL-8, antileptin, anti-IL-8 plus antileptin, or anti-IgG antibodies and then utilized to assess migration/invasion and proliferation capabilities. The graphs are representative of different experiments. (H) LeaL-10 cells were treated as indicated, and then whole-cell lysates were prepared and used to assess the levels of the indicated proteins by immunoblotting. For the immunoblots, the numbers below the bands indicate the corresponding protein expression levels relative to GAPDH. The levels of phosphorylated proteins were normalized against the total amounts of the respective nonphosphorylated proteins.
The exposure of LeaL-10 cells to the four CAA-SFCM strongly reduced the levels of the epithelial proteins E-cadherin and EpCAM, while they potently increased the levels of the mesenchymal markers N-cadherin and Twist-1 relative to SFM (Fig. 2B). On the other hand, the effects of SFCM from the corresponding TCA cells were only minimal (Fig. 2B). Similar results were obtained for the breast cancer MCF-7 cells (Fig. 2D).
Next, we sought to mimic the in vivo situation, wherein a basement membrane separates the epithelia from stromal cells. To this end, LeaL-10 cells were cultured for 48 h in 3 dimensions (3D) in a network containing a mesh (basement membrane simulator), which separated cells from CAAs, TCAs, or SFM. Real-time RT-PCR shows significant reduction in the expression of E-cadherin and clear upregulation of N-cadherin and vimentin in cells cultured with CAAs relative to controls (Fig. 2C). However, the levels of these EMT-related transcripts were only slightly affected in cells cultured with the corresponding TCAs relative to control cells (SFM) (Fig. 2C). This shows that CAA cells secrete factors that can cross the basement membrane and promote gene expression changes in luminal cells, even when cultured in 3D. To confirm these findings, LeaL-10 cells cultured in 3D and treated as described above were collected and used to assess the EMT-related proteins by immunofluorescence. Figure 2D shows that while LeaL-10 cells cultured with TCAs stained positive for E-cadherin and EpCAM relative to controls, the levels of these proteins decreased when LeaL-10 cells were exposed to CAA-SFCM. However, the level of vimentin increased when LeaL-10 cells were exposed to CAA-SFCM relative to SFM but was not affected in cells cultured in the presence of TCA-SFCM (Fig. 2D). Together, these findings indicate that CAA cells enhance the procarcinogenic/metastatic EMT process in normal breast luminal cells in a paracrine manner, which confirms their active status.
Next, we investigated the possible implication of IL-8 in the paracrine pro-EMT effect of CAA cells on luminal cells. To this end, SFCM from CAA1 and CAA2 cells were challenged with anti-IL-8 and antileptin neutralizing antibodies either separately or in combination, using IgG as control, and then applied onto LeaL-10 cells for 24 h. Figure 2E shows that the inhibition of IL-8, leptin, or both in SFCM collected from CAA1 suppressed their promigratory/invasiveness, as well as proliferative effects on LeaL-10 cells. In addition, the EMT-triggering effect of CAA-SFCM was normalized by inhibiting IL-8 or leptin, with no further inhibition when both neutralizing antibodies were combined in both CAA1- and CAA2-SFCM (Fig. 2F). This shows that, like leptin, IL-8 also mediates the paracrine procarcinogenic effects of CAA cells.
IL-8 upregulation activates breast stromal adipocytes.
To show the implication of IL-8 in the activation of breast stromal adipocytes, IL8ORF was introduced into TCA1 and TCA3 cells, while empty vector was used as a control (TCA1-IL8ORF, TCA3-IL8ORF, TCA1-Ctrl, and TCA3-Ctrl, respectively). Figure 3A shows that the level of the IL-8 protein increased in TCA-IL8ORF cells compared to their corresponding control TCA-Ctrl cells, reaching a level similar to that observed in CAA cells. Likewise, the secreted level of IL-8 was also increased upon ectopic expression of IL-8 in TCA cells (Fig. 3B). Interestingly, as in CAA cells, IL-8 upregulation was accompanied by strong increase in the levels of JAK2/P-JAK2, STAT3/P-STAT3, and AKT/P-AKT, as well as leptin, while the level of the p16 protein was reduced, compared to control cells (Fig. 3A). Furthermore, IL-8 upregulation enhanced the migration, invasion, and proliferation abilities of TCA1 cells (Fig. 3C). This suggests that IL-8 upregulation activates TCA cells.
FIG 3.

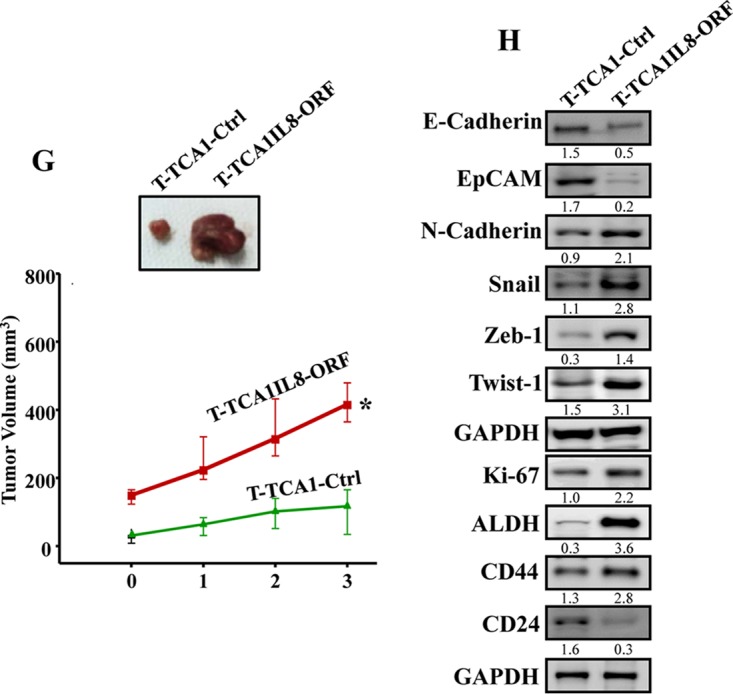
IL-8 upregulation activates breast stromal adipocytes. (A) TCA1 and TCA3 cells were transfected with a plasmid bearing IL-8 ORF (TCA1-IL8ORF and TCA3-IL8ORF, respectively; an empty plasmid was used as the control, TCA1-Ctrl and TCA3-Ctrl, respectively), and cell lysates were prepared and utilized for immunoblotting, using CAA1 and CAA3 as controls, respectively. (B) SFCM were collected from the indicated cells and utilized to assess the levels of secreted IL-8 by ELISA. Error bars represent the means ± the SD of three different experiments. *, P = 0.018. (C) Same as for Fig. 1F. (D and E) LeaL-10 cells were exposed to SFM, SFCM from TCA1-IL8ORF, or TCA1-Ctr for 24 h and then were utilized to assess migration/invasion and proliferation capabilities (D) (the graphs are representative of different experiments) and to prepare whole-cell lysates to assess the levels of the indicated proteins by immunoblotting (E). (F) LeaL-10 cells were transfected with STAT3 siRNA and then exposed to SFM, SFCM from TCA1-IL8ORF, or TCA1-Ctrl for 24 h and were then utilized to prepare whole-cell lysates to assess the levels of the indicated proteins by immunoblotting. (G) Breast cancer xenografts were created by coinjecting MDA-MB-231 cells with TCA1-IL8ORF or TCA1-Ctrl cells (n = 4/each group) under the nipples of nude mice. (Upper panel) Representative tumor size. (Lower panel) Graph showing time-dependent tumor growth. Error bars represent the means ± the SD, with values from four mice. *, P= 0.014. (H) Tumors were excised, and whole-tissue lysates were prepared and utilized for immunoblotting. For the immunoblots, the numbers below the bands indicate the corresponding protein expression levels relative to GAPDH. The levels of phosphorylated proteins were normalized against the total amounts of the respective nonphosphorylated proteins.
Next, we sought to investigate the paracrine effects of TCA1-IL8ORF on normal breast luminal cells. To this end, SFCM were collected and then applied onto LeaL-10 cells for 24 h, using SFM as a negative control. Figure 3D shows that the migration, invasion, and proliferation capacities of LeaL-10 cells were enhanced in the presence of TCA1-IL8ORF-SFCM compared to TCA1-Ctrl-SFCM (Fig. 3D). This was confirmed by showing IL-8-dependent upregulation of the proliferation marker Ki-67, as well as strong activation of the migration/invasion proteins AKT and STAT3 (Fig. 3E). In addition, SFCM from TCA1-IL8ORF upregulated the mesenchymal markers (N-cadherin, Twist-1, and ZEB1), whereas it downregulated the epithelial markers (E-cadherin and EpCAM) compared to the SFCM from controls (Fig. 3E). This indicates that IL-8-expressing adipocytes trigger EMT in luminal cells in a paracrine fashion.
IL-8-related paracrine effects of breast stromal adipocytes is STAT3 dependent.
In order to delineate the molecular pathway implicated in the IL-8-dependent paracrine effect of breast stromal adipocytes, we sought to evaluate the effect of STAT3 knockdown using specific small interfering RNA (siRNA) on such process in luminal cells. Figure 3F, upper panel, shows a 3-fold decrease in the levels of both basal and phosphorylated forms of STAT3 upon transfection of Leal-10 cells with STAT3-siRNA. Interestingly, TCA1-IL8ORF-SFCM did not induce EMT in STAT3-deficient luminal cells relative to controls. Indeed, STAT3-siRNA prevented the downregulation of epithelial markers and the upregulation of mesenchymal markers in Leal-10 luminal cells (Fig. 3F, lower panel). This indicates that the IL-8-dependent paracrine effect of breast stromal adipocytes on luminal cells is mediated through STAT3 signaling.
We have next created orthotopic breast cancer xenografts by coinjecting MDA-MB-231 cells with TCA1-IL8ORF or TCA1-Ctrl (n = 4/each group) under nude mice nipples. Although all animals developed tumors 2 weeks postinjection, tumors bearing TCA1-IL8ORF cells (T-TCA1-IL8ORF) were much bigger and reached a volume 3.3-fold larger than those containing TCA1-Ctrl cells (T-TCA1-Ctrl) (Fig. 3G). This shows that IL-8 augments the paracrine protumorigenic activity of breast stromal adipocytes in vivo.
Next, whole-cell lysates were prepared from the two tumor groups, and the levels of various EMT/stemness-related proteins were assessed by immunoblotting. Figure 3H shows that while T-TCA1-IL8ORF tumors expressed lower levels of the epithelial markers E-cadherin and EpCAM, the levels of the mesenchymal proteins N-cadherin, Snail, Zeb-1, and Twist-1 were much higher in these tumors compared to controls. Furthermore, the levels of the AKT/phospho-AKT and Ki-67 proteins were higher in T-TCA1-IL8ORF tumors relative to T-TCA1-Ctrl tumors (Fig. 3H).
The induction of EMT promotes stem cell features (17). Therefore, we decided to assess the levels of the major mammary cancer stem cell markers ALDH, CD44, and CD24. Figure 3H shows that T-TCA1-IL8ORF tumors expressed elevated levels of ALDH1 and CD44 proteins and lower level of the CD24 protein relative to T-TCA1-Ctrl tumors. This indicates that IL-8 upregulation in TCAs increases their paracrine capacity in enhancing cancer stem cell attributes in breast cancer cells in vivo.
IL-8 knockdown suppresses active breast stromal adipocytes.
Next, we sought to investigate the effect of IL-8 knockdown on the status of CAAs and their potential “normalization.” Thereby, mature CAAs were transfected with specific shRNA against IL-8 (four different sequences), and a scrambled sequence was used as negative control. Whole-cell extracts were then prepared from the generated cells (CAA1-IL8sh1, -IL8sh2, -IL8sh3, and -IL8sh4 and CAA1-Ctrl, respectively), and the level of the IL-8 protein was assessed by immunoblotting using whole-cell lysates from the corresponding TCA1 cells as a control. Figure 4A shows a clear decrease in the level of the IL-8 protein in CAA1-IL8sh3 and CAA1-IL8sh4 cells compared to controls, reaching a level similar to that observed in TCA1 cells. The level of the secreted IL-8 was also decreased in CAA1-IL8sh3 and CAA1-IL8sh4 cells compared to controls (Fig. 4B). In addition, upon downregulation of IL-8 in CAA1 cells, the migration/invasion and proliferation capabilities of these cells decreased compared to control cells and became similar to their TCA1 counterparts (Fig. 4C). Furthermore, the levels of AKT/P-AKT and STAT3/P-STAT3, as well as leptin, were also reduced in CAA1-IL8sh3 and CAA1-IL8sh4 cells compared to control cells, reaching levels similar to those observed in TCA1 cells (Fig. 4D). This implies that IL-8 downregulation suppresses the proliferative and migratory/invasive abilities of CAAs. Since downregulation of IL-8 in CAA1 significantly reduced their proliferative ability, we assessed the level of the proliferative marker PCNA, as well as the cell cycle regulator proteins p16 and p53. Figure 4D shows that while the levels of p16 and p53 increased in CAA1-IL8sh3 and CAA1-IL8sh4 cells, the levels of PCNA decreased compared to controls. These results indicate that IL-8 downregulation repressed CAA cells.
FIG 4.

IL-8 downregulation suppresses the activity features of CAA. (A) CAA1 cells were separately transfected with four different specific IL-8-shRNAs (CAA1-IL8sh1, CAA1-IL8sh2, CAA1-IL8sh3, and CAA1-IL8sh4), as well as a control plasmid bearing a scrambled sequence (CAA1-Ctrl), and then cell lysates were prepared and used for immunoblotting. (B) SFCM collected from the indicated cells were utilized to assess the levels of secreted IL-8 by ELISA. Error bars represent means ± the SD, and the values represent three independent experiments. *, P ≤ 0.000246. (C) Same as for Fig. 1F. (D) Cell lysates were prepared from the indicated cells and used for immunoblotting analysis. For the immunoblots, the numbers below the bands indicate the corresponding protein expression levels relative to GAPDH. The levels of phosphorylated proteins were normalized against the total amounts of the respective nonphosphorylated proteins. (E) LeaL-10 cells were exposed to SFM or SFCM from the indicated cells and then were utilized to assess the migration/invasion and proliferation capabilities by the RTCA-xCELLigence system. The graphs are representative of different experiments.
Next, we sought to investigate the paracrine effects of IL-8 downregulation in CAA cells on normal breast luminal cells. Figure 4E shows that the migration, invasion, and proliferation capacities of LeaL-10 cells were reduced in the presence of CAA1-IL8sh3-SFCM compared to CAA1-Ctrl-SFCM. In addition, whereas SFCM from CAA1-Ctrl cells induced the EMT process, SFCM from CAA1-IL8sh3 cells had an effect similar to SFM (Fig. 3E). Similar results were obtained for the proliferative marker Ki-67 and the proinvasive kinase P-AKT (Fig. 3E). This indicates that IL-8 downregulation suppresses the procarcinogenic effects of CAA cells.
Inhibition of CXCR1/2 persistently suppresses active adipocytes.
To further confirm the role of IL-8, the autocrine binding of IL-8 to its receptors was inhibited by treating CAA1 cells with serial concentrations of reparixin, a noncompetitive CXCR1/2 dual inhibitor (18). Therefore, CAA1 cells were either sham treated or challenged with different doses of reparixin (0, 50, and 100 nM) for 24 h, using anti-IL-8 neutralizing antibody as a control. Cells were then harvested, and whole-cell lysates were prepared for immunoblotting analysis using specific antibodies. Figure 5A shows dose-dependent downregulation of IL-8, reaching a level similar to that obtained with neutralizing anti-IL-8 antibody, compared to nontreated cells. In addition, similar to the effect of anti-IL-8, the levels of STAT3/P-STAT3 and P-AKT were also strongly decreased in response to reparixin (Fig. 5A). The migration/invasion and the proliferative capabilities of CAA1 cells were also decreased after treatment with reparixin and anti-IL-8 antibody (Fig. 5B). This indicates that targeting IL-8 receptors by treatment with reparixin inhibits the activity of CAA cells.
FIG 5.

Inhibition of CXCR1/2 persistently suppresses the CAA activity. (A) CAA1 cells were treated with different doses of reparixin for 24 h, and anti-IL-8 neutralizing antibody was used as a positive control. Cell lysates were prepared and utilized to assess the level of the indicated proteins using specific antibodies. The numbers below the bands indicate the corresponding protein expression levels relative to GAPDH. The levels of phosphorylated proteins were normalized against the total amounts of the respective nonphosphorylated proteins. (B) CAA1 cells were treated as described above and seeded in SFM in the upper wells of the CIM plates, and the migration/invasion abilities were assessed. For proliferation, pretreated CAA1 cells were seeded in complete medium in the E-plate, and the proliferation rate was assessed by the RTCA-xCELLigence system. (C) SFCM were collected from the indicated cells (either 24 h posttreatment [24 h] or 72 h postsplit [split]) and then utilized to assess the levels of secreted IL-8 by ELISA. Error bars represent means ± the SD of three different experiments. *, P ≤ 0.018. (D and E) Total RNA was prepared from the indicated adipocytes (either 72 h postsplit [split] or after the fourth split [split-4]) and then utilized to assess the levels of the indicated transcripts by qRT-PCR. Error bars represent means ± the SD, and the values represent three independent experiments. *, P ≤ 0.0015. (F) LeaL-10 cells were exposed to SFM or SFCM from CAA1 cells treated either with reparixin or anti-IL-8 antibody, and total RNA was prepared and used to assess the levels of the indicated transcripts by qRT-PCR. Error bars represent means ± the SD, and values represent three different experiments. *, P ≤ 0.0037.
Next, SFCM were collected from CAA1 treated with reparixin or anti-IL-8 antibody as described above, and then the level of secreted IL-8 was measured by ELISA. Figure 5C shows a dose-dependent decrease in the level of secreted IL-8, reaching a level similar to that obtained with anti-IL-8 relative to the nontreated cells.
We next sought to test whether the effect of reparixin is persistent. To this end, CAA1 cells were treated as described above for 24 h, and then reparixin- and anti-IL-8-containing media were removed, and the cells were washed three times with PBS. The cells were incubated in fresh medium for 24 h and then split and reincubated in fresh medium for another 72 h. Subsequently, SFCM were collected, and the cells were utilized to assess the level of the secreted IL-8 by ELISA. Figure 5C shows that, compared to 24-h treatment, the reduced level of secreted IL-8 was sustained for 72 h after splitting in fresh medium. In addition, the levels of the CXCL8 and LEP mRNAs remained also downregulated after splitting, whereas the CDKN2A mRNA remained upregulated (Fig. 5D). Similar results were obtained when cells were reparixin treated and then split four times in the absence of the drug (Fig. 5E). These data indicate the sustained effect of reparixin in suppressing CAA cell activity.
Next, the expression levels of the EMT-related genes were also assessed in LeaL-10 cells exposed to SFCM from CAA1 treated with either reparixin or anti-IL-8 neutralizing antibody for 24 h. Figure 5F shows that whereas SFCM from mock-treated CAA1 cells increased the levels of the mesenchymal markers N-cadherin, SNAI-1, TWIST-1, and ZEB1 mRNAs and decreased the levels of the epithelial marker E-cadherin mRNA relative to their levels in SFM-treated Leal-10 cells, SFCM from reparixin- or anti-IL-8-treated CAA1 decreased the levels of the mesenchymal markers N-cadherin, SNAI-1, TWIST-1, and ZEB1 mRNAs and increased the levels of the epithelial marker E-cadherin mRNA (Fig. 5F). These results indicate that the procarcinogenic effects of CAA can be permanently repressed by reparixin by downregulating IL-8.
IL-8 activates the IL-6/NF-κB/Lin28B epigenetic feedback loop to maintain the active status of breast stromal adipocytes.
To elucidate the molecular mechanism underlying the persistent effect of IL-8 inhibition on CAA, we tested the possible inhibition of the IL-6/NF-κB/Lin28B loop, which has been recently shown to play a major role in maintaining the active status of active breast cancer-associated fibroblasts (19). To this end, the total RNA prepared above was utilized to assess the levels of the components of this loop in reparixin- or anti-IL-8-treated CAA1 cells that were split four times. Figure 5E shows that the levels of the IL-6, RELA, and Lin28B mRNAs remained decreased in CAA1-treated cells compared to nontreated cells. In addition, the level of the Let-7b miRNA, which has been recently shown to be a member of the IL-6/NF-κB/Lin28B positive-feedback loop (20), was upregulated in reparixin- and anti-IL-8-treated CAA1 cells compared to controls (Fig. 5E). This indicates that transient IL-8 inhibition permanently inhibited the IL-6/NF-κB/Lin28B positive-feedback loop in CAA cells, which suggests the implication of this loop in the persistent active status of CAAs. The level of Lin28B protein was also assessed in all mature CAAs and TCAs. Indeed, the level of the Lin28B protein was higher in all CAAs (100%) compared to their corresponding TCAs (Fig. 1A). Together, these results show that IL-6/NF-κB/Lin28B positive-feedback loop is active in breast cancer-associated adipocytes and is responsible for the persistent active status of these cells.
CAA cells stimulate breast cancer xenograft growth in mice in an IL-8-dependent manner.
To elucidate the effects of IL-8 inhibition in vivo, orthotopic breast cancer xenografts were created by coinjecting MDA-MB-231 cells with CAA1-Ctrl, CAA1-IL8sh3, or CAA1 pretreated with 100 ng of reparixin for 24 h (CAA1-Rep) (n = 4/each group). Although all mice coinjected with MDA-MB-231 and adipocytes developed tumors by 2 weeks after injection, tumors containing CAA1-IL8sh3 cells (T-CAA1-IL8sh3) or CAA1 pretreated with reparixin (T-CAA1-Rep) were smaller than those containing CAA1-Ctrl cells (T-CAA1-Ctrl) (Fig. 6A). Furthermore, T-CAA1-IL8sh3 and T-CAA1-Rep tumors grew much slower than T-CAA1-Ctrl tumors (Fig. 6A). These results show that CAAs promote breast cancer formation in vivo in an IL-8-dependent manner.
FIG 6.
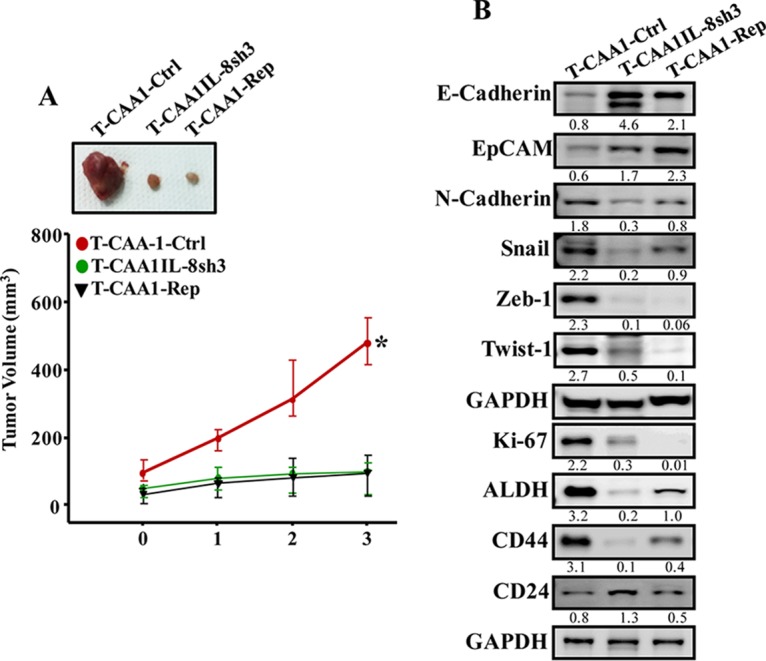
CAA cells stimulate breast cancer xenograft growth in mice in an IL-8-dependent manner. (A) Breast cancer xenografts were created by coinjecting MDA-MB-231 cells with CAA1-Ctrl, CAA1-IL8sh3 or CAA1 pretreated with reparixin (100 ng) for 24 h (CAA1-Rep) (n = 4/each group) under the nipples of nude mice. (Upper panel) Representative tumor sizes. (Lower panel) Time-dependent tumor growth. Error bars represent means ± the SD, and the values are derived from four mice. *, P = 0.014. (B) Tumors were excised, and whole tissue lysates were prepared and utilized for immunoblotting. The numbers below the bands indicate the corresponding protein expression levels relative to GAPDH.
To further elucidate the role of IL-8 in the paracrine procarcinogenic effects of CAA cells on tumor growth, whole-cell extracts were prepared from the three tumor groups, and the levels of various EMT-related proteins were assessed by immunoblotting. Figure 6B shows that while the levels of the epithelial markers E-cadherin and EpCAM were much higher in T-CAA-IL8sh3 and T-CAA1-Rep tumors compared to T-CAA-Ctrl tumor, the levels of the mesenchymal N-cadherin, Snail, Zeb-1, and Twist-1 proteins were much lower in T-CAA1-IL8sh3 and T-CAA1-Rep tumors relative to T-CAA1-Ctrl tumors. In addition, the total levels of the AKT protein and its phosphorylated form, as well as the level of the Ki-67 protein, were lower in T-CAA1-IL8sh3 and T-CAA1-Rep tumors compared to T-CAA1-Ctrl tumors (Fig. 6B), which explains the lower growth of T-CAA1-IL8sh3 and T-CAA1-Rep compared to T-CAA1-Ctrl tumor. This also shows that the pro-EMT effects of CAA cells are IL-8 related in vivo as well.
Next, we assessed the levels of the main mammary cancer stem cell markers ALDH, CD44, and CD24 by immunoblotting. Figure 6B shows that T-CAA1-IL8sh3 and T-CAA1-Rep tumors expressed low levels of the ALDH1 and CD44 proteins and higher levels of the CD24 protein compared to T-CAA1-Ctrl tumor. This indicates that IL-8 downregulation in CAAs decreased their paracrine induction of stemness (stem cell features) in orthotopic tumor xenografts.
Active adipocytes induce angiogenesis in vivo in an IL-8-dependent manner.
To study the paracrine effect of IL-8-expressing adipocytes on vascular formation in vivo, the level of CD34, an endothelial cell marker, was assessed in the orthotopic tumor xenografts by immunohistochemistry using anti-CD34 antibody. Figure 7A shows a higher density of CD34+ cells in tumors containing TCA1-IL8ORF cells compared to tumors mixed with TCA1-Ctrl cells. Interestingly, the assessment of the averaged microvascular density in all tumor xenografts showed 2.25-fold-higher levels of blood vessels in tumor containing TCA1-IL8ORF cells compared to T-TCA1-Ctrl cells (Fig. 7B). This shows the proangiogenic role of IL-8 in adipocytes in vivo. To explore the molecular mechanism(s) that underlies the proangiogenic effect of TCA1-IL8ORF cells, we assessed the level of proangiogenic proteins in the two tumors by immunoblotting. Figure 7C shows that the levels of hypoxia-inducible factor 1α (HIF-1α), vascular endothelial growth factor A (VEGF-A), and mTOR/P-mTOR, as well as IL-8, were increased in T-TCA1-IL8ORF compared to T-CAA1-Ctrl tumors. These results show IL-8-related proangiogenic role of breast stromal adipocytes in vivo through an increase in the expression of angiogenic factors in a paracrine manner. To further confirm this, the level of CD34 was assessed in tumors containing CAA1-IL8sh3 or CAA1-Rep by immunohistochemistry. Figure 7D shows lower density of CD34+ cells in tumors containing CAA1-IL8sh3 or CAA1-Rep cells than in tumors containing CAA1-Ctrl cells. Interestingly, the assessment of the averaged microvascular density in all tumor xenografts showed 3.6- and 18-fold-lower levels of blood vessels in T-CAA1-IL8sh3 and T-CAA1-Rep tumors, respectively, than in T-CAA1-Ctrl tumors (Fig. 7E). In addition, the levels of HIF-1α, VEGF-A, and mTOR/P-mTOR, as well as IL-8, were decreased in T-CAA1-IL8sh3 and T-CAA1-Rep tumors compared to T-CAA1-Ctrl tumors. Together, these data show the role of IL-8 in promoting the paracrine proangiogenic effect of breast stromal adipocytes.
FIG 7.

CAA cells induce angiogenesis in vivo in an IL-8-depednent manner. (A and D) FFPE tissues were prepared from the indicated tumors, and immunohistochemistry was carried out with anti-CD34 antibody. Scale bars, 50 µm. (B and E) Histograms show the average numbers of CD34+ microvessels observed in five different fields from the different tumors. Error bars represent means ± the SD. *, P ≤ 0.0013. (C and F) Same as in Fig. 3H and 6B. The numbers below the bands indicate the corresponding protein expression levels relative to GAPDH. The levels of phosphorylated proteins were normalized against the total amount of the respective nonphosphorylated proteins. (G) SFM or SFCM from the indicated adipocytes were collected and were added independently on HUVEC cells plated on Matrigel (96-well plate), and the differentiation into capillary-like structures was assessed after 5 h of incubation. Representative photographs of HUVEC cavities are shown. Scale bars, 30 µm. (H) A histogram shows the average numbers of microvessels counted in five different fields. Error bars represent means ± the SD. *, P ≤ 1.19 × 10−6.
Next, we sought to investigate the paracrine effect of IL-8-expressing adipocytes on endothelial cell differentiation in vitro. To this end, SFCM collected from CAA1-Ctrl, CAA1-IL8sh3, CAA1-Rep, TCA1-Ctrl, and TCA1-IL8ORF cells were added separately to 96-well plates seeded with 2 × 104 human umbilical vein endothelial cells (HUVECs) in Matrigel and used for in vitro angiogenic assay. SFM was also added as negative control. After 5 h of incubation, the number of closed cavity constructions was significantly lower in the presence of SFCM from CAA1-IL8sh3 and CAA1-Rep cells compared to SFCM from CAA1-Ctrl cells. However, SFCM from TCA1-IL8ORF significantly increased the number of closed cavity constructions compared to SFCM from TCA1-Ctrl or SFM cells (Fig. 7G and H).
DISCUSSION
The breast ductal epithelial cells are immersed in a fatty environment, and during the invasive stages of breast carcinogenesis, adipocytes come into close contact with cancer cells (21–23). Thus, adipocytes may play important roles in both the onset and the spread of breast tumors. In the present report, we have shown that breast cancer-associated adipocytes show several tumor-specific features compared to their adjacent counterparts isolated from histologically normal part of the same breast. Like tumor cells, CAAs exhibited high proliferation rate and had strong migratory and invasive capabilities. Furthermore, all CAAs expressed high levels of the cancer/inflammatory transcription factor NF-κB, as well as several of its proinflammatory targets such as leptin, IL-8, and IL-1β. In fact, the binding of NF-κB (p65) to the CXCL8 promoter was found to be more pronounced in CAAs compared to their respective TCAs. This indicates that CAAs have proinflammatory potential and thus can promote tumorigenesis. Indeed, we have shown that CAAs secrete high level of IL-8, and media conditioned with CAAs promoted the procarcinogenic EMT process in normal breast luminal cells in an IL-8/leptin-dependent manner. Together, these findings have clearly shown that breast cancer-associated adipocytes are active stromal cells that possess potent procarcinogenic potential. The presence of modified adipocytes that exhibit phenotypic changes and express high levels of matrix metalloproteinase (MMP-11), PAI-1, IL-6, IL-1β, and tumor necrosis factor alpha (TNF-α) has been previously shown by immunohistochemistry and qRT-PCR in breast tumors (1). Furthermore, breast cancer-associated adipose tissue has been shown to promote F-actin remodeling, cellular scattering, invasiveness, and spheroid reorganization of breast cancer cells in vitro (24).
In addition to their protumorigenic effects, active CAAs may also modulate tumor response to various therapeutics. Indeed, it has been shown that tumor-surrounding adipocytes have role in fostering a radioresistant phenotype in breast tumors (25). In addition, a recent study has shown that adipocytes actively catabolize the chemotherapeutic agent daunorubicin to a therapeutically less effective metabolite, daunorubicinol (26).
The observed differences between CAAs and their corresponding TCAs suggest a role of cancer cells in the activation of CAA cells. In fact, several lines of evidence suggest the ability of neoplastic cells in affecting their microenvironment and modulating gene expression in the surrounding stromal cells, either directly through secreted factors or indirectly via persistent proliferative stress (27–29). We have recently shown that IL-6, secreted from highly invasive breast cancer cells, activates breast stromal fibroblasts in a paracrine manner (30). Dirat et al. (1) have also shown that coculturing adipocytes with cancer cells altered their phenotype, decreased the levels of adipocyte markers, and promoted an activated state characterized by the overexpression of proteases such as MMP-11 and the proinflammatory cytokines IL-6 and IL-1β, as well as Ptx3 (31).
Importantly, CAA cells, like cancer-associated fibroblasts, remained active despite their separation from tumor cells and their propagation in vitro. This could result from genetic and/or epigenetic changes. Indeed, we have recently shown that cancer-associated fibroblasts remain active owing to the induction of the IL-6/STAT3/NF-κB positive-feedback loop (19). Since CAAs express active STAT3, AKT, NF-κB, and Lin28B, it is plausible that this epigenetic inflammatory loop is also responsible for the persistent activity of CAAs. Indeed, transient inhibition of the NF-κB and STAT3 target IL-8 (32) by reparixin blocked this signaling circuit through potent and sustained inhibition of IL-6, RELA, and Lin28B, as well as increased the level of Let-7b for several passages and also reverted active adipocytes to a normalized state (Fig. 5). This suggests that IL-8 mediates the positive IL-6/STAT3/NF-κB feedback loop-dependent activation of cancer-associated adipocytes, which is responsible for their sustained active status, and that reversion of the active status of CAAs by disrupting this feedback loop could be of great therapeutic value. Furthermore, we have found that Lin28B, which acts mainly as an oncogene (33), is highly expressed in 100% of CAAs relative to their respective TCAs. This suggests that Lin28B upregulation may play a major role in the activation of breast stromal adipocytes. In addition, the increase in the levels of NF-κB in CAAs may constitute a major change in these cell populations that could explain, in great part, the significant modulation in gene expression found in our present study, as well as what has been previously reported in adipocytes cultivated in vitro with cancer cells (1).
Furthermore, we have shown that IL-8 plays a major role in the activation of breast stromal adipocytes. Although the ectopic expression of IL-8 promoted the proinflammatory/carcinogenic effects of adipocytes both in vitro and in vivo, IL-8 inhibition suppressed these effects in CAA cells. This indicates that IL-8 has both cell autonomous and non-cell-autonomous protumorigenic effects. These paracrine effects on primary breast luminal cells were mediated through activation of the STAT3 signaling, which plays a major role in breast carcinogenesis. In fact, the active/phosphorylated form of STAT3 has been found highly expressed in about 40% of all breast cancers (34).
A high level of IL-8 in the sera of breast cancer patients correlates well with early dissemination, poor disease outcome, bad prognosis, and earlier distant metastasis (12, 14). Thereby, suppressing the effects of IL-8 signaling could constitute a potent therapeutic approach through targeting cancer microenvironment. Indeed, novel therapeutics aimed at inhibiting CXCR1/2 signaling may pause the progression of the disease in tumors driven by IL-8 and other CXCR1/2 ligands. We have shown here that targeting breast adipocytes CXCR1/2 by reparixin treatment deactivates CAAs and blocks their IL-8-dependent procarcinogenic effects both in vitro and in vivo. This provides a proof of principle that the active status of CAAs can be normalized, which also indicates the possible targeting of these procarcinogenic cells for more efficient treatment of breast cancer.
Similarly, coculturing adipocytes with breast cancer cells has been shown to enhance their migratory and invasive abilities in an IL-6-dependent fashion (35, 36). In addition, inhibition of adipocyte-secreted IL-6 with specific antibody reduced the proliferation, migration, and invasion abilities of breast cancer cells and reduced the expression of mesenchymal markers, including TWIST1 and SNAIL. However, an increase in the level of secreted IL-6 from adipocytes cultured with cancer cells promotes angiogenesis through the upregulation of STAT3 and VEGF, as well as Src activation. This leads to the upregulation of Sox2, c-Myc, and Nanog, which results in the emergence of stem cell traits (37). These studies indicate that, like IL-8, adipocyte-secreted IL-6 also plays important roles in the interaction between adipocytes and breast cancer cells.
In addition, we have shown that IL-8 enhances the proangiogenic effect of breast stromal adipocytes in orthotopic breast tumor xenografts. Although ectopic expression of IL-8 increased the number of microvessels in tumors, IL-8 inhibition with specific shRNA or with reparixin inhibited the formation of these vessels (Fig. 7). This could be mediated through the proangiogenic protein VEGF-A, since the expression level of this protein in tumor cells was modulated in an IL-8-dependent manner. This IL-8-dependent paracrine upregulation of VEGF-A could result from the activation of its upstream effectors mTOR and HIF-1α, whose levels were affected by the IL-8 status. In addition, a high level of IL-8 can directly promote angiogenesis in breast tumors (38); when secreted from breast cancer cells, IL-8 cooperates with VEGF-A to establish and enhance tumor neovascularization (14). On the other hand, IL-8 downregulation significantly reduced the microvessel density in breast tumors in vivo (39). These results suggest the possible use of reparixin to suppress CAA-dependent induction of angiogenesis through inhibiting the IL-8 signaling either directly or indirectly through VEGF-A. As a case in point, a reducing tumor cell proliferation, angiogenesis, apoptosis, and metastasis have been shown in CXCR-negative mice relative to wild-type animals (40).
In summary, the present data indicate that breast cancer-associated adipocytes exhibit active and proinflammatory features, in addition to cancer-promoting functions. Importantly, whereas IL-8 upregulation activates breast stromal adipocytes, the inhibition of IL-8 signaling normalizes the active phenotypes of these stromal cells. Therefore, targeting CAAs via suppression of the IL-8 signaling pathway could enhance the efficiency of breast cancer therapeutic approaches.
MATERIALS AND METHODS
Tissue collection.
Breast cancer (invasive ductal carcinoma) specimens were collected from primary tumors of 10 patients who underwent lumpectomy at King Faisal Specialist Hospital and Research Centre. Signed informed consent was obtained from all patients under Research Ethical Committee Project RAC#2140017. Processing of tissues was performed after routine examination by certified anatomical pathologist using hematoxylin-eosin-stained sections.
Generation of primary adipocytes and luminal cells from breast tissues.
The obtained tissues were immediately digested with collagenase to separate preadipocytes from the stroma vascular fraction, as previously described (41). Preadipocytes were first cultured in special medium (PAGM) containing fetal calf serum (0.05 ml/ml), endothelial cell growth supplement (0.004 ml/ml), epidermal cell growth factor (10 ng/ml), hydrocortisone (1 μg/ml), and heparin (90 μg/ml) and used until reaching confluence. Preadipocytes were then differentiated into mature adipocytes by replacement of the preadipocyte medium with serum-free preadipocytes differentiation medium, supplemented with d-biotin (8 μg/ml), insulin (0.5 μg/ml), dexamethasone (400 ng/ml), IBMX (44 μg/ml), l-thyroxine (9 ng/ml), and ciglitazone (3 μg/ml). Mature adipocytes were then cultured using adipocyte nutrition medium containing fetal calf serum (0.03 ml/ml), d-biotin (8 μg/ml), insulin (0.5μg/ml), and dexamethasone (400 ng/ml). All supplements were purchased from PromoCell GmbH, Germany. Cancer-associated adipocytes were derived from tumor areas of the samples, while tumor-counterpart adipocytes were developed from histologically noncancerous region of the breast at least 2 cm away from the outer tumor margin.
Normal breast luminal cells (LeaL-10) were developed from a tissue collected from reduction mammoplasty and were cultured as previously described (42). Signed informed consent was obtained from all patients under Research Ethical Committee Project RAC#2140017.
Cell lines, cell culture, and chemicals.
MDA-MB-231 and MCF-7 cells were obtained from the American Type Culture Collection (Manassas, VA) and were cultured according to the instructions of the company. Human CXCL8/IL-8 antagonist (MAB208), human super leptin antagonist (MBS400098), and IgG were purchased from MyBioSource. Blocking antibodies were used at 2.5 µg/ml. Reparixin was purchased from Cayman Chemical.
Cellular lysate preparation and immunoblotting.
Cellular lysate preparation and immunoblotting were performed as previously described (43). Antibodies directed against E-cadherin (4E2), N-cadherin, AKT (C73H10), P-AKT (Thr308), EpCAM (VU1D9), NF-κB (p65), STAT3, P-STAT3-Tyr705 (D3A7), JAK2 (D2E12)XP, P-JAK2 (Tyr1007/1008), Lin28B (D4H1), HIF-1α (H1α67), mTOR (7C10), P-mTOR (Ser2448), and Snail (C15D3) were purchased from Cell Signaling Technology (Danvers, MA). GAPDH (FL-335) was obtained from Santa Cruz (USA). p16 was from BD Biosciences (San Jose, CA). Leptin, vimentin (RV202), Twist-1, IL-8, Ki-67, VEGF-A, and IL-1β were from Abcam (Cambridge, MA). ZEB-1 (4C4) was obtained from Abnova (Taipei, Taiwan). p53 (DO-1), ALDH1/2 (H-85), and CD24 (C-20) were obtained from Santa Cruz Biotech (Dallas, TX), and CD44 was obtained from Sigma-Aldrich (St. Louis, MO).
RNA purification and qRT-PCR.
Total RNA was purified using an RNeasy minikit (Qiagen, UK) according to the manufacturer’s instructions and treated with RNase-free DNase. Then, 1 µg of RNA was used to synthesize cDNA utilizing an Advantage RT-PCR kit (Clontech Laboratories, Mountain View, CA). Quantitative RT-PCR was performed in triplicate using 4 µl of cDNA mixed with 2× FastStart Essential DNA Green qPCR Mastermix (Roche, New York, NY) and 0.3 µM concentrations of forward and reverse primers. Amplifications were performed utilizing a LightCycler 96 real-time PCR detection system (Roche) with the following cycle conditions: 95°C for 10 min (1 cycle), followed by 95°C for 10 s, 59°C for 20 s, and 72°C for 30 s (45 cycles). GAPDH expression levels were used for normalization, and gene expression differences were calculated using the threshold cycle (CT). The obtained values were plotted as means ± the standard deviations (SD). Three independent experiments were performed for each reaction (see Table 2 for the sequences of the primers).
TABLE 2.
Sequence of the primers utilized for qRT-PCR
| Gene or promoter | Sequence (5′–3′) |
|
|---|---|---|
| Forward | Reverse | |
| GAPDH | GAGTCCACTGGCGTCTTC | GGGGTGCTAAGCAGTTGGT |
| CDKN2A | CAACGCACCGAATAGTTACG | CAGCTCCTCAGCCAGGTC |
| LEP | TTTGGCCCTATCTTTTCTATGTCC | TGGAGGAGACTGACTGCGT |
| CXCL8 | GATCCACAAGTCCTTGTTCCA | GCTTCCACATGTCCTCACAA |
| E-cadherin | CCA GAA ACG GAG GCC TGA T | CTG GGA CTC CAC CTA CAG AAA GTT |
| N-cadherin | CCT CCA GAG TTT ACT GCC ATG AC | GTA GGA TCT CCG CCA CTG ATT C |
| Vimentin gene | TTC CAA ACT TTT CCT CCC TGA ACC | TCA AGG TCA TCG TGA TGC TGA G |
| ZEB1 | GGC AGA GAA TGA GGG AGA AG | CTT CAG ACA CTT GCT CAC TAC TC |
| Twist-1 | GGA CAA GCT GAG CAA GAT TCA GA | GTG AGC CAC ATA GCT GCA G |
| Snail | CCT CAA GAT GCA CAT CCG AAG | ACA TGG CCT TGT AGC AGC CA |
| RELA | CACGATGGTGACCTCCTTCT | GCAGGTGCGTTTCTATGACA |
| Lin28B | CCTGTTTAGGAAGTGAAAGAAGAC | CACTTCTTTGGCTGAGGAGGTAG |
| IL-6 | AGACAG CCA CTC ACC TCT TCA G | TTC TGC CAG TGC CTC TTT GCT G |
| Mature Let-7b | TGAGGTAGTAGGTTGTGTGGTT | |
| U6 | CGCAAGGATGACACGCAAATT | |
| IL-8 promoter (ChIP) | CAT CAG TTG CAA ATC GTG GA | TGC ACC CTC ATC TTT TCA TT |
| GAPDH promoter (ChIP) | TACTAGCGGTTTTACGGGCG | TCGAACAGGAGGAGCAGAGAGCGA |
Transfection/viral infection.
pGFP-C-sh-IL-8-Lenti, pLenti-C-Myc-DDK-CXCL8 (expressing IL-8; Origene), and their control plasmids were used at 1 µg/ml each for the transfection of 293FT cells. Lentiviral supernatants were collected at 48 h posttransfection. Culture media were removed from the target cells and replaced with the lentiviral supernatant, followed by incubation for 24 h in the presence of 1 µg/ml Polybrene (Sigma-Aldrich). Transduced cells were selected after 48 h with puromycin.
STAT3-siRNA transfection.
Signal silence STAT3-siRNA and control-siRNA were obtained from Cell Signaling Technology. The transfections were carried out using the High Perfect reagent (Qiagen), as recommended by the manufacturer. Cells were incubated for 3 days after transfection, recovered, and then were recultured for another 3 days before collection for subsequent experiments.
ChIP.
Cells (106) were treated with 2% formaldehyde for 10 min at room temperature to cross-link the transcription factor to DNA. The cross-linking was terminated by addition of glycine (0.125 M). After a wash with phosphate-buffered saline, the cells were collected and resuspended in sodium dodecyl sulfate (SDS) lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris [pH 8.1]) with protease inhibitors. The sonicated lysate was processed using a ChIP assay kit according to the manufacturer’s instructions (Cell Signaling Technology). ChIP experiments were performed using antibodies against NF-κB (p65) (1C12; Cell Signaling Technology), IgG, and H3. The input was an aliquot of sheared chromatin before immunoprecipitation, which represents the starting genomic DNA that was used to normalize the sample to the amount of chromatin added to each ChIP. Immunoprecipitated chromatin was analyzed by qPCR using a GAPDH promoter as the unlinked locus control. Each experiment was performed three times. The sequence of the primers used for the ChIP assay can be found in Table 2.
Immunofluorescence.
Cells were fixed with 4% paraformaldehyde for 10 min, permeabilized with 0.2% Triton X-100 for 10 min, and quenched in 100 mM glycine for 10 min at room temperature. The cells were then blocked in 10% fetal calf serum, incubated with primary antibody, washed, and incubated with the Alexa Fluor 594- or Alexa Fluor 488-conjugated antibodies (Life Technologies, Carlsbad, CA). Nuclei were stained with DAPI (4′,6′-diamidino-2-phenylindole;Vector Laboratories, Inc.), and images were captured using a Floid cell imaging station (Life Technologies). Antibodies against Ki-67 and vimentin (RV202) were purchased from Abcam (Cambridge, MA); antibodies against EpCam (D13B) and E-cadherin (HECD-1) were purchased from Cell Signaling Technology (Danvers, MA).
Immunohistochemistry.
Formalin-fixed/paraffin-embedded (FFPE) tissues were utilized for immunohistochemistry as previously described (44). Tissues were incubated overnight with anti-CD34 antibody (Abcam) at a dilution of 1:500, followed by peroxidase-conjugated antibody (Dako). Sites of antibody binding were visualized based on the deposition of a brown polymer of 3,3′-diaminobenzidine chromogen (Novocastra Laboratories, Ltd., Buffalo Grove, IL).
Cell migration, invasion, and proliferation.
These assays were performed in a label-free real-time settings using xCELLigence RTCA technology (Roche, Germany) that measures impedance changes in a meshwork of interdigitated gold microelectrodes located at the well bottom (E-plate) or at the bottom side of a microporous membrane (CIM plate 16) (45, 46). Cell migration and invasion were assessed according to the manufacturer’s instructions. In brief, 2 × 104 cells in SFM were added to the upper wells of the CIM plate coated with a thin layer of Matrigel (BD Biosciences) basement membrane matrix diluted 1:20 in SFM (invasion) or noncoated (migration). Complete medium was used as a chemoattractant in the lower chambers. Subsequently, the plates were incubated in the RTCA for 24 h, and the impedance value of each well was automatically monitored by the xCELLigence system and expressed as the cell index value, which represents the cell status based on the measured electrical impedance change divided by a background value. Each assay was performed in triplicate.
For the proliferation assay, the differentiated preadipocytes in culture (exponentially growing cells [2 × 104]) were seeded in an E-plate with complete medium according to the manufacturer’s instructions. Cell proliferation was assessed for 48 h. All data were recorded and analyzed by the RTCA software. The cell index was used to measure the change in the electrical impedance divided by background value, which represents cell status. Each assay was biologically performed in triplicate (47).
Conditioned media.
Cells were cultured in SFM for 48 h, and then the media were collected and centrifuged. Conditioned media were used either immediately or were frozen at –80°C until needed.
ELISA.
ELISA was performed according to the manufacturer’s instructions. IL-8 was obtained from Abcam (Cambridge, MA). An optical density at 450 nm was used with a standard ELISA plate reader (Bio-Rad, Hercules, CA). These experiments were performed in triplicate.
Tumor xenografts.
Animal experiments were approved by the KFSH&RC Institutional Animal Care and Use Committee and were conducted according to relevant national and international guidelines. Female nude mice were randomized into different groups, and breast cancer xenografts were created by coimplantation of the MDA-MB-231 cells (2 × 106) with adipocytes (4 × 106) under the nipple. The growth of the tumors was then monitored weekly. Tumor size was measured with a caliper according to the formula “length × width × height.” At the end of the experiments, the animals were sacrificed, and tumors were excised and weighed.
Quantification of protein expression level.
The protein signal intensity of each band was determined using ImageQuant TL software (GE Healthcare). Next, dividing the obtained value of each band by the value of the corresponding internal control allowed a correction of the loading differences.
Statistical analysis.
Statistical analysis was performed using GraphPad software. The results are presented as means ± the SD of at least three independent experiments. The statistical significance was determined using a two-tailed Student t test, and P values of ≤0.05 were considered statistically significant.
ACKNOWLEDGMENTS
We thank Ibrahim S. Al-Mssallem for great help and support.
This study was performed under RAC proposal 2140017 and was supported in part by a KACST grant under the National Comprehensive Plan for Science and Technology (KACST 14-BIO159-20).
REFERENCES
- 1.Dirat B, Bochet L, Dabek M, Daviaud D, Dauvillier S, Majed B, Wang YY, Meulle A, Salles B, Le Gonidec S, Garrido I, Escourrou G, Valet P, Muller C. 2011. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res 71:2455–2465. doi: 10.1158/0008-5472.CAN-10-3323. [DOI] [PubMed] [Google Scholar]
- 2.Wang YY, Attane C, Milhas D, Dirat B, Dauvillier S, Guerard A, Gilhodes J, Lazar I, Alet N, Laurent V, Le Gonidec S, Biard D, Herve C, Bost F, Ren GS, Bono F, Escourrou G, Prentki M, Nieto L, Valet P, Muller C. 2017. Mammary adipocytes stimulate breast cancer invasion through metabolic remodeling of tumor cells. JCI Insight 2:e87489. doi: 10.1172/jci.insight.87489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Duong MN, Geneste A, Fallone F, Li X, Dumontet C, Muller C. 2017. The fat and the bad: mature adipocytes, key actors in tumor progression and resistance. Oncotarget 8:57622–57641. doi: 10.18632/oncotarget.18038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cirillo D, Rachiglio AM, la Montagna R, Giordano A, Normanno N. 2008. Leptin signaling in breast cancer: an overview. J Cell Biochem 105:956–964. doi: 10.1002/jcb.21911. [DOI] [PubMed] [Google Scholar]
- 5.Ando S, Catalano S. 2011. The multifactorial role of leptin in driving the breast cancer microenvironment. Nat Rev Endocrinol 8:263–275. doi: 10.1038/nrendo.2011.184. [DOI] [PubMed] [Google Scholar]
- 6.Newman G, Gonzalez-Perez RR. 2014. Leptin-cytokine crosstalk in breast cancer. Mol Cell Endocrinol 382:570–582. doi: 10.1016/j.mce.2013.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Elliott CL, Allport VC, Loudon JA, Wu GD, Bennett PR. 2001. Nuclear factor-κB is essential for up-regulation of interleukin-8 expression in human amnion and cervical epithelial cells. Mol Hum Reprod 7:787–790. doi: 10.1093/molehr/7.8.787. [DOI] [PubMed] [Google Scholar]
- 8.Huang S, Mills L, Mian B, Tellez C, McCarty M, Yang XD, Gudas JM, Bar-Eli M. 2002. Fully humanized neutralizing antibodies to interleukin-8 (ABX-IL8) inhibit angiogenesis, tumor growth, and metastasis of human melanoma. Am J Pathol 161:125–134. doi: 10.1016/S0002-9440(10)64164-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Inoue K, Slaton JW, Kim SJ, Perrotte P, Eve BY, Bar-Eli M, Radinsky R, Dinney CP. 2000. Interleukin 8 expression regulates tumorigenicity and metastasis in human bladder cancer. Cancer Res 60:2290–2299. [PubMed] [Google Scholar]
- 10.Inoue K, Slaton JW, Eve BY, Kim SJ, Perrotte P, Balbay MD, Yano S, Bar-Eli M, Radinsky R, Pettaway CA, Dinney CP. 2000. Interleukin 8 expression regulates tumorigenicity and metastases in androgen-independent prostate cancer. Clin Cancer Res 6:2104–2119. [PubMed] [Google Scholar]
- 11.Waugh DJ, Wilson C. 2008. The interleukin-8 pathway in cancer. Clin Cancer Res 14:6735–6741. doi: 10.1158/1078-0432.CCR-07-4843. [DOI] [PubMed] [Google Scholar]
- 12.Benoy IH, Salgado R, Van Dam P, Geboers K, Van Marck E, Scharpe S, Vermeulen PB, Dirix LY. 2004. Increased serum interleukin-8 in patients with early and metastatic breast cancer correlates with early dissemination and survival. Clin Cancer Res 10:7157–7162. doi: 10.1158/1078-0432.CCR-04-0812. [DOI] [PubMed] [Google Scholar]
- 13.Fernando RI, Castillo MD, Litzinger M, Hamilton DH, Palena C. 2011. IL-8 signaling plays a critical role in the epithelial-mesenchymal transition of human carcinoma cells. Cancer Res 71:5296–5306. doi: 10.1158/0008-5472.CAN-11-0156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu Q, Li A, Tian Y, Wu JD, Liu Y, Li T, Chen Y, Han X, Wu K. 2016. The CXCL8-CXCR1/2 pathways in cancer. Cytokine Growth Factor Rev 31:61–71. doi: 10.1016/j.cytogfr.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Al-Khalaf HH, Amir M, Al-Mohanna F, Tulbah A, Al-Sayed A, Aboussekhra A. 2017. Obesity and p16INK4A downregulation activate breast adipocytes and promote their protumorigenicity. Mol Cell Biol 37:e00101-37. doi: 10.1128/MCB.00101-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Buettner C, Pocai A, Muse ED, Etgen AM, Myers MG Jr, Rossetti L. 2006. Critical role of STAT3 in leptin’s metabolic actions. Cell Metab 4:49–60. doi: 10.1016/j.cmet.2006.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, Yang J, Weinberg RA. 2008. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133:704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bertini R, Allegretti M, Bizzarri C, Moriconi A, Locati M, Zampella G, Cervellera MN, Di Cioccio V, Cesta MC, Galliera E, Martinez FO, Di Bitondo R, Troiani G, Sabbatini V, D’Anniballe G, Anacardio R, Cutrin JC, Cavalieri B, Mainiero F, Strippoli R, Villa P, Di Girolamo M, Martin F, Gentile M, Santoni A, Corda D, Poli G, Mantovani A, Ghezzi P, Colotta F. 2004. Noncompetitive allosteric inhibitors of the inflammatory chemokine receptors CXCR1 and CXCR2: prevention of reperfusion injury. Proc Natl Acad Sci U S A 101:11791–11796. doi: 10.1073/pnas.0402090101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hendrayani SF, Al-Harbi B, Al-Ansari MM, Silva G, Aboussekhra A. 2016. The inflammatory/cancer-related IL-6/STAT3/NF-κB positive feedback loop includes AUF1 and maintains the active state of breast myofibroblasts. Oncotarget 7:41974–41985. doi: 10.18632/oncotarget.9633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Al-Harbi B, Hendrayani SF, Silva G, Aboussekhra A. 2018. Let-7b inhibits cancer-promoting effects of breast cancer-associated fibroblasts through IL-8 repression. Oncotarget 9:17825–17838. doi: 10.18632/oncotarget.24895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Andarawewa KL, Motrescu ER, Chenard MP, Gansmuller A, Stoll I, Tomasetto C, Rio MC. 2005. Stromelysin-3 is a potent negative regulator of adipogenesis participating to cancer cell-adipocyte interaction/crosstalk at the tumor invasive front. Cancer Res 65:10862–10871. doi: 10.1158/0008-5472.CAN-05-1231. [DOI] [PubMed] [Google Scholar]
- 22.Tan J, Buache E, Chenard MP, Dali-Youcef N, Rio MC. 2011. Adipocyte is a non-trivial, dynamic partner of breast cancer cells. Int J Dev Biol 55:851–859. doi: 10.1387/ijdb.113365jt. [DOI] [PubMed] [Google Scholar]
- 23.Cozzo AJ, Fuller AM, Makowski L. 2017. Contribution of adipose tissue to development of cancer. Compr Physiol 8:237–282. doi: 10.1002/cphy.c170008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lapeire L, Hendrix A, Lambein K, Van Bockstal M, Braems G, Van Den Broecke R, Limame R, Mestdagh P, Vandesompele J, Vanhove C, Maynard D, Lehuede C, Muller C, Valet P, Gespach CP, Bracke M, Cocquyt V, Denys H, De Wever O. 2014. Cancer-associated adipose tissue promotes breast cancer progression by paracrine oncostatin M and Jak/STAT3 signaling. Cancer Res 74:6806–6819. doi: 10.1158/0008-5472.CAN-14-0160. [DOI] [PubMed] [Google Scholar]
- 25.Bochet L, Meulle A, Imbert S, Salles B, Valet P, Muller C. 2011. Cancer-associated adipocytes promotes breast tumor radioresistance. Biochem Biophys Res Commun 411:102–106. doi: 10.1016/j.bbrc.2011.06.101. [DOI] [PubMed] [Google Scholar]
- 26.Sheng X, Parmentier JH, Tucci J, Pei H, Cortez-Toledo O, Dieli-Conwright CM, Oberley MJ, Neely M, Orgel E, Louie SG, Mittelman SD. 2017. Adipocytes sequester and metabolize the chemotherapeutic daunorubicin. Mol Cancer Res 15:1704–1713. doi: 10.1158/1541-7786.MCR-17-0338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Polyak K, Haviv I, Campbell IG. 2009. Co-evolution of tumor cells and their microenvironment. Trends Genet 25:30–38. doi: 10.1016/j.tig.2008.10.012. [DOI] [PubMed] [Google Scholar]
- 28.Arendt LM, Rudnick JA, Keller PJ, Kuperwasser C. 2010. Stroma in breast development and disease. Semin Cell Dev Biol 21:11–18. doi: 10.1016/j.semcdb.2009.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu M, Polyak K. 2008. Molecular characterization of the tumour microenvironment in breast cancer. Eur J Cancer 44:2760–2765. doi: 10.1016/j.ejca.2008.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hendrayani SF, Al-Khalaf HH, Aboussekhra A. 2014. The cytokine IL-6 reactivates breast stromal fibroblasts through transcription factor STAT3-dependent up-regulation of the RNA-binding protein AUF1. J Biol Chem 289:30962–30976. doi: 10.1074/jbc.M114.594044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee J, Hong BS, Ryu HS, Lee HB, Lee M, Park IA, Kim J, Han W, Noh DY, Moon HG. 2017. Transition into inflammatory cancer-associated adipocytes in breast cancer microenvironment requires microRNA regulatory mechanism. PLoS One 12:e0174126. doi: 10.1371/journal.pone.0174126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Freund A, Jolivel V, Durand S, Kersual N, Chalbos D, Chavey C, Vignon F, Lazennec G. 2004. Mechanisms underlying differential expression of interleukin-8 in breast cancer cells. Oncogene 23:6105–6114. doi: 10.1038/sj.onc.1207815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Viswanathan SR, Powers JT, Einhorn W, Hoshida Y, Ng TL, Toffanin S, O’Sullivan M, Lu J, Phillips LA, Lockhart VL, Shah SP, Tanwar PS, Mermel CH, Beroukhim R, Azam M, Teixeira J, Meyerson M, Hughes TP, Llovet JM, Radich J, Mullighan CG, Golub TR, Sorensen PH, Daley GQ. 2009. Lin28 promotes transformation and is associated with advanced human malignancies. Nat Genet 41:843–848. doi: 10.1038/ng.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Al Zaid Siddiquee K, Turkson J. 2008. STAT3 as a target for inducing apoptosis in solid and hematological tumors. Cell Res 18:254–267. doi: 10.1038/cr.2008.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Walter M, Liang S, Ghosh S, Hornsby PJ, Li R. 2009. Interleukin 6 secreted from adipose stromal cells promotes migration and invasion of breast cancer cells. Oncogene 28:2745–2755. doi: 10.1038/onc.2009.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nieman KM, Romero IL, Van Houten B, Lengyel E. 2013. Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochim Biophys Acta 1831:1533–1541. doi: 10.1016/j.bbalip.2013.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Picon-Ruiz M, Pan C, Drews-Elger K, Jang K, Besser AH, Zhao D, Morata-Tarifa C, Kim M, Ince TA, Azzam DJ, Wander SA, Wang B, Ergonul B, Datar RH, Cote RJ, Howard GA, El-Ashry D, Torné-Poyatos P, Marchal JA, Slingerland JM. 2016. Interactions between adipocytes and breast cancer cells stimulate cytokine production and drive Src/Sox2/miR-302b-mediated malignant progression. Cancer Res 76:491–504. doi: 10.1158/0008-5472.CAN-15-0927. [DOI] [PubMed] [Google Scholar]
- 38.De Larco JE, Wuertz BR, Rosner KA, Erickson SA, Gamache DE, Manivel JC, Furcht LT. 2001. A potential role for interleukin-8 in the metastatic phenotype of breast carcinoma cells. Am J Pathol 158:639–646. doi: 10.1016/S0002-9440(10)64005-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yao C, Lin Y, Chua MS, Ye CS, Bi J, Li W, Zhu YF, Wang SM. 2007. Interleukin-8 modulates growth and invasiveness of estrogen receptor-negative breast cancer cells. Int J Cancer 121:1949–1957. doi: 10.1002/ijc.22930. [DOI] [PubMed] [Google Scholar]
- 40.Sharma B, Nannuru KC, Varney ML, Singh RK. 2015. Host Cxcr2-dependent regulation of mammary tumor growth and metastasis. Clin Exp Metastasis 32:65–72. doi: 10.1007/s10585-014-9691-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Al-Khalaf HH, Amir M, Al-Mohanna F, Tulbah A, Al-Sayed A, Aboussekhra A. 2017. Obesity and p16INK4A downregulation activate breast adipocytes and promote their protumorigenicity. Mol Cell Biol 37:e00101–e00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ghebeh H, Sleiman GM, Manogaran PS, Al-Mazrou A, Barhoush E, Al-Mohanna FH, Tulbah A, Al-Faqeeh K, Adra CN. 2013. Profiling of normal and malignant breast tissue show CD44high/CD24low phenotype as a predominant stem/progenitor marker when used in combination with Ep-CAM/CD49f markers. BMC Cancer 13:289. doi: 10.1186/1471-2407-13-289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Al-Mohanna MA, Al-Khalaf HH, Al-Yousef N, Aboussekhra A. 2006. The p16INK4a tumor suppressor controls p21WAF1 induction in response to ultraviolet light. Nucleic Acids Res 35:223–233. Epub 2006 Dec 2007. doi: 10.1093/nar/gkl1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ghebeh H, Barhoush E, Tulbah A, Elkum N, Al-Tweigeri T, Dermime S. 2008. FOXP3+ Tregs and B7-H1+/PD-1+ T lymphocytes co-infiltrate the tumor tissues of high-risk breast cancer patients: Implication for immunotherapy. BMC Cancer 8:57. doi: 10.1186/1471-2407-8-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Knopfová L, Beneš P, Pekarčíková L, Hermanová M, Masařík M, Pernicová Z, Souček K, Smarda J. 2012. c-Myb regulates matrix metalloproteinases 1/9, and cathepsin D: implications for matrix-dependent breast cancer cell invasion and metastasis. Mol Cancer 11:15. doi: 10.1186/1476-4598-11-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jurmeister S, Baumann M, Balwierz A, Keklikoglou I, Ward A, Uhlmann S, Zhang JD, Wiemann S, Sahin O. 2012. MicroRNA-200c represses migration and invasion of breast cancer cells by targeting actin-regulatory proteins FHOD1 and PPM1F. Mol Cell Biol 32:633–651. doi: 10.1128/MCB.06212-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Adams CJ, Pike AC, Maniam S, Sharpe TD, Coutts AS, Knapp S, La Thangue NB, Bullock AN. 2012. The p53 cofactor Strap exhibits an unexpected TPR motif and oligonucleotide-binding (OB)-fold structure. Proc Natl Acad Sci U S A 109:3778–3783. doi: 10.1073/pnas.1113731109. [DOI] [PMC free article] [PubMed] [Google Scholar]