

As genetic instability drives disease or loss of cell fitness, cellular safeguards have evolved to protect the genome, especially during sensitive cell cycle phases, such as DNA replication. Fission yeast Brc1 has emerged as a key factor in promoting cell survival when replication forks are stalled or collapsed.
KEYWORDS: Brc1, Smc5-Smc6, SUMO, Rad18, replication stress
ABSTRACT
As genetic instability drives disease or loss of cell fitness, cellular safeguards have evolved to protect the genome, especially during sensitive cell cycle phases, such as DNA replication. Fission yeast Brc1 has emerged as a key factor in promoting cell survival when replication forks are stalled or collapsed. Brc1 is a multi-BRCT protein that is structurally related to the budding yeast Rtt107 and human PTIP DNA damage response factors, but functional similarities appear limited. Brc1 is a dosage suppressor of a mutation in the essential Smc5-Smc6 genome stability complex and is thought to act in a bypass pathway. In this study, we reveal an unexpectedly intimate connection between Brc1 and Smc5-Smc6 function. Brc1 is required for the accumulation of the Smc5-Smc6 genome stability complex in foci during replication stress and for activation of the intrinsic SUMO ligase activity of the complex by collapsed replication forks. Moreover, we show that the chromatin association and SUMO ligase activity of Smc5-Smc6 require the Nse5-Nse6 heterodimer, explaining how this nonessential cofactor critically supports the DNA repair roles of Smc5-Smc6. We also found that Brc1 interacts with Nse5-Nse6, as well as gamma-H2A, so it can tether Smc5-Smc6 at replicative DNA lesions to promote survival.
INTRODUCTION
The maintenance of genome stability is paramount to suppress disease-priming changes to the genetic code. DNA is particularly vulnerable to damage as it is replicated, and therefore, multiple pathways have evolved to safeguard it during this sensitive process.
The multi-BRCT domain protein Brc1 has emerged as a key factor in protecting the fission yeast genome during replication (1–3). Cells lacking Brc1 are specifically hypersensitive to genotoxins that impede replication fork progression, including methyl methanesulfonate (MMS), hydroxyurea (HU), and camptothecin (CPT). Brc1 forms foci at DNA lesions through its gamma-H2A binding C-terminal BRCT domain, and disruption of this interaction with the Brc1-T672A mutation causes sensitivity to genotoxic stress (2). In response to HU, gamma-H2A and Brc1-T672A mutations are epistatic, indicating that a major role of gamma-H2A in this context is the recruitment of Brc1 to stalled forks.
Brc1 was initially identified as a dosage suppressor of smc6-74, a hypomorphic mutation in the octameric Smc5-Smc6 genome stability complex (4). Smc5-Smc6 is related to the cohesin and condensin structural maintenance of chromosomes (SMC) complexes, and all play key roles in chromosome segregation or other DNA transactions that require large-scale dynamic manipulation of the genome (5). Whereas functions for cohesin and condensin in chromosome segregation are quite well defined, those of Smc5-Smc6 are still emerging.
Smc5-Smc6 is known to prevent the accumulation of aberrant homologous recombination (HR) structures when replication fork progress is hampered, and also during meiosis (6–15). Fission yeast Smc5-Smc6 is recruited to stalled and collapsed replication forks, where it helps promote replication restart, together with Rad52 and RPA (12, 16). The loading of Smc5-Smc6 at collapsed replication forks appears to activate the SUMO ligase activity of its Nse2 (MMS21) subunit, which is required to inhibit/resolve HR intermediates (15–18). In addition, Smc5-Smc6 is involved in the separase-independent pathway that removes cohesin from chromosome arms (19). Together, unresolved HR intermediates and persistent arm cohesion cause many of the chromosome segregation defects associated with mutations in Smc5-Smc6.
The Brc1-suppressible smc6-74 mutation is close to the Smc6 ATPase head domain and causes both sensitivity to genotoxins and reduced chromatin loading of Smc5-Smc6 (4, 20, 21). However, Brc1 overexpression does not suppress the smc6-X hinge region mutation that also causes genotoxin sensitivity but exhibits normal chromatin loading of Smc5-Smc6 (4, 20–22). Therefore, smc6-74 has specific defects that can be mitigated by increased levels of Brc1. Suppression of smc6-74 by Brc1 requires the structure-specific endonucleases Slx1/Slx4 and Mus81-Eme1 and the postreplication repair factor Rad18 (3). As a result, Brc1 is thought to act in parallel to or downstream of Smc5-Smc6, thereby reducing the need for Smc5-Smc6 in the management of stalled or collapsed replication forks.
Of note, a mutation in the Nse4 (Rad62) subunit of Smc5-Smc6 is also partially suppressed by Brc1 overexpression (23), suggesting that smc6-74 and nse4-1 could share a functional defect. Nse4 is a kleisin that bridges the ATPase head groups of Smc5-Smc6 to generate a ring-like structure and could impact loading like the smc6-74 ATPase proximal mutation (21, 24). Therefore, among other roles, we speculated that Brc1 could enhance Smc5-Smc6 chromatin association to partially compensate for smc6-74 or nse4-1 defects.
In this study, we identify Brc1 as an Smc5-Smc6 interactor and key mediator of its focal accumulation at sites of replication fork stalling or collapse. The formation of Smc5-Smc6 foci requires the gamma-H2A binding BRCTs of Brc1 (2) but not the recently identified Brc1-interacting partner, Rad18 (1). In addition, we found that Brc1 is required for the MMS-induced SUMOylation of Nse4 by the Nse2 SUMO ligase subunit of Smc5-Smc6 (16). Thus, the high local concentration of Smc5-Smc6 in Brc1-dependent foci likely contributes to the MMS-induced activation of Nse2 (16, 17).
Moreover, we show that in the absence of the Smc5-Smc6 subunits, Nse5 and Nse6, the amount of the complex on chromatin at its binding hot spots is strongly reduced (16, 25). Therefore, the Nse5-Nse6 heterodimer plays a key role in the recruitment and/or chromatin loading of Smc5-Smc6, as we had proposed earlier (25). We also found that Brc1 interacts with Nse5-Nse6 in vivo; therefore, Brc1 could tether Smc5-Smc6 at damaged replication forks via gamma-H2A to enhance focus formation, local chromatin loading, and SUMO ligase activity of the complex to promote survival.
RESULTS
Replication stress-induced Smc5-Smc6 foci are Brc1 dependent.
Replication fork stalling by HU treatment induces a single subnuclear focus of Smc5-Smc6 at the clustered heterochromatic centromeres of fission yeast (16). MMS causes DNA damage that impedes replication fork progression and induces multiple Smc5-Smc6 foci, which are often perinucleolar (16).
Brc1 is intimately associated with Smc5-Smc6 function, as (i) increased Brc1 dosage can partially suppress the genome stability defects caused by smc6-74; (ii) the deletion of Brc1 is synthetically lethal with most hypomorphic Smc5-Smc6 mutants; (iii) like Smc5-Smc6 (16), Brc1 accumulates at the pericentromere (dg-dh repeats) following HU treatment and forms MMS-induced nuclear and perinucleolar foci (2, 26); and (iv) Brc1 peptides were identified by mass spectrometry in our previous purifications of Smc5-Smc6 (4, 23, 25, 27).
We therefore asked if Brc1 influences the punctate localization of Smc5-Smc6 that is induced by HU or MMS treatment. As we previously observed, Nse4-green fluorescent protein (GFP) formed a single focus in HU-treated cells and multiple foci in MMS-treated cells (Fig. 1A and B) (16). Strikingly, however, deletion of Brc1 abolished Nse4-GFP foci under both conditions (Fig. 1A and B). This reveals an unappreciated role for Brc1 in the focal accumulation of Smc5-Smc6 at collapsed and stalled replication forks and indicates that there is a more complex interplay between Brc1 and Smc5-Smc6 than thought.
FIG 1.

Replication stress-induced Nse4 focus formation depends on Brc1 but not on Rad18 (Rhp18). (A) Live-cell microscopy of endogenous Nse4-GFP upon HU or MMS treatment. Nse4-GFP foci were present in wild-type (wt) and rhp18Δ background cells but largely absent in brc1Δ and brc1Δ rhp18Δ cells. The boxed areas are enlarged below the images. (B) Quantification of the data in panel A. Mean values and standard deviations (SD) from the results of ≥2 independent experiments are shown. (C) Expression of wild-type Brc1 from a plasmid restores Nse4-GFP focus formation, unlike the empty-plasmid control or expression of Brc1-T672A, which cannot bind gamma-H2A. vector, pREP41; pBrc, pREP41-NTAP-Brc1; pBrc1-T672A, pREP41-NTAP-Brc1-T672A. (D) Quantification of the data in panel C. Mean values and SD from the results of 3 independent experiments are shown.
Rad18 is dispensable for Smc5-Smc6 replication stress-induced foci.
The suppression of smc6-74 by increased Brc1 dosage requires a noncatalytic role of Rad18 (fission yeast Rhp18) (3, 28). In addition, Brc1 was recently shown to physically interact with Rad18 via its N-terminal BRCT cluster to maintain genome stability (1). We therefore tested if the Nse4-GFP HU/MMS-induced foci were also dependent on Rad18 by generating an Nse4-GFP rad18Δ strain. Interestingly, Rad18 was not required for Nse4-GFP focus formation under either condition (Fig. 1A and B). Of note, because rad18Δ cells are sensitive to MMS, similarly to brc1Δ cells (28), this result also indicates that lesion amplification in brc1Δ cells cannot account for their defective Smc5-Smc6 focus formation.
As rad18Δ compromises postreplicative repair (PRR) (29), we wanted to test if Smc5-Smc6 focal accumulation could occur via a Brc1-independent pathway in rad18Δ cells due to the use of alternative repair pathways. To this end, we generated Nse4-GFP brc1Δ rad18Δ triple-mutant cells, treated them with MMS or HU as before, and monitored Nse4-GFP foci. In this context, brc1Δ was epistatic to rad18Δ cells, as no Nse4-GFP foci were observed (Fig. 1A and B).
Therefore, Brc1, but not the Brc1-Rad18 complex (1), is required for Smc5-Smc6 accumulation at centromeres and subtelomeres during replication stress, which is interesting in light of the Rad18 dependency of Brc1-mediated suppression of smc6-74 (see Discussion).
Brc1-T672A reduces focal accumulation of Smc5-Smc6.
The C-terminal BRCT domains of Brc1 interact with gamma-H2A, which is required for Brc1 accumulation at DNA lesions during replication (2). We therefore tested if the Brc1 gamma-H2A binding mutant, Brc1-T672A (2), can support Nse4-GFP focus formation. We monitored Nse4-GFP foci in MMS- or HU-treated brc1Δ cells carrying an empty control vector, wild-type Brc1, or Brc1-T672A. Nse4-GFP foci in MMS- and HU-treated brc1Δ cells expressing wild-type Brc1 were abundant, whereas they were strongly reduced in the vector-alone controls (Fig. 1C and D). Interestingly, Brc1-T672A-expressing brc1Δ cells were also deficient in Nse4-GFP focus formation, similar to those expressing the control vector (Fig. 1C and D). Therefore, gamma-H2A recognition by Brc1 is important for the local accumulation of Smc5-Smc6 at DNA lesions induced during replication.
Brc1 physically interacts with the Smc5-Smc6 complex.
In light of the above-mentioned data, we tested whether Brc1 could act as a physical tether on chromatin (i.e., gamma-H2A bound), recruiting Smc5-Smc6 to replication-born lesions in the quantities required to detect GFP foci. We previously detected Brc1 peptides in mass spectrometry-based approaches to identify Smc5-Smc6 complex components (25, 27). The peptide number did not reach our threshold for follow-up experiments at the time; however, a recent study revealed that endogenous Brc1 is not readily precipitated and detected by Western analysis under the native buffer conditions used in our analyses (1, 25, 27). Thus, detection of any peptides coprecipitating with Smc5-Smc6 subunits was likely more significant than we had appreciated.
Therefore, we used ectopically expressed TAP-Brc1 to assess interaction with Smc5-Smc6, a method that recently identified a physical and functional interaction between Brc1 and Rad18 in surviving replicative stress (1). In pairwise tests against endogenous epitope-tagged Smc5-Smc6 subunits, we detected an interaction of TAP-Brc1 with Nse6-myc, which is part of the nonessential Nse5-Nse6 heterodimeric subcomplex (Fig. 2A) (25). This interaction does not depend on gamma-H2A binding by the Brc1 C-terminal BRCT domains, as it was also observed between TAP–Brc1-T672A and Nse6-myc (Fig. 2A). Brc1 also interacts with the essential core complex subunit Smc5, again independently of its gamma-H2A binding C-terminal BRCT domains (Fig. 2B). Therefore, Brc1 physically interacts with both Smc5-Smc6 and gamma-H2A via nonoverlapping interfaces and thus can promote Smc5-Smc6 focus formation at DNA lesions.
FIG 2.

Brc1 physically interacts with the Smc5-Smc6 complex. (A) Amino-terminally TAP-tagged Brc1, Brc1-T672A, or empty vector (pREP41) was ectopically expressed in an Nse6-Myc strain. Wild-type cells were also transformed with pREP41-TAP-Brc1 as an additional control. All the strains were subjected to TAP pulldown followed by anti-Myc Western blotting (WB). Brc1 and Brc1-T672A specifically interacted with Nse6. (B) TAP pulldown of ectopically expressed TAP-Brc1, TAP-Brc1-T672A, or empty vector in the Smc5-Myc strain.
A role for Nse5-Nse6 and Brc1 in Smc5-Smc6 chromatin association.
We initially identified the fission yeast Nse5-Nse6 heterodimer as a cofactor for Smc5-Smc6 functions in DNA repair and genome stability (8, 25). Cells lacking Nse5-Nse6 exhibit phenotypes that overlap with those caused by mutations in the essential Smc5-Smc6 subunits, including sensitivity to various genotoxic stresses and chromosome segregation failure due to the accumulation of unprocessed homologous recombination intermediates in mitosis and meiosis (8, 12, 17, 23–25, 27, 30, 31). Importantly, because fission yeast Nse5-Nse6 is nonessential, unlike the core Smc5-Smc6 subunits, it provides a clean separation of function within the Smc5-Smc6 holocomplex (8, 18, 25).
We had speculated that Nse5-Nse6 supports the recruitment and/or loading of Smc5-Smc6 on chromatin (25). To begin to test this possibility, we assayed genotoxic-stress-induced Nse4-GFP focus formation in wild-type and nse6Δ cells, as done for brc1Δ cells. To enable clear microscopy-based quantification of Nse4-GFP foci, we used Nts1 overexpression, which specifically mitigates the relatively severe genotoxin sensitivity of nse6Δ versus brc1Δ cells (32). Strikingly, whereas wild-type cells expressing Nts1 had the expected levels of HU- and MMS-induced Nse4-GFP foci, they were almost completely absent in cells lacking Nse6 (Fig. 3A). Therefore, Brc1 and Nse5-Nse6 are both required for the formation of Smc5-Smc6 foci around DNA lesions.
FIG 3.

Nse5-Nse6 and Brc1 are involved in Smc5-Smc6 chromatin association. (A) Live-cell microscopy of endonuclear HU- or MMS-induced Nse4-GFP foci in wt and nse6Δ strains, both on a background of constitutive overexpression of Nts1. Foci were strongly reduced in the absence of Nse6. The bar graph represents mean values and SD from the results of 3 independent repeats. (B and C) Loading of Nse4 at the indicated loci is impaired in nse5Δ and nse6Δ strains and to a lesser extent in the brc1Δ strain compared to the wild-type background. (B) ChIP-qPCR of Nse4-FlagHis at centromeric loci and a tRNAAla1 site in HU-treated (+HU) and untreated (−HU) cells. (C) ChIP-qPCR of Nse4-FlagHis at telomeric loci in MMS-treated (+MMS) and untreated (−MMS) cells. (Inset) Western analysis of Nse4-FlagHis in the indicated strains using Cdc2 as a loading control.
In light of the above-mentioned results, we more directly tested the roles of Nse5-Nse6 and Brc1 in Smc5-Smc6 chromatin association. Previously, we used chromatin immunoprecipitation with microarray technology (ChIP-chip) and ChIP-quantitative PCR (qPCR) against Nse4 and Smc5 to determine the binding sites of fission yeast Smc5-Smc6 genomewide in the presence or absence of genotoxic stress (16). This revealed constitutive enrichment of Smc5-Smc6 at fission yeast transfer DNAs (tDNAs) and core centromeres, with inducible enrichment at heterochromatic centromeric loci and subtelomeres upon HU and MMS treatment, respectively. These Smc5-Smc6 binding hot spots have since been confirmed by others using different epitope-tagged subunits and ChIP-qPCR (21, 31). Overall, the six essential core subunits of the Smc5-Smc6 holocomplex operate interdependently and load as a unit at distinct chromosome loci to function (21, 23–25).
Therefore, we tested Smc5-Smc6 chromatin association in wild-type, nse5Δ, nse6Δ, and brc1Δ cells using ChIP-qPCR against Nse4 at some Smc5-Smc6 binding hot spots. In wild-type cells, we observed the anticipated HU- and MMS-induced enrichment of Nse4 at each of these Smc5-Smc6 binding hot spots (Fig. 3B and C) (16). Notably, however, deletion of either Nse5 or Nse6 strongly reduced Nse4 residence at all binding sites tested, which was most evident under conditions of genotoxic stress that stimulate de novo Smc5-Smc6 loading (Fig. 3B and C) (16).
Deletion of Brc1 also significantly reduced Smc5-Smc6 residence at its binding hot spots under conditions of genotoxic stress, albeit to a lesser extent (Fig. 3B and C). A model incorporating the different penetrances of loading defects in each background is included in the discussion. Overall, consistent with their nonessential but critical functions in DNA repair, Nse5-Nse6 and Brc1 promote the recruitment of Smc5-Smc6 to chromatin.
Brc1 and Nse5-Nse6 support Smc5-Smc6 SUMO ligase activation by MMS.
Upon MMS-induced replicative stress, several Smc5-Smc6 complex subunits are SUMOylated by the resident Nse2 SUMO E3 ligase (16, 17). For example, we previously determined that Nse4 is specifically SUMOylated by Nse2 in MMS-treated cells (16). We hypothesized that the chromatin loading and high local concentration of Smc5-Smc6 in MMS-induced foci could support its auto-SUMOylation. To test this, we monitored endogenous Nse4-TAP in wild-type, nse6Δ, brc1Δ, and rhp18Δ cells, as previously described (16).
As anticipated, the MMS-induced SUMOylation of Nse4-TAP in wild-type cells was detectable upon immunoprecipitation (IP) of Nse4 without overexpression or initial enrichment of SUMO (Fig. 4A) (16). This indicates uncommonly high stoichiometry of the Nse4 SUMO modification. Notably, however, Nse4-TAP SUMOylation was undetectable in nse6Δ cells (Fig. 4A). Our SUMO antibody also revealed a baseline level of Nse4 SUMOylation, which was strongly induced by MMS treatment in wild-type but not nse6Δ cells (Fig. 4A).
FIG 4.
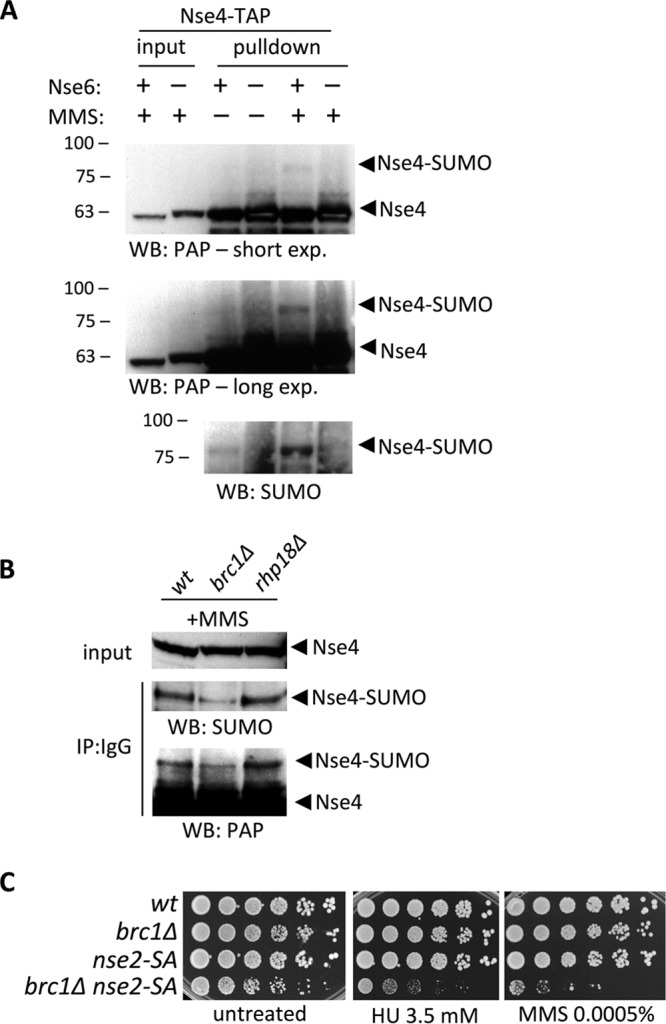
Nse6 and Brc1 support Nse4 SUMOylation. (A) IP and Western blotting of endogenously TAP-tagged Nse4 showing MMS-induced SUMOylation of Nse4-TAP in wild-type cells. The SUMOylation of Nse4 is eliminated in nse6Δ cells. exp., exposure. (B) SUMOylation of Nse4 is diminished in brc1Δ cells, but not in rhp18Δ cells, upon IP and WB of Nse4-TAP. (C) Spot assay of wt, brc1Δ, nse2-SA, and double-mutant brc1Δ nse2-SA strains. The sensitivity of the double mutant to genotoxins was higher than that of the parental single-mutant strains.
MMS-induced Nse4 SUMOylation was also reduced in brc1Δ cells but was similar to wild-type in rhp18Δ cells, which support normal Nse4-GFP focus formation (Fig. 4B). Double-mutant nse2-SA brc1Δ cells are more sensitive to genotoxins than either single mutant (Fig. 4C). Therefore, although Brc1-mediated Smc5-Smc6 focus formation supports Nse2 SUMO ligase activity, this is not the only function of Brc1.
Overall, the Nse5-Nse6- and Brc1-dependent chromatin association of Smc5-Smc6, together with its accumulation in MMS-induced foci, drives the SUMOylation of Nse4 by Nse2. Of note, the Nse4 SUMOylation defect is more pronounced in nse6Δ versus brc1Δ cells, which agrees well with their respective impacts on Smc5-Smc6 chromatin association (Fig. 3B and C).
Interestingly, we have shown that MMS, but not HU, treatment induces readily detectable Nse4 SUMOylation, despite HU promoting the formation of a single nuclear focus of Smc5-Smc6 at centromeres (16). Therefore, either the higher number of Smc5-Smc6 foci in MMS versus HU reveals Nse2 SUMO ligase activity, or there are additional DNA forms/structures at MMS lesions that activate Nse2.
A repair-defective Brc1 mutant compromises the Brc1-Nse6 interaction.
The interaction of Brc1 with Nse6 does not require its C-terminal gamma-H2A binding BRCT domains (Fig. 2A); we therefore tested if the N-terminal BRCT domains of Brc1 were required. We found that Brc1 with mutations in conserved residues of BRCT domain 4, brc1-HYP307-9GFG (1), severely compromised the interaction between Brc1 and Nse6 (Fig. 5A). In addition, the sensitivity of brc1Δ cells to MMS was only minimally rescued by brc1-HYP307-9GFG, whereas brc1-W248F, P301G, with mutations in conserved BRCT domain 3 residues, provided a full rescue (Fig. 5B).
FIG 5.

(A) Immunoprecipitation of TAP-Brc1 or TAP-Brc1-HYP307-309GFG ectopically expressed from pREP41 plasmid in the wt or Nse6-Myc strain. Mutations in Brc1 BRCT domain 4 weaken its interaction with Nse6. (B) Sensitivity of brc1Δ to MMS is rescued by brc1+ and brc1-W248F, P301G but not by a brc1-HYP307-309GFG mutant. All Brc1 variants were expressed from the pREP41 plasmid. The strains were cultured in minimal medium lacking leucine and spotted onto YES medium with or without MMS at 30°C. (C) Effect of Brc1 dosage on Smc5-Smc6 mutant genotoxin sensitivity. Ectopic expression of Brc1 in smc6-74, nse6Δ, nse5Δ, and wild-type strains was analyzed in a spot assay. Overexpression of Brc1 rescued the MMS sensitivity of smc6-74 but not nse5 or nse6 deletion. vector, pREP41; brc1+, pREP41-NTAP-Brc1. (D) Spot assay of nse6Δ, brc1Δ, and the double nse6Δ brc1Δ mutant strains. Cells were spotted onto YES medium containing the indicated concentrations of genotoxic agent at 32°C. The double-mutant nse6Δ brc1Δ strain showed higher sensitivity to genotoxic stress than the single-mutant parental strains.
Interestingly, brc1-HYP307-9GFG was recently shown to also compromise the interaction between Brc1 and Rhp18 (Rad18) (1). This is consistent with the known multiprotein interaction scaffolding of the budding yeast Brc1 orthologue Rtt107 (33, 34). Therefore, phenotypes ascribed to brc1-HYP307-9GFG are compound and can minimally be attributed to defects in the interaction of Brc1 with both Smc5-Smc6 and Rad18. In the future, once protein-protein interfaces are defined at the structural level, it will be interesting to dissect these interactions to determine their relative contributions to DNA repair.
Impact of Brc1 dosage on the growth of Smc5-Smc6 mutants.
Brc1 was first identified as a strong dosage suppressor of the smc6-74 Smc5-Smc6 complex mutant (4) and was later found to also suppress the MMS sensitivity of a hypomorphic allele of Nse4 (Rad62) (23). To further probe the functional relationship between Brc1 and Nse5-Nse6, we tested the effect of Brc1 overexpression on the MMS sensitivity of nse5Δ or nse6Δ versus smc6-74 cells. Interestingly, pBrc1 robustly suppressed the MMS hypersensitivity of smc6-74 cells, whereas it provided little benefit to cells lacking Nse5-Nse6 (Fig. 5C).
Importantly, hypomorphic alleles of any of the essential Smc5-Smc6 core subunits, i.e., Smc5, Smc6, and Nse1 through Nse4, absolutely require Brc1 for viability (4, 23, 25, 27). Given that pBrc1 failed to suppress either the nse5Δ or nse6Δ phenotype, we tested the Brc1 dependency of cells lacking Nse5-Nse6. Strikingly, compared to nse6∆ cells, nse6Δ brc1Δ double mutant cells were viable and showed little additional growth defect in the unchallenged cell cycle (Fig. 5D). This is all the more notable as nse6Δ cells are generally sicker and more sensitive to genotoxic stresses than cells with the above-mentioned Smc5-Smc6 core hypomorphic alleles, which are synthetically lethal with brc1Δ cells. The nse6Δ brc1Δ double mutants were more sensitive to replication stress than nse6Δ cells, indicating that Brc1 also has Nse5-Nse6-independent DNA repair functions (Fig. 5D).
The most economical interpretation of the above-mentioned data is that Brc1 can act in part through Nse5-Nse6 to support the functions of the Smc5-Smc6 core complex. Indeed, like brc1Δ, deletion of either Nse5 or Nse6 is lethal in combination with hypomorphic alleles of the Smc5-Smc6 hexameric core (25). This fact precludes more direct experimental testing of the Nse5-Nse6 dependence of pBrc1-mediated smc6-74 suppression.
DISCUSSION
In this study, we reveal that there is a more direct functional link between Brc1 and the Smc5-Smc6 complex than previously thought. This has significant implications for models of how Brc1 functions in replication stress tolerance and as a dosage suppressor of smc6-74. Before now, it appeared that Brc1 functions in a separate pathway to bypass smc6-74 (28), but our data indicate that the situation may not be so straightforward. That is, Brc1 reinforces Smc5-Smc6 recruitment into foci at replication-derived DNA lesions and enhances its SUMO ligase activity.
Moreover, we demonstrate a critical role for the Nse5-Nse6 subunits of the fission yeast Smc5-Smc6 complex in recruiting/loading the complex at its known binding hot spots across the genome (16). Fission yeast Nse5-Nse6 is nonessential yet still promotes key functions of Smc5-Smc6, thus providing a clean separation of function within the holocomplex (25). Our data showing that Nse5-Nse6 and Brc1 together support Smc5-Smc6 focal accumulation, chromatin association, and SUMO ligase activity now provide an explanation for the importance of Nse5-Nse6 in genome stability.
Brc1 is structurally related to budding yeast Rtt107 and human PTIP in terms of their content of 6 BRCT domains, although PTIP has a different linker arrangement than the yeast proteins (35). These three BRCT domain-containing proteins share the ability to bind gamma-H2A that is formed by the master checkpoint kinases at DNA lesions (2, 36, 37). However, Brc1, Rtt107, and PTIP appear to bind functionally distinct cofactors (1, 35, 38, 39), which likely accounts for the markedly different synthetic genetic interaction profiles of Rtt107 and Brc1 (40). Therefore, despite their common involvement in the replication stress response (2, 3, 38, 39), Brc1, Rtt107, and PTIP appeared to function in different pathways.
However, our results now reveal an apparently conserved role for Brc1 and Rtt107 in the recruitment of the Smc5-Smc6 complex to DNA lesions. During replicative stress or following the induction of DNA double-strand breaks (DSBs), Rtt107 was found to interact with the budding yeast Smc5-Smc6 complex (38, 41). Although fission yeast Smc5-Smc6 does not accumulate at DSBs, there is a clear parallel between the interaction of Brc1 and Rtt107 with Smc5-Smc6 in response to collapsed replication forks (35, 38, 42).
Thus, although genetic interrogation suggested major functional divergence of Brc1 and Rtt107 (40), physical interaction and other analyses indicate that they both can act as scaffolds for the Smc5-Smc6 complex at DNA lesions. As noted, both Brc1 and Rtt107 bind gamma-H2A that is generated in proximity to both DSBs and collapsed replication forks. Therefore, as Brc1 is specifically required for the response to replication stress but Rtt107 acts in response to both replication stress and DSBs, it will be interesting to determine how this selectivity is achieved. PTIP localizes to replication forks and regulates their stability (39), but it has not yet been implicated in regulating Smc5-Smc6 localization.
However, another complex, consisting of RAD18 and BRCTx, has been implicated in localizing Smc5-Smc6 to DNA interstrand cross-links and laser-induced damage stripes (43). BRCTx contains BRCT domains that bind RAD18 (not gamma-H2A), and it is a well-characterized RAD18 binding protein, with reports differing on its involvement in RAD18-mediated DNA repair (44, 45). As Rad18 does not localize fission yeast Smc5-Smc6 to lesions, it will be interesting to determine if RAD18-BRCTx involvement in humans represents a major divergence from yeast or if, perhaps, PTIP is also required in certain instances.
As described above, Brc1 interacts with and requires Rad18 to suppress smc6-74 (1, 28), which seems to agree with the role of human RAD18 in SMC5-SMC6 localization (43). However, as Rad18 is not required for fission yeast Smc5-Smc6 localization, we envisage a distinct role for the Brc1-Rad18 complex in smc6-74 suppression.
By binding Nse5-Nse6, Brc1 would recruit the dysfunctional Smc5–Smc6-74 complex to DNA lesions. While this may partially compensate for the lower chromatin association of smc6-74 caused by its ATPase defect (21, 46), loading of the mutant complex may also pose a challenge or “roadblock” for replication and other repair processes. Here, the Brc1-Rad18 complex could act in a parallel pathway to help circumvent such roadblocks and enhance the survival of smc6-74 cells during replication stress (1, 28).
It is known that MMS treatment strongly enhances the SUMOylation of components of the fission yeast Smc5-Smc6 complex in an Nse2-dependent manner (16, 17). However, determinants of such Smc5-Smc6-Nse2 activation have remained unidentified. From our data, one factor that contributes to MMS-induced auto-SUMOylation of Smc5-Smc6 is its Nse5-Nse6- and Brc1-mediated recruitment into foci at DNA lesions during replication. Incorporation of Smc5-Smc6 into foci may enable trans SUMOylation of its subunits by Nse2 due to the increased local concentration of the complex. Indeed, a recent in vitro study revealed that budding yeast Nse4 is SUMOylated in trans in a DNA-stimulated manner by Smc5-Smc6-Nse2 (47). Thus, the defective recruitment and/or loading of Smc5-Smc6 at DNA lesions in cells lacking Brc1 or Nse5-Nse6 can explain the strongly reduced MMS-induced Nse4 SUMOylation in these cells.
We previously showed that Nse2-dependent SUMO conjugation also contributes to the accumulation of Smc5-Smc6 in replication stress foci (16). Therefore, it appears that Brc1 and Nse2 cooperate to maximize local recruitment of Smc5-Smc6 to the region around collapsed replication forks. It will be interesting to determine if fission yeast Smc5-Smc6 subunits contain SUMO-interacting motifs (SIMs) that could contribute to such a mechanism by mediating interactions between proximal SUMO-modified Smc5-Smc6 complexes. In this regard, although clear SIMs have not yet been delineated, the Nse5 subunit of the fission and budding yeast Smc5-Smc6 complexes interacts with SUMO pathway factors (48, 49). Clearly, it is also possible that protein targets outside the Smc5-Smc6 complex are targeted by Nse2 to reinforce its recruitment.
Overall, our data are consistent with an initial recruitment of Smc5-Smc6 to DNA lesions by Nse5-Nse6 and Brc1, in part through its gamma-H2A binding BRCT domain, followed by Nse2-mediated reinforcement. A similar cooperative recruitment mechanism was revealed for the DNA damage checkpoint factor Crb2, an orthologue of human 53BP1 (50). Crb2 binds gamma-H2A and methylated histone H4-K20 via its BRCT and Tudor domains, respectively (50). Interestingly, although Crb2 DNA damage-induced foci are largely abolished in cells lacking H4-K20 methylation (e.g., H4-K20R), such cells show only a minor checkpoint defect compared to crb2Δ cells (50). This is consistent with the focal accumulation of proteins at DNA lesions functioning as a safety net or backup pool that maintains a high local concentration of key factors, even though not all of it is necessarily engaged in signaling or lesion processing.
How the nonessential fission yeast Nse5-Nse6 heterodimer mediates the DNA repair roles of Smc5-Smc6 had remained enigmatic since its discovery (8, 25). We found that Nse5-Nse6 is required for full levels of Smc5-Smc6 chromatin loading in the absence and presence of genotoxic stress at all genomic sites tested. It remains unknown if Nse5-Nse6 supports the topological loading of Smc5-Smc6, like Mis4-Ssl3 (Sc Scc2-Scc4) of the related cohesin complex (5), or if its role is restricted to localizing Smc5-Smc6 to specific genomic sites. Residual Smc5-Smc6 loading is sufficient for viability but leads to increased genomic instability and sensitivity to genotoxic stress. The function of Nse5-Nse6 in loading the core Smc5-Smc6 complex may begin to explain some of its unique features.
For example, unlike all other hypomorphic core Smc5-Smc6 alleles tested, nse5Δ and nse6Δ are viable in the absence of Brc1. This is consistent with an Nse5-Nse6-mediated role for Brc1 in recruiting the Smc5-Smc6 complex to chromatin, wherein nse5Δ or nse6Δ would be expected to be epistatic to Brc1. A requirement for Nse5-Nse6 to bridge Brc1 to the core Smc5-Smc6 complex may also explain the inability of Brc1 overexpression to suppress nse5Δ or nse6Δ cells. However, as nse6Δ brc1Δ double-mutant cells are more sensitive to genotoxins than nse6Δ or brc1Δ single mutants, each protein also has independent functions.
In conclusion, we found that Brc1 and Nse5-Nse6 together enrich the fission yeast Smc5-Smc6 complex at replication-derived DNA lesions, leading to activation of its SUMO ligase activity and promoting recovery from replication stress (Fig. 6). In general, the concentration or forced nucleation of SUMO pathway components leads to increased local activity (51–53). Thus, although not tested here due to the lack of an appropriate substrate, it is likely that other proteins at collapsed replication forks in the vicinity of Smc5-Smc6-Nse2 foci also undergo increased regulatory SUMOylation.
FIG 6.

Brc1 and the Nse5-Nse6 dimer are required for the accumulation of the Smc5-Smc6 complex in foci or on chromatin during replication stress and for the Nse2-dependent SUMOylation of its subunits, e.g., Nse4. Brc1 physically interacts with Nse5-Nse6, as well as gamma-H2A, so it can tether Smc5-Smc6 at replicative DNA lesions. It is likely that other proteins at collapsed replication forks in the vicinity of Smc5-Smc6-Nse2 foci also undergo SUMOylation.
We envisage that Brc1-mediated Smc5-Smc6 focal accumulation flanking a lesion provides a funnel that pushes the loading equilibrium toward wild-type levels. Nse5-Nse6 likely plays an additional direct role in the loading/recruitment of the Smc5-Smc6 complex via interacting factors at DNA lesions. This would explain the more penetrant defects in Smc5-Smc6 localization and SUMO ligase activity in nse6Δ versus brc1Δ cells.
Our data also reveal apparent differences between the recruitment of human and fission yeast Smc5-Smc6 proteins to lesions, e.g., Rad18 is not required in fission yeast. It will be interesting to determine if this reflects truly divergent mechanisms or if the cancer cells used in the human cell studies have coopted additional Smc5-Smc6 recruitment mechanisms.
MATERIALS AND METHODS
Yeast strains and growth conditions.
Standard yeast methods were performed as described previously (54). The strains used in this study are listed in Table 1 .
TABLE 1.
Yeast strains used in this study
| Strain | Genotypea | Source or reference |
|---|---|---|
| NB231 | h− brc1::kanMX6 | 27 |
| NB402 | h+ Smc5-Myc:kanMX6 | Laboratory stock |
| NB771 | h+ nse4-GFP:hphMX4 | 16 |
| NB780 | h+ | Laboratory stock |
| NB835 | h+ nse6::kanMX6 | 25 |
| NB848 | h+ nse6-Myc:kanMX6 | 25 |
| NB895 | h− nse5::kanMX6 | 25 |
| NB898 | h? nse4-TAP:kanMX6 nse6::ura4+ | 25 |
| NB978 | h+ nse6::ura4+ | 49 |
| NB1322 | h+ nse4-TAP:kanMX6 | 27 |
| NB1538 | h− nse4-TAP:hphMX4 | 27 |
| NB3269 | h+ nse2-SA:ura4+ ade6-704 | 17 |
| NB5931 | h+ nse4-Flag-His:kanr | 21 |
| NB5949 | h+ nse6::ura4+ brc1::kanMX6 | This study |
| NB5953 | h+ pJK148-Padh-ura4:leu1 | This study |
| NB5954 | h+ pJK148-Padh-nts1-ura4:leu1 | This study |
| NB5955 | h+ nse6::kanMX6 pJK148-Padh-ura4:leu1 | This study |
| NB5956 | h+ nse6::kanMX6 pJK148-Padh-nts1-ura4:leu1 | This study |
| NB5959 | h+ smc6-74 | 4 |
| NB5961 | h+ nse4-Flag-His:kanr nse5::ura4+ | This study |
| NB5963 | h− nse4-Flag-His:kanr nse6::natMX6 | This study |
| NB5993 | h− smc6-74 pJK148-Padh-ura4:leu1 | This study |
| NB5994 | h− smc6-74 pJK148-Padh-nts1-ura4:leu1 | This study |
| NB6164 | h? nse4-GFP:hphMX4 brc1::kanMX6 | This study |
| NB6165 | h? nse4-GFP:hphMX4 rhp18::KanMX6 | This study |
| NB6178 | h? nse4-GFP:hphMX4 brc1::hphMX | This study |
| NB6179 | h? nse4-GFP:hphMX4 brc1::hphMX rhp18::kanMX6 | This study |
| NB6188 | h? nse4-TAP:hphMX4 brc1::KanMX6 | This study |
| NB6189 | h? nse4-TAP:hphMX4 rhp18::kanMX6 | This study |
| NB6214 | h? brc1::kanMX6 nse2-SA:ura4 | This study |
All strains are of ura4-D18 leu1-32 background genotype. Double colons represent knockouts; single colons represent tagging.
Spot assays.
Cells were grown at 32°C to logarithmic phase (optical density at 600 nm [OD600], 0.6 to 0.8), spotted on yeast extract-sucrose (YES) agar plates supplemented with the relevant drug in 5-fold dilutions from a starting OD600 of 0.5, and then incubated at 30°C or 32°C for 3 to 5 days.
Fluorescence microscopy.
The cells were treated with 0.03% MMS or 20 mM HU for 5 h. GFP fusion proteins were imaged in live cells using an Eclipse E800 microscope (Nikon Metrology, Brighton, MI) with a 100× Plan-Apochromat differential interference contrast (DIC) H oil immersion objective. The images were captured through an Infinity 3 charge-coupled-device (CCD) camera using Infinity Analyze software (Lumenera Corp., Ottawa, Canada).
Western blotting.
Exponentially growing cells (15 OD600 units) were washed with stop buffer (10 mM EDTA, 50 mM NaF, 150 mM NaCl, 1 mM NaN3). The pellets were flash-frozen in liquid nitrogen, and the cells were lysed by bead beating in 200 µl of 20% trichloroacetic acid (TCA). The beads were washed with 400 µl of 5% TCA, and the total cell lysate was centrifuged at 16,000 × g for 5 min at 4°C. The pellet was washed twice with 0.1% TCA. The precipitated proteins were resuspended in 8 M urea, 50 mM Tris, pH 8.5, 150 mM NaCl; resolved on a gradient SDS-PAGE gel; and transferred to a nitrocellulose membrane. The membrane was blocked in 1% (wt/vol) nonfat milk in phosphate-buffered saline solution with 0.1% (vol/vol) Tween 20, probed with either PAP (P1291; Sigma) alone or anti-Myc (9E10; in house), anti-Flag (F3165; Sigma), anti-PSTAIRE, or SUMO in combination with goat anti-mouse horseradish peroxidase (HRP)-conjugated antibody and then detected using an ECL Dura system (Pierce).
IgG pulldown.
Cell pellets (30 OD600 units) were lysed in 400 µl of IP buffer (20 mM Tris, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 1 mM NaF, pH 7.5) supplemented with 1 mM dithiothreitol (DTT), 1 mM phenylmethylsulfonyl fluoride (PMSF), protease inhibitor cocktail (Roche), and 25 mg N-ethylmaleimide and bead beaten. The lysate was incubated with 6 mM MgCl2 and Benzonase (EMD Millipore) for 30 min at 4°C. Protein in the clarified supernatant was quantitated by measuring absorbance at OD280, and 2 mg of proteins was incubated with 50 µl of Pan mouse IgG beads (Invitrogen) for 4 h at 4°C. The beads were washed once in IP buffer, 3 times in IP2 buffer (20 mM Tris, 200 mM NaCl, 1 mM EDTA, 1% Triton X-100, 1 mM NaF, pH 7.5), and once in IP3 buffer (20 mM Tris, 150 mM NaCl, 1 mM EDTA, 0.1% Triton X-100, 1 mM NaF, pH 7.5) and eluted with 2× LDS sample loading buffer (Invitrogen).
In addition to the positive interactions of Nse6 and Smc5 with Brc1, we also tested several other subunits of the complex, including Nse5-myc, 3myc-Smc6, and Nse4-TAP. We were unable to detect interactions in these cases due to technical issues that included target band sizes overlapping with background bands on the Western blots and insufficient sensitivity of detection for TAP and 3myc (versus 13myc on Nse6 and Smc5).
Chromatin immunoprecipitation.
The ChIP method was adapted from that of Nelson et al. (55). Briefly, exponentially growing cell cultures (untreated or treated with 15 mM HU or 0.005% MMS for 6 h) were fixed with 1% formaldehyde for 25 min and quenched with glycine. The cells were broken with beads in lysis buffer (50 mM HEPES, 1 mM EDTA, 150 mM NaCl, 1% Triton X-100, 0.1% sodium deoxycholate, pH 7.6) supplemented with protease inhibitor cocktail (Roche) and 2 mM PMSF. The chromatin extract was sheared with a Bioruptor Pico (Diagenode). Immunoprecipitation was done with 10 µg of FLAG antibody (F3165; Sigma) for 2 h, followed by incubation with protein G Dynabeads (Invitrogen) for 1 h at 4°C. The Dynabeads were washed 6 times with IP buffer (50 mM Tris-HCl, 1% Triton X-100, 150 mM NaCl, 5 mM EDTA, 0.5% NP-40, pH 7.5). Input and IP samples were incubated with 10% Chelex 100 (Bio-Rad) and treated with proteinase K. Quantitative PCR was performed with the resulting DNA samples using a SensiFast SYBR No-ROX kit (Bioline). Primer sequences for dg2, cnt2, and telo2R were previously published (56), as well as tRNAAla and STE1 (16, 31). DNA recovery was calculated using the 2−ΔCT method, and data are represented as the fold enrichment over the untagged wild-type strain control.
Plasmid construction.
Unless otherwise stated, cDNAs were cloned by PCR amplification using specific primers and PrimeStar DNA polymerase (Clontech, Mountain View, CA), followed by ligation into the pREP41 series of yeast shuttle vectors (57). Details regarding plasmid construction are available upon request.
ACKNOWLEDGMENTS
We thank Jan Palecek, Matthew O’Connell, and Felicity Watts for strains. We also thank members of the Cell Cycle Groups at TSRI for their support.
This study was funded by NIH grants GM068608 and GM081840 awarded to M.N.B. Work in the laboratory of P.R. was supported by grants CA077325, CA117638, and GM059447.
M.O., M.C.G., M.N., M.C.R., O.L., P.R., and M.N.B. were all involved in study design, experiment execution, and writing of the manuscript.
None of us has a conflict of interest with regard to the paper.
REFERENCES
- 1.Reubens MC, Rozenzhak S, Russell P. 2017. Multi-BRCT domain protein Brc1 links Rhp18/Rad18 and gammaH2A to maintain genome stability during S phase. Mol Cell Biol 37:e00260-17. doi: 10.1128/MCB.00260-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Williams JS, Williams RS, Dovey CL, Guenther G, Tainer JA, Russell P. 2010. gammaH2A binds Brc1 to maintain genome integrity during S-phase. EMBO J 29:1136–1148. doi: 10.1038/emboj.2009.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sheedy DM, Dimitrova D, Rankin JK, Bass KL, Lee KM, Tapia-Alveal C, Harvey SH, Murray JM, O'Connell MJ. 2005. Brc1-mediated DNA repair and damage tolerance. Genetics 171:457–468. doi: 10.1534/genetics.105.044966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Verkade HM, Bugg SJ, Lindsay HD, Carr AM, O'Connell MJ. 1999. Rad18 is required for DNA repair and checkpoint responses in fission yeast. Mol Biol Cell 10:2905–2918. doi: 10.1091/mbc.10.9.2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Uhlmann F. 2016. SMC complexes: from DNA to chromosomes. Nat Rev Mol Cell Biol 17:399–412. doi: 10.1038/nrm.2016.30. [DOI] [PubMed] [Google Scholar]
- 6.Copsey A, Tang S, Jordan PW, Blitzblau HG, Newcombe S, Chan AC, Newnham L, Li Z, Gray S, Herbert AD, Arumugam P, Hochwagen A, Hunter N, Hoffmann E. 2013. Smc5/6 coordinates formation and resolution of joint molecules with chromosome morphology to ensure meiotic divisions. PLoS Genet 9:e1004071. doi: 10.1371/journal.pgen.1004071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Verver DE, Hwang GH, Jordan PW, Hamer G. 2016. Resolving complex chromosome structures during meiosis: versatile deployment of Smc5/6. Chromosoma 125:15–27. doi: 10.1007/s00412-015-0518-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wehrkamp-Richter S, Hyppa RW, Prudden J, Smith GR, Boddy MN. 2012. Meiotic DNA joint molecule resolution depends on Nse5-Nse6 of the Smc5-Smc6 holocomplex. Nucleic Acids Res 40:9633–9646. doi: 10.1093/nar/gks713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xaver M, Huang L, Chen D, Klein F. 2013. Smc5/6-Mms21 prevents and eliminates inappropriate recombination intermediates in meiosis. PLoS Genet 9:e1004067. doi: 10.1371/journal.pgen.1004067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ampatzidou E, Irmisch A, O'Connell MJ, Murray JM. 2006. Smc5/6 is required for repair at collapsed replication forks. Mol Cell Biol 26:9387–9401. doi: 10.1128/MCB.01335-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bermúdez-López M, Ceschia A, de Piccoli G, Colomina N, Pasero P, Aragón L, Torres-Rosell J. 2010. The Smc5/6 complex is required for dissolution of DNA-mediated sister chromatid linkages. Nucleic Acids Res 38:6502–6512. doi: 10.1093/nar/gkq546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Irmisch A, Ampatzidou E, Mizuno K, O'Connell MJ, Murray JM. 2009. Smc5/6 maintains stalled replication forks in a recombination-competent conformation. EMBO J 28:144–155. doi: 10.1038/emboj.2008.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miyabe I, Morishita T, Hishida T, Yonei S, Shinagawa H. 2006. Rhp51-dependent recombination intermediates that do not generate checkpoint signal are accumulated in Schizosaccharomyces pombe rad60 and smc5/6 mutants after release from replication arrest. Mol Cell Biol 26:343–353. doi: 10.1128/MCB.26.1.343-353.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choi K, Szakal B, Chen YH, Branzei D, Zhao X. 2010. The Smc5/6 complex and Esc2 influence multiple replication-associated recombination processes in Saccharomyces cerevisiae. Mol Biol Cell 21:2306–2314. doi: 10.1091/mbc.e10-01-0050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Branzei D, Sollier J, Liberi G, Zhao X, Maeda D, Seki M, Enomoto T, Ohta K, Foiani M. 2006. Ubc9- and mms21-mediated sumoylation counteracts recombinogenic events at damaged replication forks. Cell 127:509–522. doi: 10.1016/j.cell.2006.08.050. [DOI] [PubMed] [Google Scholar]
- 16.Pebernard S, Schaffer L, Campbell D, Head SR, Boddy MN. 2008. Localization of Smc5/6 to centromeres and telomeres requires heterochromatin and SUMO, respectively. EMBO J 27:3011–3023. doi: 10.1038/emboj.2008.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andrews EA, Palecek J, Sergeant J, Taylor E, Lehmann AR, Watts FZ. 2005. Nse2, a component of the Smc5-6 complex, is a SUMO ligase required for the response to DNA damage. Mol Cell Biol 25:185–196. doi: 10.1128/MCB.25.1.185-196.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao X, Blobel G. 2005. A SUMO ligase is part of a nuclear multiprotein complex that affects DNA repair and chromosomal organization. Proc Natl Acad Sci U S A 102:4777–4782. doi: 10.1073/pnas.0500537102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Outwin EA, Irmisch A, Murray JM, O'Connell MJ. 2009. Smc5-Smc6-dependent removal of cohesin from mitotic chromosomes. Mol Cell Biol 29:4363–4375. doi: 10.1128/MCB.00377-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fousteri MI, Lehmann AR. 2000. A novel SMC protein complex in Schizosaccharomyces pombe contains the Rad18 DNA repair protein. EMBO J 19:1691–1702. doi: 10.1093/emboj/19.7.1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zabrady K, Adamus M, Vondrova L, Liao C, Skoupilova H, Novakova M, Jurcisinova L, Alt A, Oliver AW, Lehmann AR, Palecek JJ. 2016. Chromatin association of the SMC5/6 complex is dependent on binding of its NSE3 subunit to DNA. Nucleic Acids Res 44:1064–1079. doi: 10.1093/nar/gkv1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alt A, Dang HQ, Wells OS, Polo LM, Smith MA, McGregor GA, Welte T, Lehmann AR, Pearl LH, Murray JM, Oliver AW. 2017. Specialized interfaces of Smc5/6 control hinge stability and DNA association. Nat Commun 8:14011. doi: 10.1038/ncomms14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morikawa H, Morishita T, Kawane S, Iwasaki H, Carr AM, Shinagawa H. 2004. Rad62 protein functionally and physically associates with the smc5/smc6 protein complex and is required for chromosome integrity and recombination repair in fission yeast. Mol Cell Biol 24:9401–9413. doi: 10.1128/MCB.24.21.9401-9413.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Palecek J, Vidot S, Feng M, Doherty AJ, Lehmann AR. 2006. The Smc5-Smc6 DNA repair complex bridging of the Smc5-Smc6 heads by the KLEISIN, Nse4, and non-Kleisin subunits. J Biol Chem 281:36952–36959. doi: 10.1074/jbc.M608004200. [DOI] [PubMed] [Google Scholar]
- 25.Pebernard S, Wohlschlegel J, McDonald WH, Yates JR III, Boddy MN. 2006. The Nse5-Nse6 dimer mediates DNA repair roles of the Smc5-Smc6 complex. Mol Cell Biol 26:1617–1630. doi: 10.1128/MCB.26.5.1617-1630.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee SY, Rozenzhak S, Russell P. 2013. GammaH2A-binding protein Brc1 affects centromere function in fission yeast. Mol Cell Biol 33:1410–1416. doi: 10.1128/MCB.01654-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pebernard S, McDonald WH, Pavlova Y, Yates JR III, Boddy MN. 2004. Nse1, Nse2, and a novel subunit of the Smc5-Smc6 complex, Nse3, play a crucial role in meiosis. Mol Biol Cell 15:4866–4876. doi: 10.1091/mbc.e04-05-0436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee KM, Nizza S, Hayes T, Bass KL, Irmisch A, Murray JM, O'Connell MJ. 2007. Brc1-mediated rescue of Smc5/6 deficiency: requirement for multiple nucleases and a novel Rad18 function. Genetics 175:1585–1595. doi: 10.1534/genetics.106.067801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frampton J, Irmisch A, Green CM, Neiss A, Trickey M, Ulrich HD, Furuya K, Watts FZ, Carr AM, Lehmann AR. 2006. Postreplication repair and PCNA modification in Schizosaccharomyces pombe. Mol Biol Cell 17:2976–2985. doi: 10.1091/mbc.e05-11-1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pebernard S, Perry JJ, Tainer JA, Boddy MN. 2008. Nse1 RING-like domain supports functions of the Smc5-Smc6 holocomplex in genome stability. Mol Biol Cell 19:4099–4109. doi: 10.1091/mbc.e08-02-0226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tapia-Alveal C, O'Connell MJ. 2011. Nse1-dependent recruitment of Smc5/6 to lesion-containing loci contributes to the repair defects of mutant complexes. Mol Biol Cell 22:4669–4682. doi: 10.1091/mbc.E11-03-0272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zilio N, Codlin S, Vashisht AA, Bitton DA, Head SR, Wohlschlegel JA, Bahler J, Boddy MN. 2014. A novel histone deacetylase complex in the control of transcription and genome stability. Mol Cell Biol 34:3500–3514. doi: 10.1128/MCB.00519-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hang L, Zhao X. 2016. The Rtt107 BRCT scaffold and its partner modification enzymes collaborate to promote replication. Nucleus 7:346–351. doi: 10.1080/19491034.2016.1201624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leung GP, Brown JA, Glover JN, Kobor MS. 2016. Rtt107 BRCT domains act as a targeting module in the DNA damage response. DNA Repair 37:22–32. doi: 10.1016/j.dnarep.2015.10.007. [DOI] [PubMed] [Google Scholar]
- 35.Wan B, Hang LE, Zhao X. 2016. Multi-BRCT scaffolds use distinct strategies to support genome maintenance. Cell Cycle 15:2561–2570. doi: 10.1080/15384101.2016.1218102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li X, Liu K, Li F, Wang J, Huang H, Wu J, Shi Y. 2012. Structure of C-terminal tandem BRCT repeats of Rtt107 protein reveals critical role in interaction with phosphorylated histone H2A during DNA damage repair. J Biol Chem 287:9137–9146. doi: 10.1074/jbc.M111.311860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yan W, Shao Z, Li F, Niu L, Shi Y, Teng M, Li X. 2011. Structural basis of gammaH2AX recognition by human PTIP BRCT5-BRCT6 domains in the DNA damage response pathway. FEBS Lett 585:3874–3879. doi: 10.1016/j.febslet.2011.10.045. [DOI] [PubMed] [Google Scholar]
- 38.Ohouo PY, Bastos de Oliveira FM, Almeida BS, Smolka MB. 2010. DNA damage signaling recruits the Rtt107-Slx4 scaffolds via Dpb11 to mediate replication stress response. Mol Cell 39:300–306. doi: 10.1016/j.molcel.2010.06.019. [DOI] [PubMed] [Google Scholar]
- 39.Ray Chaudhuri A, Callen E, Ding X, Gogola E, Duarte AA, Lee JE, Wong N, Lafarga V, Calvo JA, Panzarino NJ, John S, Day A, Crespo AV, Shen B, Starnes LM, de Ruiter JR, Daniel JA, Konstantinopoulos PA, Cortez D, Cantor SB, Fernandez-Capetillo O, Ge K, Jonkers J, Rottenberg S, Sharan SK, Nussenzweig A. 2016. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 535:382–387. doi: 10.1038/nature18325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sanchez A, Roguev A, Krogan NJ, Russell P. 2015. Genetic interaction landscape reveals critical requirements for Schizosaccharomyces pombe Brc1 in DNA damage response mutants. G3 5:953–962. doi: 10.1534/g3.115.017251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leung GP, Lee L, Schmidt TI, Shirahige K, Kobor MS. 2011. Rtt107 is required for recruitment of the SMC5/6 complex to DNA double strand breaks. J Biol Chem 286:26250–26257. doi: 10.1074/jbc.M111.235200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu Y, Ren JY, Zhang JM, Suo F, Fang XF, Wu F, Du LL. 2013. A proteome-wide visual screen identifies fission yeast proteins localizing to DNA double-strand breaks. DNA Repair 12:433–443. doi: 10.1016/j.dnarep.2013.04.001. [DOI] [PubMed] [Google Scholar]
- 43.Raschle M, Smeenk G, Hansen RK, Temu T, Oka Y, Hein MY, Nagaraj N, Long DT, Walter JC, Hofmann K, Storchova Z, Cox J, Bekker-Jensen S, Mailand N, Mann M. 2015. DNA repair proteomics reveals dynamic assembly of repair complexes during bypass of DNA cross-links. Science 348:1253671. doi: 10.1126/science.1253671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Adams DJ, van der Weyden L, Gergely FV, Arends MJ, Ng BL, Tannahill D, Kanaar R, Markus A, Morris BJ, Bradley A. 2005. BRCTx is a novel, highly conserved RAD18-interacting protein. Mol Cell Biol 25:779–788. doi: 10.1128/MCB.25.2.779-788.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu T, Chen H, Kim H, Huen MS, Chen J, Huang J. 2012. RAD18-BRCTx interaction is required for efficient repair of UV-induced DNA damage. DNA Repair 11:131–138. doi: 10.1016/j.dnarep.2011.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kanno T, Berta DG, Sjogren C. 2015. The Smc5/6 complex is an ATP-dependent intermolecular DNA linker. Cell Rep 12:1471–1482. doi: 10.1016/j.celrep.2015.07.048. [DOI] [PubMed] [Google Scholar]
- 47.Varejao N, Ibars E, Lascorz J, Colomina N, Torres-Rosell J, Reverter D. 2018. DNA activates the Nse2/Mms21 SUMO E3 ligase in the Smc5/6 complex. EMBO J 37:e98306. doi: 10.15252/embj.201798306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bustard DE, Ball LG, Cobb JA. 2016. Non-Smc element 5 (Nse5) of the Smc5/6 complex interacts with SUMO pathway components. Biol Open 5:777–785. doi: 10.1242/bio.018440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Prudden J, Pebernard S, Raffa G, Slavin DA, Perry JJ, Tainer JA, McGowan CH, Boddy MN. 2007. SUMO-targeted ubiquitin ligases in genome stability. EMBO J 26:4089–4101. doi: 10.1038/sj.emboj.7601838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Du LL, Nakamura TM, Russell P. 2006. Histone modification-dependent and -independent pathways for recruitment of checkpoint protein Crb2 to double-strand breaks. Genes Dev 20:1583–1596. doi: 10.1101/gad.1422606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nie M, Moser BA, Nakamura TM, Boddy MN. 2017. SUMO-targeted ubiquitin ligase activity can either suppress or promote genome instability, depending on the nature of the DNA lesion. PLoS Genet 13:e1006776. doi: 10.1371/journal.pgen.1006776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Prudden J, Perry JJ, Nie M, Vashisht AA, Arvai AS, Hitomi C, Guenther G, Wohlschlegel JA, Tainer JA, Boddy MN. 2011. DNA repair and global sumoylation are regulated by distinct Ubc9 noncovalent complexes. Mol Cell Biol 31:2299–2310. doi: 10.1128/MCB.05188-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chung I, Leonhardt H, Rippe K. 2011. De novo assembly of a PML nuclear subcompartment occurs through multiple pathways and induces telomere elongation. J Cell Sci 124:3603–3618. doi: 10.1242/jcs.084681. [DOI] [PubMed] [Google Scholar]
- 54.Moreno S, Klar A, Nurse P. 1991. Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods Enzymol 194:795–823. doi: 10.1016/0076-6879(91)94059-L. [DOI] [PubMed] [Google Scholar]
- 55.Nelson JD, Denisenko O, Bomsztyk K. 2006. Protocol for the fast chromatin immunoprecipitation (ChIP) method. Nat Protoc 1:179–185. doi: 10.1038/nprot.2006.27. [DOI] [PubMed] [Google Scholar]
- 56.Hayashi M, Katou Y, Itoh T, Tazumi A, Tazumi M, Yamada Y, Takahashi T, Nakagawa T, Shirahige K, Masukata H. 2007. Genome-wide localization of pre-RC sites and identification of replication origins in fission yeast. EMBO J 26:1327–1339. doi: 10.1038/sj.emboj.7601585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maundrell K. 1993. Thiamine-repressible expression vectors pREP and pRIP for fission yeast. Gene 123:127–130. doi: 10.1016/0378-1119(93)90551-D. [DOI] [PubMed] [Google Scholar]