

The picornavirus foot-and-mouth disease virus (FMDV) is a notorious animal pathogen that puts a major economic burden on the global livestock industry. Outbreaks have significant consequences for animal health and product safety. Like many other viruses, FMDV must manipulate antiviral host responses to establish infection. Upon infection, viral double-stranded RNA (dsRNA) is detected, which results in the activation of the RNA-dependent protein kinase (PKR)-mediated stress response, leading to a stop in cellular and viral translation and the formation of stress granules (SG), which are thought to have antiviral properties. Here, we show that FMDV can suppress SG formation via its leader protease (Lpro). Simultaneously, we observed that Lpro can cleave the SG scaffolding proteins G3BP1 and G3BP2. Understanding the molecular mechanisms of the antiviral host response evasion strategies of FMDV may help to develop countermeasures to control FMDV infections in the future.
KEYWORDS: Aphthovirus, FMDV, G3BP1, G3BP2, SGs, Stress granules
ABSTRACT
Like other viruses, the picornavirus foot-and-mouth disease virus (FMDV; genus Aphthovirus), one of the most notorious pathogens in the global livestock industry, needs to navigate antiviral host responses to establish an infection. There is substantial insight into how FMDV suppresses the type I interferon (IFN) response, but it is largely unknown whether and how FMDV modulates the integrated stress response. Here, we show that the stress response is suppressed during FMDV infection. Using a chimeric recombinant encephalomyocarditis virus (EMCV), in which we functionally replaced the endogenous stress response antagonist by FMDV leader protease (Lpro) or 3Cpro, we demonstrate an essential role for Lpro in suppressing stress granule (SG) formation. Consistently, infection with a recombinant FMDV lacking Lpro resulted in SG formation. Additionally, we show that Lpro cleaves the known SG scaffold proteins G3BP1 and G3BP2 but not TIA-1. We demonstrate that the closely related equine rhinitis A virus (ERAV) Lpro also cleaves G3BP1 and G3BP2 and also suppresses SG formation, indicating that these abilities are conserved among aphthoviruses. Neither FMDV nor ERAV Lpro interfered with phosphorylation of RNA-dependent protein kinase (PKR) or eIF2α, indicating that Lpro does not affect SG formation by inhibiting the PKR-triggered signaling cascade. Taken together, our data suggest that aphthoviruses actively target scaffolding proteins G3BP1 and G3BP2 and antagonize SG formation to modulate the integrated stress response.
IMPORTANCE The picornavirus foot-and-mouth disease virus (FMDV) is a notorious animal pathogen that puts a major economic burden on the global livestock industry. Outbreaks have significant consequences for animal health and product safety. Like many other viruses, FMDV must manipulate antiviral host responses to establish infection. Upon infection, viral double-stranded RNA (dsRNA) is detected, which results in the activation of the RNA-dependent protein kinase (PKR)-mediated stress response, leading to a stop in cellular and viral translation and the formation of stress granules (SG), which are thought to have antiviral properties. Here, we show that FMDV can suppress SG formation via its leader protease (Lpro). Simultaneously, we observed that Lpro can cleave the SG scaffolding proteins G3BP1 and G3BP2. Understanding the molecular mechanisms of the antiviral host response evasion strategies of FMDV may help to develop countermeasures to control FMDV infections in the future.
INTRODUCTION
Innate antiviral responses represent the first line of defense against viral infections. Cellular antiviral signaling is initiated upon recognition of pathogen-associated molecular patterns (PAMPs) by the host. Cytosolic viral RNA can be detected by the RIG-I like receptors (RLRs). Upon recognition of viral RNA, RLRs activate signal transduction pathways, resulting in the production and secretion of type I interferons (alpha/beta interferon [IFN-α/β]) and proinflammatory cytokines. Secreted IFN-α/β triggers the transcription of interferon-stimulated genes (ISGs), thereby generating an antiviral state in both infected and surrounding cells that limits virus spread (1).
Besides the IFN-α/β pathway, the cellular integrated stress response (ISR) has been increasingly recognized as an antiviral pathway (2). Cells can halt translation upon different kinds of cellular stress, including viral infection. Double-stranded (ds) RNA-dependent protein kinase (PKR), an ISG, detects cytosolic viral double-stranded RNA (dsRNA) and activates the stress response in virus-infected cells (3). Detection of dsRNA by PKR results in structural rearrangements in PKR leading to auto-phosphorylation. Next, activated PKR phosphorylates the alpha subunit of eIF2. Phosphorylation of eIF2α halts translation, thereby interfering with the synthesis of viral proteins and virus propagation (3). Upon halted translation, specific nucleating factors bind specific stalled mRNA-protein complexes (mRNPs) to form cytoplasmic aggregates known as stress granules (SGs). These nucleating factors include Ras GTPase-activating protein-binding proteins 1 and 2 (G3BP1 and G3BP2), T cell-restricted intracellular antigen 1 (TIA-1) and the closely related TIAR, and multiple other factors (e.g., Caprin1 and Sam68). SGs are highly dynamic cytoplasmic aggregates. Scaffold proteins continuously cycle between the cytosol and SGs, facilitating mRNA triage in order to direct translation toward resolving the cellular stress (2, 4).
SGs have been proposed to act as a platform for antiviral signaling (2, 4–7). The local enrichment of nucleating factors and mRNPs most likely constitutes a warning signal in the cell, recruiting numerous signaling molecules to the SGs (4). These include several signaling molecules involved in the innate antiviral response, such as RIG-I (8), MDA5 (9), TRAF2 (10), and PKR (5). Despite these observations, the importance and role of SGs as an antiviral signaling platform are still poorly understood. The most important argument that supports an antiviral role of SGs is the fact that many viruses have developed antagonistic mechanisms that interfere with SG assembly. These mechanisms include inhibition of PKR, cleavage of SG scaffold proteins by viral proteases, and sequestration of SG scaffold proteins by viral proteins (2, 11–17).
Picornaviruses, a large family of small nonenveloped viruses that contain single-stranded 7- to 8-kb RNA genomes of positive polarity, actively antagonize both RLR signaling and the stress response. Viruses belonging to the genus Enterovirus, best known for poliovirus, actively suppress RLR signaling through their viral proteases, 2Apro and 3Cpro, which cleave multiple proteins in the RLR signaling pathway (18–22). 2Apro and 3Cpro also cleave signaling molecules of the IFN-α/β signaling pathway to prevent the expression of ISGs (23). Enteroviruses also actively antagonize the cellular stress response, as SGs rapidly dissolve as infection progresses. This observation has been linked to the cleavage of the SG scaffold protein G3BP1 by the viral 3Cpro (24), although other viral proteins have also been suggested to affect SG formation (25). Similarly, caliciviruses encode NS6, a 3C-like protease, which also cleaves G3BP1 to prevent SG assembly (11). Members of the genus Cardiovirus (e.g., encephalomyocarditis virus [EMCV] and Theiler’s virus) encode the leader protein, one antagonist that suppresses both RLR signaling and the stress response pathway (9, 18, 26, 27). The mechanism of action of the cardiovirus leader protein, which lacks proteolytic activity, is largely unknown. Besides the leader protein, the 3Cpro of cardioviruses has been implicated in suppressing SG formation by cleaving G3BP1 (28), although this was not observed in another study (26).
Another well-known picornavirus is foot-and-mouth disease virus (FMDV), which is one of the most notorious pathogens in the global livestock industry. FMDV infections are well known for their devastating effects on animal health and product safety and can have a huge economic impact. FMDV is a member of the genus Aphthovirus, which also contains bovine rhinitis A and B viruses, and equine rhinitis A virus (ERAV). Like cardioviruses, aphthoviruses encode a leader protein. The FMDV leader protein is a papain-like protease (Lpro) that is best known for cleaving translation initiation factor eIF4G, thereby shutting off host mRNA translation (29). The efficient spread of FMDV infection in vivo relies on Lpro (30). Besides causing translational shutoff of host proteins, Lpro suppresses the induction of IFN-α/β (31). For this, several mechanisms have been proposed. Lpro causes the degradation of NF-κB subunit p65/RelA (32), decreases the levels of interferon regulatory factor 3 (IRF3) and IRF7 (33), and has been proposed to possess deubiquitinase (DUB) and deISGylase activities (34, 35). Having DUB activity is a common strategy for viruses to manipulate IFN signaling pathways (36). The 3Cpro of aphthoviruses has also been implicated in suppressing IFN-α/β production by cleaving NF-κB essential modulator (NEMO), an adaptor protein that is essential for activating NF-κB and IRF signaling pathways (37), and by interfering with the JAK/STAT signaling pathways by manipulating the STAT1/STAT2 nuclear translocation (38).
Little is known about whether and how aphthoviruses suppress the cellular stress response. Here, we demonstrate that aphthoviruses can suppress the cellular stress response through the action of the viral protease Lpro, and we identify SG scaffold proteins G3BP1 and G3BP2 as new substrates of Lpro.
RESULTS
The stress response pathway is suppressed during FMDV infection.
To investigate whether FMDV antagonizes the stress response pathway, we infected LFPK cells that stably express the integrin αvβ6 (LFPK αvβ6 cells), the entry receptor of FMDV (39), with FMDV-A12 and performed immune fluorescence staining to visualize SG formation (Fig. 1A). As a positive control, cells were treated with sodium arsenite, a known activator of the stress response pathway (Fig. 1B). SGs were visualized by anti-G3BP1 staining, while viral replication was monitored by staining for the capsid protein VP1. In LFPK αvβ6 cells, FMDV replication is relatively fast; virus production and release can be observed as early as 5 h postinfection (hpi) (40). Consistently, we observed a clear accumulation of VP1 both at 4 and 6 hpi, indicative of efficient viral replication. Notwithstanding this, only a few G3BP1 foci were detected in the infected cells. In contrast, arsenite treatment of the cells led to extensive SG formation (Fig. 1B). These data show that FMDV infection does not induce SG formation. To investigate whether the virus actively suppresses SG formation, we infected LFPK αvβ6 cells with FMDV-A12 and added sodium arsenite during the last 30 min of infection (Fig. 1C). No SGs were observed in infected cells, whereas many SGs were observed in uninfected neighboring cells. Similar observations were made using IBRS-2 cells (Fig. 1D), providing further support that FMDV actively antagonizes SG formation.
FIG 1.

FDMV suppresses SG formation. (A) LFPK αvβ6 cells were infected with FMDV-A12 and fixed at 4 and 6 hpi, and immunofluorescence staining was performed for the viral capsid protein VP1 (green) and the SG marker G3BP1 (red). (B) LFPK αvβ6 cells were treated with 500 μM sodium arsenite (NaArs) for 30 min to induce SG formation (as previously described [24, 26]), which were subsequently visualized by immunofluorescence staining for the SG marker G3BP1 (red). (C) LFPK αvβ6 cells were infected with FMDV-A12, treated with 500 μM sodium arsenite for the last 30 min to induce SG formation, and fixed at 4 hpi. Immunofluorescence staining was performed for the viral capsid protein VP1 (green) and the SG marker G3BP1 (red). (D) Similar to the image shown in panel C but with IBRS-2 cells.
FMDV and ERAV Lpro suppress SG formation when introduced into a recombinant EMCV containing an inactive stress antagonist.
To determine whether the viral proteases are involved in suppressing SG formation, we generated recombinant EMCVs harboring FMDV Lpro or 3Cpro at the start of the open reading frame (ORF) of EMCV-LZn (EMCV-FL or EMCV-F3C, respectively) (Fig. 2A). EMCV-LZn is a mutant EMCV strain (mengovirus) in which the antagonistic functions of the leader (L) protein are abolished by inactivating mutations in the zinc finger domain of L. This virus no longer suppresses the IFN-α/β or stress response (9, 18, 41, 42) and therefore provides a tool to study the antagonistic function of a heterologous viral protein (18, 43). The inserted proteases were flanked at their N termini by the first 6 amino acids of EMCV L to ensure efficient translation (44, 45) and by an EMCV 3Cpro cleavage site at their C termini for efficient viral polyprotein processing. To determine whether the function of FMDV proteases is conserved in other aphthoviruses, we also constructed chimeric viruses encoding Lpro and 3Cpro of ERAV (EMCV-EL and EMCV-E3C, respectively). After recovering these chimeric EMCVs, we characterized viral replication kinetics by reverse transcription-quantitative PCR (RT-qPCR) analysis of EMCV viral RNA (vRNA) levels at various time points (Fig. 2B). As described previously (18, 41), mutation of the zinc-finger domain of EMCV L slightly reduced vRNA levels. Heterologous expression of either FMDV or ERAV Lpro (EMCV-FL and EMCV-EL) rescued viral replication to levels similar to those of wild-type (wt) EMCV, whereas expression of 3Cpro (EMCV-F3C and EMCV-E3C) did not affect vRNA levels.
FIG 2.

Aphthovirus Lpro suppresses SG formation. (A) Schematic representation of viral genome of chimeric EMCV viruses that were constructed for this study. The endogenous stress response antagonist (leader) was inactivated by point mutations in its Zn finger domain (C19A/C22A), and subsequently, the genes encoding Lpro and 3Cpro were introduced at the 5′ end of the EMCV open reading frame. (B) HeLa R19 cells were infected at an MOI of 10 with the indicated chimeric EMCVs, and subsequently, virus growth kinetics were determined using RT-qPCR analysis with EMCV-specific primers. (C) HeLa R19 cells were infected at an MOI of 10 with the indicated chimeric EMCVs and fixed at 6 hpi. Subsequent immunofluorescence staining was performed against the SG markers G3BP1 (green), TIA-1 (red), and G3BP2 (cyan). (D) Quantification of data from panel C in which the numbers of SGs and SG sizes were analyzed for at least 50 cells per condition. One-way ANOVA with the Bonferroni post hoc test was used to determine statistical significance. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Subsequently, we infected HeLa cells with these recombinant EMCVs and visualized SGs at 6 hpi using immunofluorescence microscopy. The SGs were visualized using anti-G3BP1, anti-G3BP2, and anti-TIA-1 staining. Consistently with previous results (9), EMCV infection suppressed SG formation, while EMCV-LZn lost this ability (Fig. 2C, top two rows). Compared to that with EMCV-LZn, infection with EMCV-FL or EMCV-EL resulted in noticeably fewer and smaller SGs. In contrast, we observed no effect of 3Cpro on SG formation (Fig. 2C). Similar observations were made at 8 hpi (data not shown). Quantification of SG size and the number of SGs showed a consistent decrease in both parameters upon the heterologous expression of Lpro (EMCV-FL and EMCV-EL) but not upon the expression of 3Cpro (EMCV-F3C and EMCV-E3C) (Fig. 2D). Intriguingly, the viruses that have the ability to suppress SG formation (EMCV-FL and EMCV-EL) also replicated to levels similar to that of the wild-type (wt) EMCV, while the viruses that failed to do so (EMCV-F3C and EMCV-E3C) replicated to levels similar to that of EMCV-LZn. This suggests that the ability of Lpro to suppress SG formation, likely combined with its ability to suppress the type I IFN response, is beneficial to the replication of EMCV-FL and EMCV-EL.
To determine if the inhibitory effect of Lpro on SG formation is dependent on Lpro’s catalytic activity, we generated an EMCV encoding a catalytically inactive FMDV Lpro (EMCV-FL C51A) and investigated the ability of this virus to suppress SG formation (Fig. 3). HeLa cells were infected with EMCV-LZn, EMCV-FL, or EMCV-FL C51A, and SGs were visualized by immunofluorescence staining against G3BP1, eIF3, and G3BP2. Infection with EMCV-FL C51A resulted in SG formation (Fig. 3A), and the SGs were of usual size (Fig. 3B). Collectively, these data show that Lpro, via its catalytic activity, suppresses SG formation.
FIG 3.

Effect of Lpro on SG formation is dependent on the catalytic activity of Lpro. (A) HeLa R19 cells were infected at an MOI of 10 with chimeric EMCV expressing Lpro (EMCV-FL) or a catalytically inactive Lpro (EMCV-FL C51A) and fixed at 6 hpi. SG formation was visualized by immunofluorescence staining for the SG markers G3BP1 (green), eIF3 (red), and G3BP2 (cyan). (B) Quantification of data from panel A in which SG sizes were analyzed for at least 50 cells per condition. One-way ANOVA with the Bonferroni post hoc test was used to determine statistical significance. *, P < 0.05.
Lpro prevents SG formation in FMDV-infected cells.
To investigate whether Lpro also suppresses SG formation in aphthovirus-infected cells, we utilized a recombinant FMDV strain in which the leader-coding region was deleted (leaderless virus [LLV]) (46). We infected LFPK αvβ6 cells with wild-type FMDV or LLV and visualized SG formation at 4 hpi. Virus replication was monitored via staining for the capsid protein VP1, while SGs were visualized by anti-G3BP1 staining (Fig. 4A). Similar amounts of VP1 were observed in cells infected with wild-type FMDV or LLV, indicating efficient viral replication irrespective of the presence or absence of Lpro, consistent with previous results (46). Infection with wild-type FMDV did not induce SG formation (Fig. 1 and 4A). In contrast, infection with FMDV LLV triggered SG formation (Fig. 4A), and upon quantification, we observed a consistent difference in the number of SG-positive cells and the number of SGs per cell between wild-type FMDV-infected cells and LLV-infected cells (Fig. 4B). These data demonstrate that FMDV LLV cannot block SG formation. Although we cannot formally exclude the involvement of some RNA element in the deleted region, our data suggest that Lpro is needed to suppress SG formation during FMDV infection.
FIG 4.

Leaderless FMDV is unable to suppress SG formation. (A) LFPK αvβ6 cells were infected with FMDV-A12 or leaderless FMDV-A12 (LLV) and fixed at 4 hpi, and immunofluorescence staining was performed for the viral capsid protein VP1 (green) and the SG marker G3BP1 (red). (B) Quantification of data from panel A in which the percentages of SG-positive infected cells and the numbers of SGs per cell were determined in at least 50 cells. A one-tailed t test was used to determine statistical significance. **, P < 0.01; ***, P < 0.001.
Lpro does not affect PKR signaling.
We investigated whether aphthovirus proteases have an effect upstream of SG assembly, at the level of stress response signaling. To this end, we assessed the phosphorylation of PKR and eIF2α by using flow cytometry. Infection of HeLa cells with EMCV-LZn is known to activate PKR (43), which results in the phosphorylation of eIF2α and a subsequent halt in translation. Indeed, we observed increased levels of phosphorylated PKR and phosphorylated eIF2α in EMCV-LZn-infected cells (Fig. 5A). The recombinant EMCVs expressing Lpro (EMCV-FL or EMCV-EL) or 3Cpro (EMCV-F3C or EMCV-E3C) also failed to suppress PKR or eIF2α phosphorylation (Fig. 5B), indicating that neither Lpro nor 3Cpro affects PKR signaling, at least in HeLa cells infected with chimeric EMCV viruses.
FIG 5.
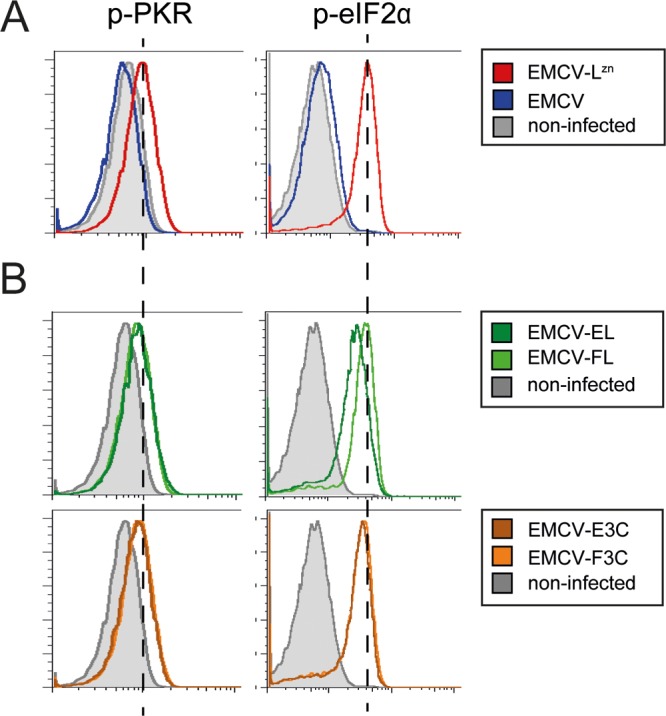
Lpro does not inhibit PKR signaling. HeLa R19 cells were infected at an MOI of 10 with EMCV or EMCV-LZn (A) or the indicated chimeric EMCVs (B) and fixed at 6 hpi. Subsequent flow cytometry staining was performed for dsRNA and p-PKR or for dsRNA and p-eIF2α. Graphs represent the levels of p-PKR or p-eIF2α in dsRNA-positive (infected) cells. Dashed lines indicate the levels of p-PKR and p-eIF2α in EMCV-LZn infected cells.
SG scaffold proteins are cleaved during aphthovirus infection.
Many viruses target SG scaffold proteins to antagonize SG formation. Therefore, we investigated whether SG scaffold proteins are cleaved during FMDV infection. LFPK αvβ6 cells were infected with wild-type FMDV-A12 or A12-LLV, and the lysates were subjected to Western blot analysis using a G3BP1 antibody. Infection with wild-type FMDV, but not LLV, resulted in the cleavage of G3BP1 (Fig. 6A). The predicted molecular weight of G3BP1 is 52 kDa, but we consistently observed the protein migrating at ∼70 kDa. Upon FMDV infection, we detected two cleavage products that migrate at apparent molecular weights of ∼65 kDa and ∼43 kDa (but which are likely smaller), but this was not observed in cells infected with FMDV LLV. An ∼65-kDa cleavage product was first detected at 4 hpi, and both cleavage products were clearly visible at 6 hpi. Collectively, our data suggest that G3BP1 is cleaved at multiple positions during FMDV infection. We also tested the lysates with antibodies against other human SG scaffold proteins, such as TIA-1 and eIF3, but unfortunately, these antibodies did not show reactivity with swine proteins.
FIG 6.
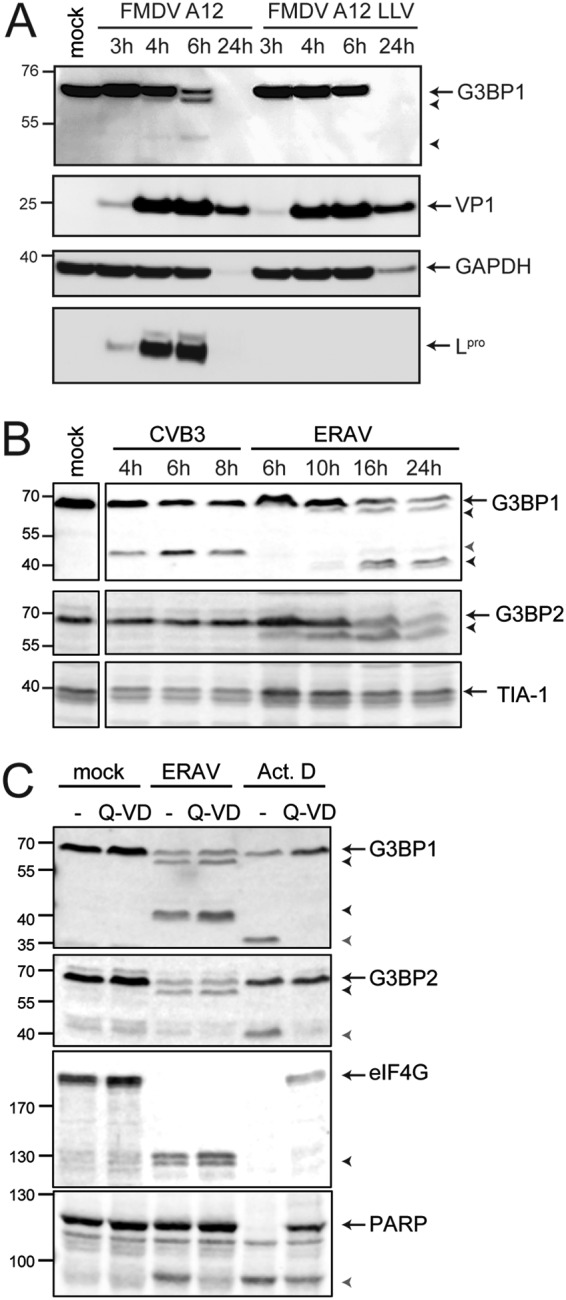
SG scaffold proteins are cleaved during aphthovirus infection. (A) LFPK αvβ6 cells were infected with FMDV-A12 or leaderless FMDV-A12 (LLV), and cells were lysed at the indicated times postinfection. Western blot analyses were performed for G3BP1, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and the viral proteins VP1 and Lpro. (B) HeLa R19 cells were infected at an MOI of 10 with CVB3 or ERAV, and the cells were lysed at the indicated times postinfection. Western blot analyses were performed for SG proteins G3BP1, G3BP2, and TIA-1. (C) HeLa R19 cells were infected with ERAV at an MOI of 10 or treated with 5 μg/ml actinomycin D. Subsequently, the cells were incubated for 16 h in the presence or absence of 10 μM Q-VD. Cell lysates were subjected to Western blot analyses for G3BP1 and G3BP2, translation initiation factor 4G (eIF4G), and PARP. Arrows indicate full-length proteins, black arrowheads indicate aphthovirus-induced cleavage products, and gray arrowheads indicate cleavage products induced upon CVB3 infection (B) or cleavage products induced upon the induction of apoptosis (C).
To investigate whether the ability to cleave SG scaffold proteins is conserved among aphthoviruses, we next studied the integrity of G3BP1, as well as G3BP2 and TIA-1, in ERAV-infected cells. To this end, HeLa cells were infected with ERAV or coxsackievirus B3 (CVB3; genus Enterovirus), a control virus that is known to cleave G3BP1 but not G3BP2 or TIA-1 (24, 25, 47) (Fig. 6B). Upon infection with ERAV, cleavages of G3BP1 and G3BP2, but not TIA-1, were observed (Fig. 6B). For G3BP1, we observed a large cleavage product that migrated at ∼65 kDa and a doublet that ran at ∼40 kDa. This ∼40-kDa doublet was not observed in FMDV-infected cells. Meanwhile, we barely detected an ∼43-kDa cleavage product in ERAV-infected cells, which we did observe in FMDV-infected cells. Thus, FMDV infection (Fig. 6A) and ERAV infection (Fig. 6B) induced similar, but not identical, cleavages of G3BP1. G3BP2, like G3BP1, consistently migrated at ∼70 kDa but has a predicted molecular weight of only 54 kDa. Upon ERAV infection, we observed one G3BP2 cleavage product, running at ∼65 kDa (although in reality, this product may be smaller).
Next, we investigated whether aphthovirus proteases cleave G3BP1 and G3BP2 directly or indirectly by triggering apoptosis and activating cellular caspases (Fig. 6C). We infected HeLa cells with ERAV and treated them with the pan-caspase inhibitor Q-VD-PH (Q-VD) to block caspase activity. While cleavage of poly(ADP-ribose) polymerase (PARP), a known caspase substrate, was inhibited by Q-VD (as demonstrated by the reduced amount of the PARP cleavage product), ERAV-induced G3BP1 and G3BP2 cleavages were unaffected. Induction of apoptosis, by treatment with actinomycin D (Act D), also induced cleavage of G3BP1 and G3BP2. Importantly, these caspase-mediated G3BP1 and G3BP2 cleavage products had different molecular weights than those observed during ERAV infection and were inhibited by the addition of Q-VD. Taken together, these results suggest that cleavage of G3BP1 and G3BP2 in aphthovirus-infected cells is the result of a direct intervention of a viral protease rather than the activation of cellular caspases.
Lpro cleaves G3BP1 and G3BP2.
It has been suggested that EMCV infection triggers G3BP1 cleavage via 3Cpro (28). This would prevent us from investigating whether aphthovirus Lpro cleaves G3BP1 and G3BP2 by infecting cells with EMCV-FL and EMCV-EL. However, the cleavage of G3BP1 by EMCV is debated and could not be reproduced in another study (26). As such, we first infected HeLa cells with EMCV, EMCV-LZn, or CVB3 (as a positive control) and determined the integrity of G3BP1 and G3BP2 via Western blot analysis (Fig. 7A). We did not detect cleavage of G3BP1 upon EMCV infection by using two different antibodies that recognize different epitopes within G3BP1. Our observations are consistent with reports that EMCV suppresses SG formation via its leader protein (9, 26).
FIG 7.

Aphthovirus Lpro and 3Cpro cleave SG proteins. (A) HeLa R19 cells were infected at an MOI of 10 with EMCV, EMCV-LZn, or CVB3, and the cells were lysed at the indicated times postinfection. Western blot analyses were performed with two different antibodies against G3BP1 (recognizing different epitopes) and G3BP2. (B) HeLa R19 cells were infected at an MOI of 10 with the indicated chimeric EMCVs or ERAV, and the cells were lysed at 8 hpi (ECMVs) or 10 hpi (ERAV). Subsequent Western blot analyses were performed for G3BP1 and G3BP2. *, additional (artificial) G3BP2 cleavage product. (C) HeLa R19 cells were infected at an MOI of 10 with EMCV-FL or EMCV-FL C51A, and the cells were lysed at the indicated time points. Subsequent Western blot analyses were performed for G3BP1 and G3BP2. (D and E) HEK293T cells were cotransfected with a Flag-tagged G3BP1 or G3BP2 and an EGFP-tagged viral protease, as indicated. At 16 h posttransfection, the cells were lysed and subjected to Western blot analyses for FLAG and EGFP. Arrows indicate full-length proteins, black arrowheads indicate Lpro-induced cleavage products, and the gray arrowhead indicates 3Cpro-induced cleavage products. (F) Schematic representation of approximate cleavage sites, based on the apparent molecular weights of the cleavage products, in G3BP1 and G3BP2 for the different aphthovirus proteases. ■, region in G3BP2 containing multiple FMDV Lpro cleavage sites.
To investigate which aphthovirus protease cleaves SG scaffold proteins, we infected HeLa cells with the EMCVs expressing Lpro and 3Cpro (EMCV-FL, EMCV-EL, EMCV-F3C, and EMCV-E3C) and performed Western blot analyses for G3BP1 and G3BP2. To compare the migration patterns of the cleavage products to those observed during aphthovirus infection, a lysate of ERAV-infected cells was run on the same gel (Fig. 7B). In agreement with the observation that leaderless FMDV failed to cleave G3BP1 (Fig. 6A), we observed that infection with EMCVs expressing Lpro (EMCV-FL and EMCV-EL) resulted in the cleavage of G3BP1 and G3BP2 (Fig. 7B). A G3BP1 cleavage product migrating at ∼65 kDa was observed during infection with EMCV-FL or EMCV-EL. A similarly sized cleavage product prominently appeared during FMDV and ERAV infections. An ∼43-kDa G3BP1 cleavage product, similar to the product that accumulated in cells infected with FMDV, also accumulated in cells infected with EMCV-FL. Such an ∼43-kDa product was barely detected in ERAV-infected cells and did not appear in cells infected with EMCV-EL. Instead, a doublet cleavage product migrating at ∼40 kDa was observed in ERAV-infected cells. We observed a similar doublet in cells infected with EMCV-F3C and EMCV-E3C, suggesting that 3Cpro can also cleave G3BP1. Intriguingly, such an ∼40-kDa doublet was not observed in FMDV-infected cells. 3Cpro can also cleave G3BP1, albeit inefficiently, and this had no effect on the integrity of SGs (Fig. 2). In ERAV-infected cells, we detected a G3BP2 cleavage product running at ∼65 kDa (Fig. 6B and Fig. 7B). We observed a similar G3BP2 cleavage product, running at ∼65 kDa, upon infections with EMCV-FL and EMCV-EL (Fig. 7B), suggesting that Lpro cleaves G3BP2. Notably, infection with EMCV-FL resulted in a number of other bands that showed reactivity with the G3BP2 antibody, ranging from ∼55 kDa to ∼45 kDa. In particular, infection with EMCV-FL and EMCV-EL induced a product of ∼45 kDa (indicated with * in Fig. 7B), that was not observed during ERAV infection. We did not observe cleavage of G3BP2 upon infection with either EMCV-F3C or EMCV-E3C. Together, these data suggest that Lpro has a major impact on the integrity of G3BP1 and G3BP2 in infected cells.
To gain more insight into the timing of G3BP1 and G3BP2 cleavage by Lpro, we next infected HeLa cells with EMCV-FL for different durations and performed Western blot analysis of G3BP1 and G3BP2 (Fig. 7C). The ∼65-kDa G3BP1 cleavage product was observed at 6 and 8 hpi. Meanwhile, we observed the ∼43-kDa fragment at 8 hpi, suggesting that the first cleavage event allows the second cleavage event to occur. This sequential cleavage was also seen in FMDV-infected cells (Fig. 6A). A G3BP2 cleavage product, running at ∼65 kDa, was first observed at 6 hpi but was more clearly visible at 8 hpi (Fig. 7C). As anticipated, infection with an EMCV encoding the catalytically inactive FMDV Lpro (EMCV-FL C51A) failed to cleave G3BP1 and G3BP2 (Fig. 7C). Collectively, our observations strongly suggest that Lpro is responsible for cleaving SG scaffold proteins in aphthovirus-infected cells.
To determine the location of the Lpro and 3Cpro cleavage sites in G3BP1, we coexpressed N-terminally Flag-tagged G3BP1 with enhanced green fluorescent protein (EGFP)-tagged Lpro or 3Cpro (Fig. 7D). Under these conditions, we observed cleavage patterns of G3BP1 that resembled those observed in FMDV- and ERAV-infected cells and upon infection with the recombinant EMCVs. As the Flag tag is located at the N terminus of G3BP1, it can be concluded that the Lpro cleavage site yielding an ∼65-kDa product is located in the C-terminal part of G3BP1. A second FMDV Lpro cleavage site, yielding an ∼43-kDa fragment, and also the 3Cpro cleavage sites, yielding an ∼40-kDa doublet, are located close to the middle of the protein but slightly more toward the C terminus. We also coexpressed Flag-tagged G3BP2 with EGFP-tagged Lpro (Fig. 7E). We observed G3BP2 cleavages that were highly similar to those observed upon infection with ERAV, EMCV-FL, and EMCV-EL. This suggests that the major Lpro cleavage site in G3BP2 is located in the C-terminal part of the protein. Notably, we did not observe the extra ∼45-kDa cleavage product that was apparent upon infection with EMCV-FL or EMCV-EL, supporting the idea that this product is an artifact. We observed multiple cleavage products migrating at ∼55 kDa upon overexpression of FMDV Lpro, similar to that in cells infected with EMCV-FL. These products were not observed upon infection with EMCV-EL (Fig. 7B) and also not upon overexpression of ERAV Lpro. The approximate Lpro and 3Cpro cleavage sites in G3BP1 and G3BP2, based on the apparent molecular weights of the cleavage products, are schematically represented in Fig. 7F. Collectively, these results indicate that the major cleavage products of G3BP1 and G3BP2, migrating at ∼65 kDa, are generated upon cleavage of the C termini of these proteins. The additional G3BP1 and G3BP2 cleavage products, produced by FMDV Lpro and migrating at ∼43 kDa and ∼55 kDa, respectively, are generated upon cleavage in the C-terminal half of the protein, close to the middle of the protein. The cleavage sequences of some substrates of Lpro are known, but we were not successful in identifying the cleavage sites in G3BP1 and G3BP2. Nevertheless, the cleavage sites of aphthovirus Lpro in G3BP1 clearly differ from those of enterovirus 3Cpro (Fig. 6B).
DISCUSSION
In recent years, SGs have increasingly been seen as important antiviral signaling platforms (2, 4–7). Several molecules involved in RLR signaling and stress response signaling have been found to localize in SGs (5, 8–10). Many viruses interfere with stress responses and the formation of SGs. Several picornaviruses belonging to the Enterovirus and Cardiovirus genera have been shown to actively suppress the stress response pathway (9, 24–26), but knowledge about members of the Aphthovirus genus is limited. Here, we provide evidence that aphthoviruses suppress SG formation through Lpro. This papain-like protease is well known for its ability to cleave translation initiation factor eIF4G and thereby to shut off host translation (29). We showed that SG formation is suppressed in cells infected with wild-type FMDV but not in a leaderless mutant FMDV. Heterologous expression of Lpro of FMDV or ERAV in a mutant EMCV lacking its own leader protein also suppressed SG formation. Lpro did not affect PKR activation and eIF2α phosphorylation. Instead, Western blot analysis showed that SG scaffold proteins G3BP1 and G3BP2 were cleaved in aphthovirus-infected cells. Similar cleavages were observed in cells infected with mutant EMCV expressing either Lpro and upon coexpression of Flag-tagged G3BP1/2 with Lpro. Collectively, our data demonstrate an essential role of Lpro in suppressing SG formation in aphthovirus-infected cells. The Lpro-mediated cleavage of SG scaffold proteins may underlie the inhibition of SG formation, although it cannot be excluded that a yet-unidentified cleavage of another cellular protein(s) involved in stress signaling or SG formation also contributes to this inhibition. Besides dsRNA-dependent and PKR-mediated SG formation, SG formation induced by sodium arsenite, which induces stress via a reactive oxygen species (ROS)-dependent mechanism through a different kinase (HRI), was inhibited. This lends support to the idea that Lpro has an inhibitory effect on the formation and/or stability of SGs by cleaving essential SG components.
Cleavage of SG components likely also underlies the inhibition of SG formation by enteroviruses. Enterovirus infection results in the formation of small SGs, which disappear as infection progresses. This observation has been attributed to the cleavage of G3BP1 by enterovirus 3Cpro (24). However, evidence that enterovirus 2Apro rather than 3Cpro is responsible for the suppression of SG formation in enterovirus-infected cells was presented recently (48). The expression of 2Apro, but not 3Cpro, of several enteroviruses was shown to inhibit SG formation induced by sodium arsenite or heat shock. Furthermore, infection by a recombinant EV-A71 in which 2Apro activity was inactivated, led to the formation of typical SGs. The mechanism by which 2Apro suppresses SG formation has not been elucidated, but these findings challenge the importance of the cleavage of G3BP1 as an antagonizing mechanism to inhibit SG formation. Enterovirus 3Cpro does not cleave G3BP2 (25). Possibly, the simultaneous cleavage of both G3BP1 and G3BP2 by Lpro is required to suppress SG formation. In line with this, it has been shown that SG formation is blocked in cells in which G3BP1 and G3BP2 are depleted but not in cells lacking only one of the G3BPs (49). Alternatively, the G3BP1 cleavage product generated by Lpro, which differs from that of enterovirus 3Cpro, may have a dominant negative effect on SG formation. The exact mechanisms used by aphthovirus Lpro and enterovirus 2Apro, both of which are considered “security proteins” (50), to suppress SG formation remains to be elucidated.
We found a strongly reduced amount of G3BP1-containing SGs in cells infected with wild-type FMDV. This observation is in agreement with previous observations that some small SGs that contain TIA-1 and Sam68, but lack G3BP1, are formed in FMDV-infected cells (51, 52). These small SGs resemble the small “atypical” SGs observed in cells expressing enterovirus 2Apro (25, 48). These atypical SGs consist of cellular mRNAs of which translation is halted due to the cleavage of eIF4G by 2Apro (25, 48). There is conflicting literature about the composition of these atypical SGs. Some studies have shown that these atypical SGs contain specific SG markers, i.e., TIA-1, TIAR, and Sam68, but lack other SG markers, G3BPs and eIFs (48, 53), whereas others have shown that G3BP1-containing SGs are formed during enterovirus infection (24, 54) or upon overexpression of enterovirus 2Apro (25). Upon infection with recombinant EMCV expressing either FMDV Lpro or ERAV Lpro, we observed small SGs containing TIA-1, G3BPs, and eIF3. Although it is unclear what the exact composition of atypical SGs is, it seems plausible that enterovirus 2Apro and aphthovirus Lpro can induce the formation of atypical SGs via their shared ability to inhibit host cell translation.
SG formation is induced upon the activation of eIF2α kinases, such as PKR and PERK. While enterovirus infection induces PKR activation, leading to an increase in both phosphorylated PKR (p-PKR) and p-eIF2α levels (55, 56), FMDV infection was shown to block the activation of this pathway (57), possibly via 3Cpro-dependent lysosomal degradation of PKR (57). It has also been reported that the titers of FMDV LLV can be rescued in PKR knockout cells (58), hinting at a possible role for Lpro in suppressing PKR signaling. We failed to observe any inhibitory effect on PKR signaling in cells infected with recombinant EMCV expressing either Lpro or 3Cpro. This may be a limit of our experimental setup to express these proteases outside the context of aphthovirus infection; e.g., it may suggest that antagonizing PKR signaling requires the actions of multiple aphthovirus proteins. Nevertheless, we observed a clear inhibitory effect of Lpro on SG formation in cells infected with the chimeric EMCV Lpro, independent of any effect on PKR signaling. Obviously, this does not rule out the possibility that the effects of Lpro and/or 3Cpro on PKR signaling also contribute to the inhibition of SG formation in aphthovirus-infected cells, where this inhibition seems to be more pronounced than in cells infected with EMCV-Lpro. Multiple complementary SG inhibitory mechanisms may coexist in aphthovirus-infected cells, one of which in mediated by FMDV Lpro.
Recently, G3BP1 was reported to bind to the internal ribosome entry site (IRES) of FMDV and function as a translation inhibitor, and it was shown that FMDV 3Cpro induced the cleavage of G3BP1 (59). Interestingly both the N-terminal and C-terminal G3BP1 cleavage products maintained the negative effect on translation (59). In our study, we observed that FMDV and ERAV 3Cpro can cleave G3BP1 to some extent when expressed through recombinant EMCV. While this cleavage did not affect SG formation, it might thus affect viral translation.
In this study, we used a recombinant EMCV lacking its stress antagonist, the leader protein, to express heterologous proteinases of FMDV and ERAV. It has been suggested by others that EMCV suppresses SG formation through the 3Cpro-mediated cleavage of G3BP1 (28). As reported previously for both EMCV and TMEV (9, 26), we show that leader-inactivated EMCV (EMCV-LZn) failed to suppress SG formation, whereas no SGs were observed in cells infected with wild-type EMCV. The leader of EMCV lacks proteolytic activity, and no cleavage of either G3BP1 or G3BP2 was observed in EMCV-infected cells. Thus, our data challenge the suggestion that EMCV 3Cpro suppresses SG formation by cleaving G3BP1.
In conclusion, in this study, we present evidence that aphthoviruses actively suppress SG formation through the proteolytic activity of Lpro, and we identified two SG scaffold proteins, G3BP1 and G3BP2, as new targets of Lpro. These results add aphthoviruses to a growing list of viruses that prevent SG assembly to antagonize the stress response. These viruses include several flaviviruses (e.g., hepatitis C virus, dengue virus, and West Nile virus), which use viral subgenomic RNA or nonstructural proteins to sequester SG proteins, such as G3BP1, TIA-1, and TIAR, to prevent SG formation (13, 14), as well as alphaviruses, such as Semliki Forest virus (SFV), which prevents SG formation by sequestrating G3BP1 and G3BP2 by its nsP3 protein (15–17). Although these examples all hint to an important antiviral role of SGs, their exact role as a platform for antiviral signaling is still poorly understood, and more studies are warranted to potentially develop new tools to fight virus infection.
MATERIALS AND METHODS
Cells and viruses.
HeLa R19, BHK21, and HEK293T cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum (FCS; vol/vol). LFPK αvβ6 cells (40) were obtained from the Foreign Animal Disease Diagnostic Laboratory (FADDL) at the PIADC. These cells were maintained in minimal essential medium (MEM) supplemented with 10% FCS (vol/vol), 1% antibiotics, and nonessential amino acids. BHK21 cells used for FMDV propagation were maintained in MEM supplemented with 10% FCS (vol/vol), 10% tryptose phosphate broth, 1% antibiotics, and nonessential amino acids. The FMDV-A12 WT (wild type) was generated from the full-length serotype A12 infectious clone pRMC35 (60), and A12-LLV2 (leaderless virus) was derived from the infectious clone lacking the Lb coding region, pRM-LLV2 (46). Viruses were propagated in BHK-21, concentrated by polyethylene glycol precipitation, titrated on BHK-21 cells, and stored at −70°C. ERAV (NM-11/67 strain; a gift from D. Rowlands and T. Tuthill [University of Leeds, Leeds, United Kingdom]) (61) was obtained after passage on HeLa R19 cells and subsequently concentrated by ultracentrifugation through a 30% sucrose cushion at 140,000 × g for 16 h in an SW32Ti rotor and stored at −80°C. Recombinant EMCVs were generated by cloning the genes of interest into the XhoI/NotI restriction sites from the pM16.1-VFETQG-Zn infectious clone that was described previously (43); the Strep2 tag was omitted in the viruses used in this study. Viruses were recovered by transfection of runoff RNA transcripts into BHK-21 cells. Upon total cytopathic effect (CPE), the viruses were concentrated by ultracentrifugation (as described for ERAV) and stored at −80°C.
Expression plasmids.
The FMDV Lpro gene was obtained by PCR from the pM16.1 Mengo-FL infectious clone (45) using the following oligonucleotides: 5′-CTCGAGCTGACACTGTACAACGGTGAG-3′ and 5′-GCGGCCGCTTTGAGCTTGCGTTGAACCTTG-3′. The FMDV 3Cpro gene was obtained by PCR using the oligonucleotides 5′-AAAAACTCGAGAGTGGTGCCCCACCG-3′ and 5′-AAAAGCGGCCGCCTCGTGGTGTGGTTCGGG-3′ and the pBind-3C-VP16 FMDV plasmid (62) as the template. The ERAV Lpro and 3Cpro genes were obtained by PCR from ERAV viral RNA (NM-11/67 strain) using the following oligonucleotides: 5′-CTCGAGATGGACAAATTCTTGCAAAAGAAAAC-3′ and 5′-GCGGCCGCCTTCTCATACCTCTGATGTAAC-3′ for Lpro and 5′-AAAACTCGAGACTGGTGTGCCAGCAACTG-3′ and 5′-AAAAAGCGGCCGCCTGTTTCTGAGGGAGAGTTCGC-3′ for 3Cpro. The oligonucleotides encode flanking XhoI and NotI restriction sites that were used to ligate the PCR products into the desired plasmids. The 3Cpro genes were ligated into the pcDNA-GFP vector described previously (11). The Lpro genes were ligated into the pIRES-EGFP-MCS plasmid. This is an pIRES2-EGFP-based vector in which the multiple-cloning site has been relocated after the EGFP, allowing expression of an N-terminal EGFP fusion protein under the expression of an EMCV IRES. The plasmids encoding Flag-G3BP1 and Flag-G3BP2 have been described elsewhere (11).
Antibodies.
The following antibodies were used for immunofluorescence analysis (IFA) staining procedures: FMDV VP1 (mouse monoclonal Ab 6HC4), anti-dsRNA (αdsRNA; J2; English & Scientific Consulting), αG3BP1 (clone 23/G3BP; BD Biosciences), αG3BP1 (ARP37713; Aviva Systems Biology), αG3BP2 (A302-040A; Bethyl Laboratories), αeIF3 (N-20; Santa Cruz Biotechnology), and αTIA-1 (C-20; Santa Cruz Biotechnology). Alexa Fluor 488-, Alexa Fluor 594-, and Alexa Fluor 647-conjugated secondary antibodies (Molecular Probes, Invitrogen) were used for detection. For flow cytometry staining, we used the αdsRNA (J2; English & Scientific Consulting), αp-PKR (E120; Abcam), and αp-eIF2α (E90; Abcam) antibodies and Alexa Fluor 488- or Alexa Fluor 647-conjugated (Molecular Probes, Invitrogen) secondary antibodies. For Western blotting, we used the antibodies described above against G3BP1, G3BP2, and TIA-1 and the additional antibodies αPARP (11835238001; Roche Diagnostics), αFlag (M2; Sigma), αGFP (OSE00003G; Invitrogen), and and α-tubulin (DM1A; Sigma). Respective IRdye680- or IRdye800-conjugated secondary antibodies (LiCor) were used for detection.
IFA.
LF-PK αvβ6 cells were grown on 12-mm glass coverslips and infected with WT FMDV or LLV (multiplicity of infection [MOI], 10) for 1 h at 37°C. After adsorption, the cells were rinsed with acidic buffer (150 mM NaCl in 20 mM morpholineethanesulfonic acid [MES]; pH 6.0), before we added MEM and proceeded with the incubation at 37°C for the durations indicated in the figure legends. The cells were fixed in 4% paraformaldehyde, permeabilized with 0.5% Triton X-100 (Sigma) in phosphate-buffered saline (PBS), blocked with blocking buffer (PBS, 2% bovine serum albumin [BSA], 5% normal goat serum, 10 mM glycine), and then incubated overnight at 4°C with primary antibodies against FMDV VP1 (mouse monoclonal Ab 6HC4) and G3BP1 (ARP37713; Aviva Systems Biology). Alexa Fluor 488- and Alexa Fluor 594-conjugated secondary Abs (Molecular Probes, Invitrogen) were used for the detection of green and red signals, respectively. Nuclei were visualized by DAPI (4′,6-diamidino-2-phenylindole) staining included in ProLong Gold antifade mounting medium (Invitrogen). The cells were examined with a fluorescence microscope, and the images were taken with a Nikon DS-Qi1 digital camera and NIS-Elements Advance Research v3.00 software (Nikon Instruments, Inc., Melville, NY). Alternatively, HeLa R19 cells were grown on 12-mm glass coverslips and infected the next day with the viruses indicated in the legends (MOI, 10). At the time points indicated in the legends, the cells were washed in PBS before being fixed in 4% paraformaldehyde for 30 min. Residual paraformaldehyde was washed away using PBS plus 10 mM glycine. The cells were permeabilized in PBS plus 0.1% Triton X-100 and subsequently incubated in blocking buffer (PBS plus 0.1% Tween 20 plus 3% BSA) for 2 h. All subsequent steps were performed in blocking buffer. The samples were incubated with primary antibody for 1 h and incubated with secondary antibodies and DAPI for 30 min. After antibody incubations, samples were washed three times with PBS plus 0.1% Tween 20 and once in Milli-Q water before being mounted on microscopy slides using FluorSave (Calbiochem). The cells were examined by confocal microscopy (Leica SPE-II) with Leica Application Suite Advanced Fluorescence software (LAS-AF).
Quantification of SGs.
We analyzed the numbers of SGs and their surface areas in ∼100 cells per condition, the combined total of 10 to 20 images, using ImageJ software. For each image, the background signal was removed by creating a blurred duplicate and subtracting it from the original image. Subsequently, remaining diffuse (cytoplasmic) SG marker signal was removed via weak blurring, adjusting the contrast settings, and applying a black-and-white threshold. In the resulting image, the numbers and average surface areas of the SGs (shown in black on a white background) were quantified. The used macro is available upon request. Subsequent statistical analyses were performed using GraphPad Prism software. Error bars represent standard deviations, and P values were calculated using one-way analyses of variance (ANOVAs) with Bonferroni’s post hoc test (infections with recombinant EMCVs) or a one-tailed t test (FMDV infections).
Flow cytometry analysis.
HeLa R19 cells were seeded in 12-wells plates (200,000 cells/well) and infected the next day with the viruses indicated in the figure legends (MOI, 10). At 6 hpi, the cells were trypsinized and resuspended in fluorescence-activated cell sorter (FACS) buffer (PBS plus 1% BSA). Cells were fixed for 30 min in 2% paraformaldehyde in FACS buffer and subsequently fixed in ice-cold methanol for 10 min. All subsequent steps were performed in FACS buffer. Cells were stained with primary antibodies for 1 h. Subsequently, cells were washed three times and incubated in secondary antibodies, in the dark, for 30 min. Cells were washed three times and kept in 1% paraformaldehyde until analysis on FACS Canto II (BD Biosciences) using BD FACS Diva software. Data analysis was performed using FlowJo software.
Western blot analysis.
HeLa R19 cells were seeded in 10-cm dishes and infected the next day (MOI, 10) with the viruses indicated in the figure legends. At the indicated time points, the cells were released using trypsin, washed once in PBS, and lysed in 100 μl lysis buffer (100 mM Tris [pH 8.0], 1 mM EDTA, 50 mM NaCl, 1% NP-40, protease inhibitor mix [Roche]). Postnuclear lysate was obtained by centrifugation at 15,000 × g at 4°C for 15 min. The amount of total protein in the lysates was determined using a bicinchoninic acid (BCA) assay (Thermo Fisher). Then, 100 μg protein from the cleared cell lysates was resolved using reducing sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to 0.2-μm nitrocellulose membranes by wet electrophoretic transfer. Membranes were incubated for 1 h in blocking buffer (PBS plus 0.1% Tween 20 plus 2% BSA) and successively incubated overnight with primary antibodies diluted in blocking buffer and then for 30 min with the respective secondary antibodies diluted in blocking buffer. Between and after the incubations, the membranes were washed three times with PBS plus 0.1% Tween 20. Finally, the membranes were washed once with PBS and scanned using an Odyssey Imager (LiCor). For the analysis of protein from FMDV-infected cells, 40 ng of protein was resolved by SDS-PAGE and then transferred for Western blotting with secondary antibodies conjugated with horseradish peroxidase (Pierce) for the detection of proteins. Following incubation with appropriate primary and secondary antibodies, protein bands were visualized using the SuperSignal West Dura extended-duration substrate (Thermo Scientific, Rockford, IL, USA) according to the manufacturer’s directions.
ACKNOWLEDGMENTS
We thank Martijn van Hemert for the kind gift of the pCMV-Flag-G3BP1 and pCMV-Flag-G3BP2 expression plasmids.
The work was supported by a Vici grant (NWO-918.12.628) from the Netherlands Organization for Scientific Research. Martijn. A. Langereis is supported by a Veni grant (NWO-863.13.008) and Linda J. Visser is supported by the NWO-graduate program for Infection and Immunity (NWO-022.004.018), both from the Netherlands Organization for Scientific Research. Gisselle N. Medina and Teresa de los Santos were supported by CRIS project no. 8064-320000-061-00D from the U.S. Department of Agriculture, Agricultural Research Service.
REFERENCES
- 1.Wu J, Chen ZJ. 2014. Innate immune sensing and signaling of cytosolic nucleic acids. Annu Rev Immunol 32:461–488. doi: 10.1146/annurev-immunol-032713-120156. [DOI] [PubMed] [Google Scholar]
- 2.McCormick C, Khaperskyy DA. 2017. Translation inhibition and stress granules in the antiviral immune response. Nat Rev Immunol 17:647–660. doi: 10.1038/nri.2017.63. [DOI] [PubMed] [Google Scholar]
- 3.Pakos‐Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, Gorman AM. 2016. The integrated stress response. EMBO Rep 17:1374–1395. doi: 10.15252/embr.201642195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kedersha N, Ivanov P, Anderson P. 2013. Stress granules and cell signaling: more than just a passing phase? Trends Biochem Sci 38:494–506. doi: 10.1016/j.tibs.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reineke LC, Lloyd RE. 2015. The stress granule protein G3BP1 recruits protein kinase R to promote multiple innate immune antiviral responses. J Virol 89:2575–2589. doi: 10.1128/JVI.02791-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reineke LC, Kedersha N, Langereis MA, van Kuppeveld FJMM, Lloyd RE. 2015. Stress granules regulate double-stranded RNA-dependent protein kinase activation through a complex containing G3BP1 and Caprin1. mBio 6:e02486-14. doi: 10.1128/mBio.02486-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reineke LC, Dougherty JD, Pierre P, Lloyd RE. 2012. Large G3BP-induced granules trigger eIF2 phosphorylation. Mol Biol Cell 23:3499–3510. doi: 10.1091/mbc.E12-05-0385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Onomoto K, Jogi M, Yoo JS, Narita R, Morimoto S, Takemura A, Sambhara S, Kawaguchi A, Osari S, Nagata K, Matsumiya T, Namiki H, Yoneyama M, Fujita T. 2012. Critical role of an antiviral stress granule containing RIG-I and PKR in viral detection and innate immunity. PLoS One 7:e43031. doi: 10.1371/journal.pone.0043031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Langereis MA, Feng Q, van Kuppeveld FJ. 2013. MDA5 localizes to stress granules, but this localization is not required for the induction of type I interferon. J Virol 87:6314–6325. doi: 10.1128/JVI.03213-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim WJ, Back SH, Kim V, Ryu I, Jang SK. 2005. Sequestration of TRAF2 into stress granules interrupts tumor necrosis factor signaling under stress conditions. Mol Cell Biol 25:2450–2462. doi: 10.1128/MCB.25.6.2450-2462.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Humoud MN, Doyle N, Royall E, Willcocks MM, Sorgeloos F, van Kuppeveld F, Roberts LO, Goodfellow IG, Langereis MA, Locker N. 2016. Feline calicivirus infection disrupts assembly of cytoplasmic stress granules and induces G3BP1 cleavage. J Virol 90:6489–6501. doi: 10.1128/JVI.00647-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garaigorta U, Heim MH, Boyd B, Wieland S, Chisari FV. 2012. Hepatitis C virus (HCV) induces formation of stress granules whose proteins regulate HCV RNA replication and virus assembly and egress. J Virol 86:11043–11056. doi: 10.1128/JVI.07101-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xia J, Chen X, Xu F, Wang Y, Shi Y, Li Y, He J, Zhang P. 2015. Dengue virus infection induces formation of G3BP1 granules in human lung epithelial cells. Arch Virol 160:2991–2999. doi: 10.1007/s00705-015-2578-9. [DOI] [PubMed] [Google Scholar]
- 14.Bidet K, Dadlani D, Garcia-Blanco MA. 2014. G3BP1, G3BP2 and Caprin1 are required for translation of interferon stimulated mRNAs and are targeted by a dengue virus non-coding RNA. PLoS Pathog 10:e1004242. doi: 10.1371/journal.ppat.1004242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Panas MD, Varjak M, Lulla A, Eng KE, Merits A, Karlsson Hedestam GB, McInerney GM. 2012. Sequestration of G3BP coupled with efficient translation inhibits stress granules in Semliki Forest virus infection. Mol Biol Cell 23:4701–4712. doi: 10.1091/mbc.E12-08-0619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scholte FEM, Tas A, Albulescu IC, Žusinaite E, Merits A, Snijder EJ, van Hemert MJ. 2015. Stress granule components G3BP1 and G3BP2 play a proviral role early in Chikungunya virus replication. J Virol 89:4457–4469. doi: 10.1128/JVI.03612-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fros JJ, Domeradzka NE, Baggen J, Geertsema C, Flipse J, Vlak JM, Pijlman GP. 2012. Chikungunya virus nsP3 blocks stress granule assembly by recruitment of G3BP into cytoplasmic foci. J Virol 86:10873–10879. doi: 10.1128/JVI.01506-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feng Q, Langereis MA, Lork M, Nguyen M, Hato SV, Lanke K, Emdad L, Bhoopathi P, Fisher PB, Lloyd RE, van Kuppeveld FJM. 2014. Enterovirus 2Apro targets MDA5 and MAVS in infected cells. J Virol 88:3369–3378. doi: 10.1128/JVI.02712-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rui Y, Su J, Wang H, Chang J, Wang S, Zheng W, Cai Y, Wei W, Gordy JT, Markham R, Kong W, Zhang W, Yu X-F. 2017. Disruption of MDA5 mediated innate immune responses by the 3C proteins of Coxsackievirus A16, Coxsackievirus A6, and Enterovirus D68. J Virol 91:e00546-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barral PM, Sarkar D, Fisher PB, Racaniello VR. 2009. RIG-I is cleaved during picornavirus infection. Virology 391:171–176. doi: 10.1016/j.virol.2009.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang B, Xi X, Lei X, Zhang X, Cui S, Wang J, Jin Q, Zhao Z. 2013. Enterovirus 71 protease 2Apro targets MAVS to inhibit anti-viral type I interferon responses. PLoS Pathog 9:e1003231. doi: 10.1371/journal.ppat.1003231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mukherjee A, Morosky SA, Delorme-Axford E, Dybdahl-Sissoko N, Oberste MS, Wang T, Coyne CB. 2011. The coxsackievirus B 3Cpro protease cleaves MAVS and TRIF to attenuate host type I interferon and apoptotic signaling. PLoS Pathog 7:e1001311. doi: 10.1371/journal.ppat.1001311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang C, Sun M, Yuan X, Ji L, Jin Y, Cardona CJ, Xing Z. 2017. Enterovirus 71 suppresses interferon responses by blocking Janus kinase (JAK)/signal transducer and activator of transcription (STAT) signaling through inducing karyopherin-α1 degradation. J Biol Chem 292:10262–10274. doi: 10.1074/jbc.M116.745729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.White JP, Cardenas AM, Marissen WE, Lloyd RE. 2007. Inhibition of cytoplasmic mRNA Stress granule formation by a viral proteinase. Cell Host Microbe 2:295–305. doi: 10.1016/j.chom.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 25.Dougherty JD, Tsai WC, Lloyd RE. 2015. Multiple poliovirus proteins repress cytoplasmic RNA granules. Viruses 7:6127–6140. doi: 10.3390/v7122922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Borghese F, Michiels T. 2011. The leader protein of cardioviruses inhibits stress granule assembly. J Virol 85:9614–9622. doi: 10.1128/JVI.00480-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Feng Q, Langereis MA, van Kuppeveld FJM. 2014. Induction and suppression of innate antiviral responses by picornaviruses. Cytokine Growth Factor Rev 25:577–585. doi: 10.1016/j.cytogfr.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ng CS, Jogi M, Yoo J-S, Onomoto K, Koike S, Iwasaki T, Yoneyama M, Kato H, Fujita T. 2013. Encephalomyocarditis virus disrupts stress granules, the critical platform for triggering antiviral innate immune responses. J Virol 87:9511–9522. doi: 10.1128/JVI.03248-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guarné A, Tormo J, Kirchweger R, Pfistermueller D, Fita I, Skern T. 1998. Structure of the foot-and-mouth disease virus leader protease: a papain-like fold adapted for self-processing and eIF4G recognition. EMBO J 17:7469–7479. doi: 10.1093/emboj/17.24.7469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brown CC, Piccone ME, Mason PW, McKenna TS, Grubman MJ. 1996. Pathogenesis of wild-type and leaderless foot-and-mouth disease virus in cattle. J Virol 70:5638–5641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Los Santos T, de Avila Botton S, Weiblen R, Grubman MJ. 2006. The leader proteinase of foot-and-mouth disease virus inhibits the induction of beta interferon mRNA and blocks the host innate immune response. J Virol 80:1906–1914. doi: 10.1128/JVI.80.4.1906-1914.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.de Los Santos T, Diaz-San Segundo F, Grubman MJ. 2007. Degradation of nuclear factor kappa B during foot-and-mouth disease virus infection. J Virol 81:12803–12815. doi: 10.1128/JVI.01467-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang D, Fang L, Luo R, Ye R, Fang Y, Xie L, Chen H, Xiao S. 2010. Foot-and-mouth disease virus leader proteinase inhibits dsRNA-induced type I interferon transcription by decreasing interferon regulatory factor 3/7 in protein levels. Biochem Biophys Res Commun 399:72–78. doi: 10.1016/j.bbrc.2010.07.044. [DOI] [PubMed] [Google Scholar]
- 34.Wang D, Fang L, Li P, Sun L, Fan J, Zhang Q, Luo R, Liu X, Li K, Chen H, Chen Z, Xiao S. 2011. The leader proteinase of foot-and-mouth disease virus negatively regulates the type I Interferon pathway by acting as a viral deubiquitinase. J Virol 85:3758–3766. doi: 10.1128/JVI.02589-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Swatek KN, Aumayr M, Pruneda JN, Visser LJ, Berryman S, Kueck AF, Geurink PP, Ovaa H, van Kuppeveld FJM, Tuthill TJ, Skern T, Komander D. 2018. Irreversible inactivation of ISG15 by a viral leader protease enables alternative infection detection strategies. Proc Natl Acad Sci U S A 115:2371–2376. doi: 10.1073/pnas.1710617115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bailey-Elkin BA, Knaap RCM, Kikkert M, Mark BL. 2017. Structure and function of viral deubiquitinating enzymes. J Mol Biol 429:3441–3470. doi: 10.1016/j.jmb.2017.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang D, Fang L, Li K, Zhong H, Fan J, Ouyang C, Zhang H, Duan E, Luo R, Zhang Z, Liu X, Chen H, Xiao S. 2012. Foot-and-mouth disease virus 3C protease cleaves NEMO to impair innate immune signaling. J Virol 86:9311–9322. doi: 10.1128/JVI.00722-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Du Y, Bi J, Liu J, Liu X, Wu X, Jiang P, Yoo D, Zhang Y, Wu J, Wan R, Zhao X, Guo L, Sun W, Cong X, Chen L, Wang J. 2014. 3Cpro of foot-and-mouth disease virus antagonizes the interferon signaling pathway by blocking STAT1/STAT2 nuclear translocation. J Virol 88:4908–4920. doi: 10.1128/JVI.03668-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Monaghan P, Gold S, Simpson J, Zhang Z, Weinreb PH, Violette SM, Alexandersen S, Jackson T. 2005. The αvβ6 integrin receptor for foot-and-mouth disease virus is expressed constitutively on the epithelial cells targeted in cattle. J Gen Virol 86:2769–2780. doi: 10.1099/vir.0.81172-0. [DOI] [PubMed] [Google Scholar]
- 40.LaRocco M, Krug PW, Kramer E, Ahmed Z, Pacheco JM, Duque H, Baxt B, Rodriguez LL. 2013. A continuous bovine kidney cell line constitutively expressing bovine αvβ6 integrin has increased susceptibility to foot-and-mouth disease virus. J Clin Microbiol 51:1714–1720. doi: 10.1128/JCM.03370-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hato SV, Ricour C, Schulte BM, Lanke KHW, de Bruijn M, Zoll J, Melchers WJG, Michiels T, van Kuppeveld FJM. 2007. The mengovirus leader protein blocks interferon-alpha/beta gene transcription and inhibits activation of interferon regulatory factor 3. Cell Microbiol 9:2921–2930. doi: 10.1111/j.1462-5822.2007.01006.x. [DOI] [PubMed] [Google Scholar]
- 42.Feng Q, Hato SVV, Langereis MA, Zoll J, Virgen-Slane R, Peisley A, Hur S, Semler BLL, van Rij RP, van Kuppeveld FJM. 2012. MDA5 detects the double-stranded RNA replicative form in picornavirus-infected cells. Cell Rep 2:1187–1196. doi: 10.1016/j.celrep.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rabouw HH, Langereis MA, Knaap RCM, Dalebout TJ, Canton J, Sola I, Enjuanes L, Bredenbeek PJ, Kikkert M, de Groot RJ, van Kuppeveld FJM. 2016. Middle East respiratory coronavirus accessory protein 4a inhibits PKR-mediated antiviral stress responses. PLoS Pathog 12:e1005982. doi: 10.1371/journal.ppat.1005982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kaminski A, Belsham GJ, Jackson RJ. 1994. Translation of encephalomyocarditis virus RNA: parameters influencing the selection of the internal initiation site. EMBO J 13:1673–1681. doi: 10.1002/j.1460-2075.1994.tb06431.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hato SV, Sorgeloos F, Ricour C, Zoll J, Melchers WJG, Michiels T, van Kuppeveld FJM. 2010. Differential IFN-α/β production suppressing capacities of the leader proteins of mengovirus and foot-and-mouth disease virus. Cell Microbiol 12:310–317. doi: 10.1111/j.1462-5822.2009.01395.x. [DOI] [PubMed] [Google Scholar]
- 46.Piccone ME, Rieder E, Mason PW, Grubman MJ. 1995. The foot-and-mouth disease virus leader proteinase gene is not required for viral replication. J Virol 69:5376–5382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fung G, Ng CS, Zhang J, Shi J, Wong J, Piesik P, Han L, Chu F, Jagdeo J, Jan E, Fujita T, Luo H. 2013. Production of a dominant-negative fragment due to G3BP1 cleavage contributes to the disruption of mitochondria-associated protective stress granules during CVB3 infection. PLoS One 8:e79546. doi: 10.1371/journal.pone.0079546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang X, Hu Z, Fan S, Zhang Q, Zhong Y, Guo D, Qin Y, Chen M. 2018. Picornavirus 2A protease regulates stress granule formation to facilitate viral translation. PLoS Pathog 14:e1006901. doi: 10.1371/journal.ppat.1006901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kedersha N, Panas MD, Achorn CA, Lyons S, Tisdale S, Hickman T, Thomas M, Lieberman J, McInerney GM, Ivanov P, Anderson P. 2016. G3BP-Caprin1-USP10 complexes mediate stress granule condensation and associate with 40S subunits. J Cell Physiol 212:845–860. doi: 10.1083/jcb.201508028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Agol VI, Gmyl AP. 2010. Viral security proteins: counteracting host defences. Nat Rev Microbiol 8:867–878. doi: 10.1038/nrmicro2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rai DK, Lawrence P, Kloc A, Schafer E, Rieder E. 2015. Analysis of the interaction between host factor Sam68 and viral elements during foot-and-mouth disease virus infections. Virol J 12:224. doi: 10.1186/s12985-015-0452-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lawrence P, Schafer EA, Rieder E. 2012. The nuclear protein Sam68 is cleaved by the FMDV 3C protease redistributing Sam68 to the cytoplasm during FMDV infection of host cells. Virology 425:40–52. doi: 10.1016/j.virol.2011.12.019. [DOI] [PubMed] [Google Scholar]
- 53.Zhu Y, Wang B, Huang H, Zhao Z. 2016. Enterovirus 71 induces anti-viral stress granule-like structures in RD cells. Biochem Biophys Res Commun 476:212–217. doi: 10.1016/j.bbrc.2016.05.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang H, Chen N, Li P, Pan Z, Ding Y, Zou D, Li L, Xiao L, Shen B, Liu S, Cao H, Cui Y. 2016. The nuclear protein Sam68 is recruited to the cytoplasmic stress granules during enterovirus 71 infection. Microb Pathog 96:58–66. doi: 10.1016/j.micpath.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 55.Black TL, Safer B, Hovanessian A, Katze MG. 1989. The cellular 68,000-Mr protein kinase is highly autophosphorylated and activated yet significantly degraded during poliovirus infection: implications for translational regulation. J Virol 63:2244–2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chang Y-H, Lau KS, Kuo R-L, Horng J-T. 2017. dsRNA binding domain of PKR is proteolytically released by enterovirus A71 to facilitate viral replication. Front Cell Infect Microbiol 7:284. doi: 10.3389/fcimb.2017.00284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li C, Zhu Z, Du X, Cao W, Yang F, Zhang X, Feng H, Li D, Zhang K, Liu X, Zheng H. 2017. Foot-and-mouth disease virus induces lysosomal degradation of host protein kinase PKR by 3C proteinase to facilitate virus replication. Virology 509:222–231. doi: 10.1016/j.virol.2017.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chinsangaram J, Koster M, Grubman MJ. 2001. Inhibition of L-deleted foot-and-mouth disease virus replication by alpha/beta interferon involves double-stranded RNA-dependent protein kinase. J Virol 75:5498–5503. doi: 10.1128/JVI.75.12.5498-5503.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Galan A, Lozano G, Piñeiro D, Martinez-Salas E. 2017. G3BP1 interacts directly with the FMDV IRES and negatively regulates translation. FEBS J 284:3202–3217. doi: 10.1111/febs.14184. [DOI] [PubMed] [Google Scholar]
- 60.Rieder E, Bunch T, Brown F, Mason PW. 1993. Genetically engineered foot-and-mouth disease viruses with poly(C) tracts of two nucleotides are virulent in mice. J Virol 67:5139–5145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.van der Schaar HM, Leyssen P, Thibaut HJ, de Palma A, van der Linden L, Lanke KHW, Lacroix C, Verbeken E, Conrath K, MacLeod AM, Mitchell DR, Palmer NJ, van de Poël H, Andrews M, Neyts J, van Kuppeveld FJM. 2013. A novel, broad-spectrum inhibitor of enterovirus replication that targets host cell factor phosphatidylinositol 4-kinase III-beta. Antimicrob Agents Chemother 57:4971–4981. doi: 10.1128/AAC.01175-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Van Der Linden L, Ulferts R, Nabuurs SB, Kusov Y, Liu H, George S, Lacroix C, Goris N, Lefebvre D, Lanke KHW, De Clercq K, Hilgenfeld R, Neyts J, Van Kuppeveld FJM. 2014. Application of a cell-based protease assay for testing inhibitors of picornavirus 3C proteases. Antiviral Res 103:17–24. doi: 10.1016/j.antiviral.2013.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]