

PRV is a pathogen of great economic and animal welfare importance in many parts of the world. PRV causes neurological, respiratory, and reproductive disorders, often resulting in mortality of young and immunocompromised animals. Mortality, decreased production, and trade restrictions result in significant financial losses for the agricultural industry. Understanding the molecular mechanisms utilized by PRV to enter host cells is an important step in identifying novel strategies to prevent infection and spread. A thorough understanding of these mechanisms will contribute to a broader understanding of alphaherpesvirus entry. Here, we demonstrate PRV entry into multiple model cell lines via a low-pH endocytosis pathway. Together, these results provide a framework for elucidating the early events of the PRV replicative cycle.
KEYWORDS: endocytosis, herpesviruses, low pH, pseudorabies virus, viral entry
ABSTRACT
The alphaherpesvirus pseudorabies virus (PRV) is the causative agent of pseudorabies, a disease of great economic and welfare importance in swine. Other alphaherpesviruses, including herpes simplex virus (HSV), utilize low-pH-mediated endocytosis to enter a subset of cell types. We investigated whether PRV used this entry pathway in multiple laboratory model cell lines. Inhibition of receptor-mediated endocytosis by treatment with hypertonic medium prevented PRV entry. PRV entry into several cell lines, including porcine kidney (PK15) cells and African green monkey kidney (Vero) cells, was inhibited by noncytotoxic concentrations of the lysosomotropic agents ammonium chloride and monensin, which block the acidification of endosomes. Inactivation of virions by acid pretreatment is a hallmark of viruses that utilize a low-pH-mediated entry pathway. Exposure of PRV virions to pH 5.0 in the absence of host cell membranes reduced entry into PK15 and Vero cells by >80%. Together, these findings suggest that endocytosis followed by fusion with host membranes triggered by low endosomal pH is an important route of entry for PRV.
IMPORTANCE PRV is a pathogen of great economic and animal welfare importance in many parts of the world. PRV causes neurological, respiratory, and reproductive disorders, often resulting in mortality of young and immunocompromised animals. Mortality, decreased production, and trade restrictions result in significant financial losses for the agricultural industry. Understanding the molecular mechanisms utilized by PRV to enter host cells is an important step in identifying novel strategies to prevent infection and spread. A thorough understanding of these mechanisms will contribute to a broader understanding of alphaherpesvirus entry. Here, we demonstrate PRV entry into multiple model cell lines via a low-pH endocytosis pathway. Together, these results provide a framework for elucidating the early events of the PRV replicative cycle.
INTRODUCTION
Pseudorabies virus (PRV) is an alphaherpesvirus and the causative agent of pseudorabies, responsible for severe economic losses to the swine industry worldwide (1). PRV is an excellent model for the study of alphaherpesvirus biology (2, 3). PRV infection in pigs is characterized by a variety of signs, including neurological disorders, severe respiratory syndromes, reproductive failure, and decreased growth rates in survivors (4, 5). PRV disease is associated with high morbidity and mortality rates in piglets. Infection of nonporcine domestic hosts, such as cats, dogs, and ruminants, commonly results in severe neurological signs and death (6–8). Similar to the prototype alphaherpesvirus herpes simplex virus 1 (HSV-1), PRV establishes primary infection at mucosal and epithelial surfaces, followed by lifelong latent infection in neurons of the peripheral nervous system. Periodic reactivation over the lifetime of the host results in spread of infection (2). Consistent with other alphaherpesviruses, PRV demonstrates a broad host range, particularly in experimental infections and cell culture (9). HSV-1 and other alphaherpesviruses utilize multiple entry routes in a cell-specific manner, which may contribute to their broad tropism (10–13). HSV entry into epithelial cells occurs via endocytosis, whereby mildly acidic pH in cellular endosomes is required for entry (12, 14). HSV enters several cell lines, including neurons, via pH-independent methods (10, 15–18).
Thorough study of PRV entry is important for construction of a cohesive model of the viral replicative cycle and for drug development. The majority of animal viruses, including herpesviruses, enter cells by an endocytosis mechanism. PRV entry via endocytosis has not been described. We investigated the pathway utilized by PRV for entry into several cell types. Utilizing multiple experimental approaches, we demonstrate that lysosomotropic chemicals that block acidification of endosomes inhibit entry of PRV into PK15 and MDBK cells, which are widely used to propagate and study the virus in cell culture. Elevated endosomal pH also inhibits entry of PRV into Vero and SK-N-SH cells, both of which support pH-neutral entry of HSV-1 (12, 14, 19). PRV relied on host proteasomal activity for efficient entry. Acid pretreatment of PRV in the absence of a target membrane inhibited entry, consistent with entry via a low-pH mechanism (12, 20–22). These findings indicate that low-pH-mediated endocytosis is an important route of PRV entry into some cell types.
RESULTS AND DISCUSSION
Inhibition of endocytic uptake from the cell surface blocks PRV entry into PK15 cells.
Treatment of cells with hypertonic medium inhibits receptor-mediated endocytosis (23–26). Hypertonic treatment selectively inhibits viral entry via endocytosis, while nonendocytic entry of HSV-1 into Vero cells is unaffected (12, 27, 28). PRV BeBlue (lacZ positive) was prebound to PK15 cells at 4°C for 1 h. Cells were shifted to 37°C and treated with 0.3 M sucrose for 30 min prior to inactivation of noninternalized virus. β-Galactosidase activity at 6 h postinfection (p.i.) was measured as an indicator of viral entry and infection. PRV infectivity in PK15 cells was inhibited by >50% in the presence of hypertonic medium, which is suggestive of impaired entry (Fig. 1). HSV-1 entry into Vero cells was not impaired by sucrose treatment, as expected. This result suggests that PRV can utilize an endocytic pathway to enter host cells.
FIG 1.
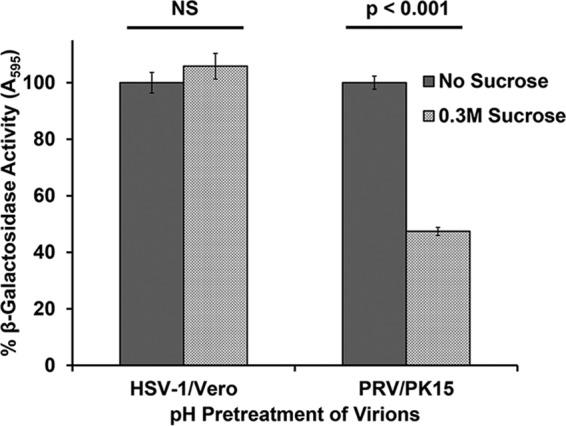
Inhibition of endocytic uptake of PRV from the cell surface. PRV BeBlue was prebound to cells for 1 h at 4°C. Cells were then treated with medium containing 0.3 M sucrose or complete DMEM and incubated at 37°C for 30 min. Cells were washed with PBS and treated with medium buffered to pH 4.7 to inactivate noninternalized virions. Infection proceeded for 6 h in the presence of normal culture medium. β-Galactosidase expression was calculated as a percentage of the activity in mock-treated cells. Values for β-galactosidase activity are the means of data from three independent experiments, each with quadruplicate samples, with standard errors. Treatment with 0.3 M sucrose was noncytotoxic under the experimental conditions, with lactate dehydrogenase (LDH) leakage values below 1% for both Vero and PK15 cells. The P values were determined using Student’s t test. NS, not significant (P > 0.05).
Lysosomotropic agents inhibit PRV entry into several cell lines.
Following internalization via endocytosis, many viruses require exposure to an acidified compartment for successful penetration and infection (12, 21, 29–32). Treatment of cells with lysosomotropic agents, such as ammonium chloride and monensin, prevents acidification and inhibits entry of viruses that require exposure to acidic pH for entry (33). Ammonium chloride is a weak base that elevates the pH of acidic cellular compartments. Monensin is a carboxylic ionophore that prevents endosomal acidification. To determine the role of low pH in endocytic entry of PRV, cells were treated with various concentrations of ammonium chloride or monensin for 20 min, followed by infection with PRV BeBlue in the continued presence of a lysosomotropic agent. β-Galactosidase expression at 6 h postinfection was an indicator of viral entry and infection. Entry of PRV into PK15 cells was inhibited by monensin (Fig. 2A) or ammonium chloride (Fig. 2B) in a concentration-dependent manner. Monensin was less effective at inhibiting infection at a higher multiplicity of infection (MOI) for reasons that are not clear. If the lysosomotropic agents are indeed altering the endosomal pH, then their effect is expected to be MOI independent. It follows that similar results were obtained at a high MOI of 5 and a lower MOI of 1. The experimental concentrations were noncytotoxic, as determined by a lactate dehydrogenase (LDH) release assay (Fig. 2, dashed lines). Together with our finding that PRV enters these cells via endocytosis (Fig. 1), the results suggest that PRV requires endosomal acidification for successful infection of PK15 cells.
FIG 2.

Effect of lysosomotropic agents on PRV entry into PK15 cells. PK15 cells were treated with monensin (A) or ammonium chloride (B) at the indicated concentrations for 20 min at 37°C. Cells were infected with PRV BeBlue at an MOI of 1 or 5 for 6 h in the continued presence of the agents. β-Galactosidase activity in the absence of the agent was set to 100%. LDH activity as a measure of cytotoxicity is reported as a percentage of the value for the detergent-lysed sample. Values for β-galactosidase activity are the means of data from three independent experiments, each with quadruplicate samples, with standard errors. Values for cytotoxicity are the means and standard errors of data from three independent experiments with triplicate samples. The P values relative to those with no drug treatment at the same MOI were determined with Student’s t test (*, P < 0.05; **, P < 0.01). For an MOI of 1, monensin treatment at 0.1 μM and ammonium chloride treatment at 1 mM were also significant (P < 0.01).
PRV is a promiscuous virus that can enter many cell types in culture. Primates, including humans, are not hosts for PRV. However, Vero cells, as well as MDBK cells, have been utilized for studies of PRV biology (34–38). Vero cells and the human neuroblastoma cell line SK-N-SH are important for comparisons with HSV-1, the prototype alphaherpesvirus. The role of low pH in endocytic entry of PRV into these additional cell lines was tested. Cells were treated with various concentrations of ammonium chloride or monensin for 20 min, followed by infection with PRV BeBlue (MOI of 1) in the continued presence of the agent for 6 h. As a control for nonendocytic entry, Vero cells were treated with lysosomotropic agents and infected with HSV-1 KOS tk12 (MOI of 5) (12). Ammonium chloride or monensin treatment of Vero, MDBK, or SK-N-SH cells inhibited PRV BeBlue entry in a dose-dependent manner (Fig. 3). HSV-1 entry into Vero cells was not inhibited at any concentration, as reported previously (12). Host cell triggers such as low intracellular pH or receptor binding are often required to surmount the energy barrier to membrane fusion (39, 40). We demonstrate here that PRV relies on low-pH exposure for entry into PK15, Vero, MDBK, and SK-N-SH cell lines.
FIG 3.

Effect of lysosomotropic agents on PRV entry into Vero, SK-N-SH, and MDBK cells. Vero (A and B), SK-N-SH (C and D), or MDBK (E and F) cells were treated with monensin or ammonium chloride at the indicated concentrations for 20 min at 37°C. Cells were infected with PRV BeBlue (MOI of 1) or HSV-1 KOS tk12 (MOI of 5) for 6 h in the continued presence of agents. β-Galactosidase activity of untreated, infected cells was set to 100%. LDH activity as a measure of cytotoxicity is reported as a percentage of the value for the detergent-lysed sample. Values for β-galactosidase activity are the means of data from three independent experiments, each with quadruplicate samples, with standard errors. Values for cytotoxicity are the means and standard errors of data from three independent experiments with triplicate samples. The P values relative to those with no drug treatment were determined using Student’s t test (*, P < 0.01).
Both monensin and ammonium chloride inhibited >50% of entry at the highest concentrations tested, suggesting that low-pH-mediated endocytosis is a major route of entry for PRV into these cells. This contrasts with the exclusively pH-independent entry of HSV-1 into Vero cells, neurons, and neuroblastoma cell lines (14, 16, 41).
Lysosomotropic agents act at an early time point in PRV infection.
A time-of-addition assay was performed to determine whether lysosomotropic agents inhibit PRV at an earlier or later step of infection. PRV BeBlue was bound to PK15 cells at 4°C for 1 h, and cultures were then shifted to 37°C. At 0 to 6 h p.i., noninternalized virus was inactivated with sodium citrate buffer, followed by the addition of 50 mM ammonium chloride or 30 μM monensin. The later the treatment, the less of an inhibitory effect on PRV entry was detected. This suggested that monensin and ammonium chloride act on an early step in PRV infection (Fig. 4). The penetration of PRV into host cells is rapid, with a half time (t1/2) of approximately 10 min (42). Since inhibition was measured relative to controls exposed to sodium citrate at the same time points, these results also suggest that the inhibitory effect on entry was at a postinternalization step of entry. Together, the results suggest that these agents act at an early, postbinding step in PRV entry, such as penetration of the endosomal membrane.
FIG 4.

Time course of lysosomotropic agent effects on PRV entry. PRV BeBlue was bound to confluent PK15 cells in 96-well plates at 4°C for 1 h. Noninternalized virus was inactivated with sodium citrate buffer, and 30 μM monensin (A) or 50 mM NH4Cl (B) was added at time points from 0 to 6 h p.i. Infection proceeded for a total of 6 h. Control wells for each time point were treated with sodium citrate buffer, and untreated medium was added to the cells. β-Galactosidase expression was calculated as a percentage of the activity in cells in control wells for each time point. Values for β-galactosidase activity are the means of data from three independent experiments, each with quadruplicate samples, with standard errors. Values for cytotoxicity are the means and standard errors of data from three independent experiments with triplicate samples.
Effect of mildly acidic pH pretreatment on infectivity of PRV.
Acid pretreatment of virions that enter cells via a low-pH pathway inactivates their infectivity (43–46). Exposure to low-pH buffers in the absence of a host target membrane is thought to prematurely trigger fusion glycoproteins of virions to change conformation in an irreversible manner, rendering the virus incapable of fusion with host membranes (43, 47). To determine if low-pH pretreatment inactivates PRV, isolated virions were exposed to acidic conditions prior to infection of cells. PRV BeBlue virions were incubated at pH 7.2, 6.5, 6.0, 5.5, or 5.0 for 30 min; neutralized to pH 7.2; and then added to PK15 or Vero cells to assess infectivity. Entry into both cell types was inhibited by low-pH pretreatment, with a >80% reduction in virus-induced β-galactosidase activity at pH 5.0 (Fig. 5A). Control experiments (Fig. 5B) suggested that similar pretreatment of HSV-1 particles with low pH inhibited entry into both cell types, as expected. Inactivation of HSV-1 destroys virion fusion activity without affecting attachment to cells or binding to gD receptors (43, 47). Notably, acid pretreatment of HSV-1 inhibits entry into all cell types, regardless of the pathway. These results are consistent with an intracellular low pH serving as an important activator during successful PRV infection of PK15 and Vero cells. We propose that low pH induces a conformational change in one or more glycoproteins, possibly gB, as is the case for HSV-1, that is necessary for fusion with endosomal membranes (43, 47, 48). Infectivity was not restored by neutralization of the treated virus, suggesting that inactivation is irreversible under these conditions. This also rules out effects of pH on the cell, such as on cell surface receptors.
FIG 5.

Effect of mildly acidic pH on PRV infectivity. PRV BeBlue (A) or HSV-1 tk12 (B) was incubated at pHs ranging from 7.2 to 5.0 at 37°C for 30 min and then neutralized to pH 7.2. Treated virions were added to confluent PK15 or Vero cells in 96-well plates for 6 h. β-Galactosidase activity was measured to indicate virus entry, with maximum infectivity set to 100%. Values for β-galactosidase activity are the means of data from three independent experiments, each with quadruplicate samples, with standard errors. The P values relative to those with no drug treatment were determined using Student’s t test (*, P < 0.05).
Results presented here contribute to our understanding of PRV entry by elucidating the role of endocytosis and exposure to low pH in several cell types. The specific endocytic pathway traversed by PRV is of great interest. Future studies will determine the dependence of PRV entry on host factors, including clathrin, caveolin, dynamin, and indicators of macropinocytosis. As with many herpesviruses, it is likely that PRV can enter different pathophysiologically relevant cell types using distinct mechanisms. PRV utilizes a nonendocytic pathway to enter RK13 cells, Chinese hamster ovary cells, and peripheral neurons (49, 50). Endocytic entry routes taken by PRV in primary cells remain to be determined. Regardless of the pathway of entry, fusion of the viral envelope with host membranes must occur. PRV entry requires viral envelope glycoproteins gB, gD, gH, and gL (51). PRV gD interacts with host cell nectin-1 or poliovirus receptor (Pvr), which is thought to trigger membrane fusion (34, 52). PRV gB, gH, and gL serve as the core fusion machinery (53). We propose that endosomal low pH is an important trigger for fusion during PRV entry into some cell types.
Role of the host cell proteasome in PRV entry.
The eukaryotic proteasome system is one mechanism by which the host cells degrade proteins and regulate cellular pathways. The 26S proteasome plays a role in HSV-1 entry at a postpenetration step mediated by tegument ICP0 (54, 55). Proteasome inhibition also reduces bovine herpesvirus 1 (BoHV-1) entry into MDBK cells in vitro (13). This suggests that a conserved proteasome-dependent mechanism may be utilized by alphaherpesviruses, although the precise mechanisms have yet to be elucidated. MG132 is a peptide aldehyde that binds to the active site of the 20S proteasome to act as a competitive inhibitor of proteasome degradative activity. To further characterize the entry pathway of PRV, we assessed the effect of MG132 on viral entry into PK15 cells. MG132 treatment resulted in a concentration-dependent inhibition of PRV-induced β-galactosidase activity (Fig. 6). Inhibition was incomplete, which suggests that PRV may also use proteasome-independent mechanisms to enter PK15 cells. This result suggests that PRV relies on proteasome-mediated proteolysis for efficient entry.
FIG 6.
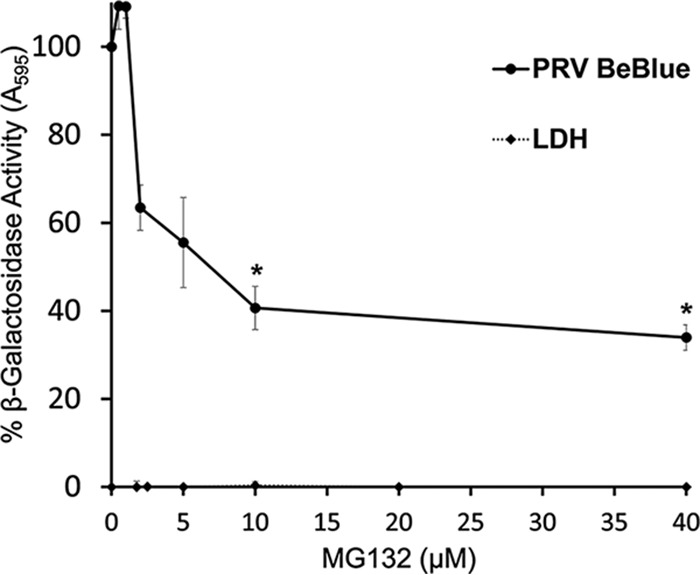
Effect of MG132, a proteasome inhibitor, on PRV entry. PK15 cells were treated with MG132 for 20 min at 37°C. PRV BeBlue was added (MOI of 1) in the continued presence of MG132. At 6 h p.i., β-galactosidase activity of untreated, infected cells was set to 100%. Cytotoxicity is shown as percent LDH activity. Each experiment was performed with quadruplicate (entry) or triplicate (LDH) samples. Values are the means and standard errors of data from three independent experiments. The P values relative to those with no drug treatment were determined using Student’s t test (*, P < 0.05).
MATERIALS AND METHODS
Cells and viruses.
PK15 cells (provided by Matthew Taylor, Montana State University), MDBK cells, and Vero cells (American Type Culture Collection, Manassas, VA) were propagated in Dulbecco’s modified Eagle’s medium (DMEM; ThermoFisher Scientific, Grand Island, NY) supplemented with 10% (PK15 and Vero) or 5% (MDBK) fetal bovine serum (FBS; Atlanta Biologicals, Atlanta, GA). SK-N-SH cells (American Type Culture Collection) were propagated in Eagle’s minimal essential medium supplemented with 10% fetal bovine serum, 1 mM sodium pyruvate, 0.1 mM nonessential amino acids, and Earle’s salts (American Type Culture Collection). PRV BeBlue (provided by Lynn Enquist, Princeton University), a PRV Becker strain derivative with the Escherichia coli lacZ gene inserted into the gG locus (56), was propagated on PK15 cells. HSV-1 strain KOS tk12 (provided by Patricia Spear, Northwestern University), which contains the lacZ gene inserted into the thymidine kinase gene under the control of the HSV-1 infected-cell protein 4 (ICP4) promoter (57), was propagated on Vero cells.
Cell viability.
A colorimetric assay for quantification of lactate dehydrogenase (LDH) release was used as a biomarker for cell cytotoxicity (58). Confluent cell monolayers grown in 96-well plates were treated with experimental conditions and concentrations of drugs. As positive controls, cells were lysed with 1% SDS for 30 min. All concentrations of drugs were tested in triplicate wells. LDH leakage was determined using a Pierce LDH cytotoxicity assay kit (Thermo Scientific, Rockford, IL) according to the manufacturer’s instructions.
β-Galactosidase reporter assay of viral entry.
Confluent cell monolayers in 96-well plates were infected with PRV BeBlue or HSV-1 KOS tk12 at the indicated MOIs for 6 h at 37°C. Cells were lysed with 0.5% Igepal (Sigma-Aldrich, St. Louis, MO) followed by one freeze-thaw cycle. Chlorophenol red-β-d-galactopyranoside (Roche Diagnostics, Indianapolis, IN) was added, and β-galactosidase activity was determined spectrophotometrically by measuring the absorbance at 595 nm with a microtiter plate reader (BioTek Instruments, Winooski, VT).
Inhibition of endocytic uptake of PRV from the cell surface.
Confluent PK15 or Vero monolayers were grown on collagen-coated 96-well plates. PRV BeBlue (MOI of 2) or HSV-1 KOS tk12 (MOI of 10) was prebound to cells on ice at 4°C for 1 h. Cells were then shifted to 37°C, and medium was replaced with DMEM (ThermoFisher Scientific) containing 0.3 M sucrose (J. T. Baker, Center Valley, PA) or complete DMEM for 30 min. Cells were washed twice with warm phosphate-buffered saline (PBS). Noninternalized virus was inactivated by treatment for 2 min with bicarbonate-free DMEM buffered to pH 4.7. Cells were washed twice with PBS. Warmed complete DMEM was added. At 6 h p.i., cells were lysed, and β-galactosidase activity was assessed.
Effect of lysosomotropic agents on PRV entry.
Stock solutions consisted of 1.5 M ammonium chloride (Sigma-Aldrich) in water and 100 μM monensin (Sigma-Aldrich) in ethanol. Confluent cell monolayers grown in 96-well plates were incubated with lysosomotropic agents at various concentrations for 20 min at 37°C. Virus was added, and cells were incubated in the continued presence of the agent for 6 h. At 6 h p.i., cells were lysed, and β-galactosidase activity was determined as described above.
Time-of-addition experiments with lysosomotropic agents.
Confluent PK15 cell monolayers grown on collagen-coated 96-well plates were preincubated with binding medium comprised of serum-free, sodium bicarbonate-free DMEM containing 20 mM HEPES (ThermoFisher Scientific) and 0.2% bovine serum albumin (BSA; ThermoFisher Scientific). PRV BeBlue (MOI of 2) was prebound to cells on ice at 4°C for 1 h. Cells were washed twice with ice-cold PBS to remove unbound virus. Warmed DMEM supplemented with 10% FBS was added, and cultures were shifted to 37°C. At hourly intervals from 0 to 6 h, noninternalized virus was inactivated by exposure to sodium citrate buffer (pH 3.0) for 5 min. After inactivation, cells were washed twice with PBS and incubated in the presence of 50 mM ammonium chloride or 30 μM monensin at 37°C for a total of 6 h.
Low-pH treatment of PRV particles.
PRV BeBlue or HSV-1 KOS tk12 virions were diluted in serum-free, sodium bicarbonate-free DMEM containing 5 mM HEPES, 5 mM 2-(N-morpholino)ethanesulfonic acid (MES; Sigma-Aldrich), 5 mM succinate, and 0.2% BSA. Pretitrated amounts of HCl were added directly to virus preparations to achieve final pHs ranging from 7.2 to 5.0. Samples were incubated at 37°C for 30 min and then neutralized to pH 7.2 by the addition of pretitrated amounts of NaOH. Confluent cell monolayers grown in 96-well plates were infected with treated and mock-treated virus (MOI of 1), and infectivity was determined by a beta-galactosidase reporter assay.
MG132 inhibition of PRV entry.
A 40 mM stock solution of MG132 (Sigma-Aldrich) in dimethyl sulfoxide (DMSO) was diluted in DMEM to achieve 0 to 40 μM concentrations. PK15 cells were grown in 96-well plates until confluent monolayers were observed. MG132 was added to confluent PK15 cells in 96-well plates for 20 min at 37°C. PRV BeBlue was added (MOI of 1) to cells in the presence of MG132 for 6 h at 37°C. β-Galactosidase activity was measured as an indication of viral entry and infectivity.
ACKNOWLEDGMENTS
We thank Lynn Enquist, Patricia Spear, and Matthew Taylor for generous gifts of reagents.
This work was supported by Public Health Service grants AI119159 (A.V.N.) and GM008336 (D.J.W.), a Washington State University veterinary research fellowship (J.L.M.), and the Boehringer Ingelheim Veterinary Scholars Program (J.L.M.).
REFERENCES
- 1.Muller T, Hahn EC, Tottewitz F, Kramer M, Klupp BG, Mettenleiter TC, Freuling C. 2011. Pseudorabies virus in wild swine: a global perspective. Arch Virol 156:1691–1705. doi: 10.1007/s00705-011-1080-2. [DOI] [PubMed] [Google Scholar]
- 2.Pomeranz LE, Reynolds AE, Hengartner CJ. 2005. Molecular biology of pseudorabies virus: impact on neurovirology and veterinary medicine. Microbiol Mol Biol Rev 69:462–500. doi: 10.1128/MMBR.69.3.462-500.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Enquist LW. 1999. Life beyond eradication: veterinary viruses in basic science. Arch Virol Suppl 15:87–109. [DOI] [PubMed] [Google Scholar]
- 4.Lee JYS, Wilson MR. 1979. A review of pseudorabies (Aujeszky’s disease) in pigs. Can Vet J 20:65–69. [PMC free article] [PubMed] [Google Scholar]
- 5.Mettenleiter TC. 1996. Immunobiology of pseudorabies (Aujeszky’s disease). Vet Immunol Immunopathol 54:221–229. doi: 10.1016/S0165-2427(96)05695-4. [DOI] [PubMed] [Google Scholar]
- 6.Marcaccini A, Peña ML, Quiroga MI, Bermúdez R, Nieto JM, Alemañ N. 2008. Pseudorabies virus infection in mink: a host-specific pathogenesis. Vet Immunol Immunopathol 124:264–273. doi: 10.1016/j.vetimm.2008.03.013. [DOI] [PubMed] [Google Scholar]
- 7.Tong W, Liu F, Zheng H, Liang C, Zhou YJ, Jiang YF, Shan TL, Gao F, Li GX, Tong GZ. 2015. Emergence of a pseudorabies virus variant with increased virulence to piglets. Vet Microbiol 181:236–240. doi: 10.1016/j.vetmic.2015.09.021. [DOI] [PubMed] [Google Scholar]
- 8.Dee SA. 2016. Overview of pseudorabies In Aiello SE, Moses MA (ed), Merck veterinary manual, 11th ed Merck & Co, Inc, Whitehouse Station, NJ: https://www.merckvetmanual.com/nervous-system/pseudorabies/overview-of-pseudorabies. [Google Scholar]
- 9.Mettenleiter TC. 1994. Pseudorabies (Aujeszky’s disease) virus: state of the art. August 1993. Acta Vet Hung 42:153–177. [PubMed] [Google Scholar]
- 10.Nicola AV. 2016. Herpesvirus entry into host cells mediated by endosomal low pH. Traffic 17:965–975. doi: 10.1111/tra.12408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frampton AR, Stolz DB, Uchida H, Goins WF, Cohen JB, Glorioso JC. 2007. Equine herpesvirus 1 enters cells by two different pathways, and infection requires the activation of the cellular kinase ROCK1. J Virol 81:10879–10889. doi: 10.1128/JVI.00504-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nicola AV, McEvoy AM, Straus SE. 2003. Roles for endocytosis and low pH in herpes simplex virus entry into HeLa and Chinese hamster ovary cells. J Virol 77:5324–5332. doi: 10.1128/JVI.77.9.5324-5332.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pastenkos G, Lee B, Pritchard SM, Nicola AV. 2018. Bovine herpesvirus 1 entry by a low-pH endosomal pathway. J Virol 92:e00839-18. doi: 10.1128/JVI.00839-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nicola AV, Hou J, Major EO, Straus SE. 2005. Herpes simplex virus type 1 enters human epidermal keratinocytes, but not neurons, via a pH-dependent endocytic pathway. J Virol 79:7609–7616. doi: 10.1128/JVI.79.12.7609-7616.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morgan C, Rose HM, Mednis B. 1968. Electron microscopy of herpes simplex virus. I. Entry. J Virol 2:507–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fuller AO, Spear PG. 1987. Anti-glycoprotein D antibodies that permit adsorption but block infection by herpes simplex virus 1 prevent virion-cell fusion at the cell surface. Proc Natl Acad Sci U S A 84:5454–5458. doi: 10.1073/pnas.84.15.5454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lycke E, Hamark B, Johansson M, Krotochwil A, Lycke J, Svennerholm B. 1988. Herpes simplex virus infection of the human sensory neuron. An electron microscopy study. Arch Virol 101:87–104. doi: 10.1007/BF01314654. [DOI] [PubMed] [Google Scholar]
- 18.Wittels M, Spear PG. 1991. Penetration of cells by herpes simplex virus does not require a low pH-dependent endocytic pathway. Virus Res 18:271–290. doi: 10.1016/0168-1702(91)90024-P. [DOI] [PubMed] [Google Scholar]
- 19.Koyama AH, Uchida T. 1987. The mode of entry of herpes simplex virus type 1 into Vero cells. Microbiol Immunol 31:123–130. doi: 10.1111/j.1348-0421.1987.tb03075.x. [DOI] [PubMed] [Google Scholar]
- 20.Dollery SJ, Lane KD, Delboy MG, Roller DG, Nicola AV. 2010. Role of the UL45 protein in herpes simplex virus entry via low pH-dependent endocytosis and its relationship to the conformation and function of glycoprotein B. Virus Res 149:115–118. doi: 10.1016/j.virusres.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Komala Sari T, Pritchard SM, Cunha CW, Wudiri GA, Laws EI, Aguilar HC, Taus NS, Nicola AV. 2013. Contributions of herpes simplex virus 1 envelope proteins to entry by endocytosis. J Virol 87:13922–13926. doi: 10.1128/JVI.02500-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Siekavizza-Robles CR, Dollery SJ, Nicola AV. 2010. Reversible conformational change in herpes simplex virus glycoprotein B with fusion-from-without activity is triggered by mildly acidic pH. Virol J 7:352. doi: 10.1186/1743-422X-7-352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mellman I. 1996. Endocytosis and molecular sorting. Annu Rev Cell Dev Biol 12:575–625. doi: 10.1146/annurev.cellbio.12.1.575. [DOI] [PubMed] [Google Scholar]
- 24.Heuser JGW, Anderson R. 1989. Hypertonic media inhibit receptor-mediated endocytosis by blocking clathrin-coated pit formation. J Cell Biol 108:389–400. doi: 10.1083/jcb.108.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carpentier JL, Sawano F, Geiger D, Gorden P, Perrelet A, Orci L. 1989. Potassium depletion and hypertonic medium reduce “non-coated” and clathrin-coated pit formation, as well as endocytosis through these two gates. J Cell Physiol 138:519–526. doi: 10.1002/jcp.1041380311. [DOI] [PubMed] [Google Scholar]
- 26.Daukas G, Zigmond SH. 1985. Inhibition of receptor-mediated but not fluid-phase endocytosis in polymorphonuclear leukocytes. J Cell Biol 101:1673–1679. doi: 10.1083/jcb.101.5.1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Simon M, Johansson C, Mirazimi A. 2009. Crimean-Congo hemorrhagic fever virus entry and replication is clathrin-, pH- and cholesterol-dependent. J Gen Virol 90:210–215. doi: 10.1099/vir.0.006387-0. [DOI] [PubMed] [Google Scholar]
- 28.Pu Y, Zhang X. 2008. Mouse hepatitis virus type 2 enters cells through a clathrin-mediated endocytic pathway independent of Eps15. J Virol 82:8112–8123. doi: 10.1128/JVI.00837-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sieczkarski SB, Whittaker G. 2002. Dissecting virus entry via endocytosis. J Gen Virol 83:1535–1545. doi: 10.1099/0022-1317-83-7-1535. [DOI] [PubMed] [Google Scholar]
- 30.Skehel JJ, Wiley DC. 2000. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu Rev Biochem 69:531–569. doi: 10.1146/annurev.biochem.69.1.531. [DOI] [PubMed] [Google Scholar]
- 31.Barrow E, Nicola AV, Liu J. 2013. Multiscale perspectives of virus entry via endocytosis. Virol J 10:177. doi: 10.1186/1743-422X-10-177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nicola AV, Aguilar HC, Mercer J, Ryckman B, Wiethoff CM. 2013. Virus entry by endocytosis. Adv Virol 2013:469583. doi: 10.1155/2013/469538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Duve CD, Barsy TD, Poole B, Trouet A, Tulkens P, Hoof FV. 1974. Lysosomotropic agents. Biochem Pharmacol 23:2495–2531. doi: 10.1016/0006-2952(74)90174-9. [DOI] [PubMed] [Google Scholar]
- 34.Milne RS, Connolly SA, Krummenacher C, Eisenberg RJ, Cohen GH. 2001. Porcine HveC, a member of the highly conserved HveC/nectin 1 family, is a functional alphaherpesvirus receptor. Virology 281:315–328. doi: 10.1006/viro.2000.0798. [DOI] [PubMed] [Google Scholar]
- 35.Karger A, Schmidt J, Mettenleiter TC. 1998. Infectivity of a pseudorabies virus mutant lacking attachment glycoproteins C and D. J Virol 72:7341–7348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Skiba M, Glowinski F, Koczan D, Mettenleiter TC, Karger A. 2010. Gene expression profiling of pseudorabies virus (PrV) infected bovine cells by combination of transcript analysis and quantitative proteomic techniques. Vet Microbiol 143:14–20. doi: 10.1016/j.vetmic.2010.02.009. [DOI] [PubMed] [Google Scholar]
- 37.Wang X, Zhang M-M, Yan K, Tang Q, Wu Y-Q, He W-B, Chen H-C, Liu Z-F. 2018. The full-length microRNA cluster in the intron of large latency transcript is associated with the virulence of pseudorabies virus. Virology 520:59–66. doi: 10.1016/j.virol.2018.05.004. [DOI] [PubMed] [Google Scholar]
- 38.Lee WC, Fuller AO. 1993. Herpes simplex virus type 1 and pseudorabies virus bind to a common saturable receptor on Vero cells that is not heparan sulfate. J Virol 67:5088–5097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dimitrov DS. 2004. Virus entry: molecular mechanisms and biomedical applications. Nat Rev Microbiol 2:109–122. doi: 10.1038/nrmicro817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ben-Porat T, Kaplan AS. 1985. Molecular biology of pseudorabies virus, p 105–173. In Roizman B. (ed), The herpesviruses, vol 3 Springer US, Boston, MA. [Google Scholar]
- 41.Fuller A, E Santos R, Spear P. 1989. Neutralizing antibodies specific for glycoprotein H of herpes simplex virus permit viral attachment to cells but prevent penetration. J Virol 63:3435–3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kopp A, Mettenleiter T. 1992. Stable rescue of a glycoprotein gII deletion mutant of pseudorabies virus by glycoprotein gI of bovine herpesvirus 1. J Virol 66:2754–2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weed DJ, Pritchard SM, Gonzalez F, Aguilar HC, Nicola AV. 2017. Mildly acidic pH triggers an irreversible conformational change in the fusion domain of herpes simplex virus 1 glycoprotein B and inactivation of viral entry. J Virol 91:e02123-16. doi: 10.1128/JVI.02123-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bron R, Wahlberg JM, Garoff H, Wilschut J. 1993. Membrane fusion of Semliki Forest virus in a model system: correlation between fusion kinetics and structural changes in the envelope glycoprotein. EMBO J 12:693–701. doi: 10.1002/j.1460-2075.1993.tb05703.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Edwards J, Mann E, Brown DT. 1983. Conformational changes in Sindbis virus envelope proteins accompanying exposure to low pH. J Virol 45:1090–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rosenthal KS, Killius J, Hodnichak CM, Venetta TM, Gyurgyik L, Janiga K. 1989. Mild acidic pH inhibition of the major pathway of herpes simplex virus entry into HEp-2 cells. J Gen Virol 70:857–867. doi: 10.1099/0022-1317-70-4-857. [DOI] [PubMed] [Google Scholar]
- 47.Dollery SJ, Wright CC, Johnson DC, Nicola AV. 2011. Low-pH-dependent changes in the conformation and oligomeric state of the prefusion form of herpes simplex virus glycoprotein B are separable from fusion activity. J Virol 85:9964–9973. doi: 10.1128/JVI.05291-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dollery SJ, Delboy MG, Nicola AV. 2010. Low pH-induced conformational change in herpes simplex virus glycoprotein B. J Virol 84:3759–3766. doi: 10.1128/JVI.02573-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nixdorf R, Schmidt J, Karger A, Mettenleiter TC. 1999. Infection of Chinese hamster ovary cells by pseudorabies virus. J Virol 73:8019–8026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Smith GA, Pomeranz L, Gross SP, Enquist LW. 2004. Local modulation of plus-end transport targets herpesvirus entry and egress in sensory axons. Proc Natl Acad Sci U S A 101:16034–16039. doi: 10.1073/pnas.0404686101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mettenleiter TC. 1994. Initiation and spread of α-herpesvirus infections. Trends Microbiol 2:2–4. doi: 10.1016/0966-842X(94)90335-2. [DOI] [PubMed] [Google Scholar]
- 52.Geraghty RJ, Krummenacher C, Cohen GH, Eisenberg RJ, Spear PG. 1998. Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science 280:1618–1620. doi: 10.1126/science.280.5369.1618. [DOI] [PubMed] [Google Scholar]
- 53.Klupp BG, Nixdorf R, Mettenleiter TC. 2000. Pseudorabies virus glycoprotein M inhibits membrane fusion. J Virol 74:6760–6768. doi: 10.1128/JVI.74.15.6760-6768.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Delboy MG, Roller DG, Nicola AV. 2008. Cellular proteasome activity facilitates herpes simplex virus entry at a postpenetration step. J Virol 82:3381–3390. doi: 10.1128/JVI.02296-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Delboy MG, Nicola AV. 2011. A pre-immediate-early role for tegument ICP0 in the proteasome-dependent entry of herpes simplex virus. J Virol 85:5910–5918. doi: 10.1128/JVI.00267-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Banfield BW, Yap GS, Knapp AC, Enquist LW. 1998. A chicken embryo eye model for the analysis of alphaherpesvirus neuronal spread and virulence. J Virol 72:4580–4588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Warner MS, Geraghty RJ, Martinez WM, Montgomery RI, Whitbeck JC, Xu R, Eisenberg RJ, Cohen GH, Spear PG. 1998. A cell surface protein with herpesvirus entry activity (HveB) confers susceptibility to infection by mutants of herpes simplex virus type 1, herpes simplex virus type 2, and pseudorabies virus. Virology 246:179–189. doi: 10.1006/viro.1998.9218. [DOI] [PubMed] [Google Scholar]
- 58.Korzeniewski C, Callewaert DM. 1983. An enzyme-release assay for natural cytotoxicity. J Immunol Methods 64:313–320. doi: 10.1016/0022-1759(83)90438-6. [DOI] [PubMed] [Google Scholar]