

Using a model examining the establishment of EBV infection in HPV-immortalized tissues, we showed an HPV-induced interruption of the normal EBV life cycle reminiscent of a latent EBV infection. Our data support the notion that a persistent EBV epithelial infection depends upon preexisting cellular alterations and suggest the ability of HPV to promote such changes. More importantly, these findings introduce a model for how EBV coinfection may influence HPV-positive (HPV-pos) OSCC pathogenesis. Latently EBV-infected epithelial cells, as well as other EBV-associated head-and-neck carcinomas, exhibit oncogenic phenotypes commonly seen in HPV-pos OSCC. Therefore, an HPV-induced shift in the EBV life cycle toward latency would not only facilitate EBV persistence but also provide additional viral oncogene expression, which can contribute to the rapid progression of HPV-pos OSCC. These findings provide a step toward defining a role for EBV as a cofactor in HPV-positive oropharyngeal tumors.
KEYWORDS: EBV, HPV, replication, latency, organotypic, Epstein-Barr virus, viral replication
ABSTRACT
Epstein-Barr virus (EBV) is implicated in the pathogenesis of human papillomavirus (HPV)-associated oropharyngeal squamous cell carcinoma (OSCC). EBV-associated cancers harbor a latent EBV infection characterized by a lack of viral replication and the expression of viral oncogenes. Cellular changes promoted by HPV are comparable to those shown to facilitate EBV latency, though whether HPV-positive cells support a latent EBV infection has not been demonstrated. Using a model of direct EBV infection into HPV16-immortalized tonsillar cells grown in organotypic raft culture, we showed robust EBV replication in HPV-negative rafts but little to no replication in HPV-immortalized rafts. The reduced EBV replication was independent of immortalization, as human telomerase-immortalized normal oral keratinocytes supported robust EBV replication. Furthermore, we observed reduced EBV lytic gene expression and increased expression of EBER1, a noncoding RNA highly expressed in latently infected cells, in the presence of HPV. The use of human foreskin keratinocyte rafts expressing the HPV16 E6 and/or E7 oncogene(s) (HPV E6 and E7 rafts) showed that E7 was sufficient to reduce EBV replication. EBV replication is dependent upon epithelial differentiation and the differentiation-dependent expression of the transcription factors KLF4 and PRDM1. While KLF4 and PRDM1 levels were unaltered, the expression levels of KLF4 transcriptional targets, including late differentiation markers, were reduced in HPV E6 and E7 rafts compared to their levels in parental rafts. However, the HPV E7-mediated block in EBV replication correlated with delayed expression of early differentiation markers. Overall, this study reveals an HPV16-mediated block in EBV replication, through E7, that may facilitate EBV latency and long-term persistence in the tumor context.
IMPORTANCE Using a model examining the establishment of EBV infection in HPV-immortalized tissues, we showed an HPV-induced interruption of the normal EBV life cycle reminiscent of a latent EBV infection. Our data support the notion that a persistent EBV epithelial infection depends upon preexisting cellular alterations and suggest the ability of HPV to promote such changes. More importantly, these findings introduce a model for how EBV coinfection may influence HPV-positive (HPV-pos) OSCC pathogenesis. Latently EBV-infected epithelial cells, as well as other EBV-associated head-and-neck carcinomas, exhibit oncogenic phenotypes commonly seen in HPV-pos OSCC. Therefore, an HPV-induced shift in the EBV life cycle toward latency would not only facilitate EBV persistence but also provide additional viral oncogene expression, which can contribute to the rapid progression of HPV-pos OSCC. These findings provide a step toward defining a role for EBV as a cofactor in HPV-positive oropharyngeal tumors.
INTRODUCTION
The incidence of oropharyngeal squamous cell carcinoma (OSCC) has dramatically increased in a number of developed countries, reaching epidemic rates over the past several decades (1). Decreasing trends in the consumption of alcohol and tobacco, traditional risk factors for OSCC, led to the identification of human papillomavirus (HPV) as an important etiological factor in a subset of OSCCs. Over 90% of HPV-positive (HPV-pos) OSCCs are infected with HPV16 (2).
HPV has been strongly associated with cancers of the anogenital tract, especially cervical cancer, of which HPV is a causal factor in over 95% of cases. Interestingly, HPV-pos OSCC exhibits distinct characteristics compared to HPV-associated anogenital tumors. HPV-pos OSCC shows a more rapid progression than HPV-pos cervical cancers, as HPV-pos OSCC patients frequently present with nodal involvement and metastatic disease and lack an HPV-pos precancerous lesion (3–5). The unique characteristics observed in HPV-pos OSCC compared to other HPV-linked tumors suggest the possibility of additional factors in tumor progression.
One candidate cofactor in HPV-pos OSCC is the oncogenic herpesvirus Epstein-Barr virus (EBV). The oropharynx serves as the site of EBV persistence, as the virus infects B lymphocytes and epithelial cells within the lymphoid-rich tonsils and base of tongue (BOT), anatomical sites where the majority of OSCCs occur (6, 7). We have shown that HPV-pos OSCCs are often coinfected with EBV, with 25% and 70% of tonsillar and BOT carcinomas exhibiting coinfection, respectively (8). In contrast to HPV, a role for EBV in OSCC pathogenesis remains controversial, given its highly variable rates of detection in oropharyngeal tumors (9–15). However, EBV may contribute to early stages of tumor progression and be lost at later stages. We have shown that EBV can epigenetically reprogram epithelial cells, resulting in tumorigenic phenotypes that remain after loss of EBV genomes from cells. Epithelial cells either persistently or transiently infected with EBV exhibit enhanced invasiveness and delayed differentiation (16–18). EBV could therefore contribute to the invasive potential and poorly differentiated status often observed in HPV-pos OSCCs through virally induced epigenetic alterations as EBV transits through the epithelium.
The oncogenic potential of EBV in carcinomas is linked to interruption of the normal viral life cycle. EBV normally replicates its genome and produces progeny virions specifically in the differentiating suprabasal layers of stratified epithelia, with viral replication requiring expression of the differentiation-specific transcription factors Kruppel-like factor 4 (KLF4) and PR domain zinc finger protein 1 (PRDM1) (19–21). However, EBV-associated carcinomas exhibit a latent EBV infection characterized by a restricted viral gene expression program in the absence of viral replication. EBV latent gene products, such as the EBV nuclear antigens (EBNA1, -2, -3A, -3B, and -3C), the latent membrane proteins (LMP1 and -2), the EBV-encoded RNAs (EBERs), and BamHI rightward transcripts (BARTs), have oncogenic potential (reviewed in reference 22). In EBV-associated nasopharyngeal carcinoma (NPC), EBV infection is thought to occur secondary to preexisting genetic alterations that facilitate latent infection (23–25). Nasopharyngeal epithelial cells, which otherwise undergo growth arrest and senescence in the presence of EBV, support a latent EBV infection upon overexpression of cyclin D1 or loss of p16 (26). Such genetic alterations are frequently observed in premalignant lesions in the nasopharynx (23–25).
The cellular alterations shown to facilitate EBV latency are reminiscent of the cell cycle-promoting effects of HPV immortalization. HPV induces an aberrant S-phase-like environment via expression of the viral E6 and E7 oncogenes. HPV E6 facilitates degradation of p53 to bypass the DNA damage cell cycle checkpoint, whereas E7 inactivates Rb to release E2F for transcriptional activation of genes involved in cellular DNA replication. Associated with EBV latency, overexpression of cyclin D1 and loss of p16 also induce Rb hyperphosphorylation and release of E2F for induction of S-phase genes. Although HPV-associated cancers show overexpression of p16 and low levels of cyclin D1 expression, these factors become dispensable in the absence of Rb (27–29). However, whether HPV-mediated changes to Rb would promote latent EBV infection has not been established.
HPV E6 and E7 have also been shown to interfere with epithelial differentiation, a process required for efficient EBV replication. Epithelial tissues expressing E7 exhibit delayed early differentiation marker expression (30, 31), and both HPV E6 and E7 have been shown to alter KLF4, a cellular transcription factor and regulator of epithelial differentiation also required for HPV replication (32–34). HPV E6 induces certain KLF4 posttranslational modifications, while HPV E7 increases KLF4 levels through suppression of microRNA-145 (miRNA-145) (35). KLF4 is required for EBV lytic replication in epithelial cells (20). In B cells, which do not express detectable levels of KLF4, forced expression of KLF4 results in EBV reactivation (20). HPV alterations of KLF4 support EBV reactivation in immortalized normal oral keratinocytes carrying a latent EBV infection when grown in organotypic raft culture (36). However, it is not known whether HPV-mediated changes to KLF4 and/or differentiation would influence the establishment of an incoming EBV infection in coinfected oropharyngeal tissues.
Here, we examined whether HPV immortalization would alter the outcome of a newly established EBV infection in organotypic raft culture. We observed that EBV infection of HPV-immortalized keratinocytes in raft culture resulted in a block in EBV replication, for which HPV E7 was sufficient. A block in EBV lytic gene expression and an increase in EBER1 expression were observed in HPV-immortalized tonsillar keratinocytes. Reduced EBV replication correlated with HPV E7 mediating an early delay in the expression of epithelial differentiation markers. We also observed a reduction in the expression of late differentiation markers, particularly those under the control of KLF4, in keratinocytes immortalized with E6 and E7. Importantly, KLF4 and PRDM1 levels remained unchanged between parental and E7-expressing keratinocytes, suggesting that E7’s interference with EBV replication is upstream of KLF4/PRDM1. Thus, HPV E7 may help to identify additional regulators or posttranslational modifications that regulate KLF4/PRDM1 activity and EBV replication. In sum, HPV-immortalized tissues, through E7, block EBV lytic replication of an incoming EBV infection and facilitate the establishment of a latent and persistent EBV infection.
RESULTS
EBV replication is blocked in HPV-immortalized tonsillar epithelial rafts following EBV infection.
To examine the outcome of the EBV life cycle upon infection of HPV-immortalized tissues, primary human tonsillar epithelial (HTE) cells immortalized by transfection of the HPV16 genome were grown in organotypic raft culture. In organotypic raft culture, cell monolayers are lifted to an air-liquid interface to promote differentiation and the formation of stratified epithelial tissue (Fig. 1A) (37). The HPV episomal genome is known to frequently integrate in cell culture. Using a molecular assay based on the susceptibility of integrated HPV to exonuclease digestion (38), we identified HTE cell lines that carried either episomal or integrated HPV following raft culture (Fig. 1B). Rafts of primary HPV-negative (HPV-neg) and HPV-immortalized (HPV-pos) HTE cells were infected with EBV 4 days postlifting by coculture of EBV-positive Akata BX1 Burkitt’s lymphoma (BL) cells at the apical surface (Fig. 1A). The addition of EBV-negative Akata BL cells served as a mock-treatment control. In rafts derived from HTE cells carrying either episomal or integrated HPV, we observed significantly lower EBV DNA levels than in HPV-neg rafts. The EBV DNA levels in HPV-neg rafts were reduced in the presence of acyclovir, a herpesvirus-specific nucleoside analog that inhibits EBV replication, signifying robust EBV replication in primary HTE rafts, consistent with previous findings (39). Acyclovir-treated HPV-neg HTE rafts showed EBV DNA levels similar to those observed in untreated HPV-pos rafts, suggesting similar infection levels between the HPV-pos and -neg rafts. Furthermore, acyclovir treatment did not affect the EBV DNA levels in HPV-pos rafts (Fig. 1C). To ensure that detection of EBV reflected epithelial infection and not contamination of EBV-positive BL cells, CD20 RNA levels were examined as a specific B cell marker. CD20 RNA levels in EBV-infected raft tissues were below the detection limit of 1 Akata BL cell in 105 293TT epithelial cells in reverse transcription-quantitative PCR (RT-qPCR) (data not shown), confirming the absence of residual EBV-positive BL in the infected rafts. To determine if the reduction in EBV replication was specific to HPV rather than a general effect of immortalization, human telomerase reverse transcriptase (hTERT)-immortalized normal oral keratinocytes (NOK) were grown in raft culture and infected with EBV. Robust EBV replication similar to that seen in HPV-neg HTE rafts, including sensitivity to acyclovir treatment, was observed in NOK rafts (Fig. 1D). Together, these results suggest that HPV-immortalized cells grown in raft tissue block the replication of a de novo EBV infection.
FIG 1.

Reduced EBV replication in HPV-immortalized rafts following EBV infection. (A) Schematic of EBV infection of HPV-immortalized human tonsillar epithelial (HPV-pos HTE) cells in raft culture. (B) HTE raft DNA harboring either episomal or integrated HPV was analyzed for exonuclease digestion resistance by qPCR. DNA from HPV-infected 293TT cells served as an episomal control. qPCR was used to detect HPV16 DNA (E6), mitochondrial DNA (Mito), and 18S ribosomal DNA. (C) EBV genome copy numbers per hCRP copy number in primary HTE (HPV-neg; n = 6) and HPV-pos HTE rafts harboring episomal (n = 3) or integrated (n = 6) HPV genomes were quantified by qPCR. Shown are the average values and standard errors of the means. *, P < 0.05 relative to the results for HPV-neg HTE rafts. (D) EBV genome copy numbers per hCRP copy number in NOK rafts were quantified by qPCR. Shown are the average values and standard errors of the means. *, P < 0.05 relative to the results for EBV. Rafts were infected in the presence or absence of acyclovir (ACV; 50 μg/ml).
EBV replicative gene expression is decreased in HPV-immortalized tonsillar epithelial rafts.
EBV lytic replication is initiated upon the expression of immediate early genes, which act as transactivators for the EBV early genes that function in replicating the viral genome. To determine whether HPV interferes with EBV replicative gene expression, we compared EBV immediate early, early, and latent gene expression levels between HPV-neg and HPV-pos HTE rafts. The gene expression analysis was carried out on acyclovir-treated samples, which harbored similar levels of EBV genomes between HPV-neg and HPV-pos rafts, to allow for a more accurate comparison of gene expression. We observed a significant reduction in immediate early (BZLF1 and BRLF1) and early (BALF5 and BMRF1) gene expression in HPV-pos compared to that in HPV-neg rafts (Fig. 2). LMP1, whose differentiation-dependent expression has been shown to enhance EBV lytic replication in stratified epithelia (19), was also significantly reduced in the presence of HPV. In contrast, we detected no change in the expression of the EBNA1 and EBNA2 latent genes, which may be expressed in the context of both EBV latency and lytic replication (39). However, we observed a significant increase in the expression of EBER1, a noncoding RNA highly expressed in latently EBV-infected cells, in HPV-pos versus HPV-neg HTE rafts (Fig. 2). Altogether, these data support an EBV gene expression pattern favoring latency over lytic replication in HPV-immortalized cells.
FIG 2.

EBV gene expression profile in HPV-immortalized rafts. Acyclovir-treated HPV-neg and HPV-pos HTE raft cultures were analyzed by RT-qPCR for mRNA levels of EBV genes. The BZLF1 expression level in HPV-neg rafts was arbitrarily set to 1. Shown are the average relative expression levels and standard errors of the means from a minimum of 3 rafts per group. *, P < 0.05 relative to the results for HPV-neg HTE rafts.
KLF4 target gene expression levels are reduced in HPV-immortalized tonsillar epithelial rafts.
EBV immediate early gene expression and genome replication rely on epithelial differentiation, a process HPV is known to delay. Indeed, hematoxylin and eosin (H&E) staining confirmed a disorganized tissue architecture in HPV-pos versus HPV-neg HTE rafts. HPV-pos rafts were characterized by an expansion of proliferating basal cells into suprabasal layers and the lack of a “basket weave” appearance in the upper layers that was seen in HPV-neg rafts (Fig. 3A). In addition, while the early differentiation marker involucrin was expressed throughout the suprabasal layers of HPV-neg HTE rafts, involucrin was restricted to the uppermost layers of HPV-pos HTE rafts (Fig. 3B). Differentiated cells express KLF4 and PRDM1, cellular transcription factors shown to facilitate EBV replication by activating the expression of immediate early genes and LMP1 (19–21). Although differentiation was delayed in HPV-pos HTE rafts, the KLF4 and PRDM1 mRNA levels were not significantly reduced (Fig. 3C). Furthermore, the percentages of cells expressing KLF4 were similar in HPV-neg and HPV-pos rafts (Fig. 3D and E). However, HPV has been shown to alter KLF4 activity (35). Indeed, we detected a significant reduction in the expression of the KLF4 transcriptional target WNT5A in HPV-pos versus HPV-neg HTE rafts (Fig. 3C). The late differentiation markers filaggrin (FLG) and loricrin (LOR) are also KLF4 transcriptional targets. Though FLG was expressed at relatively low levels in tonsillar rafts, a significant decrease in FLG transcript levels was observed in HPV-pos rafts compared to the levels in HPV-neg rafts (Fig. 3C). LOR was not detected in either HPV-neg or HPV-pos rafts (data not shown). Together, these data indicate that the inhibition of EBV replication in HPV-immortalized HTE rafts correlated to a delay in epithelial differentiation and altered KLF4 activity.
FIG 3.

Delayed epithelial differentiation in HPV-immortalized rafts. (A, B) H&E staining (A) and involucrin (Inv) immunofluorescent (IF) staining (B) of HPV-neg and HPV-pos HTE rafts. (C) HPV-neg and HPV-pos rafts were analyzed by RT-qPCR for mRNA levels of KLF4, PRDM1, WNT5A and filaggrin (FLG). Shown are the average normalized expression levels and standard errors of the means from a minimum of three rafts per group. *, P < 0.05 relative to the results for HPV-neg HTE rafts. (D) IF staining of KLF4 in HPV-neg and HPV-pos rafts. (E) Amounts of KLF4-positive cells were quantified as the percentages of total cells. Shown are the average values and standard errors of the means. (A, B, D) Basal layers are delineated by dotted lines. Scale bars represent 50 μm.
HPV16 E6E7 reduce EBV replication in HFK rafts.
The ability of HPV to interrupt epithelial differentiation is linked to expression of its oncogenes E6 and E7. Given that epithelial differentiation is also required for EBV replication, we speculated that E6 and E7 were sufficient to abrogate EBV replication. Primary human foreskin keratinocytes (HFK) or NOK stably expressing either HPV16 E6E7 or an empty vector were rafted and infected with EBV. The expression of HPV16 E6 and E7 was confirmed by RT-qPCR (Fig. 4A and B). As with HPV-pos HTE rafts, EBV exhibited reduced genome levels in E6E7-expressing keratinocytes compared to the levels in vector controls (Fig. 4C). Keeping E6E7-expressing HFK (HFK E6E7) in raft culture for 15 days post-EBV infection did not stimulate EBV replication to the levels observed in parental controls (Fig. 4D), suggesting an HPV-mediated block of EBV replication rather than a delay. HFK rafts supported the replication of cell-free EBV, although the replication was not as robust as with infection following coculture with EBV-positive Akata BL cells (Fig. 4E) (39). Despite the lower replication levels in HPV-neg rafts, a block in replication was also observed in E6E7-expressing HFK rafts following infection with cell-free EBV.
FIG 4.

Abrogated EBV replication in HPV16 E6E7-expressing rafts. (A, B) HPV oncogene expression confirmed by RT-qPCR in NOK (n = 1) (A) and HFK (n = 3) (B) cell lines expressing E6, E7, or both (E6E7). E6 expression levels in NOK E6E7 and HFK E6E7 were arbitrarily set to 1. (B) Shown are the average values and standard errors of the means. (C) NOK or HFK expressing E6E7 (n = 5) or carrying an empty vector (Vec; NOK [n = 7], HFK [n = 2]) were rafted, infected with EBV, and analyzed for EBV genome copy numbers per hCRP copy number by qPCR. *, P < 0.05 relative to the results for NOK Vec. (D) Parental HFK and HFK E6E7 rafts (n = 5) were analyzed for EBV genome copy numbers per hCRP copy number at 15 days postinfection (dpi). Shown are the average values and standard errors of the means. (E) HFK and HFK E6E7 rafts infected with cell-free EBV were analyzed for EBV genome copy numbers per hCRP copy number at 6 dpi. Shown are the average values and standard errors of the means from a minimum of 3 rafts per group. *, P < 0.05 relative to the results for parental HFK.
Inhibition of EBV early gene expression in HPV16 E6E7-expressing HFK rafts.
Immunofluorescence analysis of cells expressing the EBV early protein EA-D (BMRF1 gene product) showed significantly fewer EA-D-positive cells in the presence of E6E7 than in parental HFK (Fig. 5A and C). Surprisingly, a similar percentage of Z (BZLF1 gene product)-positive cells was observed in HFK E6E7 versus parental controls (Fig. 5A and B), in contrast with the reduced BZLF1 expression previously noted in the HPV-pos HTE rafts (Fig. 2). Such effects on Z expression may reflect the reduced differentiation potential of HTE cells versus HFK in raft culture, noted by the reduced levels of filaggrin and loricrin expression in HTE rafts. Examination of the EBV late gene product gp350 showed expression throughout the upper layers of parental HFK rafts, while no gp350 was detected in HFK E6E7 rafts (Fig. 5D). Together, these data suggest that HPV16 E6E7 interfere with EBV EA-D expression, with effects on EBV replication and late gene expression.
FIG 5.

Loss of EBV early and late proteins in HPV16 E6E7-expressing rafts. (A to C) IF staining (A) and quantification of EBV Z (B) and EA-D (C) in HFK and HFK E6E7 rafts infected with EBV. Three independent rafts per group were analyzed. Signal-positive cells were quantified as the percentages of total cells. (B, C) Shown are the average values and standard errors of the means. **, P < 0.01 relative to the results for parental HFK. (A) Scale bars represent 50 μm. (D) IF staining of EBV gp350 in EBV-infected HFK and HFK E6E7 rafts. White arrows indicate cells exhibiting a prominent signal. Scale bar represents 15 μm. (A, D) Basal layers are delineated by dotted lines.
TGF-β treatment does not rescue HPV inhibition of EBV replication.
As we have observed that HPV inhibition of EBV replication may involve alterations in the expression and/or activity of EBV immediate early proteins, we examined whether HPV suppression of transforming growth factor β (TGF-β) signaling was responsible for blocking EBV replication. TGF-β has been shown to increase the expression and activity of the EBV immediate early Z protein. The BZLF1 promoter has several Smad-binding sites, and TGF-β signaling leads to binding of Smad proteins to the promoter, resulting in activation of BZLF1 expression (40, 41). In addition, TGF-β has been shown to suppress MYC expression, with c-Myc known to act as a negative regulator of Z transactivator function (42, 43). HPV has been shown to interfere with TGF-β signaling through various mechanisms (44–46). HPV-immortalized keratinocytes have increased c-Myc levels, as E7 blocks TGF-β suppression of MYC expression (47). Thus, we reasoned that HPV suppression of TGF-β signaling may block EBV lytic replication by either reducing BZLF1 transactivation by Smad proteins or inducing c-Myc to negatively regulate Z transactivator activity. As expected, HFK E6E7 showed a statistically significant increase in MYC transcript levels compared to the levels in parental HFK rafts (Fig. 6A). Treatment of HFK E6E7 rafts with exogenous TGF-β1, added at the time of lifting cells to the air-liquid interface, returned the MYC transcript levels to those seen in parental HFK rafts, reversing the effect of E7 on TGF-β signaling (Fig. 6A). We next examined the outcome of TGF-β1 treatment for EBV replication in both parental HFK and HFK E6E7. Compared to the levels in the vehicle control, EBV DNA levels in the parental rafts increased approximately 8-fold (Fig. 6B). TGF-β1 treatment of HFK E6E7 rafts also increased EBV DNA levels approximately 5-fold over the levels in the vehicle control and did not rescue EBV replication to the DNA levels observed in the TGF-β-treated parental HFK rafts (Fig. 5B). Importantly, the increased EBV DNA levels in HFK E6E7 rafts were insensitive to acyclovir treatment (Fig. 6B). TGF-β1 treatment has been reported to enhance EBV infection of primary and immortalized nasopharyngeal epithelial cells (48). Thus, the lack of response to acyclovir of TGF-β1-treated HFK E6E7 rafts suggested that increased EBV DNA levels resulted from enhanced infection rather than increased DNA replication.
FIG 6.
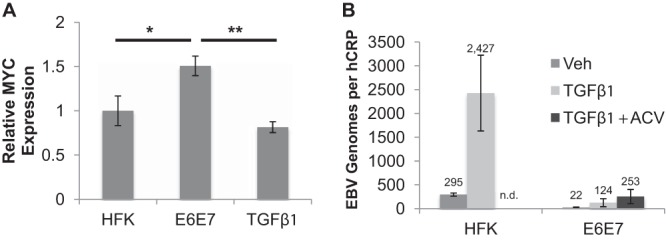
TGF-β1 effect on EBV replication in HFK E6E7 rafts. (A) Parental HFK (n = 6) and HFK E6E7 (n = 5) rafts and HFK E6E7 rafts treated with TGF-β1 (10 ng/ml; n = 5) were analyzed for MYC expression by RT-qPCR. MYC expression in HFK was arbitrarily set to 1. Shown are the average relative expression levels and standard errors of the means. *, P < 0.05; **, P < 0.01. (B) Rafts were infected with EBV and treated with TGF-β1 and acyclovir (ACV; 100 μg/ml) or vehicle (Veh). EBV genome copy numbers per hCRP copy number were quantified by qPCR. n.d., not done. Shown are the average values and standard errors of the means.
HPV16 E7 is sufficient to reduce EBV replication in human foreskin keratinocyte rafts.
To determine if HPV16 E6 or E7 alone would be sufficient to abrogate EBV replication, HFK expressing either oncogene individually were grown in raft culture and infected with EBV. The expression of HPV16 E6 or E7 was confirmed by RT-qPCR (Fig. 4B). Analysis of EBV DNA showed that HFK expressing E6 alone (HFK E6) replicated EBV to levels similar to those in parental HFK. Treatment with acyclovir reduced the EBV DNA levels in HFK E6 and parental HFK, indicating that the EBV DNA levels in the rafts reflected EBV DNA replication (Fig. 7). However, HFK expressing HPV16 E7 alone (HFK E7) exhibited low EBV DNA levels, similar to those in HFK E6E7 and the parental HFK treated with acyclovir (Fig. 7). These results indicate that HPV16 E7 is sufficient to reduce EBV replication in organotypic rafts.
FIG 7.
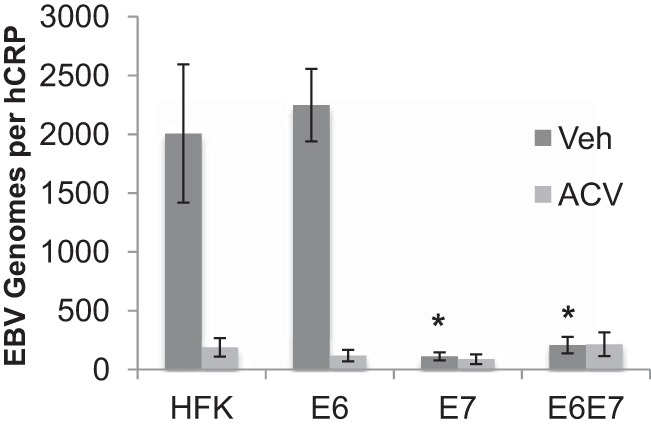
Decreased EBV replication in E7-expressing rafts. Parental HFK rafts (n = 8) and HFK rafts expressing E6 (n = 7), E7 (n = 6), or both (E6E7) (n = 10) were analyzed for EBV genome copy numbers per hCRP copy number in the presence of acyclovir (ACV; 100 μg/ml) or vehicle (Veh). Shown are the average values and standard errors of the means. *, P < 0.05 relative to the results for parental HFK.
Reduced EBV replication correlates with delayed expression of early epithelial differentiation markers.
Given that HPV E6 supported EBV replication, distinct effects of E6 and E7 on epithelial differentiation would reveal additional requirements for EBV lytic replication. Using HFK expressing E6 and/or E7, we examined the expression of early and late epithelial differentiation markers. Parental and HFK E6 rafts expressed involucrin throughout the suprabasal layers (Fig. 8A). In contrast, HFK E7 rafts showed involucrin only at the uppermost layers, while involucrin was nearly absent in HFK E6E7 rafts (Fig. 8A). Similar results were observed regarding the early differentiation marker cytokeratin 10 (K10) (Fig. 8B). Immunofluorescence analysis of the late differentiation marker filaggrin showed uniform expression throughout several epithelial layers in parental HFK rafts. Filaggrin was not detected in HFK E6 rafts, while HFK E7 rafts showed reduced, less uniform filaggrin expression only in the uppermost layers, similar to HFK E6E7 rafts (Fig. 8C). Thus, the ability of HPV to block EBV replication correlated with interference of early rather than late differentiation markers.
FIG 8.

Delayed differentiation in E7-expressing HFK rafts. (A to E) IF staining of involucrin (Inv) (A), cytokeratin 10 (K10) (B), filaggrin (Flg) (C), KLF4 (D), and PRDM1 (E) in parental HFK rafts and HFK rafts expressing E6 and/or E7. Basal layers are delineated by dotted lines. Scale bars represent 50 μm. (F) Percentages of KLF4- and PRDM1-positive cells were quantified as the percentages of total cells in rafts. Shown are the average values and standard errors of the means. (G) HFK and E6E7 rafts (n = 3) were analyzed by RT-qPCR for mRNA levels of KLF4 and PRDM1. KLF4 expression level in HFK was arbitrarily set to 1. (H) Rafts were analyzed by RT-qPCR for mRNA levels of FLG, LOR, and WNT5A. (G, H) Shown are the average relative expression levels and standard errors of the means from a minimum of three rafts per group. *, P < 0.05 relative to the results for parental HFK.
As filaggrin (FLG) is a transcriptional target of KLF4, we examined KLF4 expression. Using immunofluorescence, we observed similar percentages of KLF4-positive cells among all cell lines in raft cultures (Fig. 8D and F). The percentage of PRDM1-expressing cells also remained unchanged across cell lines (Fig. 8E and F), and both KLF4 and PRDM1 RNA levels were similar between parental HFK and HFK E6E7 (Fig. 8G). HFK expressing E6, E7, or both showed significant reductions in the RNA levels of various KLF4 transcriptional targets, which included FLG, LOR, and WNT5A, compared to the levels in parental controls (Fig. 8H). Despite similar transcriptional inhibition of KLF4 target genes by E6 and E7, only E7 affected EBV replication, despite the presence of KLF4 in differentiated keratinocytes. Thus, E7 inhibition of EBV replication may be upstream of KLF4 and involve additional regulators or alterations to KLF4 activity that differ from E6.
DISCUSSION
HPV and EBV share an epithelial tropism for oropharyngeal cells, which can manifest as a coinfection in a subset of oral cancers. Using a model of an incoming EBV infection into HPV-immortalized epithelial rafts, we have shown a block in EBV replication and reduced lytic gene expression in HPV-immortalized keratinocytes (Fig. 1 and 2). hTERT-immortalized normal oral keratinocyte (NOK) rafts supported robust EBV replication similar to that observed in primary HTE rafts, implicating a specific factor in HPV and not immortalization per se in reducing EBV replication (Fig. 1D). In addition, reduced EBV replication occurred in the context of an episomal or integrated HPV genome (Fig. 1B and C). We observed that HPV16 E7 was sufficient to reduce EBV replication in HFK raft culture (Fig. 7).
HPV E7 is a potent transforming protein that reprograms several host cell pathways for viral replication (49). Among the many processes that E7 is known to regulate, we focused on the ability of HPV E7 to delay differentiation and affect the expression/activity of the differentiation-dependent transcription factors KLF4 and PRDM1. Cooperatively with PRDM1, KLF4 activates the expression of the EBV BZLF1 and BRLF1 immediate early genes (20). The transcription factor KLF4 is highly expressed in embryonic stem cells to maintain pluripotency, and overexpression of KLF4 can induce pluripotency in somatic cells (50, 51). In addition, KLF4 directs epithelial terminal differentiation, and the ability of KLF4 to balance stemness and differentiation is dependent upon posttranslational modifications capable of altering KLF4 activity (52, 53). KLF4 is one of a group of transcription factors that activates the expression of a cluster of genes known as the epidermal differentiation complex (EDC) (54). Members of the EDC targeted by KLF4 include differentiation markers that contribute to the formation of the epidermal cornified envelope, such as loricrin and filaggrin (32, 33). A recent study demonstrated that WNT5A, a noncanonical Wnt ligand, is a transcriptional target of KLF4 and suggests a role for WNT5A as a mediator of KLF4 regulation of epithelial differentiation (55). While no differences in KLF4 or PRMD1 expression or the percentages of KLF4-/PRDM1-positive cells were noted, we observed significant reductions in several KLF4 target genes, including the WNT5A, FLG, LOR, BZLF1, BRLF1, and LMP1 genes, in HPV or E6E7-immortalized keratinocytes (Fig. 2, 3, and 8). These observations suggested an alteration in KLF4’s activity, with HPV known to regulate KLF4. HPV E7 suppresses a negative regulator of KLF4 (miRNA-145), while E6 inhibits KLF4 sumoylation and phosphorylation (35). Interestingly, HPV E6 did not affect EBV replication, suggesting that HPV E6-induced posttranslational modifications of KLF4 permitted EBV replication (Fig. 7). In contrast, HPV E7 inhibited EBV replication. Common to both HPV oncogenes were reductions in the late differentiation markers loricrin and filaggrin and in WNT5A, suggesting that the effect of E7 was upstream of KLF4 (Fig. 8). Indeed, the early differentiation markers involucrin and K10 were dramatically reduced in the presence of E7- but not E6-expressing HFK (Fig. 8A and B). As the KLF4 levels in HFK E7 were similar to those in parental HFK and posttranslational modifications influence KLF4 activity, the E7 early block in epithelial differentiation may induce a set of KLF4 modifications distinct from that observed for HPV E6. For example, KLF4 acetylation is also important for KLF4 transactivation activity (56); E7 can interact with both histone acetyltransferases (HATs) and histone deacetylases (HDACs) (57–60) and has been shown to alter the acetylation patterns of cellular proteins (61, 62). Alternatively, E7 may interfere with another factor required for EBV lytic replication.
Central to the immortalization phenotype of HPV E7 is the ability to bind and degrade the retinoblastoma (Rb) family of pocket proteins, pRB, p107, and p130 (63). Herpesviruses, including EBV, also encode viral factors that interact with Rb proteins during latent and lytic infection. For EBV, the EBV nuclear antigens (EBNAs) EBNA-LP and EBNA3C are latent proteins that interact with Rb (64, 65), while BRLF1 is an immediate early protein that interacts with Rb during EBV lytic replication (66). Following the paradigm observed for HPV, efficient EBV infection may also require loss of Rb. However, the role of Rb in herpesvirus infection appears to be more complex. Consistent with our observations that E7 can block EBV replication, the loss of Rb was shown to dramatically reduce human cytomegalovirus (HCMV) viral DNA replication, late gene expression, and virion production (67). In addition to promoting S-phase progression, Rb can transcriptionally repress pluripotency genes, with the observation that Rb inactivation reprogrammed differentiated cells back into pluripotency (68). Distinct interactions of HPV E7 with Rb can promote S phase in differentiating cells or delay epithelial differentiation (30). Future studies will explore how HPV E7 inhibits EBV replication.
While EBV normally replicates in epithelial tissues, the EBV life cycle is altered in epithelial tumors, where it undergoes a shift toward latency. In NPC, early dysplastic changes have been shown to precede EBV infection, and loss of p16 and increased cyclin D1 expression promote EBV latency (23, 26). Thus, HPV immortalization may facilitate a latent EBV infection. Herpesvirus latency is defined as the absence of viral replication and expression of lytic genes with the ability of the virus to reactivate from a latent state. Indeed, the lack of EBV replication and reduced lytic gene expression in HPV-immortalized cells support a latent infection, though we have yet to demonstrate EBV reactivation in HPV-immortalized tissues. Further supporting latency, we detected a significant increase in EBER1 transcript levels in the presence of HPV (Fig. 2). EBERs are found highly expressed in latently infected cells and EBV-positive tumors and possess oncogenic activities. In Burkitt’s lymphomas, EBERs confer resistance to apoptosis (69). In NPC and gastric carcinomas, EBERs induce insulin-like growth factor 1 (IGF-1), which can act in an autocrine manner to stimulate cell growth (70, 71). In the tumor context, a latent EBV infection following HPV immortalization would allow persistent EBV oncogene expression to promote the rapid progression observed in HPV-pos OSCC.
The influence of EBV/HPV coinfection on viral life cycles may differ depending on the timing of infection. Makielski et al. showed that EBV-infected, hTERT-immortalized NOK transfected with the HPV genome exhibit increased EBV lytic replication in raft culture compared to that in HPV-negative NOK (36). We have shown that EBV-infected NOK exhibit a predominant latent gene expression profile (16). Importantly, we have reported that EBV-infected NOK undergo an epigenetic reprogramming, evidenced by DNA hypermethylation and altered gene expression that are retained following loss of the EBV genome from cells (16, 17). Phenotypically, EBV-infected NOK and transiently infected cells showed delayed responses to differentiation induction and increased invasiveness (16, 17). Changes to the Wnt pathway were observed, with increased expression of lymphoid enhancer factor 1 (LEF1) and WNT5A in EBV-infected NOK compared to their expression in parental controls (17). WNT5A is a transcriptional target of KLF4 that can rescue epithelial stratification in the absence of KLF4 (55). Thus, the establishment of latent EBV prior to HPV infection may have distinct effects on epithelial differentiation compared to the effects of HPV infection alone. In support of this notion, EBV-infected NOK exhibited a block in HPV amplification and late gene expression (36), which are steps in the HPV life cycle dependent on epithelial differentiation. Furthermore, EBV replication in latently infected NOK involves viral reactivation from a chromatinized EBV genome, which is not the case for de novo EBV infection. In our model, increased EBV DNA levels were not evident through 15 days following infection (Fig. 4). Thus, EBV reactivation from latency may not be as robust as the DNA amplification that occurs during the establishment of infection.
In this study, we demonstrate an alteration in the EBV life cycle upon infection of HPV-immortalized epithelial tissues. Our data demonstrate HPV-induced inhibition of EBV lytic replication and a possible shift toward EBV latency. As the definition of latency involves an ability for the virus to reactivate, whether EBV is truly latent in HPV-immortalized tissues remains to be determined. HPV E7 was sufficient to block EBV lytic replication, uncovering potential requirements for Rb, altered KLF4 activity, and/or additional regulators in the reduction of EBV replication in HPV-immortalized cells. In oral keratinocytes latently infected with EBV, we previously demonstrated an enhanced invasive phenotype and delayed differentiation (16, 17). Taking these data together, we propose a model where HPV-induced cellular changes facilitate the establishment of a latent EBV infection. Such latent infections would allow for long-term expression of EBV oncogenes and EBV-induced epigenetic reprogramming that contribute to the progression of HPV-pos OSCC.
MATERIALS AND METHODS
Epithelial cell isolation.
Human tonsils without patient identifiers were collected from routine tonsillectomies and classified as an exempt study by our Institutional Review Board. Tonsils were washed with phosphate-buffered saline (PBS) supplemented with nystatin (240 units/ml) and gentamicin (50 μg/ml). Necrotic tissue and fat were removed, leaving behind epithelial tissues which were scraped and rinsed to remove lymphocytes with PBS supplemented with nystatin and gentamicin. Tissues were then submerged in 2 ml 0.25× trypsin/EDTA in a conical vial, vortexed, and incubated at 37°C in 5% CO2 for 20 min, with intermittent vortexing. Trypsin was then removed and inactivated with E medium (74) containing 5% fetal bovine serum (FBS). Harvested cells (human tonsillar epithelial [HTE] cells) were plated in 5% FBS–E medium. The tissue was treated with trypsin twice more, and harvested cells were combined. HTE cells were cocultured with mitomycin C-treated NIH 3T3 J2 fibroblasts in E medium supplemented with 5% FBS and 10 μM Rho kinase inhibitor Y-27632 (Tocris). Cells from four different tissue donors were used to make HTE cell lines.
Cell culture.
To establish HPV-positive HTE cell lines, plasmid containing the HPV16 genome was transfected as described previously for human foreskin keratinocytes (HFK) (72, 73). Briefly, HTE cells were seeded at 2 to 3 million cells per 10-cm dish. HTE cells were cotransfected (using polyethyleneimine [PEI; Polysciences]) with a plasmid harboring the HPV16 genome flanked by LoxP sites, a Cre recombinase-expressing vector, and a neomycin resistance plasmid for 6 to 8 h at 37°C. The medium was then changed, and the following day, G418 selection was used to select for stable cell lines, with 1 mg/ml G418 for 4 days and 500 μg/ml G418 for an additional 4 days. Resistant colonies were expanded in the absence of G418. HTE cells from four different tissue donors were transfected with HPV. HFK were cultured in E medium supplemented with 5% FBS and cocultured with mitomycin C-treated NIH 3T3 J2 fibroblast feeders as described previously (74). E6 and/or E7 was expressed in HFK via retroviral vectors as described previously (73, 75, 76). Cells from at least two different tissue donors were used to make each HFK cell line. A clonal population of human telomerase reverse transcriptase (hTERT)-immortalized normal oral keratinocytes (NOK; gifted by Karl Munger [77]) was propagated in keratinocyte serum-free medium (KSFM) supplemented with bovine pituitary extract and epidermal growth factor (Life Technologies). E6E7-expressing NOK were produced via transfection of an E6E7-expressing vector. NOK were seeded at 4 × 105 cells/well in a 6-well culture dish. The next day, a mixture of plasmid (pLXSN with E6E7 or pLXSN vector alone), Dharmafect 1 (5 μl/ml; Dharmacon), and medium (600 μl total) was incubated on ice for 15 min and then added to plated cells for 5 h. Following transfection, E6E7-expressing NOK were selected with 350 μg/ml G418. The Akata BX1 Burkitt’s lymphoma (BL) cell line was cultured in RPMI supplemented with 10% FBS. Akata BX1 BL cells were induced in 1% RPMI and 100 μg/ml anti-IgG for 48 h. 3T3 J2 fibroblasts were cultivated in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% calf serum. Cells were propagated in a 37°C humidified incubator with 5% CO2. For use as feeders, 3T3 J2 fibroblasts were treated with 8 μg/ml mitomycin C (Santa Cruz) in 10% FBS-supplemented DMEM for 2 to 4 h, washed three times with 1× PBS, and stored at 37°C in a humidified 5% C02 incubator. Mitomycin C-treated J2 fibroblasts were used within 2 weeks.
Three-dimensional tissue culture.
Organotypic rafts were cultured as previously described (78). Briefly, 6.25 × 105 3T3 J2 fibroblasts were suspended in a 2.5 ml collagen plug composed of 80% collagen, 10% 10× DMEM, 10% reconstitution buffer (0.05 M NaOH containing 2.2% NaHCO3 and 4.8% HEPES), and 0.24% 10 M NaOH. Plugs were made in a 6-well culture dish and allowed to solidify for at least 30 min prior to adding E medium supplemented with 5% FBS on top of each plug. One to 4 days later, 1 million epithelial cells in 2 ml E medium with 5% FBS were seeded onto each plug. Prior to seeding, J2 fibroblast feeders were removed from epithelial cultures by incubation in a 1:3 dilution of 0.25% trypsin–EDTA in 1× PBS, followed by trypsinization of epithelial cells in undiluted trypsin. Once confluent, plugs were washed twice with 1× PBS and lifted from the 6-well culture dish onto a stainless steel metal grid, such that the plug was exposed to the air from above and fed from below with E medium supplemented with 5% FBS. Medium was changed every 2 days. Four days postlifting, the apical surface of the rafts was scored and overlaid with 2.5 × 106 Akata BX1 BL cells induced with anti-human IgG for 48 h prior to raft infection, as previously described (39). Anti-human IgG-treated, EBV-negative Akata BL cells were used as a mock-treatment control. For cell-free virus infections, scored rafts were overlaid with ∼4 × 109 EBV particles isolated from the medium of productively replicating Akata BX-1 BL cells. Acyclovir (Tocris) or 1 N hydrochloric acid (HCl) as a vehicle control was added to the medium 1 day post-EBV infection. TGF-β1 (R&D) or 4 mM HCl containing 1 mg/ml bovine serum albumin as a vehicle control was added throughout the time of rafting. At the time of harvest, each raft was washed with 1× PBS and cut into separate pieces for DNA/RNA isolation and tissue processing/paraffin embedding.
DNA and RNA isolation.
To isolate DNA, raft epithelial tissues were lysed in 200 μl Qiagen ATL buffer plus 2 mg/ml proteinase K overnight at 56°C. DNA from lysed tissues or cells was purified using the QIAamp DNA minikit (Qiagen). RNA was harvested from raft tissues using the Qiagen QIAcube via the miRNeasy aqueous-phase protocol, while RNA from monolayer cultures was harvested in RNA STAT60 (Tel-Test, Friendswood, TX) and isolated as instructed by the manufacturer. One microgram of RNA was digested with the TURBO DNA-free kit (Invitrogen) and converted to cDNA by random priming using SuperScript IV reverse transcriptase (Invitrogen) following the manufacturer’s protocol. cDNA was subjected to real time reverse transcription-quantitative PCR (RT-qPCR).
qPCR.
EBV DNA levels in raft cultures were quantified using TaqMan-based real-time quantitative PCR (qPCR) with primer and probe sets specific to the EBV BHRF1 gene and human C-reactive protein (hCRP) as a cellular copy number control. Serial dilutions of DNA from Namalwa, a BL cell line containing two integrated copies of EBV, were used in standard curve analysis to extrapolate EBV and hCRP copy numbers in raft samples (79). Relative expression levels of transcripts were estimated by RT-qPCR. Serial dilutions of Akata BX1 BL cell cDNA were used to generate a standard curve for estimation of expression, which was normalized to cyclophilin A. Normalized expression refers to expression levels relative to cyclophilin A. In relative expression, the normalized expression was compared to a sample being arbitrarily set to 1. All qPCRs were performed in 15-μl reaction mixtures using the Applied Biosystems 7500 Fast thermocycler. PCRs consisted of 10 to 100 ng DNA or 50 ng cDNA, 300 nM primer sets, 200 nM probe (for TaqMan assays), and Power SYBR or TaqMan (Applied Biosystems). All PCR primer sequences are available upon request.
HPV16 genome configuration.
An amount of 100 ng of DNA from raft culture was treated with exonuclease V (RecBCD; NEB) or left untreated at 37°C for 1 h, followed by 95°C heat inactivation for 10 min. DNA from HPV16-infected 293TT cells served as an episomal control. An amount of 10 ng of DNA was quantified by qPCR with primers to detect HPV16 E6, human mitochondrial DNA (episomal internal control), and human 18S ribosomal DNA (multicopy linear DNA internal control). Standard curve analysis based on serially diluted UMSCC47 DNA was used to calculate relative DNA amounts in digested and undigested samples. Resistance to exonuclease digestion was calculated as the percentages of the DNA amounts in digested versus undigested samples (38).
Immunofluorescent staining.
At the time of harvest, raft tissues were fixed for 30 min in 4% paraformaldehyde at 4°C, washed three times (15 min) in cold 1× PBS and once in cold 70% ethanol, and stored in fresh cold 70% ethanol at 4°C prior to processing and paraffin embedding. Antigen retrieval was performed using a pressure cooker for 10 min in 10 mM sodium citrate (pH 6.0) plus 0.05% Tween. Tissues were permeabilized with Triton X-100 for 45 min, blocked in 5% BSA, and probed at 4°C overnight with antibodies specific for Z (sc-53904, 1:200; Santa Cruz), EA-D (EBV-108-48180, 1:10; Capricorn), gp350 (clone 72A1, 13 μg/ml), involucrin (sc-28557, 1:50; Santa Cruz), KLF4 (HPA002926, 1:100; Sigma), PRDM1 (9115, 1:50; Cell Signaling), K10 (sc-52318, 1:50; Santa Cruz), and filaggrin (sc-25896, 1:50; Santa Cruz). For cellular protein detection, tissues were probed with secondary antibodies conjugated to a fluorophore. For viral protein detection, tissues were probed with a poly-HRP secondary antibody (Invitrogen) and subsequently subjected to tyramide signal amplification (TSA) according to the manufacturer’s instructions (Life Technologies). For TSA amplification, tissues were incubated with 10% hydrogen peroxide for thirty minutes prior to blocking, to quench background peroxidase activity. Sections were mounted and stained with 4′,6-diamidino-2-phenylindole (DAPI). Photographic images were obtained using an Olympus BX-50 microscope with MetaView software and edited with ImageJ. For quantification, at least six images from three separate rafts were analyzed. The percentage of positive cells was calculated as the ratio of signal-positive cells to individual nuclei (DAPI), both of which were counted manually.
Data analysis.
Data from at least 3 rafts per group were averaged, and the standard errors of the means calculated. Student’s t test was used to calculate P values.
ACKNOWLEDGMENTS
We thank R. Ellen Friday, Christi Eugene, Yali Jia, and Aimee Vozenilek for technical support.
This research was supported by NIH COBRE grant P30GM110703 from the National Institute of General Medical Sciences and NIH grant R01 DE025565 from the National Institute of Dental and Craniofacial Research to R.S.S. and a Carroll Feist predoctoral fellowship from the LSUHSC-S Feist-Weiller Cancer Center to J.T.G.
REFERENCES
- 1.Gooi Z, Chan JY, Fakhry C. 2016. The epidemiology of the human papillomavirus related to oropharyngeal head and neck cancer. Laryngoscope 126:894–900. doi: 10.1002/lary.25767. [DOI] [PubMed] [Google Scholar]
- 2.Ramqvist T, Dalianis T. 2010. Oropharyngeal cancer epidemic and human papillomavirus. Emerg Infect Dis 16:1671–1677. doi: 10.3201/eid1611.100452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dahlstrom KR, Adler-Storthz K, Etzel CJ, Liu Z, Dillon L, El-Naggar AK, Spitz MR, Schiller JT, Wei Q, Sturgis EM. 2003. Human papillomavirus type 16 infection and squamous cell carcinoma of the head and neck in never-smokers: a matched pair analysis. Clin Cancer Res 9:2620–2626. [PubMed] [Google Scholar]
- 4.Gillison ML, Koch WM, Capone RB, Spafford M, Westra WH, Wu L, Zahurak ML, Daniel RW, Viglione M, Symer DE, Shah KV, Sidransky D. 2000. Evidence for a causal association between human papillomavirus and a subset of head and neck cancers. J Natl Cancer Inst 92:709–720. doi: 10.1093/jnci/92.9.709. [DOI] [PubMed] [Google Scholar]
- 5.Husain N, Neyaz A. 2017. Human papillomavirus associated head and neck squamous cell carcinoma: controversies and new concepts. J Oral Biol Craniofac Res 7:198–205. doi: 10.1016/j.jobcr.2017.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Licitra L, Bernier J, Grandi C, Merlano M, Bruzzi P, Lefebvre JL. 2002. Cancer of the oropharynx. Crit Rev Oncol Hematol 41:107–122. doi: 10.1016/S1040-8428(01)00129-9. [DOI] [PubMed] [Google Scholar]
- 7.Dahlstrand HM, Dalianis T. 2005. Presence and influence of human papillomaviruses (HPV) in tonsillar cancer. Adv Cancer Res 93:59–89. doi: 10.1016/S0065-230X(05)93002-9. [DOI] [PubMed] [Google Scholar]
- 8.Jiang R, Ekshyyan O, Moore-Medlin T, Rong X, Nathan S, Gu X, Abreo F, Rosenthal EL, Shi M, Guidry JT, Scott RS, Hutt-Fletcher LM, Nathan CA. 2015. Association between human papilloma virus/Epstein-Barr virus coinfection and oral carcinogenesis. J Oral Pathol Med 44:28–36. doi: 10.1111/jop.12221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cruz I, Van den Brule AJ, Steenbergen RD, Snijders PJ, Meijer CJ, Walboomers JM, Snow GB, Van der Waal I. 1997. Prevalence of Epstein-Barr virus in oral squamous cell carcinomas, premalignant lesions and normal mucosa—a study using the polymerase chain reaction. Oral Oncol 33:182–188. doi: 10.1016/S0964-1955(96)00054-1. [DOI] [PubMed] [Google Scholar]
- 10.D’Costa J, Saranath D, Sanghvi V, Mehta AR. 1998. Epstein-Barr virus in tobacco-induced oral cancers and oral lesions in patients from India. J Oral Pathol Med 27:78–82. [DOI] [PubMed] [Google Scholar]
- 11.Kobayashi I, Shima K, Saito I, Kiyoshima T, Matsuo K, Ozeki S, Ohishi M, Sakai H. 1999. Prevalence of Epstein-Barr virus in oral squamous cell carcinoma. J Pathol 189:34–39. doi:. [DOI] [PubMed] [Google Scholar]
- 12.Sand LP, Jalouli J, Larsson PA, Hirsch JM. 2002. Prevalence of Epstein-Barr virus in oral squamous cell carcinoma, oral lichen planus, and normal oral mucosa. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 93:586–592. doi: 10.1067/moe.2002.124462. [DOI] [PubMed] [Google Scholar]
- 13.Tsang NM, Chang KP, Lin SY, Hao SP, Tseng CK, Kuo TT, Tsai MH, Chung TC. 2003. Detection of Epstein-Barr virus-derived latent membrane protein-1 gene in various head and neck cancers: is it specific for nasopharyngeal carcinoma? Laryngoscope 113:1050–1054. doi: 10.1097/00005537-200306000-00025. [DOI] [PubMed] [Google Scholar]
- 14.Iamaroon A, Khemaleelakul U, Pongsiriwet S, Pintong J. 2004. Co-expression of p53 and Ki67 and lack of EBV expression in oral squamous cell carcinoma. J Oral Pathol Med 33:30–36. doi: 10.1111/j.1600-0714.2004.00192.x. [DOI] [PubMed] [Google Scholar]
- 15.Bagan JV, Jiménez Y, Murillo J, Poveda R, Díaz JM, Gavaldá C, Margaix M, Scully C, Alberola TM, Torres Puente M, Pérez Alonso M. 2008. Epstein-Barr virus in oral proliferative verrucous leukoplakia and squamous cell carcinoma: a preliminary study. Med Oral Patol Oral Cir Bucal 13:E110–E113. [PubMed] [Google Scholar]
- 16.Birdwell CE, Queen KJ, Kilgore PC, Rollyson P, Trutschl M, Cvek U, Scott RS. 2014. Genome-wide DNA methylation as an epigenetic consequence of Epstein-Barr virus infection of immortalized keratinocytes. J Virol 88:11442–11458. doi: 10.1128/JVI.00972-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Birdwell CE, Prasai K, Dykes S, Jia Y, Munroe TGC, Bienkowska-Haba M, Scott RS. 2018. Epstein-Barr virus stably confers an invasive phenotype to epithelial cells through reprogramming of the WNT pathway. Oncotarget 9:10417–10435. doi: 10.18632/oncotarget.23824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Queen KJ, Shi M, Zhang F, Cvek U, Scott RS. 2013. Epstein-Barr virus-induced epigenetic alterations following transient infection. Int J Cancer 132:2076–2086. doi: 10.1002/ijc.27893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nawandar DM, Ohashi M, Djavadian R, Barlow E, Makielski K, Ali A, Lee D, Lambert PF, Johannsen E, Kenney SC. 2017. Differentiation-dependent LMP1 expression is required for efficient lytic Epstein-Barr virus reactivation in epithelial cells. J Virol 91:e02438-16. doi: 10.1128/JVI.02438-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nawandar DM, Wang A, Makielski K, Lee D, Ma S, Barlow E, Reusch J, Jiang R, Wille CK, Greenspan D, Greenspan JS, Mertz JE, Hutt-Fletcher L, Johannsen EC, Lambert PF, Kenney SC. 2015. Differentiation-dependent KLF4 expression promotes lytic Epstein-Barr virus infection in epithelial cells. PLoS Pathog 11:e1005195. doi: 10.1371/journal.ppat.1005195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reusch JA, Nawandar DM, Wright KL, Kenney SC, Mertz JE. 2015. Cellular differentiation regulator BLIMP1 induces Epstein-Barr virus lytic reactivation in epithelial and B cells by activating transcription from both the R and Z promoters. J Virol 89:1731–1743. doi: 10.1128/JVI.02781-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kang MS, Kieff E. 2015. Epstein-Barr virus latent genes. Exp Mol Med 47:e131. doi: 10.1038/emm.2014.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lo KW, Chung GT, To KF. 2012. Deciphering the molecular genetic basis of NPC through molecular, cytogenetic, and epigenetic approaches. Semin Cancer Biol 22:79–86. doi: 10.1016/j.semcancer.2011.12.011. [DOI] [PubMed] [Google Scholar]
- 24.Lo KW, Huang DP, Lau KM. 1995. p16 gene alterations in nasopharyngeal carcinoma. Cancer Res 55:2039–2043. [PubMed] [Google Scholar]
- 25.Hui AB, Or YY, Takano H, Tsang RK, To KF, Guan XY, Sham JS, Hung KW, Lam CN, van Hasselt CA, Kuo WL, Gray JW, Huang DP, Lo KW. 2005. Array-based comparative genomic hybridization analysis identified cyclin D1 as a target oncogene at 11q13.3 in nasopharyngeal carcinoma. Cancer Res 65:8125–8133. doi: 10.1158/0008-5472.CAN-05-0648. [DOI] [PubMed] [Google Scholar]
- 26.Tsang CM, Yip YL, Lo KW, Deng W, To KF, Hau PM, Lau VM, Takada K, Lui VW, Lung ML, Chen H, Zeng M, Middeldorp JM, Cheung AL, Tsao SW. 2012. Cyclin D1 overexpression supports stable EBV infection in nasopharyngeal epithelial cells. Proc Natl Acad Sci U S A 109:E3473–E3482. doi: 10.1073/pnas.1202637109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lukas J, Muller H, Bartkova J, Spitkovsky D, Kjerulff AA, Jansen DP, Strauss M, Bartek J. 1994. DNA tumor virus oncoproteins and retinoblastoma gene mutations share the ability to relieve the cell’s requirement for cyclin D1 function in G1. J Cell Physiol 125:625–638. doi: 10.1083/jcb.125.3.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li W, Thompson CH, Cossart YE, O’Brien CJ, McNeil EB, Scolyer RA, Rose BR. 2004. The expression of key cell cycle markers and presence of human papillomavirus in squamous cell carcinoma of the tonsil. Head Neck 26:1–9. doi: 10.1002/hed.10335. [DOI] [PubMed] [Google Scholar]
- 29.Klussmann JP, Gultekin E, Weissenborn SJ, Wieland U, Dries V, Dienes HP, Eckel HE, Pfister HJ, Fuchs PG. 2003. Expression of p16 protein identifies a distinct entity of tonsillar carcinomas associated with human papillomavirus. Am J Pathol 162:747–753. doi: 10.1016/S0002-9440(10)63871-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Collins AS, Nakahara T, Do A, Lambert PF. 2005. Interactions with pocket proteins contribute to the role of human papillomavirus type 16 E7 in the papillomavirus life cycle. J Virol 79:14769–14780. doi: 10.1128/JVI.79.23.14769-14780.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Flores ER, Allen-Hoffmann BL, Lee D, Lambert PF. 2000. The human papillomavirus type 16 E7 oncogene is required for the productive stage of the viral life cycle. J Virol 74:6622–6631. doi: 10.1128/JVI.74.14.6622-6631.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Patel S, Xi ZF, Seo EY, McGaughey D, Segre JA. 2006. Klf4 and corticosteroids activate an overlapping set of transcriptional targets to accelerate in utero epidermal barrier acquisition. Proc Natl Acad Sci U S A 103:18668–18673. doi: 10.1073/pnas.0608658103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boxer LD, Barajas B, Tao S, Zhang J, Khavari PA. 2014. ZNF750 interacts with KLF4 and RCOR1, KDM1A, and CTBP1/2 chromatin regulators to repress epidermal progenitor genes and induce differentiation genes. Genes Dev 28:2013–2026. doi: 10.1101/gad.246579.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Segre JA, Bauer C, Fuchs E. 1999. Klf4 is a transcription factor required for establishing the barrier function of the skin. Nat Genet 22:356–360. doi: 10.1038/11926. [DOI] [PubMed] [Google Scholar]
- 35.Gunasekharan VK, Li Y, Andrade J, Laimins LA. 2016. Post-transcriptional regulation of KLF4 by high-risk human papillomaviruses is necessary for the differentiation-dependent viral life cycle. PLoS Pathog 12:e1005747. doi: 10.1371/journal.ppat.1005747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Makielski KR, Lee D, Lorenz LD, Nawandar DM, Chiu YF, Kenney SC, Lambert PF. 2016. Human papillomavirus promotes Epstein-Barr virus maintenance and lytic reactivation in immortalized oral keratinocytes. Virology 495:52–62. doi: 10.1016/j.virol.2016.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Asselineau D, Prunieras M. 1984. Reconstruction of “simplified” skin: control of fabrication. Br J Dermatol 111(Suppl 27):219–222. doi: 10.1111/j.1365-2133.1984.tb15608.x. [DOI] [PubMed] [Google Scholar]
- 38.Bienkowska-Haba M, Luszczek W, Myers JE, Keiffer TR, DiGiuseppe S, Polk P, Bodily JM, Scott RS, Sapp M. 2018. A new cell culture model to genetically dissect the complete human papillomavirus life cycle. PLoS Pathog 14:e1006846. doi: 10.1371/journal.ppat.1006846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Temple RM, Zhu J, Budgeon L, Christensen ND, Meyers C, Sample CE. 2014. Efficient replication of Epstein-Barr virus in stratified epithelium in vitro. Proc Natl Acad Sci U S A 111:16544–16549. doi: 10.1073/pnas.1400818111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liang CL, Chen JL, Hsu YP, Ou JT, Chang YS. 2002. Epstein-Barr virus BZLF1 gene is activated by transforming growth factor-beta through cooperativity of Smads and c-Jun/c-Fos proteins. J Biol Chem 277:23345–23357. doi: 10.1074/jbc.M107420200. [DOI] [PubMed] [Google Scholar]
- 41.Iempridee T, Das S, Xu I, Mertz JE. 2011. Transforming growth factor beta-induced reactivation of Epstein-Barr virus involves multiple Smad-binding elements cooperatively activating expression of the latent-lytic switch BZLF1 gene. J Virol 85:7836–7848. doi: 10.1128/JVI.01197-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lin Z, Yin Q, Flemington E. 2004. Identification of a negative regulatory element in the Epstein-Barr virus Zta transactivation domain that is regulated by the cell cycle control factors c-Myc and E2F1. J Virol 78:11962–11971. doi: 10.1128/JVI.78.21.11962-11971.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pietenpol JA, Holt JT, Stein RW, Moses HL. 1990. Transforming growth factor beta 1 suppression of c-myc gene transcription: role in inhibition of keratinocyte proliferation. Proc Natl Acad Sci U S A 87:3758–3762. doi: 10.1073/pnas.87.10.3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hypes MK, Pirisi L, Creek KE. 2009. Mechanisms of decreased expression of transforming growth factor-beta receptor type I at late stages of HPV16-mediated transformation. Cancer Lett 282:177–186. doi: 10.1016/j.canlet.2009.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee DK, Kim BC, Kim IY, Cho EA, Satterwhite DJ, Kim SJ. 2002. The human papilloma virus E7 oncoprotein inhibits transforming growth factor-beta signaling by blocking binding of the Smad complex to its target sequence. J Biol Chem 277:38557–38564. doi: 10.1074/jbc.M206786200. [DOI] [PubMed] [Google Scholar]
- 46.French D, Belleudi F, Mauro MV, Mazzetta F, Raffa S, Fabiano V, Frega A, Torrisi MR. 2013. Expression of HPV16 E5 down-modulates the TGFbeta signaling pathway. Mol Cancer 12:38. doi: 10.1186/1476-4598-12-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pietenpol JA, Stein RW, Moran E, Yaciuk P, Schlegel R, Lyons RM, Pittelkow MR, Munger K, Howley PM, Moses HL. 1990. TGF-beta 1 inhibition of c-myc transcription and growth in keratinocytes is abrogated by viral transforming proteins with pRB binding domains. Cell 61:777–785. doi: 10.1016/0092-8674(90)90188-K. [DOI] [PubMed] [Google Scholar]
- 48.Tsang CM, Zhang G, Seto E, Takada K, Deng W, Yip YL, Man C, Hau PM, Chen H, Cao Y, Lo KW, Middeldorp JM, Cheung AL, Tsao SW. 2010. Epstein-Barr virus infection in immortalized nasopharyngeal epithelial cells: regulation of infection and phenotypic characterization. Int J Cancer 127:1570–1583. doi: 10.1002/ijc.25173. [DOI] [PubMed] [Google Scholar]
- 49.Roman A, Munger K. 2013. The papillomavirus E7 proteins. Virology 445:138–168. doi: 10.1016/j.virol.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Takahashi K, Yamanaka S. 2006. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 51.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. 2007. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 52.Li HX, Han M, Bernier M, Zheng B, Sun SG, Su M, Zhang R, Fu JR, Wen JK. 2010. Kruppel-like factor 4 promotes differentiation by transforming growth factor-beta receptor-mediated Smad and p38 MAPK signaling in vascular smooth muscle cells. J Biol Chem 285:17846–17856. doi: 10.1074/jbc.M109.076992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim MO, Kim SH, Cho YY, Nadas J, Jeong CH, Yao K, Kim DJ, Yu DH, Keum YS, Lee KY, Huang Z, Bode AM, Dong Z. 2012. ERK1 and ERK2 regulate embryonic stem cell self-renewal through phosphorylation of Klf4. Nat Struct Mol Biol 19:283–290. doi: 10.1038/nsmb.2217. [DOI] [PubMed] [Google Scholar]
- 54.Kypriotou M, Huber M, Hohl D. 2012. The human epidermal differentiation complex: cornified envelope precursors, S100 proteins and the “fused genes” family. Exp Dermatol 21:643–649. doi: 10.1111/j.1600-0625.2012.01472.x. [DOI] [PubMed] [Google Scholar]
- 55.Tetreault MP, Weinblatt D, Shaverdashvili K, Yang Y, Katz JP. 2016. KLF4 transcriptionally activates non-canonical WNT5A to control epithelial stratification. Sci Rep 6:26130. doi: 10.1038/srep26130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Evans PM, Zhang W, Chen X, Yang J, Bhakat KK, Liu C. 2007. Kruppel-like factor 4 is acetylated by p300 and regulates gene transcription via modulation of histone acetylation. J Biol Chem 282:33994–34002. doi: 10.1074/jbc.M701847200. [DOI] [PubMed] [Google Scholar]
- 57.Avvakumov N, Torchia J, Mymryk JS. 2003. Interaction of the HPV E7 proteins with the pCAF acetyltransferase. Oncogene 22:3833–3841. doi: 10.1038/sj.onc.1206562. [DOI] [PubMed] [Google Scholar]
- 58.Bernat A, Avvakumov N, Mymryk JS, Banks L. 2003. Interaction between the HPV E7 oncoprotein and the transcriptional coactivator p300. Oncogene 22:7871–7881. doi: 10.1038/sj.onc.1206896. [DOI] [PubMed] [Google Scholar]
- 59.Brehm A, Nielsen SJ, Miska EA, McCance DJ, Reid JL, Bannister AJ, Kouzarides T. 1999. The E7 oncoprotein associates with Mi2 and histone deacetylase activity to promote cell growth. EMBO J 18:2449–2458. doi: 10.1093/emboj/18.9.2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Huang SM, McCance DJ. 2002. Down regulation of the interleukin-8 promoter by human papillomavirus type 16 E6 and E7 through effects on CREB binding protein/p300 and P/CAF. J Virol 76:8710–8721. doi: 10.1128/JVI.76.17.8710-8721.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang B, Laribee RN, Klemsz MJ, Roman A. 2004. Human papillomavirus type 16 E7 protein increases acetylation of histone H3 in human foreskin keratinocytes. Virology 329:189–198. doi: 10.1016/j.virol.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 62.Richards KH, Wasson CW, Watherston O, Doble R, Blair GE, Wittmann M, Macdonald A. 2015. The human papillomavirus (HPV) E7 protein antagonises an imiquimod-induced inflammatory pathway in primary human keratinocytes. Sci Rep 5:12922. doi: 10.1038/srep12922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dyson N, Howley PM, Munger K, Harlow E. 1989. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science 243:934–937. doi: 10.1126/science.2537532. [DOI] [PubMed] [Google Scholar]
- 64.Parker GA, Crook T, Bain M, Sara EA, Farrell PJ, Allday MJ. 1996. Epstein-Barr virus nuclear antigen (EBNA)3C is an immortalizing oncoprotein with similar properties to adenovirus E1A and papillomavirus E7. Oncogene 13:2541–2549. [PubMed] [Google Scholar]
- 65.Szekely L, Selivanova G, Magnusson KP, Klein G, Wiman KG. 1993. EBNA-5, an Epstein-Barr virus-encoded nuclear antigen, binds to the retinoblastoma and p53 proteins. Proc Natl Acad Sci U S A 90:5455–5459. doi: 10.1073/pnas.90.12.5455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zacny VL, Wilson J, Pagano JS. 1998. The Epstein-Barr virus immediate-early gene product, BRLF1, interacts with the retinoblastoma protein during the viral lytic cycle. J Virol 72:8043–8051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.VanDeusen HR, Kalejta RF. 2015. The retinoblastoma tumor suppressor promotes efficient human cytomegalovirus lytic replication. J Virol 89:5012–5021. doi: 10.1128/JVI.00175-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kareta MS, Gorges LL, Hafeez S, Benayoun BA, Marro S, Zmoos A-F, Cecchini MJ, Spacek D, Batista LFZ, O’Brien M, Ng Y-H, Ang CE, Vaka D, Artandi SE, Dick FA, Brunet A, Sage J, Wernig M. 2015. Inhibition of pluripotency networks by the Rb tumor suppressor restricts reprogramming and tumorigenesis. Cell Stem Cell 16:39–50. doi: 10.1016/j.stem.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Komano J, Maruo S, Kurozumi K, Oda T, Takada K. 1999. Oncogenic role of Epstein-Barr virus-encoded RNAs in Burkitt’s lymphoma cell line Akata. J Virol 73:9827–9831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Iwakiri D, Sheen TS, Chen JY, Huang DP, Takada K. 2005. Epstein-Barr virus-encoded small RNA induces insulin-like growth factor 1 and supports growth of nasopharyngeal carcinoma-derived cell lines. Oncogene 24:1767–1773. doi: 10.1038/sj.onc.1208357. [DOI] [PubMed] [Google Scholar]
- 71.Iwakiri D, Takada K. 2010. Role of EBERs in the pathogenesis of EBV infection. Adv Cancer Res 107:119–136. doi: 10.1016/S0065-230X(10)07004-1. [DOI] [PubMed] [Google Scholar]
- 72.Wilson R, Laimins LA. 2005. Differentiation of HPV-containing cells using organotypic “raft” culture or methylcellulose. Methods Mol Med 119:157–169. doi: 10.1385/1-59259-982-6:157. [DOI] [PubMed] [Google Scholar]
- 73.Bodily JM, Mehta KP, Cruz L, Meyers C, Laimins LA. 2011. The E7 open reading frame acts in cis and in trans to mediate differentiation-dependent activities in the human papillomavirus type 16 life cycle. J Virol 85:8852–8862. doi: 10.1128/JVI.00664-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Meyers C, Laimins LA. 1994. In vitro systems for the study and propagation of human papillomaviruses. Curr Top Microbiol Immunol 186:199–215. [DOI] [PubMed] [Google Scholar]
- 75.Bodily JM, Mehta KP, Laimins LA. 2011. Human papillomavirus E7 enhances hypoxia-inducible factor 1-mediated transcription by inhibiting binding of histone deacetylases. Cancer Res 71:1187–1195. doi: 10.1158/0008-5472.CAN-10-2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hebner CM, Wilson R, Rader J, Bidder M, Laimins LA. 2006. Human papillomaviruses target the double-stranded RNA protein kinase pathway. J Gen Virol 87:3183–3193. doi: 10.1099/vir.0.82098-0. [DOI] [PubMed] [Google Scholar]
- 77.Piboonniyom SO, Duensing S, Swilling NW, Hasskarl J, Hinds PW, Munger K. 2003. Abrogation of the retinoblastoma tumor suppressor checkpoint during keratinocyte immortalization is not sufficient for induction of centrosome-mediated genomic instability. Cancer Res 63:476–483. [PubMed] [Google Scholar]
- 78.Anacker D, Moody C. 2012. Generation of organotypic raft cultures from primary human keratinocytes. J Vis Exp 2012:e3668. doi: 10.3791/3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wagner HJ, Scott RS, Buchwald D, Sixbey JW. 2004. Peripheral blood lymphocytes express recombination-activating genes 1 and 2 during Epstein-Barr virus-induced infectious mononucleosis. J Infect Dis 190:979–984. doi: 10.1086/423211. [DOI] [PubMed] [Google Scholar]