

IFITM proteins have been associated with the sequestration of incoming virions in endosomes (target cell protection) and with the production of virion particles that incorporate IFITMs and exhibit decreased infectivity (negative imprinting of virion infectivity). How the latter is regulated and whether these two antiviral properties are related remain unknown. By examining the behavior of a large panel of IFITM3 mutants against HIV-1, we determined that IFITM3 mutants are essentially packaged into virions proportionally to their intracellular levels of expression. However, even in the context of an IFITM-susceptible virus, IFITM3 packaging is not sufficient for the antiviral effects. Most mutations were found to concomitantly affect both antiviral properties of IFITM3, but one CTD mutant exhibited a divergent behavior, possibly highlighting a novel regulatory role for this domain. These findings thus advance our comprehension of how this class of broad antiviral restriction factors acts.
KEYWORDS: IFITM, IFITM3, interferon, restriction factor, human immunodeficiency virus, viral resistance
ABSTRACT
The interferon-induced transmembrane proteins (IFITMs) are a family of highly related antiviral factors that affect numerous viruses at two steps: in target cells by sequestering incoming viruses in endosomes and in producing cells by leading to the production of virions that package IFITMs and exhibit decreased infectivity. While most studies have focused on the former, little is known about the regulation of the negative imprinting of virion particle infectivity by IFITMs and about its relationship with target cell protection. Using a panel of IFITM3 mutants against HIV-1, we have explored these issues as well as others related to the biology of IFITM3, in particular virion packaging, stability, the relation to CD63/multivesicular bodies (MVBs), the modulation of cholesterol levels, and the relationship between negative imprinting of virions and target cell protection. The results that we have obtained exclude a role for cholesterol and indicate that CD63 accumulation does not directly relate to an antiviral behavior. We have defined regions that modulate the two antiviral properties of IFITM3 as well as novel domains that modulate protein stability and that, in so doing, influence the extent of its packaging into virions. The results that we have obtained, however, indicate that, even in the context of an IFITM-susceptible virus, IFITM3 packaging is not sufficient for negative imprinting. Finally, while most mutations concomitantly affect target cell protection and negative imprinting, a region in the C-terminal domain (CTD) exhibits a differential behavior, potentially highlighting the regulatory role that this domain may play in the two antiviral activities of IFITM3.
IMPORTANCE IFITM proteins have been associated with the sequestration of incoming virions in endosomes (target cell protection) and with the production of virion particles that incorporate IFITMs and exhibit decreased infectivity (negative imprinting of virion infectivity). How the latter is regulated and whether these two antiviral properties are related remain unknown. By examining the behavior of a large panel of IFITM3 mutants against HIV-1, we determined that IFITM3 mutants are essentially packaged into virions proportionally to their intracellular levels of expression. However, even in the context of an IFITM-susceptible virus, IFITM3 packaging is not sufficient for the antiviral effects. Most mutations were found to concomitantly affect both antiviral properties of IFITM3, but one CTD mutant exhibited a divergent behavior, possibly highlighting a novel regulatory role for this domain. These findings thus advance our comprehension of how this class of broad antiviral restriction factors acts.
INTRODUCTION
The interferon-induced transmembrane proteins (IFITMs) are a family of highly related proteins characterized by the presence of two hydrophobic transmembrane domains (TM1 and TM2) separated by a conserved intracellular loop (CIL) and variable N- and C-terminal domains (NTD and CTD, respectively) (1, 2). In humans, this family is composed of five expressed members (IFITM1, -2, -3, -5, and -10), three of which are interferon regulated and endowed with broad antiviral properties (IFITM1, -2, and -3 [collectively referred to as IFITMs in the present manuscript]) (3–5).
A number of studies have firmly established that IFITMs target a large spectrum of distinct viral families (5–33), although viruses that resist IFITMs, or use them as cofactors, such as human coronavirus OC43, have been identified (6, 34–36).
For viruses, such as HIV-1, that are susceptible to these restriction factors, IFITMs appear to act according to a bimodal inhibition mode, with which they are able to target two different steps of the viral life cycle (15, 36, 37).
The first and most studied antiviral mechanism described for IFITMs takes place in target cells, where IFITMs arrest incoming virion particles in endosomes, leading to their subsequent degradation (6, 11, 12, 16, 18, 20–22, 24, 26, 31, 38). This inhibition mode targets pH-dependent viruses that undergo viral-to-cellular membrane fusion after endosomal internalization but also pH-independent viruses that either fuse directly at the plasma membrane or, as for HIV-1, appear to use endosomes as an additional, nonmandatory entry pathway. The second mechanism, first described for HIV-1 by our and other laboratories (15, 23, 37) and more recently extended to other viruses (36), occurs in virion-producing cells. In this setting, IFITMs coalesce with viral structural proteins at sites of assembly, leading to the production of virion particles that package IFITMs and display reduced infectivity. We refer to the latter property as the negative imprinting of virion particle infectivity by IFITMs (23, 36).
At present, it is unknown whether this decrease in infectivity is due to the physical incorporation of IFITMs in virion particles or if it is the result of an action of IFITMs on unknown cellular functions that in turn influence virion assembly. Over the years, several studies raised the possibility that IFITM3 could act by either modulating the intracellular content of cholesterol (during target cell protection) (39) or commandeering a decrease in the amount of mature envelope glycoproteins incorporated into HIV-1 virions (37). However, both of these findings remain debated, as further studies failed to obtain evidence of changes in either intracellular cholesterol levels (31, 40) or glycoprotein incorporation upon IFITM expression (16, 23, 36, 41, 42).
Despite the fact that the exact molecular mechanism through which IFITMs act remains unclear, virions produced in the presence of IFITMs exhibit an impaired ability to undergo fusion with cellular membranes, the same step that hinders fusion between the membranes of incoming virions and IFITM-decorated endosomes during target cell infection (15, 23, 37, 40, 43–45). In the case of HIV-1, partial to complete resistance to this antiviral effect has been shown to be conferred by R5-tropic envelope proteins that use the CCR5 chemokine receptor to access target cells (16, 46, 47). It appears, therefore, that the antiviral effects that IFITMs have on membrane fusion can be countered through the use of specific envelope glycoproteins or through the engagement of specific coreceptors on target cells.
A number of studies have revealed domains that have important regulatory functions on the antiviral properties of the different IFITM members, influencing their ability to recruit multiple cellular partners and to mediate protection of target cells from viral challenge (19, 33, 43, 48–55). However, little is known about domains that specify the negative imprinting properties of IFITMs.
In this study, we have directly addressed this question by analyzing the behavior of a panel of 16 different mutants spanning the entire length of IFITM3 against HIV-1 (19). The relatively large number of mutants examined allowed us to also explore the functional relationship between antiviral behavior and several parameters that have been associated with the biology of IFITM3, including their incorporation into virions, their stability, their reported ability to increase the levels of formation of CD63-positive vesicles and multivesicular bodies (MVBs) in the cell, as well as their ability to affect cholesterol levels. To complete these analyses, we have also determined the antiviral activities of these mutants during target cell protection, establishing for the first time a comprehensive analysis of the relationship between these two antiviral properties.
The data that we have obtained in our study highlight novel regions of IFITM3 that modulate the negative imprinting properties of IFITM3, its ability to mediate target cell protection, as well as its stability in the cell. The latter parameter seems to be the main driver of IFITM3 incorporation into virions, as most mutants were found to be incorporated into virions proportionally to their level of expression in the cell. The extent of IFITM3 incorporation into virions, however, does not correlate with the antiviral phenotype, allowing us to conclude that IFITM3 virion packaging is not sufficient for its antiviral functions. Finally, we noted that mutations throughout IFITM3 exerted a concomitant modulation of both target cell protection and negative imprinting of virions. Only two domains made an exception to this rule, the first located in the CIL that we believe to be a novel gain-of-function IFITM3 mutant and the second located in the CTD that may represent a region involved in the differential regulation of the two antiviral properties ascribed to IFITM3.
RESULTS
Presentation of the IFITM3 mutants used in this study.
To comprehensively identify domains regulating the negative effects of IFITM3 against HIV-1, we took advantage of a collection of mutants spanning the entire length of IFITM3 (6 amino acids changed to alanines per mutant, named by the number of the first and last substituted residues) (19) (Fig. 1A). A few mutants were excluded from the analysis due to unreliable expression or a lack of detection with the antibodies used here, leaving a total of 16 mutants. Given that only seven of these mutants had been previously examined by confocal microscopy (19), a complete qualitative analysis was carried out for all of them (Fig. 1B). Several of the IFITM3 mutants were found to exhibit a more peripheral (mutants 8-13 and 19-24) or a more internal (mutants 79-84, 85-90, 103-108, 115-120, and 127-132) distribution than the wild type (WT) after confocal microscopy analysis. A more restricted analysis of the colocalization of relevant IFITM3 mutants with the endosomal marker CD63 is provided below.
FIG 1.

IFITM3 mutants used here. (A) IFITM3 mutants used in this study. Mutants that could not be reliably detected upon transient DNA transfection of HEK293T cells were excluded from our study and are not presented here. FACS, fluorescence-activated cell sorter. (B) The intracellular localization of these mutants was analyzed by confocal microscopy, and representative pictures are displayed. Of note, this analysis is meant to provide a qualitative assessment of the intracellular distribution of the different IFITM3 mutants and does not reflect their relative expression levels, which instead were quantified by WB.
Identification of mutations that modulate the ability of IFITM3 to negatively imprint the infectivity of HIV-1 virion particles.
In previous studies, we determined that the effects of IFITMs on the infectivity of virion particles derived from HIV-1, but also from many other viruses, are apparent in all cell types tested, ranging from primary macrophages and blood lymphocytes to established cell lines (23, 36). As such, we used HEK293T cells as a validated model cell line in which to comprehend the roles of distinct regions of IFITM3 in HIV-1 infectivity. IFITM3 mutants were expressed transiently at levels that we have previously shown to be comparable to those observed in primary dendritic cells stimulated with interferon alpha (IFN-α) (as schematically presented in Fig. 2A) (23, 36). Cells were cotransfected with DNAs encoding the IFITM3 variants, the NL4-3 proviral clone, and a miniviral HIV-1-based genome bearing the green fluorescent protein (GFP) reporter and a self-inactivating long terminal repeat (LTR). This setup allowed us to examine replication-competent HIV-1 virions (in which all viral proteins are expressed at natural levels from their cognate promoter) on the basis of a single round of infection, as the GFP-bearing viral genome can be mobilized only once during infection, from virus-producing cells to target ones. Virion particles were purified by ultracentrifugation through a 25% sucrose cushion and then normalized by the exogenous reverse transcriptase (exo-RT) activity, which in our hands provides results identical to those of p24-based normalization (23). Cellular and viral lysates were then analyzed by Western blotting (WB), and the amounts of Gag, Env, and IFITM3 proteins were quantified by densitometry (see Fig. 2B for a representative example). All IFITM3 mutants were detectable in cell lysates albeit with notable variations that required enhancement of the signal in the case of the most drastically affected mutant (8-13 [this mutant was retained due to its interesting phenotype {see below}]). The structural viral proteins Gag p55 and Env gp160 were also detected at similar levels, although small variations could be observed across experiments. Upon purification, the amounts of virion particles produced under the different conditions varied little in comparison to the control, with the exception of a small yet significant decrease upon the expression of the IFITM3 mutants 79-84, 85-90, and 103-108 (55 to 56% of the total) (Fig. 2C). Given that virions were normalized for further analyses, these differences were not investigated further. When exo-RT-normalized virion particles were analyzed by WB, no major variations were observed in the levels of mature Env gp120 incorporated in virions produced in the presence of the different IFITM3 mutants, with the exception of mutant 85-90, whose expression resulted in a near loss of Env detection (Fig. 2B). The expression of WT IFITM3 had been previously associated with the loss of mature Env incorporation in HIV (37). However, this finding remains controversial, as other studies have not observed such changes in the HIV-1 glycoprotein (16, 23, 41, 42) or for other viral glycoproteins (36), so at present, the reasons for this discrepancy remain unclear. Given that under the experimental conditions used here, no major changes were observed in the amounts of mature Env incorporated into HIV-1 virions upon the expression of the WT or of most IFITM3 mutants, we believe that 85-90 most likely represents a gain-of-function mutant.
FIG 2.

Effects of the expression of the different IFITM3 mutants on the production and infectivity of HIV-1 virion particles. (A) Experimental scheme used to further characterize the antiviral effects of IFITM3 mutants during the negative imprinting of virion particle infectivity. Briefly, HIV-1 virion particles were produced by transient DNA transfection of HEK293T cells with DNAs coding for the different IFITM3 mutants along with the NL4-3 proviral clone and an HIV-1-based miniviral genome bearing a self-inactivating LTR and an expression cassette for GFP. Two days afterwards, cells were lysed, and virion particles were purified by ultracentrifugation through a 25% sucrose cushion, prior to normalization by exo-RT activity and further analyses. (B) Western blot panels presenting typical results obtained for both cell and viral lysates. Mutant 19-24 was the sole mutant whose detection required hybridization with a distinct anti-IFITM3 antibody compared to the remaining mutants. (C) Quantification of the amounts of virion particles produced in the presence of the different IFITM3 mutants by exo-RT. The graph presents data obtained from 6 experiments. *, P < 0.05 as determined by a Student t test upon comparison to WT IFITM3 (two tailed, unpaired). (D) Normalized amounts of virions were used to challenge target HeLaP4 cells, and the extent of infection was measured 2 to 3 days afterwards by flow cytometry. The graph presents data obtained with 4 to 8 independent experiments. *, P < 0.05 as determined by a Student t test for comparisons between the indicated mutant and WT IFITM3 (two tailed, unpaired). All mutants displayed statistically significant differences over the control according to the same test (not displayed on the graph for simplicity).
All IFITM3 mutants copurified with HIV-1 virions, although to widely different extents (including mutant 13-18 after enhancement of the WB signal), indicating that none of the mutations analyzed here prevented the incorporation of IFITM3 into HIV-1 virions.
The infectivity of normalized virion particles was then measured upon viral challenge of target HeLaP4 cells, prior to flow cytometry analysis 2 to 3 days afterwards (Fig. 2D). Under conditions in which the expression of wild-type IFITM3 resulted in a 55% decrease in infectivity in single-round infection assays, all IFITM3 mutants tested exerted measurable and variable effects on the infectivity of virion particles with respect to the control, and four exhibited substantially distinct behaviors with respect to the WT (Fig. 2D). Specifically, the 19-24 and the 85-90 IFITM3 mutants imparted greater infectivity defects (70% and 85% lower infectivity than the control, respectively), while the 73-78 and the 109-114 mutants displayed less infectivity defects (29% and 25%). Among these four mutants, the infectivity defect of IFITM3 mutant 85-90 was expected in light of the major impairment in gp120 incorporation observed upon its expression, and the one of the 19-24 mutant was also expected due to previous studies (41). In search of mutants that could completely relieve the antiviral effects of IFITM3 on the production of infectious virion particles, the two relevant mutations were combined (73-78/109-114) (Fig. 3). However, this double mutant was still proficient in altering virion infectivity (exhibiting wild-type antiviral activities rather than decreased ones), indicating that the effects of these mutations cannot be functionally combined into a single IFITM3 molecule. The reasons why the combination of two relieving mutations in TM1 and TM2 result in the reacquisition of the antiviral phenotype remain unclear.
FIG 3.
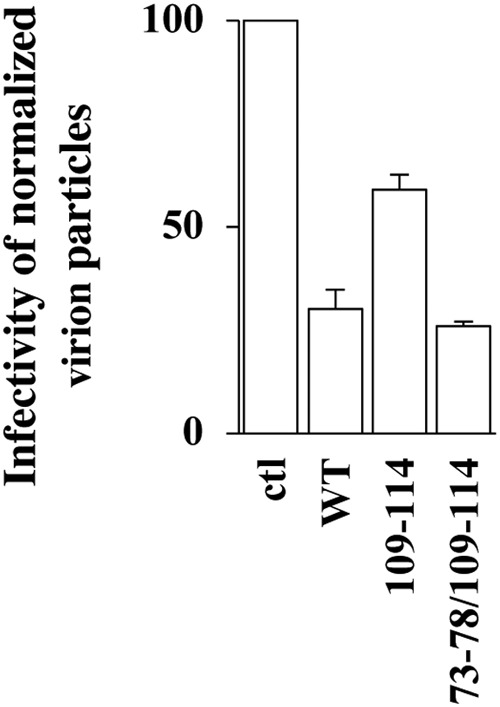
Two mutations that individually relieve the negative imprinting properties of IFITM3 on virion particles fail to cooperatively alleviate the antiviral phenotype. The antiviral activity of the double IFITM3 mutant 73-78/109-114 was assessed as described in the legend to Fig. 2. The graph presents the infectivity of normalized amounts of virion particles produced in the presence of this mutant during the challenge of target HeLaP4 cells (averages and standard errors of the means [SEM] from two independent experiments).
Several conserved domains in IFITM3 influence the protein’s stability.
Differently from an approach based on stable expression and selection of IFITM3-expressing cells (19), transient transfection allowed us to more directly appreciate differences in IFITM3 protein levels and therefore to highlight novel domains that affected the steady-state levels of accumulation of IFITM3 (Fig. 4A). Several mutations led to a statistically significant decrease in the accumulation of IFITM3 in the cell: in the NTD (13-18 and 37-42), in TM1 (73-78 and 79-84), and in the CIL (85-90 and 103-108). The involvement of these amino acid stretches in the stability of IFITM3 has not been appreciated previously, with the exception of mutant 13-18 (13NSGQPP18) adjacent to the region 17PPNYEML23, which acts as a docking site for E3 ubiquitin ligase neural precursor cell expressed developmentally downregulated protein 4 (NEDD4), adaptor protein 2 (AP2), as well as the Fyn kinase (10, 18, 51, 54, 55). Mining of public databases for “immune IFITMs” (i.e., IFITM1, -2, and -3) indicated that the TM1-CIL region and in particular amino acids 72 to 91 and 103 to 108, which affect the protein’s stability, have been strictly conserved during million years of primate evolution (Fig. 4B), while, as expected from previous studies, the NTD exhibited higher variability (30, 41, 56–58). In contrast to mutants 72-91 and 103-108, the evolutionary history of the domains spanning residues 13 to 18 and 37 to 42 (both contained within the NTD and affecting protein stability) shows signs of divergence within different primate clades, given that the region spanning residues 37 to 42 is well conserved among New and Old World monkeys, while the one spanning residues 13 to 18 has undergone more-drastic diversification.
FIG 4.

Identification of conserved IFITM3 domains that influence the intracellular levels of accumulation of IFITM3 and modulate the extent of IFITM3 incorporation into virions. (A) Domains influencing the intracellular levels of accumulation of IFITM3. The amounts of IFITM3 mutants present in intracellular lysates were determined after WB analysis following densitometry-based quantification and normalization to WT values. (B) Evolutionary conservation of domains influencing IFITM3 levels. Shown are primate IFITM1, -2, and -3 with sequence logos showing amino acids (probability is plotted on the y axis) corresponding to the different domains of interest for each primate gene clade. The number of sequences used for the analysis is indicated (seq, sequences) (see Materials and Methods for details). Color-coding is the default in WebLogo3. (C) Domains influencing IFITM3 incorporation into virion particles (as in panel A), after quantification of the levels of IFITM3 mutants present in exo-RT-normalized virion particles. (D) Comparison between the intracellular levels of expression and virion incorporation of the different IFITM3 mutants (log scale) following densitometry-based quantification and normalization to WT values. The graph also displays the ratio of the amount of IFITM3 protein incorporated into virion particles with respect to their intracellular levels of expression as a virion incorporation index, after normalization to WT IFITM3. *, P < 0.05 as determined by a Student t test for comparison between the indicated mutant and the WT (two tailed, unpaired).
Determinants of IFITM3 incorporation into virion particles.
Our mutagenesis study failed to reveal a single mutation that completely abrogated the ability of IFITM3 to be incorporated into virion particles. However, several mutations were identified that decreased the absolute levels of IFITM3 molecules incorporated into normalized amounts of virion particles, in particular 13-18, 37-42, 73-78, 79-84, and 127-132 (Fig. 4C). To determine whether this decrease was due to the loss of a specific virion-packaging signal, or whether it was linked to lower levels of expression in the cell, we compared the levels of accumulation of the different IFITM3 mutants in cells and in purified virions (Fig. 4D [the normalized protein levels in virions and cells as well as their ratio, defined here as an IFITM3 virion incorporation index, are displayed]). This analysis strongly indicated that IFITM3 mutants were incorporated into virions proportionally to their levels of expression in the cell. The sole exception was represented by mutant 103-108, which was less stable than the wild type in cells yet was incorporated as well as WT IFITM3 into virions (therefore displaying a virion incorporation index of 2.25). This result indicates that mutation of the corresponding amino acid stretch (103AKCLNI108) at the border between the CIL and TM2 somehow exerts a double effect, by reducing the protein’s stability on one hand and by leading to higher levels of incorporation into virions on the other.
Overall, the results obtained here indicate (i) that several regions control the stability of IFITM3 in the cell that, based on their evolutionary conservation/divergence across primates, may act by influencing the overall structural conservation of IFITMs (TM1-CIL) or more-specific regulatory functions (NTD) and (ii) that an important determinant of IFITM3 incorporation into virions is the intracellular level of its expression.
The levels of incorporation of IFITM3 into virions do not correlate with their ability to negatively affect HIV-1 virion infectivity.
Given that an open question in the field is to determine whether the physical presence of IFITM3 in virion particles is instrumental for their ability to decrease virion infectivity, we used our results to determine the possible existence of a correlation between incorporation levels of IFITM3 and the potency of the antiviral phenotype (Fig. 5). Overall, the results that we have obtained indicate no correlation between antiviral properties of virion particle infectivity and either the extent of IFITM3 incorporation into virions or intracellular levels of expression. These data therefore suggest that the incorporation of IFITM3 into HIV-1 particles is not sufficient for the negative imprinting phenotype, as previously suggested by other studies (16), as well as by the existence of viruses that resist IFITMs despite robust incorporation (for instance, Rift Valley fever virus) (36). The question of whether IFITM3 incorporation is necessary for the antiviral phenotype remains open at present due to the fact that we could not isolate an IFITM3 mutant completely excluded from virion particles. In light of the fact that approximately one-third of IFITM3 is composed of transmembrane regions, such a mutant may be difficult to obtain.
FIG 5.
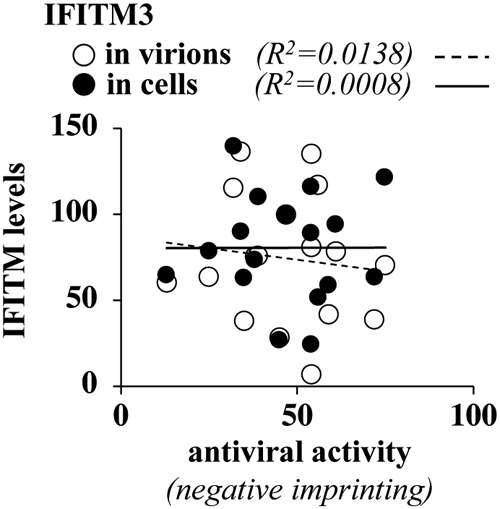
The antiviral activities of the different IFITM3 mutants do not correlate with their intracellular expression levels or with the extent of their incorporation into virion particles. Shown are correlative analyses between the levels of accumulation of the different IFITM3 mutants in the cell, their incorporation into virions, and their antiviral activity in the negative imprinting of HIV-1 virion particles.
The ability of IFITM3 to decrease virion particle infectivity does not correlate with the accumulation of CD63-positive vesicles in cells, and IFITM3 does not affect cholesterol homeostasis.
A commonly observed phenotype associated with the expression of IFITM3 is an increase in the levels of the endosomal marker CD63, which may be indicative of increased vesicular trafficking or multivesicular body (MVB) formation (11, 20, 38, 39, 48, 59) and, by extension, of higher lysosomal activity, which may be pertinent for the antiviral effects of IFITM3, at least during viral entry. However, it is unclear whether this observation is functionally linked to the antiviral functions of IFITM3. The expression of WT IFITM3 was clearly accompanied by an increase in the proportion of CD63-positive cells (from 35% to 70%) (Fig. 6A and B), in agreement with previous studies. When a similar analysis was conducted on a restricted number of IFITM3 mutants, the proportion of cells displaying higher-level accumulation of CD63+ vesicles varied across mutants. Under the same experimental conditions, IFITM3-expressing cells displayed higher proportions of discernible MBVs following electron microscopy than did control cells (from 9% to 25%) (Fig. 6C presents MVBs from WT IFITM3-expressing cells), and similarly to CD63, this proportion varied among the different IFITM3 mutants (Fig. 6C, graph). Supporting the notion that higher CD63 positivity relates to higher MVB activity, a near-perfect correlation was observed between these two parameters for the different mutants analyzed (Fig. 6D). However, neither correlated with the antiviral behavior of IFITM3 mutants (Fig. 6E [CD63 accumulation]), indicating that this phenotype does not directly relate to the ability of IFITM3 to decrease virion particle infectivity.
FIG 6.

IFITM3 mutants modulate the extent of accumulation of CD63-positive vesicles as well as MVBs in cells, but these properties do not correlate with their antiviral behaviors. (A and B) Cells expressing or not expressing a short list of IFITM3 mutants were analyzed by confocal microscopy using the endosomal marker CD63. (C) Same as above, but cells were analyzed by electron microscopy. A representative picture of a cell expressing WT IFITM3 is displayed, while the number of scored MVB-positive cell sections in the different mutants is presented in the graph (please note the different scale). (D and E) Correlative analyses, as indicated. The graphs present data obtained in 2 independent experiments (electron microscopy) or 3 independent experiments (immunofluorescence analysis) with over 100 scored cells under each condition.
An increase in the intracellular levels of cholesterol in A549 cells was previously hypothesized to be at the basis of the defect imparted by IFITM3 at membrane fusion. However, the role of cholesterol homeostasis in IFITM3-mediated restriction remains controversial (31, 39, 40, 60). To explore this issue, we measured the amount of cholesterol-laden lipid droplets after Nile Red staining by confocal microscopy in HEK293T cells expressing or not expressing WT IFITM3 (see Fig. 7A for representative pictures and Fig. 7B for averages of data from multiple experiments). Given that the antiviral effects of IFITM3 are manifest in this cell type, changes in cholesterol levels should also appear here if they are functionally relevant. Cells treated with either methyl-β-cyclodextrin (mβCD) or oleic acid (OA) presented low to undetectable and increased intracellular cholesterol levels, respectively, as measured by Nile Red staining and in agreement with their well-known effects on cholesterol homeostasis.
FIG 7.
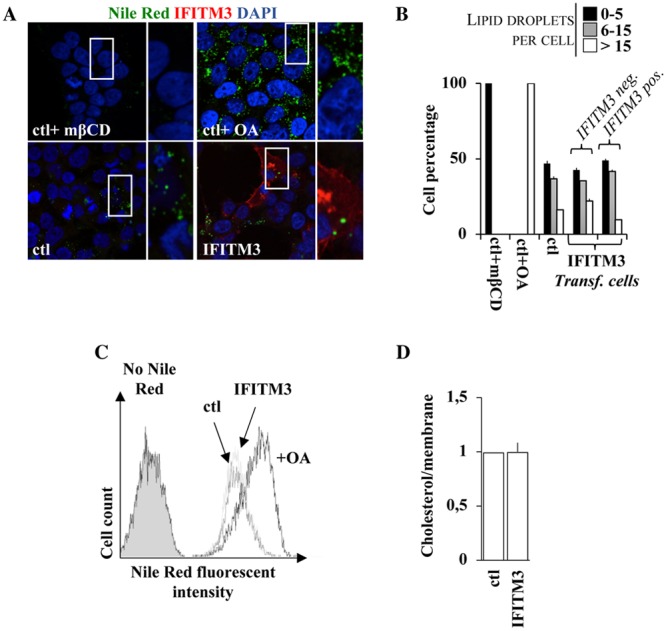
IFITM3 does not affect cholesterol levels in cells or in purified virion particles. (A and B) HEK293T cells were analyzed by confocal fluorescence microscopy following staining of intracellular lipid droplets by Nile Red (0.25 μg/ml). Control cells were treated with either methyl-β-cyclodextrin (1 mM) or oleic acid (100 μM), well known to either deplete or increase the intracellular levels of cholesterol. Representative pictures are presented in panel A, while the number of lipid droplets per cell is presented in panel B for over 75 cells analyzed under each condition. (C) Same as panel A, but the accumulation of Nile Red was quantified by flow cytometry. (D) Virions produced in the presence or absence of WT IFITM3 were produced and purified as described above. In this case, viral preparations were normalized by their membrane content upon R18 labeling. Cholesterol levels were measured according to the cholesterol/cholesteryl ester quantification kit (Abcam). The graph presents averages and SEM of data from three independent experiments.
Instead, when control cells were compared to cells expressing WT IFITM3, no qualitative or quantitative differences were observed. Similarly, no differences in median fluorescence intensities (MFIs) were observed when the extent of Nile Red accumulation was assessed through a distinct assay (flow cytometry) (Fig. 7C).
While these results indicate that IFITM3 is unlikely to affect cholesterol homeostasis in the cells tested in this study, they do not formally exclude the possibility that the infectivity defect of virion particles produced in the presence of IFITM3 could be due to a specific enrichment of cholesterol in virion particle membranes. To address this issue, HIV-1 virions were produced and purified by ultracentrifugation through a 25% sucrose cushion from cells expressing or not expressing IFITM3. The virion preparations were then normalized by either exo-RT activity or total lipid contents (using R18, a fluorescent lipophilic dye that intercalates linearly in membranes and allows their quantification, prior to the quantification of cholesterol).
Under these conditions, no changes were observed in the amounts of cholesterol under the two conditions (Fig. 7D). Despite the fact that this purification procedure cannot separate bona fide virion particles from exosomal vesicles, these data, in addition to the lack of changes in cholesterol levels in cells, strongly suggest that the membrane fusion defect ascribed to IFITM3 does not involve cholesterol changes. Due to these results, the extent of cholesterol modification for the different mutants was not explored further.
Identification of mutations that modulate the ability of IFITM3 to protect target cells from HIV-1 infection.
Although the IFITM3 mutants used in this study have been analyzed against other viruses during target cell protection (19), their activities against HIV-1 have not been explored. To directly compare the antiviral activities of IFITM3 mutants at entry and in virion particle imprinting, HEK293T cells were transiently transfected with the different IFITM3 mutants along with CD4 and CXCR4, and their respective expression levels were assessed by WB and flow cytometry, respectively (as schematically presented in Fig. 8A, with typical results shown in Fig. 8B). No notable differences were observed in the cell surface expression of both CD4 and CXCR4 in the presence of the different IFITM3 mutants (Fig. 8B). These cells were used as targets for viral challenge (NL4-3-pseudotyped HIV-1 encoding GFP used at multiplicities of infection [MOIs] of between 0.1 and 0.5), and the extent of infection was measured 2 to 3 days afterwards by flow cytometry (Fig. 8C). As expected, cells expressing WT IFITM3 displayed increased resistance to viral challenge (70% decrease in infection rates, compared to control cells). The susceptibility of cells expressing the different IFITM3 mutants varied from 85 to 15% (all statistically significant compared to control cells [not marked in Fig. 8]), but despite variations in target cell susceptibility, only two mutants significantly relieved the antiviral properties of IFITM3 compared to the wild type, mutant 13-18 and mutant 37-42, both located in the protein N terminus (Fig. 8C). The partial loss of the antiviral activity of these mutants may be related to their lower expression levels than the WT, although on a more global scale, the intracellular levels of expression of the different IFITM3 mutants did not correlate with their antiviral activity in target cell protection (Fig. 8D).
FIG 8.

Behaviors of the different IFITM3 mutants during target cell protection. (A) Experimental scheme used here to evaluate the abilities of the different IFITM3 mutants to protect target cells from HIV-1 infection. HEK293T cells were used as target cells due to their efficient transfection rates, following transient transfection with DNAs coding for the different IFITM3 mutants along with CD4 and CXCR4. (B) Two days after transfection, cells were analyzed by either WB or flow cytometry to determine the levels of expression of the different proteins. (C) Cells were then challenged with an equal dose of GFP-encoding HIV-1 (NL4-3 at an MOI of between 0.1 and 0.5), prior to flow cytometry analysis 2 to 3 days afterwards. The panels present typical results obtained, while the graph presents averages and SEM obtained from 5 to 7 independent experiments. *, P < 0.05 as determined by a Student t test for comparisons between the indicated mutant and WT IFITM3 (two tailed, unpaired). All mutants display P values of <0.05 when the same test is carried out in comparison to control cells (asterisks are not displayed for simplicity). (D) Correlative analysis between the extent of incorporation of the different IFITM3 mutants into virion particles and their levels of expression in cells.
Overall comparison of the behaviors of IFITM3 mutants with respect to their ability to mediate negative imprinting of virion particles and target cell protection and to affect protein stability.
To reveal potential links between the abilities of the different IFITM3 mutants to act at both steps of the viral life cycle, as well as to appreciate the relationship between these antiviral properties and IFITM3 stability, the effects of individual mutations on these three parameters were normalized to those of the WT and compared (Fig. 9), as indicated.
FIG 9.

Overall comparison of the activities of individual mutants to affect negative imprinting of virion particles, target cell protection, as well as the intracellular levels of accumulation. (A) The two antiviral activities of the different IFITM3 mutants as well as their relative stability were normalized to those of WT and overlaid to highlight the behavior of individual mutants that deviated from the wild type, as indicated.
On a general level, with two exceptions that are discussed in more detail below, most mutants presented similar trends in their ability to influence negative imprinting of virion particles and target cell protection. The magnitudes of such variations were generally similar for both antiviral phenotypes, with the exception of mutants 13-18 and 37-42, in which target cell protection was relieved 2-fold more than negative imprinting. These two IFITM3 mutants are less stable than the WT, which would suggest that target cell protection is more sensitive than negative imprinting to the intracellular levels of IFITM3. However, although protein stability can influence the behavior of specific mutants, this parameter does not simply explain the protein’s phenotypes, because no correlation could be drawn between the protein levels of the different IFITM3 mutants and their antiviral phenotypes.
Although the concomitant variations observed for most IFITM3 mutants in their two antiviral properties could suggest a common antiviral mechanism, mutants 85-90 and 127-132 display a divergent behavior, a finding that may support a model in which two distinct antiviral mechanisms are at play. Mutant 85-90 behaves as the WT during target cell protection, but its expression in virion-producing cells leads to a drastic decrease in Env incorporation into virions, which readily explains their accrued infectivity defect.
Interference with Env maturation and incorporation has been reported previously for WT IFITM3. However, this finding remains controversial, and similarly to other studies, here we have not observed changes in Env incorporation for WT IFITM3 as well as most mutants. As a consequence, we believe that mutant 85-90 gained a novel function, and as such, it is unlikely to represent a protein domain that can disconnect the two antiviral properties. Instead, the behavior of IFITM3 mutant 127-132 in the CTD may suggest that this region differentially regulates target cell protection and negative imprinting of virions.
DISCUSSION
In this study, we have systematically examined the behavior of a large panel of IFITM3 mutants with respect to their ability to interfere with the production of infectious HIV-1 virions. We have done so in a widely used model cell line that reproduces the infectivity defects imparted by IFITMs on different viruses and in different cell types (both primary and established). This approach allowed us to gather novel insights into the mechanisms regulating the negative imprinting properties of IFITM3 as well as to explore their relationship with other issues of interest in the biology of this antiviral factor. For the sake of clarity, these topics are described in separate subsections, below.
Novel domains that regulate IFITM3 stability.
Making use of stable cell lines, a previous study identified regions of IFITM3 that strongly influenced protein stability, and accordingly, these mutants could not be used here (19). However, the use of transient transfection allowed us to identify additional domains involved in protein stability: mutants 13-18 and 37-42 in the NTD; 73-78 in TM1; and 79-84, 85-90, and 103-108 in the CIL. The behavior of mutant 13-18 (13NSGGQPP18) is not unexpected, as it overlaps a region that acts as a docking site for several proteins (17PPNYEML23): the E3 ubiquitin ligase Nedd4 (PPNY), AP2 (YEML), and the tyrosine kinase Fyn (Y) (10, 18, 51, 54, 55). Mutation of the two proline residues in mutant 13-18 may favor the association with AP2, leading to higher levels of internalization of IFITM3 (this mutant indeed displays an increased intracellular distribution) and higher levels of lysosomal degradation. Instead, the destabilizing role of mutations in the CIL is of high interest because this region is extremely conserved among members of the IFITM family, and its role in the biology of IFITM3 remains relatively less well studied than other regions. Our results therefore suggest that the CIL plays a novel regulatory role in IFITM3 protein stability and/or trafficking that deserves further investigation.
IFITM3 incorporation in and negative imprinting properties on HIV-1 virion particles.
Perhaps unsurprisingly for a protein composed for one-third of transmembrane regions, our analysis did not reveal a single 6-amino-acid-stretch mutation capable of excluding IFITM3 from virions. However, the comparison between IFITM3 levels in cells and in virions allowed us to appreciate that the main driver of the incorporation of mutant into virion particles seems to be their degree of expression, in agreement with similar findings obtained with endogenous IFITMs in primary macrophages undergoing spreading HIV-1 infection. Under the experimental conditions used here, differences in the localization of IFITM3 mutants did not have a major influence, in line with similar findings for IFITM1, -2, and -3, which were also equally well incorporated into virion particles despite distinct intracellular distributions (15, 16, 23, 36). Although we have extensively determined in previous studies that IFITMs copurifying with viral fractions are packaged into virions and not in exosomes in cells undergoing active virion assembly (by CD45 depletion and immunogold electron microscopy [23, 36]), it can be argued that IFITMs could in principle also be incorporated into virion particles in other cellular vesicles (for instance, CD45-negative cells).
The results obtained here allow us to determine that IFITM3 incorporation is not sufficient for its effect on virions and that the potency of the antiviral phenotype can be disconnected from the levels of IFITM3 packaging. Indeed, several mutants endowed with wild-type antiviral activities are incorporated at low to undetectable levels in virion particles, as best illustrated by the mutant 13-18, which is barely detectable in HIV-1 virion particles. However, our results do not allow us to determine whether the physical presence of IFITM3 in virion particles is absolutely necessary for the antiviral phenotype, because we have not identified a single IFITM3 mutant completely excluded from HIV-1 virions (this mutant may be impossible to obtain without disrupting most of the protein structure). Hence, two possibilities still exist to explain how IFITM3 may influence virion infectivity: either very low levels of IFITM3 are sufficient to impart a negative effect on virion infectivity or the physical presence of IFITM3 in virion particles is not important for the antiviral phenotype. At present, we favor the latter hypothesis, as it would explain not only the lack of a correlation between the antiviral activities of several IFITM3 mutants and their degree of incorporation into particles but also a previous observation from our laboratory indicating that IFITM1 can decrease the infectivity of Dugbe virus (DUGV) (a member of the Bunyaviridae family) without being detectably incorporated into virion particles (36). An important corollary of this hypothesis would be that IFITM3 acts on the cell during virion assembly to modify an important component of virion particles that results in the membrane fusogenicity defect.
Relationship between antiviral phenotypes, accumulation of CD63-positive vesicles, and cholesterol.
In agreement with previous studies, we also found here that the expression of several IFITM3 mutants leads to higher CD63 and MVB contents (11, 19, 20, 38, 39, 48, 59). However, the analysis of a number of IFITM3 mutants indicated that this parameter does not correlate with antiviral activities, as, for instance, mutant 43-48 retained robust antiviral activities despite a clear inability to induce CD63 vesicle accumulation. Therefore, our analysis does not grant a direct functional relevance to this widely observed phenotype during the antiviral activities of IFITM3, although it remains possible that qualitative rather than quantitative differences in the induction of CD63-MVBs may have been missed in our analyses.
The data that we have obtained here also exclude a role of cholesterol changes in the antiviral effects of IFITM3, as we have observed no modulation of this lipid in either cells or virion particles expressing or not expressing this restriction factor using different techniques. However, cholesterol is only one of the many lipids present in membranes, and it remains possible that IFITM3 acts by affecting the homeostasis of other lipids (for instance, phospholipids) that in turn affect the behavior of membranes in a more functional manner.
Mechanistic insights into the bimodal inhibitory activity of IFITM3.
Our analysis reveals that two regions in the NTD (spanning residues 13 to 18 and 37 to 42) are important for the antiviral effects of IFITM3 against HIV-1 during target cell protection. These regions do not play major roles in the case of influenza A virus (IAV) and dengue virus (DENV), viruses that are instead influenced by nonoverlapping regions in the NTD as well as by mutations in TM1 and the CIL (19). The fact that different viruses display distinct susceptibilities to the same IFITM3 mutant had been noted previously after comparison of IAV and DENV. If such differences exist in the case of viruses that at least share a pH-dependent mechanism to enter target cells, it is highly plausible that other portions of IFITM3 regulate HIV-1 entry, as this virus uses a pH-independent mechanism to fuse at the plasma membrane and likely also in endosomes (61). In this respect, it will be of interest to determine whether other pH-independent viruses display susceptibility to specific mutations in IFITM3 similar to that of HIV-1 or not.
In contrast to target cell protection, no exhaustive study has examined the abilities of different regions of IFITM3 to modulate virion particle infectivity, so the results that we have obtained in the case of HIV-1 cannot be directly compared to those of other studies. Our analysis highlights four modulatory regions in IFITM3: two regions in TM1 and TM2 (residues 73 to 78 and 109 to 114) that partially relieve the infectivity defect on newly produced virion particles and two in the NTD and CIL that interestingly increase it (residues 19 to 24 and 85 to 90).
At first sight, therefore, the domains involved in the two antiviral properties of IFITM3 do not overlap each other. However, when the respective activities of each mutant are normalized, an interesting link between these antiviral effects becomes more readily apparent, in that most IFITM3 mutations that alter negative imprinting concomitantly affect target cell protection albeit with slightly different magnitudes. This would therefore indicate that the antiviral activities against HIV-1 ascribed to IFITM3 may be similarly modulated by the same regions. While the decreased stability of certain IFITM3 mutants could be called into play to explain the behavior of certain mutants, on a more global scale, IFITM3 stability is not the main parameter influencing the antiviral activities.
At present, only two mutants diverge from this common trend, and at least one of them (127-132) may represent a region that differentially controls target cell protection over negative imprinting. Mutant 85-90 in the CIL represents in our view a gain-of-function mutant that interferes with the trafficking and/or incorporation of HIV-1 envelope glycoproteins into virions. Env modulation by IFITM3 remains a controversial subject (16, 23, 36, 37, 41), and it remains possible that the relative expression levels of both IFITM3 and HIV-1 Env determine whether this effect is observable or not. However, under the experimental conditions used here, IFITM3 variants exerted their antiviral effects without modulating the extent of HIV-1 Env incorporation into virions. As such, the 85-90 mutant should be considered to have a gain-of-function phenotype. How this unique activity occurs has not been further investigated here, but it is intuitive that this mechanism would affect more deeply the infectivity of newly produced virion particles rather than target cell protection.
Instead, mutant 127-132 presents wild-type behavior with respect to virion particle infectivity but is less active during target cell protection. The CTD underwent strong diversifying evolution between members of the IFITM family (30, 41, 56–58), and given that this region has been described to insert into lysosomal membranes where it is processed (1), it is possible that mutations in this domain affect more strongly their ability to sequester incoming virion particles in endolysosomal vesicles during target cell infection rather than the production of infectious virion particles.
In conclusion, the results that we have gathered here provide novel insights into the regions of IFITM3 that intervene in the production of infectious virion particles, in target cell protection, as well as in IFITM3 protein stability. More specifically, our study highlighted the role of two regions of interest supporting the need for further investigations on the role of the CIL and CTD in the stability and antiviral inhibition associated with this broad antiviral restriction factor.
MATERIALS AND METHODS
Cells, plasmids, antibodies, and reagents.
HEK293T and HeLaP4 cells (stably expressing the CD4/CXCR4 receptors in addition to an HIV-1-LTR-β-galactosidase reporter cassette) were maintained in complete Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum (both cell lines were obtained from the Cellulonet repository of SFR Biosciences in Lyon-Gerland). The different IFITM3 mutants were described previously (19). IFITM3 variants are expressed in the context of the QCXIP vector (Clontech) in the absence of any epitope tag. Double mutants were engineered in the same backbone using standard molecular biology techniques. HIV-1 virion particles were produced by transient DNA cotransfection of HEK293T cells in the presence of IFITM3, the proviral clone NL4-3, and an HIV-1-based miniviral genome bearing a self-inactivating LTR and coding for GFP (pRRL-CMV-GFP) (23, 36). This allowed us to precisely quantify the infectivity of otherwise replication-competent virion particles on the basis of a single cycle of infection. CD4- and CXCR4-encoding DNAs were obtained from the AIDS Reagent and Reference Program of the NIH. Antibodies were as follows: antitubulin (Sigma), anti-Gag/p24 (clone 183-H12-5C from the AIDS Reagents Program of the NIH), antiserum to HIV-1 gp120 (clone 288 from the AIDS Reagents Program of the NIH), anti-NTD IFITM3 (catalog number AP1153a; Abgent) or anti-IFITM3 (rabbit polyclonal) (catalog number 11714-1-AP; Proteintech), anti-CD63 (catalog number 556019; BD biosciences), and anti-CD4 and anti-CXCR4 (catalog numbers 551980 and 551968, respectively; Becton, Dickinson). Nile Red, methyl-β-cyclodextrin (MβCD), and oleic acid (OA) were purchased from Sigma (catalog numbers 19123, C4555, and O1008, respectively). The 19-24 mutant did not cross-react with the anti-NTD antibody, so this mutant was quantified separately using the polyclonal anti-IFITM3 antibody mentioned above.
Quantification of the different proteins analyzed here was carried out after densitometric analysis of results obtained after WB (ImageJ software). For IFITM3 mutants expressed at low levels, quantifications were carried out on overexpressed signals. Pilot comparative experiments were carried out with digital-fluorescence-based WB quantification (Odyssey CLx imaging system; LiCor), but given the high level of concordance of the results, Odyssey quantification was not used further.
To measure the amount of cholesterol in virion-associated membranes, supernatants obtained after transient DNA transfection of HEK293T cells were divided into two aliquots and either left untreated or incubated with 1 μg/ml of octadecyl rhodamine B chloride (R18; Invitrogen), a fluorescent lipophilic dye that intercalates linearly in membranes (62). Virions were then purified for both R18-labeled and unlabeled aliquots by ultracentrifugation through a 25% (wt/vol) sucrose cushion. The R18 fluorescence incorporated into viral membranes was quantified on a Tecan plate reader and used to normalize for cholesterol levels measured on the specular aliquot of virion particles (cholesterol/cholesteryl ester quantification kit, catalog number ab65359; Abcam). Cholesterol levels are therefore normalized here for the total amount of lipids present in each sample.
Viral production and infection.
HIV-1 virion particles were produced by calcium phosphate DNA transfection of HEK293T cells in the presence of the different IFITM3 mutants or control DNAs with a DNA ratio of 8:4:3 for NL4-3, pRRL-CMV-GFP, and IFITM3, respectively (for a total of 15 μg of DNA per 10-cm plate). We had previously determined that these concentrations of IFITM3 were comparable to those measured in primary dendritic cells stimulated with type I interferons. Forty-eight hours after DNA transfection, cells were lysed, supernatants were filtered through a 0.45-μm syringe filter, and virions were then purified by ultracentrifugation at 28,000 rpm for 75 min through a 25% (wt/vol) sucrose cushion. After ultracentrifugation, the pellet was resuspended in 1× phosphate-buffered saline (PBS) and normalized by exo-RT activity. Viral infectivity was determined 2 to 3 days after challenge of HeLaP4 cells by flow cytometry. To measure the effects of the different IFITM3 mutants on target cell protection, HEK293T cells were transiently transfected with DNAs coding for CD4 and CXCR4 along with IFITM3 mutants (ratio of 0.2:0.2:1, respectively, for a total of 1.4 μg DNA per well of a 6-well plate). Two days after transfection, cells were used as targets for infection with WT GFP-HIV-1, prior to flow cytometry analysis 2 to 3 days afterwards.
Confocal microscopy.
HEK293T cells were grown on 0.01% poly-l-lysine-coated coverslips and analyzed 36 h after transfection with DNAs coding for wild-type IFITM3 or mutants. For DNA transfections, Lipofectamine 2000 (Invitrogen) was used according to the manufacturer’s instructions. Cells were fixed with 4% paraformaldehyde for 20 min, quenched with 50 mM NH4Cl for 10 min, and permeabilized with PBS–0.5% Triton X-100 for 5 min. After a blocking step in PBS–5% milk, cells were incubated with primary antibodies overnight, followed by incubation for 2 h with a donkey anti-rabbit IgG–Alexa Fluor 594 conjugate (catalog number A-21207; Life Technologies) and/or a donkey anti-mouse IgG–Alexa Fluor 488 conjugate (catalog number A-21202; Life Technologies). A 4′,6-diamidino-2-phenylindole (DAPI)-containing mounting medium was used (Southern Biotech). Images were acquired using a spectral Zeiss LSM710 confocal microscope and analyzed with Fiji software. Where indicated, Nile Red was added during the last three 5-min washes with PBS at 0.25 μg/ml before mounting of the coverslips (confocal microscopy) or added directly to the medium at the same concentration during the last 30 min of culture, prior to cell harvest.
Electron microscopy.
HEK293T cells transfected as described above were fixed for 24 h in 4% paraformaldehyde (PFA) and 1% glutaraldehyde in 0.1 M phosphate buffer (pH 7.2). Samples were then washed in PBS and postfixed by incubation with 2% osmium tetroxide for 1 h. Samples were then fully dehydrated in a graded series of ethanol solutions and propylene oxide. The impregnation step was performed with a mixture (1:1) of propylene oxide-Epon resin, and the samples were then left overnight in pure resin. Cells were then embedded in Epon resin, which was allowed to polymerize for 48 h at 60°C. Ultrathin sections (90 nm) of these blocks were obtained with an EM UC7 ultramicrotome (Leica Microsystems, Wetzlar, Germany). Sections were stained with 2% uranyl acetate and 5% lead citrate, and observations were made with a transmission electron microscope (catalog number 1011; JEOL, Tokyo, Japan).
Host phylogenetic analyses.
Homologous sequences of the primate IFITM1 to -3 genes were retrieved from publicly available databases using NCBI tBLASTn with human IFITM1 or human IFITM3 as the query. Multiple duplication events in ancient and recent times during primate evolution make phylogenetic analyses of the IFITM1 to -3 genes in primates challenging (41). For this study, a conservative approach was used in which all sequences annotated as being noncoding (XR/NR identification) in the NCBI database were discarded so that the analysis was performed on the following sequences: 29× for IFITM1, corresponding to 24 species; 8× for nonsimian primate IFITM3, corresponding to 4 species; 10× for New World monkey IFITM3, corresponding to 4 species; and 48× for hominoid and New World monkey (Catarrhini) IFITM2/3, corresponding to 25 species. Amino acid sequences were aligned with Muscle. The motifs of interest were subsequently isolated and separated according to gene phylogeny. A sequence logo was then produced for each motif and each group using WebLogo3 (http://weblogo.threeplusone.com/).
Data availability.
All relevant data are within the paper.
ACKNOWLEDGMENTS
We thank the NIH AIDS Reagent Program for DNAs and antibodies. We are also indebted to the CelluloNet and PLATIM facilities of UMS3444 Biosciences Gerland. We thank all the contributors of freely available genome sequences.
Work in the laboratory of A.C. received the support of grants from the Agence Nationale de Recherche sur le Sida (ANRS), Sidaction, Finovi, and the ENS-L. L.E. is supported by the CNRS and by grants from amfAR (Mathilde Krim phase II fellowship 109140-58-RKHF), the Fondation pour la Recherche Médicale (FRM) (Projet Innovant, ING20160435028), Finovi (“recently settled scientist” grant), the ANRS (ECTZ19143), and the ANR LABEX ECOFECT (ANR-11-LABX-0048 of Université de Lyon, within the program Investissements d’Avenir [ANR-11-IDEX-0007] operated by the French National Research Agency). G.S. and A.B. were supported by the Burroughs Wellcome Investigators in the Pathogenesis of Infectious Disease Fund, the Bill and Melinda Gates Foundation, and Gilead Sciences, Inc. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
REFERENCES
- 1.Bailey CC, Kondur HR, Huang IC, Farzan M. 2013. Interferon-induced transmembrane protein 3 is a type II transmembrane protein. J Biol Chem 288:32184–32193. doi: 10.1074/jbc.M113.514356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weston S, Czieso S, White IJ, Smith SE, Kellam P, Marsh M. 2014. A membrane topology model for human interferon inducible transmembrane protein 1. PLoS One 9:e104341. doi: 10.1371/journal.pone.0104341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Friedman RL, Manly SP, McMahon M, Kerr IM, Stark GR. 1984. Transcriptional and posttranscriptional regulation of interferon-induced gene expression in human cells. Cell 38:745–755. doi: 10.1016/0092-8674(84)90270-8. [DOI] [PubMed] [Google Scholar]
- 4.Farber CR, Reich A, Barnes AM, Becerra P, Rauch F, Cabral WA, Bae A, Quinlan A, Glorieux FH, Clemens TL, Marini JC. 2014. A novel IFITM5 mutation in severe atypical osteogenesis imperfecta type VI impairs osteoblast production of pigment epithelium-derived factor. J Bone Miner Res 29:1402–1411. doi: 10.1002/jbmr.2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perreira JM, Chin CR, Feeley EM, Brass AL. 2013. IFITMs restrict the replication of multiple pathogenic viruses. J Mol Biol 425:4937–4955. doi: 10.1016/j.jmb.2013.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brass AL, Huang IC, Benita Y, John SP, Krishnan MN, Feeley EM, Ryan BJ, Weyer JL, van der Weyden L, Fikrig E, Adams DJ, Xavier RJ, Farzan M, Elledge SJ. 2009. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell 139:1243–1254. doi: 10.1016/j.cell.2009.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM. 2011. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472:481–485. doi: 10.1038/nature09907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anafu AA, Bowen CH, Chin CR, Brass AL, Holm GH. 2013. Interferon-inducible transmembrane protein 3 (IFITM3) restricts reovirus cell entry. J Biol Chem 288:17261–17271. doi: 10.1074/jbc.M112.438515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Everitt AR, Clare S, McDonald JU, Kane L, Harcourt K, Ahras M, Lall A, Hale C, Rodgers A, Young DB, Haque A, Billker O, Tregoning JS, Dougan G, Kellam P. 2013. Defining the range of pathogens susceptible to Ifitm3 restriction using a knockout mouse model. PLoS One 8:e80723. doi: 10.1371/journal.pone.0080723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Everitt AR, Clare S, Pertel T, John SP, Wash RS, Smith SE, Chin CR, Feeley EM, Sims JS, Adams DJ, Wise HM, Kane L, Goulding D, Digard P, Anttila V, Baillie JK, Walsh TS, Hume DA, Palotie A, Xue Y, Colonna V, Tyler-Smith C, Dunning J, Gordon SB, GenISIS Investigators, MOSAIC Investigators, Smyth RL, Openshaw PJ, Dougan G, Brass AL, Kellam P. 2012. IFITM3 restricts the morbidity and mortality associated with influenza. Nature 484:519–523. doi: 10.1038/nature10921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang IC, Bailey CC, Weyer JL, Radoshitzky SR, Becker MM, Chiang JJ, Brass AL, Ahmed AA, Chi X, Dong L, Longobardi LE, Boltz D, Kuhn JH, Elledge SJ, Bavari S, Denison MR, Choe H, Farzan M. 2011. Distinct patterns of IFITM-mediated restriction of filoviruses, SARS coronavirus, and influenza A virus. PLoS Pathog 7:e1001258. doi: 10.1371/journal.ppat.1001258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu J, Pan Q, Rong L, He W, Liu SL, Liang C. 2011. The IFITM proteins inhibit HIV-1 infection. J Virol 85:2126–2137. doi: 10.1128/JVI.01531-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mudhasani R, Tran JP, Retterer C, Radoshitzky SR, Kota KP, Altamura LA, Smith JM, Packard BZ, Kuhn JH, Costantino J, Garrison AR, Schmaljohn CS, Huang IC, Farzan M, Bavari S. 2013. IFITM-2 and IFITM-3 but not IFITM-1 restrict Rift Valley fever virus. J Virol 87:8451–8464. doi: 10.1128/JVI.03382-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bailey CC, Huang IC, Kam C, Farzan M. 2012. Ifitm3 limits the severity of acute influenza in mice. PLoS Pathog 8:e1002909. doi: 10.1371/journal.ppat.1002909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Compton AA, Bruel T, Porrot F, Mallet A, Sachse M, Euvrard M, Liang C, Casartelli N, Schwartz O. 2014. IFITM proteins incorporated into HIV-1 virions impair viral fusion and spread. Cell Host Microbe 16:736–747. doi: 10.1016/j.chom.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Foster TL, Wilson H, Iyer SS, Coss K, Doores K, Smith S, Kellam P, Finzi A, Borrow P, Hahn BH, Neil SJD. 2016. Resistance of transmitted rounder HIV-1 to IFITM-mediated restriction. Cell Host Microbe 20:429–442. doi: 10.1016/j.chom.2016.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gorman MJ, Poddar S, Farzan M, Diamond MS. 2016. The interferon-stimulated gene Ifitm3 restricts West Nile virus infection and pathogenesis. J Virol 90:8212–8225. doi: 10.1128/JVI.00581-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jia R, Xu F, Qian J, Yao Y, Miao C, Zheng YM, Liu SL, Guo F, Geng Y, Qiao W, Liang C. 2014. Identification of an endocytic signal essential for the antiviral action of IFITM3. Cell Microbiol 16:1080–1093. doi: 10.1111/cmi.12262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.John SP, Chin CR, Perreira JM, Feeley EM, Aker AM, Savidis G, Smith SE, Elia AE, Everitt AR, Vora M, Pertel T, Elledge SJ, Kellam P, Brass AL. 2013. The CD225 domain of IFITM3 is required for both IFITM protein association and inhibition of influenza A virus and dengue virus replication. J Virol 87:7837–7852. doi: 10.1128/JVI.00481-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Muñoz-Moreno R, Cuesta-Geijo MÁ, Martínez-Romero C, Barrado-Gil L, Galindo I, García-Sastre A, Alonso C. 2016. Antiviral role of IFITM proteins in African swine fever virus infection. PLoS One 11:e0154366. doi: 10.1371/journal.pone.0154366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Narayana SK, Helbig KJ, McCartney EM, Eyre NS, Bull RA, Eltahla A, Lloyd AR, Beard MR. 2015. The interferon-induced transmembrane proteins, IFITM1, IFITM2, and IFITM3 inhibit hepatitis C virus entry. J Biol Chem 290:25946–25959. doi: 10.1074/jbc.M115.657346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Savidis G, Perreira JM, Portmann JM, Meraner P, Guo Z, Green S, Brass AL. 2016. The IFITMs inhibit Zika virus replication. Cell Rep 15:2323–2330. doi: 10.1016/j.celrep.2016.05.074. [DOI] [PubMed] [Google Scholar]
- 23.Tartour K, Appourchaux R, Gaillard J, Nguyen XN, Durand S, Turpin J, Beaumont E, Roch E, Berger G, Mahieux R, Brand D, Roingeard P, Cimarelli A. 2014. IFITM proteins are incorporated onto HIV-1 virion particles and negatively imprint their infectivity. Retrovirology 11:103. doi: 10.1186/s12977-014-0103-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weston S, Czieso S, White IJ, Smith SE, Wash RS, Diaz-Soria C, Kellam P, Marsh M. 2016. Alphavirus restriction by IFITM proteins. Traffic 17:997–1013. doi: 10.1111/tra.12416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang W, Zhang L, Zan Y, Du N, Yang Y, Tien P. 2015. Human respiratory syncytial virus infection is inhibited by IFN-induced transmembrane proteins. J Gen Virol 96:170–182. doi: 10.1099/vir.0.066555-0. [DOI] [PubMed] [Google Scholar]
- 26.Poddar S, Hyde JL, Gorman MJ, Farzan M, Diamond MS. 2016. The interferon-stimulated gene IFITM3 restricts infection and pathogenesis of arthritogenic and encephalitic alphaviruses. J Virol 90:8780–8794. doi: 10.1128/JVI.00655-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qian J, Le Duff Y, Wang Y, Pan Q, Ding S, Zheng YM, Liu SL, Liang C. 2015. Primate lentiviruses are differentially inhibited by interferon-induced transmembrane proteins. Virology 474:10–18. doi: 10.1016/j.virol.2014.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Warren CJ, Griffin LM, Little AS, Huang IC, Farzan M, Pyeon D. 2014. The antiviral restriction factors IFITM1, 2 and 3 do not inhibit infection of human papillomavirus, cytomegalovirus and adenovirus. PLoS One 9:e96579. doi: 10.1371/journal.pone.0096579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weidner JM, Jiang D, Pan XB, Chang J, Block TM, Guo JT. 2010. Interferon-induced cell membrane proteins, IFITM3 and tetherin, inhibit vesicular stomatitis virus infection via distinct mechanisms. J Virol 84:12646–12657. doi: 10.1128/JVI.01328-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wilkins J, Zheng YM, Yu J, Liang C, Liu SL. 2016. Nonhuman primate IFITM proteins are potent inhibitors of HIV and SIV. PLoS One 11:e0156739. doi: 10.1371/journal.pone.0156739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wrensch F, Winkler M, Pohlmann S. 2014. IFITM proteins inhibit entry driven by the MERS-coronavirus spike protein: evidence for cholesterol-independent mechanisms. Viruses 6:3683–3698. doi: 10.3390/v6093683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McMichael TM, Zhang Y, Kenney AD, Zhang L, Zani A, Lu M, Chemudupati M, Li J, Yount JS. 15 June 2018. IFITM3 restricts human metapneumovirus infection. J Infect Dis doi: 10.1093/infdis/jiy361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chesarino NM, Compton AA, McMichael TM, Kenney AD, Zhang L, Soewarna V, Davis M, Schwartz O, Yount JS. 2017. IFITM3 requires an amphipathic helix for antiviral activity. EMBO Rep 18:1740–1751. doi: 10.15252/embr.201744100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao X, Guo F, Liu F, Cuconati A, Chang J, Block TM, Guo JT. 2014. Interferon induction of IFITM proteins promotes infection by human coronavirus OC43. Proc Natl Acad Sci U S A 111:6756–6761. doi: 10.1073/pnas.1320856111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao X, Sehgal M, Hou Z, Cheng J, Shu S, Wu S, Guo F, Le Marchand SJ, Lin H, Chang J, Guo JT. 2018. Identification of residues controlling restriction versus enhancing activities of IFITM proteins on entry of human coronaviruses. J Virol 92:e01535-17. doi: 10.1128/JVI.01535-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tartour K, Nguyen XN, Appourchaux R, Assil S, Barateau V, Bloyet LM, Burlaud Gaillard J, Confort MP, Escudero-Perez B, Gruffat H, Hong SS, Moroso M, Reynard O, Reynard S, Decembre E, Ftaich N, Rossi A, Wu N, Arnaud F, Baize S, Dreux M, Gerlier D, Paranhos-Baccala G, Volchkov V, Roingeard P, Cimarelli A. 2017. Interference with the production of infectious viral particles and bimodal inhibition of replication are broadly conserved antiviral properties of IFITMs. PLoS Pathog 13:e1006610. doi: 10.1371/journal.ppat.1006610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu J, Li M, Wilkins J, Ding S, Swartz TH, Esposito AM, Zheng YM, Freed EO, Liang C, Chen BK, Liu SL. 2015. IFITM proteins restrict HIV-1 infection by antagonizing the envelope glycoprotein. Cell Rep 13:145–156. doi: 10.1016/j.celrep.2015.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Feeley EM, Sims JS, John SP, Chin CR, Pertel T, Chen LM, Gaiha GD, Ryan BJ, Donis RO, Elledge SJ, Brass AL. 2011. IFITM3 inhibits influenza A virus infection by preventing cytosolic entry. PLoS Pathog 7:e1002337. doi: 10.1371/journal.ppat.1002337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Amini-Bavil-Olyaee S, Choi YJ, Lee JH, Shi M, Huang IC, Farzan M, Jung JU. 2013. The antiviral effector IFITM3 disrupts intracellular cholesterol homeostasis to block viral entry. Cell Host Microbe 13:452–464. doi: 10.1016/j.chom.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Desai TM, Marin M, Chin CR, Savidis G, Brass AL, Melikyan GB. 2014. IFITM3 restricts influenza A virus entry by blocking the formation of fusion pores following virus-endosome hemifusion. PLoS Pathog 10:e1004048. doi: 10.1371/journal.ppat.1004048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Compton AA, Roy N, Porrot F, Billet A, Casartelli N, Yount JS, Liang C, Schwartz O. 2016. Natural mutations in IFITM3 modulate post-translational regulation and toggle antiviral specificity. EMBO Rep 17:1657–1671. doi: 10.15252/embr.201642771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shi G, Schwartz O, Compton AA. 2017. More than meets the I: the diverse antiviral and cellular functions of interferon-induced transmembrane proteins. Retrovirology 14:53. doi: 10.1186/s12977-017-0377-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li K, Markosyan RM, Zheng YM, Golfetto O, Bungart B, Li M, Ding S, He Y, Liang C, Lee JC, Gratton E, Cohen FS, Liu SL. 2013. IFITM proteins restrict viral membrane hemifusion. PLoS Pathog 9:e1003124. doi: 10.1371/journal.ppat.1003124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gerlach T, Hensen L, Matrosovich T, Bergmann J, Winkler M, Peteranderl C, Klenk HD, Weber F, Herold S, Pohlmann S, Matrosovich M. 2017. pH optimum of hemagglutinin-mediated membrane fusion determines sensitivity of influenza A viruses to the interferon-induced antiviral state and IFITMs. J Virol 91:e00246-17. doi: 10.1128/JVI.00246-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Desai TM, Marin M, Mason C, Melikyan GB. 2017. pH regulation in early endosomes and interferon-inducible transmembrane proteins control avian retrovirus fusion. J Biol Chem 292:7817–7827. doi: 10.1074/jbc.M117.783878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang Y, Pan Q, Ding S, Wang Z, Yu J, Finzi A, Liu SL, Liang C. 2017. The V3 loop of HIV-1 Env determines viral susceptibility to IFITM3 impairment of viral infectivity. J Virol 91:e02441-16. doi: 10.1128/JVI.02441-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yu J, Liu SL. 2018. The inhibition of HIV-1 entry imposed by interferon inducible transmembrane proteins is independent of co-receptor usage. Viruses 10:E413. doi: 10.3390/v10080413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yount JS, Karssemeijer RA, Hang HC. 2012. S-palmitoylation and ubiquitination differentially regulate interferon-induced transmembrane protein 3 (IFITM3)-mediated resistance to influenza virus. J Biol Chem 287:19631–19641. doi: 10.1074/jbc.M112.362095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shan J, Zhao B, Shan Z, Nie J, Deng R, Xiong R, Tsun A, Pan W, Zhao H, Chen L, Jin Y, Qian Z, Lui K, Liang R, Li D, Sun B, Lavillette D, Xu K, Li B. 2017. Histone demethylase LSD1 restricts influenza A virus infection by erasing IFITM3-K88 monomethylation. PLoS Pathog 13:e1006773. doi: 10.1371/journal.ppat.1006773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McMichael TM, Zhang L, Chemudupati M, Hach JC, Kenney AD, Hang HC, Yount JS. 2017. The palmitoyltransferase ZDHHC20 enhances interferon-induced transmembrane protein 3 (IFITM3) palmitoylation and antiviral activity. J Biol Chem 292:21517–21526. doi: 10.1074/jbc.M117.800482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chesarino NM, McMichael TM, Hach JC, Yount JS. 2014. Phosphorylation of the antiviral protein interferon-inducible transmembrane protein 3 (IFITM3) dually regulates its endocytosis and ubiquitination. J Biol Chem 289:11986–11992. doi: 10.1074/jbc.M114.557694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu WL, Grotefend CR, Tsai MT, Wang YL, Radic V, Eoh H, Huang IC. 2017. Delta20 IFITM2 differentially restricts X4 and R5 HIV-1. Proc Natl Acad Sci U S A 114:7112–7117. doi: 10.1073/pnas.1619640114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jia R, Ding S, Pan Q, Liu SL, Qiao W, Liang C. 2015. The C-terminal sequence of IFITM1 regulates its anti-HIV-1 activity. PLoS One 10:e0118794. doi: 10.1371/journal.pone.0118794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jia R, Pan Q, Ding S, Rong L, Liu SL, Geng Y, Qiao W, Liang C. 2012. The N-terminal region of IFITM3 modulates its antiviral activity by regulating IFITM3 cellular localization. J Virol 86:13697–13707. doi: 10.1128/JVI.01828-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chesarino NM, McMichael TM, Yount JS. 2015. E3 ubiquitin ligase NEDD4 promotes influenza virus infection by decreasing levels of the antiviral protein IFITM3. PLoS Pathog 11:e1005095. doi: 10.1371/journal.ppat.1005095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sällman Almén M, Bringeland N, Fredriksson R, Schiöth HB. 2012. The dispanins: a novel gene family of ancient origin that contains 14 human members. PLoS One 7:e31961. doi: 10.1371/journal.pone.0031961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang Z, Liu J, Li M, Yang H, Zhang C. 2012. Evolutionary dynamics of the interferon-induced transmembrane gene family in vertebrates. PLoS One 7:e49265. doi: 10.1371/journal.pone.0049265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hickford D, Frankenberg S, Shaw G, Renfree MB. 2012. Evolution of vertebrate interferon inducible transmembrane proteins. BMC Genomics 13:155. doi: 10.1186/1471-2164-13-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wee YS, Roundy KM, Weis JJ, Weis JH. 2012. Interferon-inducible transmembrane proteins of the innate immune response act as membrane organizers by influencing clathrin and v-ATPase localization and function. Innate Immun 18:834–845. doi: 10.1177/1753425912443392. [DOI] [PubMed] [Google Scholar]
- 60.Lin TY, Chin CR, Everitt AR, Clare S, Perreira JM, Savidis G, Aker AM, John SP, Sarlah D, Carreira EM, Elledge SJ, Kellam P, Brass AL. 2013. Amphotericin B increases influenza A virus infection by preventing IFITM3-mediated restriction. Cell Rep 5:895–908. doi: 10.1016/j.celrep.2013.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Miyauchi K, Kim Y, Latinovic O, Morozov V, Melikyan GB. 2009. HIV enters cells via endocytosis and dynamin-dependent fusion with endosomes. Cell 137:433–444. doi: 10.1016/j.cell.2009.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mangeot PE, Dollet S, Girard M, Ciancia C, Joly S, Peschanski M, Lotteau V. 2011. Protein transfer into human cells by VSV-G-induced nanovesicles. Mol Ther 19:1656–1666. doi: 10.1038/mt.2011.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All relevant data are within the paper.