

Abstract
Spina bifida (SB) is a complex disorder of failed neural tube closure during the first month of human gestation, with a suspected etiology involving multiple gene and environmental interactions. GPR161 is a ciliary G-protein coupled receptor that regulates Sonic Hedgehog (Shh) signaling. Gpr161 null and hypomorphic mutations cause neural tube defects (NTDs) in mouse models. Herein we show that several genes involved in Shh and Wnt signaling were differentially expressed in the Gpr161 null embryos using RNA-seq analysis. To determine whether there exists an association between GPR161 and SB in humans, we performed direct Sanger sequencing on the GPR161 gene in a cohort of 384 SB patients and 190 healthy controls. We identified six rare variants of GPR161 in six SB cases, of which two of the variants were novel and did not exist in any databases. Both of these variants were predicted to be damaging by SIFT and/or PolyPhen analysis. The novel GPR161 rare variants mislocalized to the primary cilia, dysregulated Shh and Wnt signaling and inhibited cell proliferation in vitro. Our results demonstrate that GPR161 mutations cause NTDs via dysregulation of Shh and Wnt signaling in mice, and novel rare variants of GPR161 can be risk factors for SB in humans.
Introduction
Neural tube defects (NTDs) are the second most common structural birth defect in humans, with a post-folate fortification prevalence of 0.3–1 per 1000 live births in the USA (1,2). NTDs result from the failure of neural tube closure (NTC) during neurulation in early embryonic development. Typically we observe three NTD subtypes: anencephaly, spina bifida (SB) and craniorachischisis, depending on the region of brain and spinal cord that failed to close (3). NTC is the developmental process used to form the neural tube, starting with the elevation and bending of the neural plate towards the dorsal midline. The process is completed when the neural folds elevate, bend and fuse in the midline, closing the tube to form the nascent brain and spinal cord. It is a highly complex process, and several cellular events including cell proliferation, movement (convergent extension), differentiation and patterning occur in a spatiotemporal fashion within neuroectodermal, non-neuroectodermal and mesodermal tissues to create and close the neural tube (4).
The etiology of human NTDs is multifactorial, including genetic and environmental causations or combinations thereof (5,6). As the genetic factors are known to contribute significantly to the population burden of human NTDs (7–9), a large number of NTD candidate genes were identified using current advanced sequencing techniques. Mutants in over 300 genes were found in mouse NTD models (10,11), and they are highly clustered on the developmentally relevant signaling pathways, including folate-one carbon metabolism, Wnt-PCP, Sonic Hedgehog (Shh), bone morphogenetic protein (BMP) and retinoic acid (RA) signaling pathways (5).
The Shh signaling pathway involves the patterning of the dorsoventral axis in the neural tube and bending of the neural plate during early stages of NTC (12–14). Shh is secreted from the notochord and floor plate to initiate the patterning of the ventral neural tube, whereas Wnt and BMP are secreted from the dorsal roof plate of the neural tube (15). Those mutant mice with increased Shh signaling are primarily associated with NTDs, primarily due to the ventralized neural tube patterning, whereas the loss of Shh signaling typically does not cause NTDs in mouse models (13). For instance, the depletion of the negative regulators of Shh signaling such as Pka or Tulp3 caused exencephaly or widened posterior neuropores in mice (16,17). However, the Shh and Smo loss of function mutant mice did not express any NTD phenotypes (13). As the input of Shh signaling is tightly connected to the primary cilia, those genes related to ciliogenesis, such as kinesin motor proteins, intraflagellar transport proteins (13) or Fuz (18), also regulate the activity of Shh signaling, and several of these mutant mice do present with NTDs (13,19).
Single nucleotide polymorphisms (SNPs) in the negative regulators of Shh signaling, such as SUFU, PTCH1 and PKA, were identified in select SB patients (20–23), suggesting that the activation of Shh signaling might well be pathogenic and contribute to the etiology of human NTDs. However, the genetic information on Shh signaling variants in human NTD cohorts has not yet been extensively investigated.
Gpr161 was first identified from a vacuolated lens hypomorphic mutant mouse, which encodes a C-terminal tail deletion mutation of an orphan G-protein coupled receptor (GPCR). Gpr161vl mice present with lumbar-sacral SB, as well as embryonic lethality, and all surviving Gpr161vl adult mice have congenital cataract (24). Recently, it was suggested that Gpr161vl regulates Wnt and RA signaling during neurulation in Gpr161vl mutant mice (25). Mukhopadhyay et al. (26) reported that Gpr161 is a ciliary GPCR and a negative regulator of Shh signaling. It is trafficked to the primary cilia in a Tulp3/Intraflagellar complex-A–dependent manner, and it regulates Gli3 processing in a PKA-dependent fashion (26). Depletion of Gpr161 causes rostral and caudal NTDs in the mouse, which is believed to have resulted from the ventralized neural tube patterning due to increased Shh signaling (26). However, the genetic association between GPR161 and human NTDs has not yet been carefully evaluated.
Herein, we identified novel rare variants of GPR161 from SB infants and determined their functional relevance in the Shh and Wnt signaling pathways, utilizing the Gpr161 mutant mouse and animal cell culture models.
Results
The NTD phenotypes of Gpr161 knock out (KO) embryos and their transcriptomic analysis
We observed that the Gpr161 KO embryos presented with open fore/mid/hind brains and extensive craniofacial abnormalities, widely open posterior neuropores and branchial arch defects at E9.5 (Fig. 1A–F). The somite numbers were similar between WT and KO embryos (data not shown), suggesting that the open rostral and caudal neural tube was not the result of developmental delay in the KO embryos. Among the 16 litters we observed, all 25 Gpr161 KO embryos showed NTD phenotypes, whereas 32 WTs and 71 Hets were grossly phenotypically normal (Table 1). The expression of Gpr161 was significantly diminished in the Gpr161 KO embryos, whereas the expression of Ptch1 and Gli1 was increased in these embryos (Supplementary Material, Fig. S1), demonstrating increased Shh signaling activity when Gpr161 is inactivated in embryos.
Figure 1.
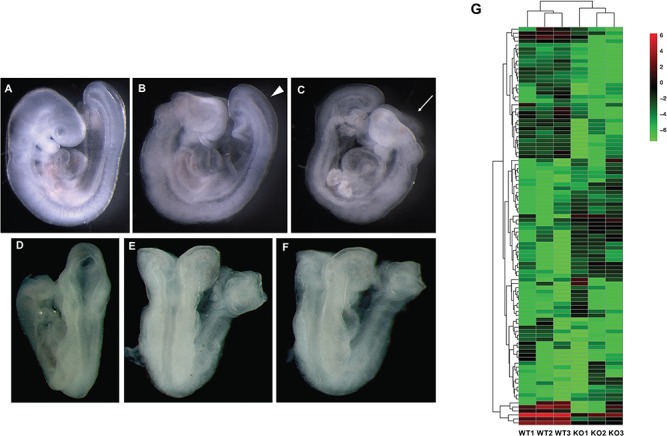
The NTD phenotypes and transcriptome analysis of Gpr161 KO embryos. (A–F) The gross morphology of Gpr161 WT and KO embryos at E9.5. The lateral (A) and dorsal (D) views of Gpr161 WT embryos at E9.5. The lateral (B and C) and dorsal (E and F) views of Gpr161 KO embryos at E9.5. Arrow head in (B) indicates the widely open posterior neuropore. Arrow in (C) indicates open mid/hind brain. (G) The Heat map from three WT and Gpr161 KO embryos demonstrating the differential gene expression profile between WT and KO embryos at E9.5.
Table 1.
NTD prevalence in Gpr161−/− embryos in E9.5
| Litters | Gpr161 +/+ | Gpr161+/− | Gpr161−/− | Resorption | |
|---|---|---|---|---|---|
| No. of embryos | 16 | 32 | 71 | 25 | 9 |
| No. of NTDs | 0 | 0 | 25 | ||
| p-HWE | 0.2022 |
To identify any differential gene expression underlying abnormal phenotypes in Gpr161 KO embryos, we performed an RNA-seq analysis with three WT and three Gpr161 KO embryos at E9.5. The RNA-seq assay revealed a total of 4460 upregulated genes and 732 downregulated genes in Gpr161 KO embryos when compared to WT embryos (Fig. 1G and Supplementary Material, Fig. S2). Target genes Ptch1, Ptch2, Hhip1 and Gli1 in the Shh signaling pathway were all upregulated more than 2-fold, and several genes in Shh signaling, including Shh, Gli3 and Gas1, were dysregulated as well (Table 2). Consistent with the ventralized neural tube patterning in Gpr161 KO embryos (26), the expression of select transcription factors (Foxa2, Nkx6.1, Nkx2.2 and Olig2), which are primarily expressed on the ventral aspect of the neural tube, was upregulated. In contrast, the expression of Pax6, Pax7 and Lbx1, which are expressed in the dorsal aspect of the neural tube, was significantly downregulated (Supplementary Material, Table S1).
Table 2.
The dysregulation of genes in Shh and Wnt signaling pathways in Gpr161 KO embryos at E.9.5
| Gene symbol | Name | FC | P-value | q-value |
|---|---|---|---|---|
| Hhip1 | Hedgehog interacting protein 1 | 4.01 | 0.00005 | 0.00289066 |
| Shh | Sonic Hedgehog | 3.55 | 0.00005 | 0.00289066 |
| Ptch1 | Patched 1 | 3.11 | 0.00005 | 0.00289066 |
| Gli1 | Glioma-associated oncogene homolog 1 | 2.29 | 0.00005 | 0.00289066 |
| Ptch2 | Patched 2 | 2.19 | 0.0008 | 0.0313625 |
| Cxxc4 | CXXC-type zinc finger protein 4 | 1.78 | 0.00025 | 0.0117547 |
| Dkk3 | Dickkopf 3 | 1.72 | 0.00005 | 0.00289066 |
| Fzd5 | Frizzled receptor 5 | 1.66 | 0.00005 | 0.00289066 |
| Fzd4 | Frizzled receptor 4 | 1.33 | 0.0006 | 0.0245587 |
| Fzd2 | Frizzled receptor 2 | 1.28 | 0.0008 | 0.0313625 |
| Daam2 | Dishevelled associated activator of morphogenesis 2 | −1.32 | 0.0011 | 0.0406562 |
| Gli3 | Glioma-associated oncogene homolog 3 | −1.56 | 0.00005 | 0.00289066 |
| Fzd10 | Frizzled receptor 10 | −1.66 | 0.00005 | 0.00289066 |
| Sfrp2 | Secreted frizzled related protein 2 | −1.87 | 0.00005 | 0.00289066 |
| Gas1 | Growth arrest specific protein 1 | −1.99 | 0.00005 | 0.00289066 |
| Wnt3a | Wnt3a | −2.07 | 0.00005 | 0.00289066 |
| Zic2 | Zinc finger protein 2 | −2.41 | 0.00005 | 0.00289066 |
| Wnt7b | Wnt7b | −2.83 | 0.00005 | 0.00289066 |
| Wnt9a | Wnt9a | −2.88 | 0.00005 | 0.00289066 |
| Wnt8b | Wnt8b | −4.62 | 0.00005 | 0.00289066 |
| Rspo4 | R-spondin 4 | −5.9 | 0.00005 | 0.00289066 |
FC: Fold Change, q-value: FDR-adjusted P-value, FDR: false discovery rate.
The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis was performed for functional enrichment identified genes in novel pathways (Neuroactive ligand-receptor interaction, cytokine–cytokine receptor interaction, Calcium signaling pathway, proteoglycans in cancer, Axon guidance, cGMP-PKG signaling pathways, pertussis), as well as genes in known pathways related to Gpr161 (cAMP signaling pathway, pathways in cancer and cell adhesion molecules) (Supplementary Material, Fig. S3).
In addition, we identified several dysregulated genes in the Wnt signaling pathway in Gpr161 KO embryos (Table 2 and Supplementary Material, Fig. S3). The upregulated Wnt genes included Cxxc4, Dkk3, Fzd5, Fzd4 and Fzd2, whereas the downregulated Wnt genes included Fzd10, Sfrp2, Wnt3a, Wnt7b, Wnt9a, Wnt8b and Rspo4 (Table 2).
GPR161 rare variants were identified in SB infants
To examine the association of GPR161 mutations in the etiology of human NTDs, we sequenced 384 newborn screening bloodspot-derived DNA samples from SB infants and 190 bloodspot DNA samples from non-malformed control infants ascertained by the California Birth Defects Monitoring Program (CBDMP). Five rare variants (minor allele frequency < 1%) (p.Val7Ile, p.Trp202Gly, p.Leu428Phe, p.Ala329Val and p.Ser282Pro) of GPR161 were identified from SB infants that were never observed in control infants (Table 3 and Supplementary Material, Table S2), and three of the variants were not found in publicly available databases such as dbSNP, EVS, ExAC and GnomAD. Of these variants, two (p.Trp202Gly and p.Leu428Phe) were predicted as probably damaging in PolyPhen and/or SIFT. The p.Leu19Gln variant, which was predicted as probably damaging in PolyPhen, was found in an SB infant, as well as in a control infant. All six rare variants from the control group of infants were found in the ExAC database. Five of them were predicted as synonymous or benign by PolyPhen or SIFT, whereas one of them (p.Thr413Ser) was predicted as possibly damaging or deleterious. The p.Trp202Gly and p.Leu428Phe variants are novel, and their amino acids substitutions were predicted to affect GPR161 functionality, thus these became the focus of our studies.
Table 3.
Resequencing of GPR161 in 384 SB infants and 190 control infants
| DNA change1 | AA change2 | rs# | SIFT | PolyPhen | Count in NTDs | Count in controls | ExAC3 MAF | gnomAD4 MAF |
|---|---|---|---|---|---|---|---|---|
| c.19G > A | p.Val7Ile | rs60383097 | Tolerated | Benign | 1/384 | ND5 | 1.72E-03 | 0.001674 |
| c.604 T > G | p.Trp202Gly | NA 6 | Damaging | Probably damaging | 1/384 | ND | Not found | Not found |
| c.1283C > T | p.Leu428Phe | NA | Tolerated | Probably damaging | 1/384 | ND | Not found | Not found |
| c.986 C > T | p.Ala329Val | NA | Tolerated | Benign | 1/384 | ND | Not found | Not found |
| c.844 T > C | p.Ser282Pro | rs576055321 | Tolerated | Benign | 1/384 | ND | 2.47E-05 | 3.25E-05 |
| c.56 T > A | p.Leu19Gln | rs200635937 | Tolerated | Probably damaging | 1/384 | 1/190 | 3.35E-03 | 2.70E-03 |
| c.126C > T | p.Gly42Gly | rs34499666 | NA | NA | ND | 1/190 | 2.09E-03 | 2.22E-03 |
| c.292G > A | p.Val98Met | rs147802262 | Tolerated | Benign | ND | 1/190 | 8.24E-05 | 4.87E-05 |
| c.720C > G | p.Arg240Arg | rs201970960 | NA | NA | ND | 1/190 | 4.95E-05 | 5.78E-05 |
| c.742G > A | p.Val248Ile | rs143799014 | Tolerated | Benign | ND | 1/190 | 4.78E-04 | 4.70E-04 |
| c.1237A > T | p.Thr413Ser | rs773362589 | Possibly damaging | Deleterious | ND | 1/190 | 8.55E-06 | 1.23E-05 |
| c.1473G > A | p.Glu491Glu | rs112752784 | NA | NA | ND | 1/190 | 1.64E-03 | 1.79E-03 |
1GPR161 nucleotide accession number: NM_001267609.
2GPR161 protein accession number: NP_001254538.
3Exome Aggregation Consortium data from http://exac.broadinstitute.org/.
4gnomAD data from http://gnomad.broadinstitute.org/.
5ND: not detected.
6NA: not available.
7The variants which were not identified in ExAC database were highlighted in bold font.
Both tryptophan (Trp) amino acid at position 202 and leucine (Leu) amino acid at position 428 of the GPR161 protein are evolutionarily conserved (Fig. 2A). The p.Trp202Gly variant localizes in the second extracellular loop of GPR161 (Fig. 2B). The amino acid substitution of Trp to glycine (Gly) loses two aromatic side chains, although both have non-polar characteristics. The p.Leu428Phe variant sits in the C-terminal intracellular tail region of GPR161 (Fig. 2B). The amino acid substitution of Leu to phenylalanine (Phe) did not change their non-polar and hydrophobic characteristics, although Phe has a benzyl side chain, whereas Leu does not.
Figure 2.
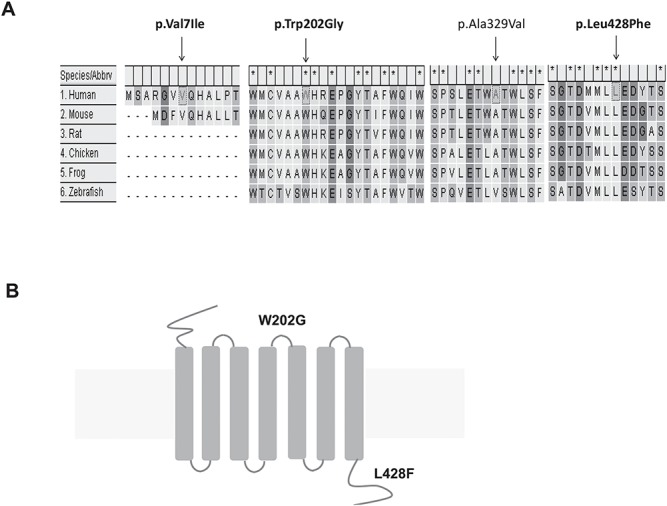
The rare variants of GPR161 were identified in human SB infants. (A) The gene alignment between GPR161 orthologs. (B) The schematic diagram of GPR161 SNVs.
The functional effects of GPR161 variants identified in NTDs
Increased Shh signaling activity contributes to the etiology of NTDs in Gpr161 KO embryos, and the ciliary localization of GPR161 might be critical for its inhibitory effect on Shh signaling. Therefore, we evaluated the ciliary localization of the GPR161 rare variants. We initially checked the protein expression of WT and variants of GPR161 to rule out the possibility that the effect resulted from a differential protein expression level. The protein amount expressed from WT or the six variants of GPR161 identified in our studies did not differ significantly in 293T cells, which are widely used for overexpression assays as they are cilited (27) (Fig. 3A). The rationale to select the four variants to test their ciliary localization was based on the following criteria: 1) they are novel variants found only in SB infants; 2) two of the variants (p.Trp202Gly and p.Leu428Phe) are predicted to be probably damaging from PolyPhen/SIFT. The ciliary localization of the p.Trp202Gly variant was dramatically decreased compared to WT, while that of p.Leu428Phe variant was only slightly decreased. The p.Val7Ile and p.Ala329Val variants did not change their ciliary localization compared to WT (Fig. 3B). Intriguingly, while the p.Trp202Gly variant was, for the most part, not localized in the primary cilia, the formation of cilia was not affected by this variant (Fig. 3B). This suggests that the p.Trp202Gly variant did not adversely affect ciliogenesis.
Figure 3.
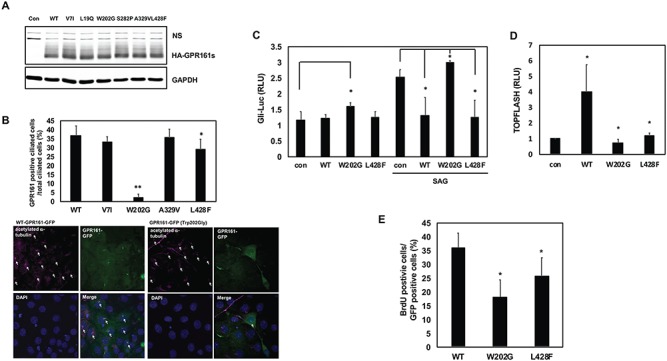
The functional assessment of GPR161 rare variants. (A) The protein expression pattern of GPR161 rare SNVs in 293 T cells. The cells were transfected with WT and six variants GPR161, and western blot analysis was performed with anti-HA and GAPDH antibodies. ns indicates a nonspecific band. (B) The ciliary localization of GPR161 rare SNVs in NIH3T3 cells. Upper panel shows the ratio of GPR161 positive cilia. GPR161 positive primary cilia were counted up to 200 cells from three independent experiments. Lower panel shows the ciliary localization with WT and p.Trp202Gly GPR161 (**P < 0.005, *P < 0.05). The arrows in the acetylated α-tubulin panels indicate the total ciliated cells, and the arrows in the merge panels indicate the GPR161 positive cilia. (C) The effect of GPR161 rare SNVs on Gli-responsive luciferase activity in C3H10T1/2 cells. The cells were transfected with Gli-luc and SV40-Renilla along with GPR161 (WT and SNVs) expression vectors and were treated with/without SAG for 24 h prior to lysis. Relative Luciferase Unit (RLU) indicates the relative Gli-luciferase activity over renilla luciferase (*P < 0.05). (D) The effect of GPR161 rare SNVs on the TOPFLASH luciferase activity in 293 T cells. The cells were transfected with TOPFLASH and pTK-Renilla along with GPR161 (WT and SNVs) expression vectors. RLU indicates the relative TOPFLASH luciferase activity over renilla luciferase (*P < 0.05). (E) The effect of GPR161 rare SNVs on the cell proliferation in Gpr161 KO MEF cells. The cells were transfected with GPR161 (WT and SNVs) expression vectors and were treated with BrdU for 12 h before fixation. The BrdU positive cells were counted and divided by the GFP positive cells (*P < 0.05).
The ciliary localization of GPR161 impacts its regulatory function in Shh signaling. As such, we next examined how GPR161 variants regulate Shh signaling activity using the Gli responsive luciferase (Gli-Luc) (28) reporter system in the presence of WT GPR161. The p.Trp202Gly variant further activated Smoothened Agonist (SAG)–induced Gli-luc activity, whereas WT and p.Leu428Phe variant inhibited it. In addition, p.Trp202Gly variant slightly activated Gli-luc activity without SAG treatment (Fig. 3C). This result suggests that p.Trp202Gly variant adversely regulates Shh signaling activity compared to WT-GPR161.
Clearly Wnt signaling is regulated in the Gpr161vl mutant mouse NTD model (25), and our transcriptome data also revealed the involvement of Wnt signaling during neurulation in Gpr161 KO embryos. Hence, we further investigated the role of GPR161 rare variants in Wnt signaling. WT-GPR161 overexpression increased TOPFLASH reporter activity (Fig. 3D and Supplementary Material, Fig. S4A), which represents Wnt signaling activity by measuring the T Cell Factor (TCF)-dependent transcription activity. In contrast, both p.Trp202Gly-GPR161 and p.Leu428Phe-GPR161 rare variants did not activate TOPFLASH activity (Fig. 3D). The role of GPR161 in Wnt signaling was further supported by the GPR161 knockdown experiment (Supplementary Material, Fig. S4B) and the measurement of target gene expression (Axin2 and c-myc) in Gpr161 WT and KO embryos (Supplementary Material, Fig. S4C). The results suggest that WT-GPR161 activates basal Wnt signaling, whereas the two rare variants (p.Trp202Gly and p.Leu428Phe) do not.
As GPR161 is known to regulate cell proliferation in several cancer cell types (29,30) and the cell proliferation in Gpr161 KO mouse embryonic fibroblast (MEF) cells was reduced (Supplementary Material, Fig. S5), we investigated the role of two GPR161 rare variants with respect to cell proliferation. To measure the kinetics of cell proliferation, we performed the bromodeoxyuridine (BrdU) incorporation assay. Both p.Trp202Gly-GPR161 and p.Leu428Phe-GPR161 rare variants reduced the numbers of BrdU positive cells compared to WT (Fig. 3E), indicating that the two GPR161 rare variants adversely impact cell proliferation.
Discussion
Two Gpr161 mutant mouse models have been previously described (24,26), including the Gpr161vl and Gpr161 KO mice. Both Gpr161vl and Gpr161 KO mouse models are associated with an increased prevalence of NTDs via downregulation of RA and Wnt signaling and increased Shh signaling, respectively. Our transcriptome analysis revealed that several genes of the Shh signaling pathway were upregulated, whereas genes encoding the secreted Wnt ligands were downregulated in the Gpr161 KO embryos (Table 2). This result is consistent with previous observations as Shh signaling is involved in the patterning of the ventral neural tube, while Wnt and BMP signaling regulates dorsal neural tube patterning (31). In addition, the Gpr161 KO embryos had smaller heads, specifically at the rostral portion of the neural tube (Fig. 1A–F), compared to WT embryos. This is an indication of the apparent dysregulation of cell proliferation or apoptosis in the developing neural tubes of the mutant embryos. Taken together, our results and studies by others suggest that there might be underlying cellular dysregulation involving Shh and Wnt signaling for the expression of the NTD phenotypes observed in the Gpr161 mutant mice.
Although two Gpr161 mutant mouse models are well known to be associated with NTD phenotypes, any genetic association and knowledge of the underlying molecular mechanism of GPR161 in the etiology of human NTDs remains lacking. We have now identified three novel rare variants of GPR161 in human SB infants that were not observed in a control group of infants without congenital malformations, nor do they exist in any database, including ExAC and GnomAD. Two (p.Trp202Gly and p.Leu428Phe) of the novel variants were predicted to be damaging based on in silico analysis. We also identified a rare variant (p.Leu19Gln), which is predicted to be damaging, with conflicting interpretations of pathogenicity recorded in the ClinVar database in both cases and controls (32). Only one rare missense variant (p.Thr413Ser) was identified in our control cohort that was not observed in any SB infants. Although it was predicted to be deteterious by both SIFT and PolyPhen, it has a frequency of 1.23 × 10−5 in gnomAD database, which indicates that this variant may not disturb normal NTC in human populations.
Functional analyses of the GPR161 rare variants identified in our human NTD cohorts support the hypothesis that dysfunctional Wnt and Shh signaling underlie the pathogenic mechanisms associated with this gene. The mislocalization of p.Trp202Gly Single Nucleotide Variant (SNV) in the primary cilia could lead to the increased Shh signaling activity, potentially creating the ventralized neural tube patterning and resulting in NTDs. Although ciliary localization of p.Trp202Gly was significantly decreased, its dysregulation of Shh and Wnt signaling activity is moderate (Fig. 3). These results further suggest the decreased ciliary localization of p.Trp202Gly may impact other signaling pathways. As the p.Trp202Gly variant does not locate within the ciliary localization signal within the third intracellular loop (26), it does not cause the mislocalization observed in the p.Trp202Gly variant. Rather, p.Trp202Gly variant might be less stable at the cell surface due to the amino acid substitution at the third extracellular loop that is critical for proper agonist binding or receptor activation, as is commonly observed for GPCRs (33).
In addition, the p.Leu428Phe SNV differentially regulates Shh and Wnt signaling, whereas the p.Trp202Gly SNV showed similar adverse effects on both signaling pathways. These data suggest that p.Trp202Gly SNV and p.Leu428Phe SNV might adversely impact NTC by differentially regulating Shh and Wnt signaling. Intriguingly, the dysregulation of cell proliferation by the two SNVs was more relevant to the change of Wnt signaling than that of Shh signaling (Fig. 3D and 3E). This data suggests that Gpr161 regulated cell proliferation might be co-mediated by Wnt signaling. It remains to be determined just how the other cellular processes, such as apoptosis and/or differentiation, are regulated by these SNVs. Furthermore, how Shh and Wnt signaling are interregulated during NTC process remains an unresolved question.
Disruption of ciliogenesis appears to be associated with the etiology of NTDs in both humans and in mice. For example, Fuz mutant mice with NTDs were found to have too few or shortened primary cilia (18), and the FUZ SNVs identified from human SB patients also affected ciliogenesis in vitro when examined using functional assays (34). However, GPR161 SNVs (p.Trp202Gly and p.Leu428Phe) do not affect ciliogenesis, rather, they are either mislocalized or less densely localized in the primary cilia. A previous report (26) also supports the finding that Gpr161 KO MEF cells did not differ with respect to ciliogenesis from WT MEFs. Taken together, mislocalization of Shh signaling molecules in primary cilia, as well as the dysregulated ciliogenesis, causes abnormal Shh signaling activity, which results in abnormal NTC during neurulation.
In this study, we identified novel rare SNVs of GPR161 in human SB patients and characterized the functional role of two GPR161 SNVs on Shh and Wnt signaling and cell proliferation. Our transcriptome analysis further supported the involvement of these signaling pathways during neurulation in Gpr161 KO embryos. A lack of familial genetic information was a limitation of this study, a limitation which could be remedied in future studies interrogating affected-NTD trios to better understand the genetic origins of GPR161 SNVs. Finally, the functional role of GPR161 in Wnt signaling during neurulation appears to be a promising area for future investigations based on our transcriptomic and functional data.
Materials and Methods
Human subjects
Human newborn screening bloodspot samples were collected from a case–control study conducted by the CBDMP (35). Included for this study were 384 singleton infants with SB (cases) and 190 non-malformed infants (controls) born during the period of 1990–1999. Case and control infants were linked to their newborn bloodspot. DNA was extracted from bloodspot using the Puregene DNA Extraction Kit (Qiagen, Valencia, CA) and amplified using the GenomiPhi Kit (GE Healthcare, Marlborough, MA). All samples were obtained with approval from the State of California Health and Welfare Agency Committee for the Protection of Human Subjects. This study was approved by University of Texas at Austin (IRB approval number: 2014120041).
DNA Resequencing of GPR161
The genomic structure of human GPR161 was determined using the NCBI GenBank and Ensemble (NM_001267609/ENST00000537-209). The coding region of GPR161 was amplified by polymerase chain reactions (PCRs) using primers flanking exon-intron junctions. Primer sequences are presented on Supplementary Material, Table S3. The PCR products were sequenced using the Prism Bigdye Terminator Kit (v3) on an ABI 3730XL DNA analyzer (Life Technologies, Carlsbad, CA). Both case and control samples were sequenced with either a specific forward or reverse primer, except for Exon4, which was sequenced with both primers from forward direction and reverse direction. The detected variants were confirmed by repeating the PCR and resequencing from both directions.
Mouse strains
Gpr161 knockout mutant mice were generated by Dr Saikat Mukhopadhyay (UT Southwestern, Dallas), and the detailed information was previously reported (36). All mice were housed and handled according to the guidelines approved by the Institutional Animal Care and Use Committee of The University of Texas at Austin. The genotypes of mice and embryos were evaluated with PCR-based genotyping (36).
RNA extraction and quantitative RT-PCR (qRT-PCR)
Total RNA was extracted with Trizol reagent (Sigma, St. Louis, MO) from Gpr161 embryos at E9.5. For qRT-PCR, 1 ug of RNA was used to synthesize cDNA using iScript reverse transcription Supermix kit (Bio-Rad, Hercules, CA). The qRT-PCR was performed using SsoAdvanced™ Universal SYBR® Green Supermix (Bio-Rad) according to manufacturer’s instruction. The primers for qRT-PCR are as below. Gpr161 (Forward: 5′-TCGGTGGAGTTTGATGAGTTCA-3′; Reverse: 5′-CCGTAGCACACTAGCATGATGA-3′), GPR161 (Forward: 5′-CGTCATCGTGGAGGAGGATG-3′; Reverse: 5′-GGAAGTCCTCTGTC-GTTGCA-3′), Gli1 (Forward: 5′-CCAAGCCAACTTTATGTCAGGG-3′; Reverse: 5′-AGCCCGCTTCTTTGTTAATTTGA-3′), Ptch1 (Forward: 5′-AAAGAACTGCGGCAAGTTTTTG-3′; Reverse: 5′-CTTCTCCTAT-CTTCTGACGGGT-3′), Axin2 (Forward: 5′-AAGTGTCTCTACCTCAT-TTTCCG-3′; Reverse: 5′-TCCAGTTTCAGTTTCTCCAGC-3′), c-myc (Forward: 5′-AAAACGACAAGAGGCGGACA-3′; Reverse: 5′-ATTCAGGGATCTGGTCACGC-3′) and β-actin (Forward: 5′-CCACCATGTACCCAGGCATT-3′; Reverse: 5′-AGGGTGTAAAACGC-AGCTCA-3′).
RNA sequencing analysis
Total RNA was extracted with Trizol reagent (Sigma) from Gpr161 embryos at E9.5. The concentration and integrity of the RNA was measured by Nanodrop (Thermofisher, Waltham, MA) and Bioanalyzer (Agilent Technologies). The library was prepared with NEBNext Ultra RNA with Poly-A selection and was sequenced on an Illumina Hi-Seq 4000 (Admera Health LLC, South Plainfield, NJ). The differential gene expression was determined with fold change >1.5 and P < 0.05 genes with <1 count per million. Any gene with a P-value greater than False Discovery Rate (FDR), after Benjamini–Hochberg correction for multi-testing, was deemed significantly differentially expressed in the test condition as compared to the control. The data set was analyzed by the KEGG.
Plasmids
HA-tagged human GPR161 clone was purchased from Gene-copoeia. EGFP-tagged human GPR161 was cloned into p-EGFP-N1 vector with XhoI/BamHI. The mutations of GPR161 with GPR161-HA and GPR161-GFP were generated using QuickChange Site-Directed mutagenesis kit (Agilent, Santa Clara, CA). GPR161 shRNA was purchased from Genecopoeia, Rockville, MD, (Catalog number: HSH069156-CH1). Gli-luciferase and SV40-Renilla plasmids were kindly provided from Xiaoyan Zheng (37) (Georgetown University, Washington DC). TOPFLASH and pTK-Renilla plasmids were obtained from Addgene (Cambridge, MA).
Immunocytochemistry
NIH3T3 cells were plated onto the cover glass and were transfected with GPR161-EGFP constructs using TransIT X2 (Mirus, Atlanta, GA). For primary cilia staining, cells were serum-starved for 24 h before fixation. The cells were fixed and stained with anti-acetylated α-tubulin (Sigma) and anti-mouse DyLight 650 antibodies. The images were obtained with a Leica Confocal Microscope (Leica, Buffalo Grove, IL).
Western blot analysis
293 T cells were transfected with GPR161-HA (WT or variants) constructs using Lipofectamine 2000 (Invitrogen, Waltham, MA). The cells were lysed with digitonin lysis buffer as described (38). The proteins were immunoblotted with anti-HA (Santa Cruz Biotechnology, Dallas, TX), anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Cell Signaling, Danvers, MA), 1RDye® 800CW goat anti-rabbit IgG secondary antibodies (LI-COR, Cambridge, UK) and 1RDye® 680CW goat anti-mouse IgG secondary antibodies (LI-COR). The images were captured by Odyssey® (LI-COR).
Luciferase assay
C3H10T1/2 cells were transfected with Gli-Luc and SV40-Renilla plasmids along with GPR161-HA (WT or mutants) using Lipofectamine LTX (Invitrogen). 293 T cells were transfected with TOPFLASH and pTK-Renilla plasmids along with GPR161-HA (WT or mutants) using Lipofectamine 2000 (Invitrogen). The cells were lysed 48 h post-transfection, and the luciferase activity was measured using a Dual Luciferase Assay Kit (Promega, Madison, WI). The Biotek-2 plate reader was used to read the luminescence activity.
BrdU incorporation assay
The Gpr161 KO MEF cells were isolated from E10.5 Gpr161 KO embryos. The cells were plated on the cover glass and were transfected with GPR161-EGFP (WT or mutants) constructs using TransIT X2 (Mirus). The cells were treated with 20-μM BrdU for 12 h, were fixed and were stained with anti-BrdU and anti-mouse DyLight 650 antibodies. The images were captured by All-In-One Fluorescence microscope (Keyence, Itasca, IL) and up to 80 cells of GPR161-EGFP expressed cells were counted from three independent experiments for the quantitation.
Statistical analysis
To evaluate the genotyping frequencies in Gpr161 KO mice, we performed Hardy–Weinberg Equilibrium analysis and a X2 value was calculated. The data was analyzed by the Standard Deviation with student t-test for comparing groups.
Supplementary Material
Acknowledgements
This project was supported by grants from National Institutes of Health (HD081216, HD083809, and HD067244 to R.H.F. and M.E.R. and 1R01GM113023 to S.M.) and from Centers for Disease Control and Prevention (C U01DD001033 to G.M.S.). We thank the California Department of Public Health Maternal Child and Adolescent Health Division for providing data and Dr Xiaoyan Zheng for providing Gli1-luciferase and SV40-Renilla plasmids. The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the California Department of Public Health.
Conflict of Interest statement. None declared.
References
- 1. Mitchell L.E. (2005) Epidemiology of neural tube defects. Am. J. Med. Genet. C Semin. Med. Genet., 135C, 88–94. [DOI] [PubMed] [Google Scholar]
- 2. Wallingford J.B., Niswander L.A., Shaw G.M. and Finnell R.H. (2013) The continuing challenge of understanding, preventing, and treating neural tube defects. Science, 339, 1222002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Greene N.D. and Copp A.J. (2014) Neural tube defects. Annu. Rev. Neurosci., 37, 221–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nikolopoulou E., Galea G.L., Rolo A., Greene N.D. and Copp A.J. (2017) Neural tube closure: cellular, molecular and biomechanical mechanisms. Development, 144, 552–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wilde J.J., Petersen J.R. and Niswander L. (2014) Genetic, epigenetic, and environmental contributions to neural tube closure. Annu. Rev. Genet., 48, 583–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Greene N.D., Stanier P. and Copp A.J. (2009) Genetics of human neural tube defects. Hum. Mol. Genet., 18, R113–R129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Leck I. (1974) Causation of neural tube defects: clues from epidemiology. Br. Med. Bull., 30, 158–163. [DOI] [PubMed] [Google Scholar]
- 8. Holmes L.B., Driscoll S.G. and Atkins L. (1976) Etiologic heterogeneity of neural-tube defects. N. Engl. J. Med., 294, 365–369. [DOI] [PubMed] [Google Scholar]
- 9. Carter C.O. and Evans K. (1973) Spina bifida and anencephalus in greater London. J. Med. Genet., 10, 209–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Harris M.J. and Juriloff D.M. (2007) Mouse mutants with neural tube closure defects and their role in understanding human neural tube defects. Birth Defects Res. A Clin. Mol. Teratol., 79, 187–210. [DOI] [PubMed] [Google Scholar]
- 11. Harris M.J. and Juriloff D.M. (2010) An update to the list of mouse mutants with neural tube closure defects and advances toward a complete genetic perspective of neural tube closure. Birth Defects Res. A Clin. Mol. Teratol., 88, 653–669. [DOI] [PubMed] [Google Scholar]
- 12. Ybot-Gonzalez P., Cogram P., Gerrelli D. and Copp A.J. (2002) Sonic hedgehog and the molecular regulation of mouse neural tube closure. Development, 129, 2507–2517. [DOI] [PubMed] [Google Scholar]
- 13. Murdoch J.N. and Copp A.J. (2010) The relationship between sonic Hedgehog signaling, cilia, and neural tube defects. Birth Defects Res. A Clin. Mol. Teratol., 88, 633–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wilson L. and Maden M. (2005) The mechanisms of dorsoventral patterning in the vertebrate neural tube. Dev. Biol., 282, 1–13. [DOI] [PubMed] [Google Scholar]
- 15. Ulloa F. and Briscoe J. (2007) Morphogens and the control of cell proliferation and patterning in the spinal cord. Cell Cycle, 6, 2640–2649. [DOI] [PubMed] [Google Scholar]
- 16. Patterson V.L., Damrau C., Paudyal A., Reeve B., Grimes D.T., Stewart M.E., Williams D.J., Siggers P., Greenfield A. and Murdoch J.N. (2009) Mouse hitchhiker mutants have spina bifida, dorso-ventral patterning defects and polydactyly: identification of Tulp3 as a novel negative regulator of the Sonic hedgehog pathway. Hum. Mol. Genet., 18, 1719–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huang Y., Roelink H. and McKnight G.S. (2002) Protein kinase A deficiency causes axially localized neural tube defects in mice. J. Biol. Chem., 277, 19889–19896. [DOI] [PubMed] [Google Scholar]
- 18. Gray R.S., Abitua P.B., Wlodarczyk B.J., Szabo-Rogers H.L., Blanchard O., Lee I., Weiss G.S., Liu K.J., Marcotte E.M., Wallingford J.B. et al. (2009) The planar cell polarity effector Fuz is essential for targeted membrane trafficking, ciliogenesis and mouse embryonic development. Nat. Cell Biol., 11, 1225–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Goetz S.C. and Anderson K.V. (2010) The primary cilium: a signalling centre during vertebrate development. Nat. Rev. Genet., 11, 331–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang Z., Wang L., Shangguan S., Lu X., Chang S., Wang J., Zou J., Wu L., Zhang T. and Luo Y. (2013) Association between PTCH1 polymorphisms and risk of neural tube defects in a Chinese population. Birth Defects Res. A Clin. Mol. Teratol., 97, 409–415. [DOI] [PubMed] [Google Scholar]
- 21. Wang Z., Shangguan S., Lu X., Chang S., Li R., Wu L., Bao Y., Niu B., Wang L. and Zhang T. (2013) Association of SMO polymorphisms and neural tube defects in the Chinese population from Shanxi Province. Int. J. Clin. Exp. Med., 6, 960–966. [PMC free article] [PubMed] [Google Scholar]
- 22. Wu J., Lu X., Wang Z., Shangguan S., Chang S., Li R., Wu L., Bao Y., Niu B., Wang L. et al. (2013) Association between PKA gene polymorphism and NTDs in high risk Chinese population in Shanxi. Int. J. Clin. Exp. Pathol., 6, 2968–2974. [PMC free article] [PubMed] [Google Scholar]
- 23. Zhu H., Lu W., Laurent C., Shaw G.M., Lammer E.J. and Finnell R.H. (2005) Genes encoding catalytic subunits of protein kinase A and risk of spina bifida. Birth Defects Res. A Clin. Mol. Teratol., 73, 591–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Matteson P.G., Desai J., Korstanje R., Lazar G., Borsuk T.E., Rollins J., Kadambi S., Joseph J., Rahman T., Wink J. et al. (2008) The orphan G protein-coupled receptor, Gpr161, encodes the vacuolated lens locus and controls neurulation and lens development. Proc. Natl. Acad. Sci. USA, 105, 2088–2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li B.I., Matteson P.G., Ababon M.F., Nato A.Q. Jr., Lin Y., Nanda V., Matise T.C. and Millonig J.H. (2015) The orphan GPCR, Gpr161, regulates the retinoic acid and canonical Wnt pathways during neurulation. Dev. Biol., 402, 17–31. [DOI] [PubMed] [Google Scholar]
- 26. Mukhopadhyay S., Wen X., Ratti N., Loktev A., Rangell L., Scales S.J. and Jackson P.K. (2013) The ciliary G-protein-coupled receptor Gpr161 negatively regulates the Sonic hedgehog pathway via cAMP signaling. Cell, 152, 210–223. [DOI] [PubMed] [Google Scholar]
- 27. Gerdes J.M., Liu Y., Zaghloul N.A., Leitch C.C., Lawson S.S., Kato M., Beachy P.A., Beales P.L., DeMartino G.N., Fisher S. et al. (2007) Disruption of the basal body compromises proteasomal function and perturbs intracellular Wnt response. Nat. Genet., 39, 1350–1360. [DOI] [PubMed] [Google Scholar]
- 28. Kim J., Hsia E.Y., Brigui A., Plessis A., Beachy P.A. and Zheng X. (2015) The role of ciliary trafficking in Hedgehog receptor signaling. Sci. Signal., 8, ra55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Feigin M.E., Xue B., Hammell M.C. and Muthuswamy S.K. (2014) G-protein-coupled receptor GPR161 is overexpressed in breast cancer and is a promoter of cell proliferation and invasion. Proc. Natl. Acad. Sci. USA, 111, 4191–4196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shimada I.S., Hwang S.H., Somatilaka B.N., Wang X., Skowron P., Kim J., Kim M., Shelton J.M., Rajaram V., Xuan Z. et al. (2018) Basal suppression of the Sonic Hedgehog pathway by the G-protein-coupled receptor Gpr161 restricts medulloblastoma pathogenesis. Cell Rep., 22, 1169–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dessaud E., McMahon A.P. and Briscoe J. (2008) Pattern formation in the vertebrate neural tube: a sonic hedgehog morphogen-regulated transcriptional network. Development, 135, 2489–2503. [DOI] [PubMed] [Google Scholar]
- 32. Karaca E., Buyukkaya R., Pehlivan D., Charng W.L., Yaykasli K.O., Bayram Y., Gambin T., Withers M., Atik M.M., Arslanoglu I. et al. (2015) Whole-exome sequencing identifies homozygous GPR161 mutation in a family with pituitary stalk interruption syndrome. J. Clin. Endocrinol. Metab., 100, E140–E147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lawson Z. and Wheatley M. (2004) The third extracellular loop of G-protein-coupled receptors: more than just a linker between two important transmembrane helices. Biochem. Soc. Trans., 32, 1048–1050. [DOI] [PubMed] [Google Scholar]
- 34. Seo J.H., Zilber Y., Babayeva S., Liu J., Kyriakopoulos P., De Marco P., Merello E., Capra V., Gros P. and Torban E. (2011) Mutations in the planar cell polarity gene, Fuzzy, are associated with neural tube defects in humans. Hum. Mol. Genet., 20, 4324–4333. [DOI] [PubMed] [Google Scholar]
- 35. Croen L.A., Shaw G.M., Jensvold N.G. and Harris J.A. (1991) Birth defects monitoring in California: a resource for epidemiological research. Paediatr. Perinat. Epidemiol., 5, 423–427. [DOI] [PubMed] [Google Scholar]
- 36. Hwang S.H., White K.A., Somatilaka B.N., Shelton J.M., Richardson J.A. and Mukhopadhyay S. (2018) The G protein-coupled receptor Gpr161 regulates forelimb formation, limb patterning and skeletal morphogenesis in a primary cilium-dependent manner. Development, 145, dev154054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hsia E.Y.C., Zhang Y., Tran H.S., Lim A., Chou Y.H., Lan G., Beachy P.A. and Zheng X. (2017) Hedgehog mediated degradation of Ihog adhesion proteins modulates cell segregation in Drosophila wing imaginal discs. Nat. Commun., 8, 1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pal K., Badgandi H. and Mukhopadhyay S. (2015) Studying G protein-coupled receptors: immunoblotting, immunoprecipitation, phosphorylation, surface labeling, and cross-linking protocols. Methods Cell. Biol., 127, 303–322. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.