

Abstract
Rett syndrome (RTT) is a neurodevelopmental disorder caused by mutations or deletions in Methyl-CpG-binding Protein 2 (MeCP2), a brain-enriched transcriptional regulator. MeCP2 is highly expressed during neuronal maturation and its deficiency results in impaired dendritic morphogenesis and reduced dendritic spine numbers in developing neurons. However, whether MeCP2 deficiency impacts the integration of new neurons has not been directly assessed. In this study, we developed a modified rabies virus-mediated monosynaptic retrograde tracing method to interrogate presynaptic integration of MeCP2-deficient new neurons born in the adult hippocampus, a region with lifelong neurogenesis and plasticity. We found that selective deletion of MeCP2 in adult-born new neurons impaired their long-range connectivity to the cortex, whereas their connectivity within the local hippocampal circuits or with subcortical regions was not significantly affected. We further showed that knockdown of MeCP2 in primary hippocampal neurons also resulted in reduced network integration. Interestingly, (1–3) insulin-like growth factor-1 (IGF-1), a small peptide under clinical trial testing for RTT, rescued neuronal integration deficits of MeCP2-deficient neurons in vitro but not in vivo. In addition, (1–3) IGF-1 treatment corrected aberrant excitability and network synchrony of MeCP2-deficient hippocampal neurons. Our results indicate that MeCP2 is essential for immature neurons to establish appropriate network connectivity.
Introduction
Rett syndrome (RTT), an X-linked dominant progressive neurodevelopmental disorder that mainly affects girls, is characterized by stereotypic hand movements, deficits in motor coordination, speech disorders and autistic behavior (1). Approximately 90% of reported cases of RTT result from mutations of the Methyl-CpG-binding protein 2 (MECP2) gene. MeCP2 is a multifunctional protein that has been shown to interpret DNA methylation, regulate chromatin architecture, activate and repress gene transcription and modulate RNA splicing (2). Extensive studies have shown that MeCP2 is critical for neuronal maturation (3–5). MeCP2-deficient neurons exhibit impaired dendritic morphogenesis and reduced dendritic spine density (6–8). These structural defects are associated with alterations in synaptic transmission and neural network activity (9). Therefore, it is proposed that RTT is a disorder of impaired neural circuits as a result of dysfunctional synaptic connectivity (10). However, whether MeCP2 deficiency in neurons directly affects their circuit integration has not been fully assessed.
MeCP2 is expressed widely in neurons throughout the brain, including the hippocampus, a brain region important for learning, memory and cognitive plasticity (11). MeCP2 deficiency significantly affects the physiology of hippocampal neurons including long-term potentiation, long-term depression, neurotransmission and altered expression of neurotransmitter receptors (12–15). In addition, a unique feature of the hippocampus is lifelong neurogenesis where new granule neurons are produced from adult neural stem cells (NSCs) residing in the dentate gyrus (DG) (16). The developmental process of adult-born new neurons recapitulates that of early brain. Similar to neurons born during embryonic development, adult-born new neurons must develop the dendrites, spines and axons that allow them to connect with the brain’s network. Adult new neurons are believed to fulfill unique functions, which may depend on the way they are connected (16,17). Several studies have assessed the role of MeCP2 in adult neurogenesis (4,18–20). MeCP2 is expressed at all developmental stages of new adult DG neurons (4). It has been shown that MeCP2 phosphorylation regulates proliferation of adult NSCs through modulating Notch signaling pathway (18). In addition, MeCP2 transgenic mice with elevated MeCP2 protein levels have increased quiescence of adult NSCs leading to reduced number of neuroblasts (19). Furthermore, we have shown that newborn DG neurons in young adult MeCP2 null mice exhibit reduced dendritic complexity, spine density and impaired transition from doublecortin-expressing immature stage to NeuN-expressing mature stage (4,20). Therefore, MeCP2 plays important roles at multiple stages of adult new neuron development. However how these structural changes of adult-born new neurons impact the integration and connectivity of these neurons has not been directly assessed.
The treatment of RTT represents an unmet therapeutic need with only symptomatic treatments currently available to patients (1). Gene therapy for neurologic ailments in human patients has been difficult to achieve. Therefore, it is desirable to identify drugs that can emolliate deficits associated with RTT. Insulin-like growth factor-1 (IGF-1) and its active tripeptide (1–3) IGF-1 have emerged as potential therapeutic approaches (21). Endogenous IGF-1 is primarily released in the serum by hepatocytes in response to growth hormone, and it can be cleaved to yield an N-terminal glycine-proline-glutamate [GPE tripeptide or (1–3) IGF-1] and a truncated IGF-1 form called des-N-(1–3)-IGF-1. IGF-1 is a crucial pleiotropic growth factor and widely expressed in the central nervous system (CNS) during normal development (22,23). IGF-1 strongly promotes neuronal cell survival and synaptic maturation (24,25) and facilitates the maturation of functional plasticity in the developing cortex (26). It has been proposed that IGF-1 may improve functional brain network connectivity in the context of neurodevelopmental disorders (27). Full-length recombinant IGF-1 can reverse RTT-like symptoms and phenotypes in MeCP2 mutant mice and neurons differentiated from RTT patient-derived induced pluripotent stem cells (28,29). Moreover, clinical pilot studies and early reports have supported preliminary efficacy of IGF-1 and related compounds in the treatment of RTT (21). Tripeptide (1–3) IGF-1 exhibits similar rescue effects of MeCP2 mutant mice as well as effects on spine density, excitatory synaptic transmission, excitatory synaptic markers such as synapsin 1 and post-synaptic density 95 and cortical circuit plasticity (30). However, the cellular targets and underlying mechanism of (1–3) IGF-1 treatment remain unclear.
In the current study we assessed the impact of MeCP2 deficiency on neuronal maturation and integration. Using a modified cell-type-specific rabies virus (RABV)-based monosynaptic retrograde tracing method, we found that selective deletion of MeCP2 from adult-born DG neurons specifically impacts long-range connections to cortical neurons. We then used a combination of RABV and multielectrode array (MEA) to show that MeCP2 deletion from cultured hippocampal neurons leads to alterations in neuronal connectivity and network excitability. We assessed the effect of (1–3) IGF-1 and found that while (1–3) IGF-1 exhibited beneficial effects on neonatal MeCP2-deficient hippocampal neurons, it showed no rescue effect on adult-born MeCP2-deficient DG new neurons. Our work demonstrates the important role of MeCP2 in neuronal integration and our assessment of (1–3) IGF-1 may shed light on treatment activity or a lack thereof for this RTT clinical trial drug.
Results
Development and validation of the dual-virus monosynaptic retrograde tracing method for targeting adult new neurons
Integration into neural circuit is a critical step of neuronal maturation. To determine whether specific deletion of MeCP2 in adult-born new neurons affect their connectivity, we modified a pseudotyped RABV-based monosynaptic retrograde tracing method (31,32) to create a new cell-type-specific dual-viral approach (Fig. 1A and B). We created two retroviruses with one expressing nuclear GFP and Cre recombinase (Retro-syn-nuclearGFP-Cre) and the other expressing Cre-dependent avian tumor virus receptor A (TVA) and rabies glycol protein (oG) [Retro-syn-double-floxed inverse open reading frame (DIO)-(TVA-oG)]. The transgenes in these viral vectors were driven by the synapsin promoter allowing for neuron-specific gene expression. The two viruses were injected together into the DG of adult MeCP2 conditional knockout (cKO) mice dependent on cre-loxp (Mecp2 f/y) or wild-type (WT) mice (Fig. 1C). Retroviruses only infect the dividing cells including dividing NSCs and progenitor cells in the DG. Only NSCs infected by both retroviruses were GFP+, expressed TVA and rabies oG and had MeCP2 deletion in MeCP2 cKO mice. To assess the effect of (1–3) IGF-1 on neuronal integration, (1–3) IGF-1 or bovine serum albumin (BSA) was administrated through intraperitoneal (i.p.) injection from the second to the fourth week after the initial retroviral injection during the period of neuronal maturation.
Figure 1.
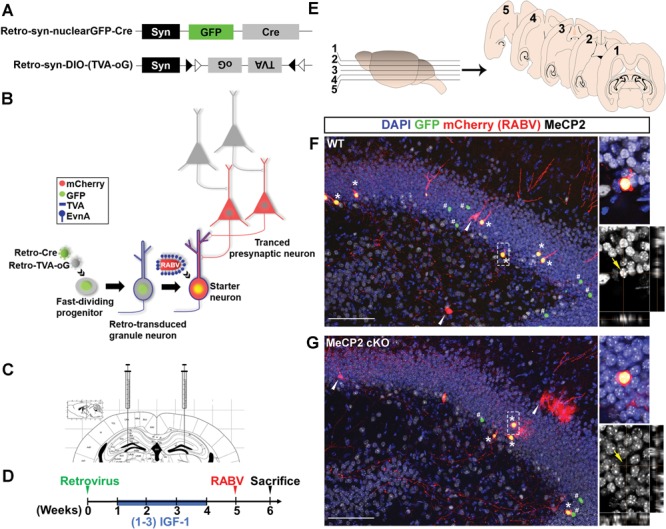
Modified RABV-monosynaptic retrograde tracing of adult new DG neurons with targeted MeCP2 gene deleted. (A) Schematic illustration of dual-retroviral vectors for targeting Cre and Cre-dependent TVA (receptor for EnvA) and oG (rabies packaging protein) specifically to adult new neurons in the DG. (B) Schematic illustration of pseudotyped RABV-mediated monosynaptic retrograde tracing to map the presynaptic inputs to newborn neurons in the DG. Retrovirus infects only proliferating NSCs and progenitor cells in the adult DG and synapsin (syn) promoter ensures the transgenes are only expressed when NSCs differentiate into neurons. Retro-syn-nulcearGFP-Cre knocked out the MeCP2 gene in NSCs of cKO mice and labeled these cells with GFP. Retro-syn-DIO-(TVA-oG) expressed TVA and oG in Cre-dependent manner. TVA is the EnvA receptor, and EnvA is the envelope protein used for pseudotyping RABV. oG is required for RABV packaging and trans-synaptic spread. When pseudotyped RABV expressing mCh is injected, they can only infect the TVA+ cells. In these cells, the pseudotyped RABV replicated and trans-complementation with oG resulting in the production of mCh+ RABV that spread trans-synaptically to presynaptic (traced) neurons. Since presynaptic neurons do not have oG, no RABV production or further trans-synaptic transfer will occur. Therefore, starter cells are marked with both GFP and mCh while presynaptic neurons express only mCh. (C) Schematic illustration of virus injection into the DG. (D) Timeline of the RABV-mediated monosynaptic retrograde tracing experiment in vivo. (E) Schematic illustration of horizontal brain sections used for quantitative analysis. (F and G) Representative confocal images of a brain section containing the hippocampal region. Asterisks indicate the starter neurons (nuclear GFP+, mCh+). White arrows indicate traced presynaptic neurons (mCh+). Pounds indicate neurons that were infected by retrovirus but not RABV (GFP+). Immunohistological analysis shows that the starter neurons in DG of a WT mouse were positive for MeCP2 protein (F, yellow arrow), but the ‘starter neurons’ in a MeCP2 cKO mouse had no detectable MeCP2 signal (G, yellow arrows). Scale bar, 100 μm.
At 5 weeks after retroviral injection, when new neurons were actively integrating into neural circuitry, mice were injected with chicken viral glycoprotein (EnvA)-pseudotyped RABV (EnvA-ΔG-mCh), in which oG was replaced with mCherry (mCh; Fig. 1D) (33). In RABV-infected neurons, EnvA-ΔG-mCh complemented with oG, crosses synapses and labels the presynaptic neurons with mCh. Because of the absence of oG in the presynaptic neurons, this virus could not spread further. Our previous study has shown that a control retrovirus missing oG does not cross synapses (31). At 7 days after the RABV injection, the mouse brains were collected, and horizontal sections of the brain were analyzed. As expected, we found neurons in the DG that expressed both GFP and mCh (GFP+ mCh+) that represented RABV-infected new neurons, which we named ‘starter neurons’. The neurons that expressed only mCh (mCh+) were presynaptic ‘traced neurons’ that were connected to ‘starter neurons’ (Fig. 1E–G). We further confirmed that ‘starter neurons’ in the DG of MeCP2 cKO mice had MeCP2 deletion (Fig. 1G; Supplementary Material, Fig. S1). Therefore, our new dual-viral targeting method allows for cell-type-specific gene deletion and connectivity assessment of ‘starter neurons’.
MeCP2-deficient adult-born DG granule neurons exhibit impaired long-range connectivity
It has been shown that adult-born DG granule neurons can form both local connection within the hippocampus and long-range connections to both cortical and subcortical regions (32,34,35). We therefore systematically quantified the number of ‘traced neurons’ throughout the brain and utilized the ratio of traced neurons over starter neurons to indicate the connectivity of ‘starter neurons’. About 30–40 starter neurons were analyzed in each mouse (Supplementary Material, Fig. S2). We first assessed whether MeCP2 deficiency or (1–3) IGF-1 treatment affects connectivity of ‘starter neurons’ within neurons in hippocampal DG, CA3, molecular layer (ML) and hilus (HL) (Fig. 2; Supplementary Material, Fig. S3). However, there was no statistical significance among groups in connectivity with DG (Fig. 2G), ML (Fig. 2H), CA3 (Fig. 2I) and HL (Fig. 2J). Moreover, (1–3) IGF-1 treatment did not show obvious rescue.
Figure 2.
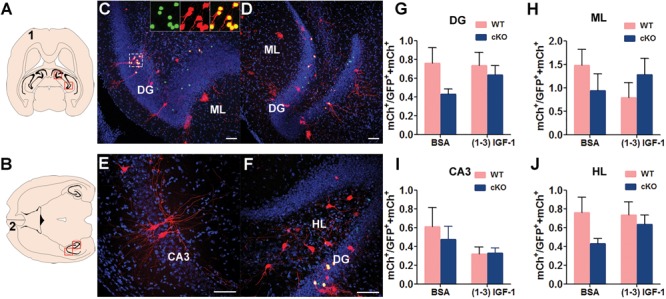
MeCP2 deletion in new DG neurons does not have significant effect on local hippocampal connection. (A and B) Schematic illustration of horizontal brain sections containing the hippocampus. (C–F) Representative confocal images of the red outlined area in (A) and (B) showing the connection between MeCP2-deficient DG new neurons with presynaptic neurons in DG, ML, CA3 and HL. Scale bar, 100 μm. (G–J) The ratio of traced cells (mCh+) to starter cells (GFP+ mCh+) in DG, ML, CA3 and HL were compared among experimental conditions: WT, n = 18; WT+ (1–3) IGF-1, n = 12; MeCP2 cKO, n = 14; MeCP2 cKO+ (1–3) IGF-1, n = 15. Two-way ANOVA with Tukey’s post hoc analysis for multiple comparisons; data were presented as mean ± SEM. There was no any significant difference in groups.
Consistent with literature (34,36), we found that the DG neurons in both WT and MeCP2 cKO mice received long-distance input from several subcortical regions, including the nucleus of the horizontal limb of the diagonal band (HDB) and the medial septal (MS) (Fig. 3A–C, E–G; Supplementary Material, Fig. S4). MeCP2 deficiency did not significantly impact the connectivity of ‘starter neurons’ with MS and HDB neurons (Fig. 3I and J). Interestingly, (1–3) IGF-1 treatment significantly reduced the connectivity to HDB neurons of WT but not MeCP2-deleted ‘starter neurons’ (Fig. 3J).
Figure 3.
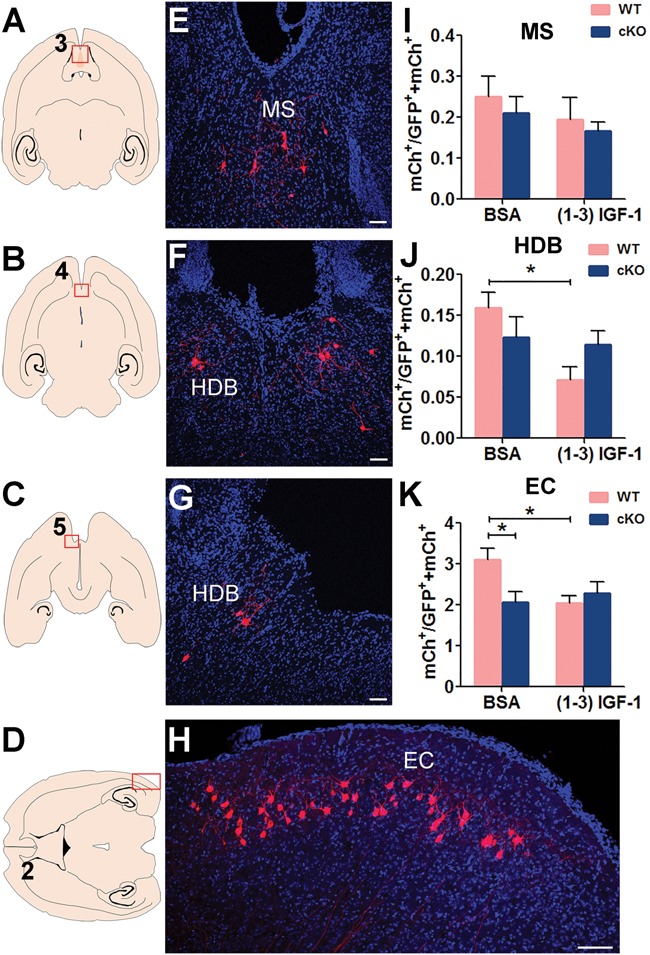
MeCP2-deleted new DG neurons have reduced long-range connectivity with the EC. (A–D) Schematic illustration of horizontal brain section through MS, HDB and EC. (E–H) Representative confocal images of the red outlined area in (A–D) showing the connection between MeCP2-deficient DG new neurons with presynaptic neurons in MS (I) and HDB (J) EC (K). Scale bar, 100 μm. (I–K) The ratio of traced cells (mCh+) to starter cells (GFP+ mCh+) in MS, HDB and EC were compared among conditions: MeCP2 deficiency in adult-born DG granule neurons reduced the ratio in EC (K, F (1, 55) = 2.299, P = 0.0467). (1–3) IGF-1 induced an obvious damage in normal newborn DG granule neurons in HDB (J, F (1, 55) = 5.958, P = 0.0149) and EC (K, F (1, 55) = 2.125, P = 0.0384). Two-way ANOVA with Tukey’s post hoc analysis for multiple comparisons; the numbers of animals used for quantitative analysis were WT, n = 18; WT+ (1–3) IGF-1, n = 12; MeCP2 cKO, n = 14; MeCP2 cKO+ (1–3) IGF-1, n = 15. Data were presented as mean ± SEM. *P < 0.05.
The major cortical input to DG granule neurons is from the Layer II/III of the entorhinal cortex (EC). We discovered that MeCP2 deletion in DG starter neurons specifically impaired this long-range connectivity of ‘starter neurons’ with the EC neurons (P < 0.05). (1–3) IGF-1 treatment did not show any rescue of this impairment but rather reduced the connectivity between WT ‘starter neurons’ and EC neurons (Fig. 3D, H and K; Supplementary Material, Fig. S4). Therefore, MeCP2 deletion in starter DG neurons had no significant impact on their connectivity to local hippocampal circuits or with subcortical regions but specifically impairs their long-range connectivity with the cortex, which cannot be rescued by (1–3) IGF-1 treatment.
Selective deletion of MeCP2 in adult-born DG granule neurons leads to impaired dendritic morphogenesis
We have previously shown that new neurons born in the postnatal DG of MeCP2 null mice exhibit impaired dendritic maturation and reduced spine density (4). This deficit could be because of either intrinsic MeCP2 deletion in new DG neurons or extrinsic MeCP2 deficiency in the surrounding mature neurons. We therefore examined the dendritic morphology of the ‘starter neurons’ that have intrinsic MeCP2 deletion (Fig. 4A). MeCP2-deleted starter neurons displayed shorter dendrites (Fig. 4B), fewer branching points (Fig. 4C), fewer dendritic ends (Fig. 4D) and less complexity compared with WT neurons (Fig. 4E) but did not impact the soma size (Supplementary Material, Fig. S5). (1–3) IGF-1 treatment did not show significant effect (Fig. 4B–E). Therefore, MeCP2 deletion in new neurons born in the adult DG impairs their dendritic development, indicating an important intrinsic role of MeCP2 in neuronal maturation.
Figure 4.
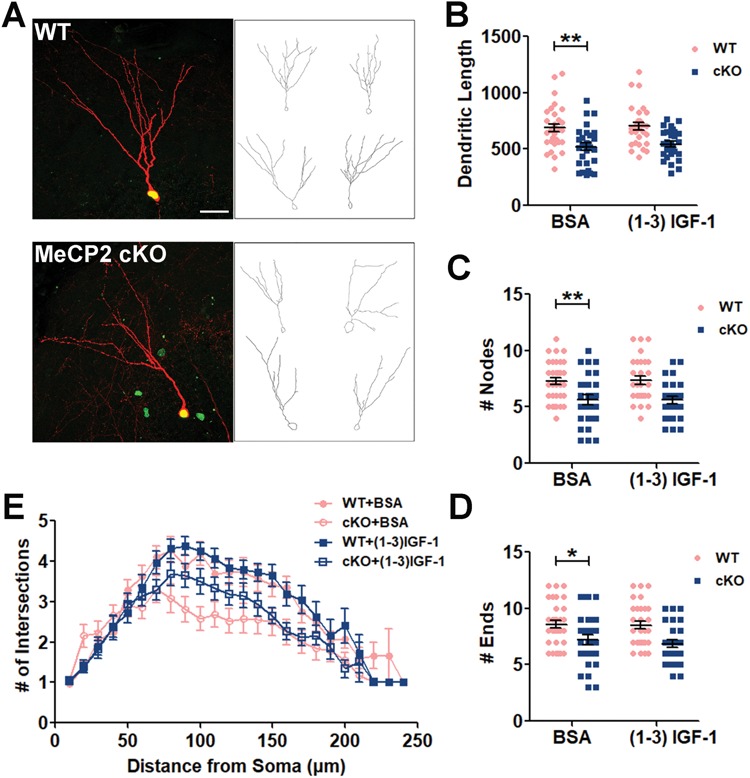
MeCP2 deletion in adult-born new neurons impairs dendritic morphogenesis. (A) Representative images (left) and sample traces generated by Neurolucida (right) of starter neurons. Scale bars, 100 μm. (B–E) MeCP2 deficiency leads to reduced total dendritic length (B, F (1, 113) = 26.27, P = 0.0017), dendritic branching points (C, F (1, 113) = 22.14, P = 0.0077) and dendritic ends (D, F (1, 113) = 16.95, P = 0.0422) dendritic complexity (E, MANOVA, F (1, 59) = 11.569, P = 0.001), compared with WT control. (1–3) IGF-1 did not rescue the impaired dendritic morphogenesis. Two-way ANOVA with Tukey’s post hoc analysis for multiple comparisons; WT, n = 32 (from 14 mice); WT+ (1–3) IGF-1, n = 29 (from 11 mice) MeCP2 cKO, n = 28 (from 11 mice); MeCP2 cKO+ (1–3) IGF-1, n = 28 (from 11 mice). Data were presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.0001.
(1–3) IGF-1 improves connectivity of MeCP2-deficient primary hippocampal neurons in vitro
Since MeCP2 deficiency impaired connectivity of immature neurons with neural circuit in vivo, we next developed a RABV-based in vitro monosynaptic tracing assay that could potentially be used to test therapeutic agents. We transfected primary hippocampal neurons with DNA plasmids expressing shRNA against MeCP2 (shMeCP2) and GFP, along with RABV packaging plasmid (TVA and oG) (Fig. 5A), on day 4 of culture (4 DIVs) when these neurons were undergoing dendritic and axonal morphogenesis. On 10 DIVs, RABV (EnvA-ΔG-mCh) were added for 3 days, followed by quantitative analysis (Fig. 5B). Although RABV has been used for in vivo neuronal connectivity assays, only very few studies have used it for in vitro connectivity analysis and all of these studies are relatively immature human stem cell differentiated neurons (37–40). The main reason is that cultured mouse neurons can establish a wide connection with large numbers of other neurons, therefore the number of ‘starter neurons’ must be kept in low number for effective analysis. We first confirmed that the control plasmid missing oG did not spread to the presynaptic neuron (P < 0.001, Fig. 5C and D). We performed pilot studies and selected a very low transfection efficiency (∼1% neurons) for labeling starter neurons to ensure that the number of traced neurons does not reach saturation (Fig. 5E and F). Using our optimized protocol, we assessed the effect of MeCP2 deletion and (1–3) IGF1 treatment on neuronal connectivity. We found that MeCP2-deficient neurons (shMeCP2) had a significantly decreased connectivity (P < 0.05). Surprisingly, unlike our observation of adult-born DG neurons (Figs. 2 and 3), treatment with (1–3) IGF-1 enhanced the connectivity of MeCP2-deficient neurons without affecting WT neurons (P < 0.05, Fig. 5G–I). Therefore, MeCP2 is important for neuronal integration of cultured hippocampal neurons and (1–3) IGF-1 rescued the impaired integration of MeCP2-deficient hippocampal neurons.
Figure 5.
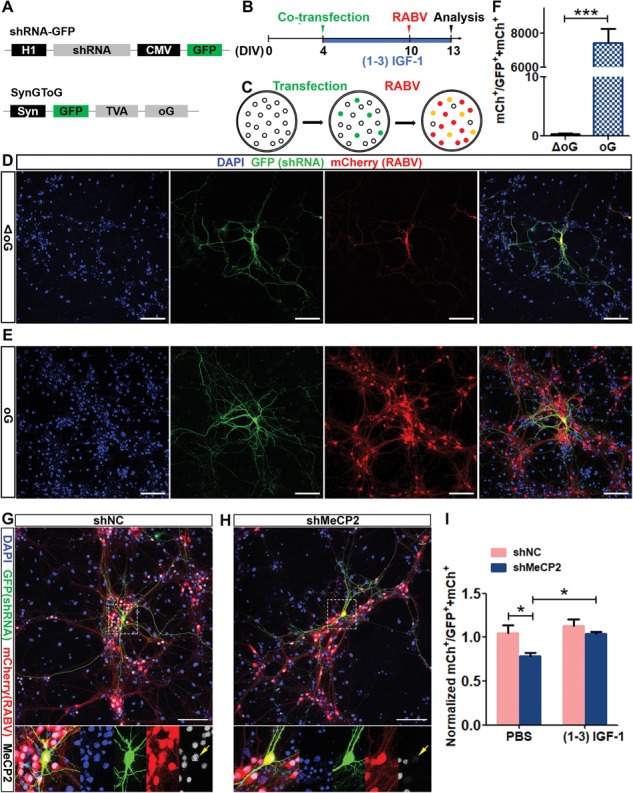
MeCP2 deficiency impairs connectivity of primary hippocampus neurons in vitro, which is rescued by (1–3) IGF-1.(A) Schematic illustration of the expression vectors expressing shMeCP2 and TVA (receptor for EnvA) and oG (rabies packaging protein). (B) Timeline of the RABV-mediated monosynaptic retrograde tracing experiment in vitro. (C) Schematic illustration of pseudotyped RABV-mediated monosynaptic retrograde tracing in vitro. (D and E) Representative confocal images of cultured primary hippocampal neurons transfected with control plasmid missing oG and TG (D) or with plasmids shown in A (E). Scale bars, 100 μm. (F) Compared with experimental vectors shown in A, the control vector missing oG did not mediate RABV spread to the presynaptic neuron (Student’s t-test, P < 0.0001). (G and H) Representative confocal images of cultured primary hippocampal neurons transfected with shNC (G) and shMeCP2 (H). Scale bars, 100 μm. (I) Normalized ratio of traced cells (mCh+) to starter cells (GFP+ mCh+). MeCP2 deficiency induced a significant decrease in the ratio of traced/starter neurons (F (1, 20) = 8.016, P = 0.0375), and (1–3) IGF-1 treatment enhanced the ratio (F (1, 20) = 7.142, P = 0.0474). Two-way ANOVA with Tukey’s post hoc analysis for multiple comparisons; n = 6 separate experiments; data were presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.0001.
(1–3) IGF-1 reduces network hyperexcitability of MeCP2-deficient primary hippocampus neurons
Despite having a relatively simple network organization, primary hippocampal neuron cultures also display similar network-wide synchronous bursting activity and are hence attractive experimental models for studying the interplay between individual neuronal activity, synaptic connectivity and network activity (41,42). To investigate whether MeCP2 deficiency affects functional maturation, primary hippocampal neurons were plated on top of MEA chips and were allowed to spontaneously form neural networks. On 4 DIVs, neurons were infected with lentivirus-shMeCP2 to knock down MeCP2 in a majority of the neurons, and (1–3) IGF-1 treatment was included. About 60% of the neurons were infected by lentivirus and there was no significant different in neuron versus glia ratio between MeCP2 knockdown (shMeCP2) and control (shNC) conditions (Supplementary Material, Fig. S6). We recorded spontaneous and evoked extracellular field activity on 13 DIVs (Fig. 6B–E). The rates of spontaneous (baseline) bursts generated by neurons firing action potentials were similar between MeCP2 knockdown and control (shNC) groups as well as (1–3) IGF-1 treatment group. However, upon addition of depolarizing KCl, MeCP2-deficient neurons showed significantly higher (evoked) burst rate compared with control (shNC) neurons (P < 0.05). (1–3) IGF-1 treatment decreased the burst rate of MeCP2 knockdown (shMeCP2) neurons compared with vehicle-treated MeCP2 knockdown neurons, suggesting that (1–3) IGF-1 partially reversed the high burst rate of MeCP-deficient neurons (Fig. 6F). Synaptic connections between neurons in a population may lead to synchronized action potentials, called network burst, which were measured as simultaneous bursts detected by all 12 electrodes within the well. We found that at baseline level, MeCP2-deficient neurons exhibited elevated network burst rate (Fig. 6G). Interestingly, treatment with depolarizing reagent KCl drastically increased network burst rate of MeCP2-deficient neurons without affecting control neurons (P < 0.0001, Fig. 6G). (1–3) IGF-1 significantly decreased the rates in KCl-evoked conditions (P < 0.0001, Fig. 6G). Therefore, these data suggest that MeCP2 deficiency leads to synchronized network activities, which can be rescued by (1–3) IGF-1.
Figure 6.
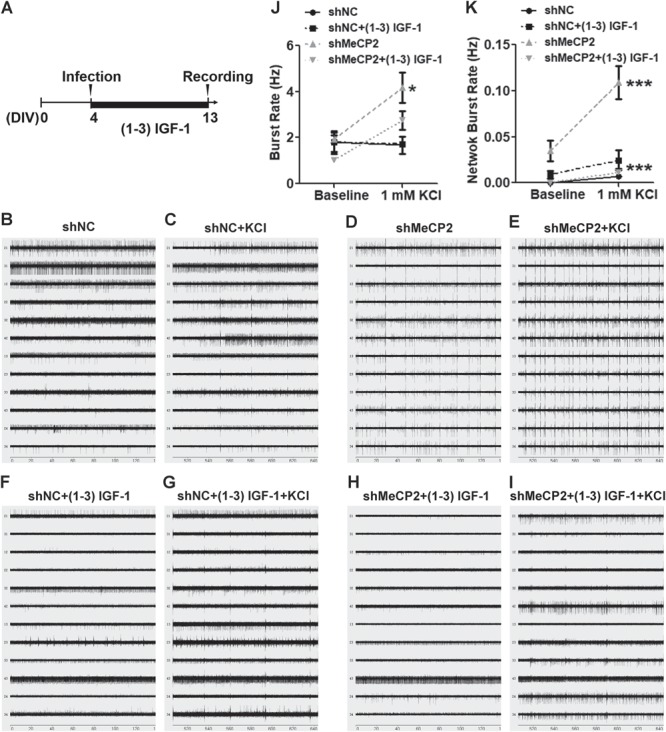
MeCP2 deficiency leads to elevated activity and network synchrony of hippocampal neurons in vitro which can be rescued by (1–3) IGF-1. (A) Timeline of MeCP2 knockdown and MEA assay in vitro. (B–I) Raw data traces for 12 electrodes from a representative well in different conditions. (J) The burst rates during 5 min recordings at baseline level and after KCl activation were compared among conditions. MeCP2-deficient neurons showed higher burst rate than negative control neurons both before and after KCl stimulation (F (1, 16) = 6.719, P = 0.0109). (K) The network burst rate (simultaneous burst detected among all electrode in the same well) during 5 min recordings were compared. MeCP2-deficient neurons exhibited higher network burst rate compared with control neurons, which was further elevated by KCl activation (F (1, 16) = 21, P < 0.0001). (1–3) IGF-1 significantly decreased the network burst rates in MeCP2-deficient neurons under KCl-stimulated conditions (F (1, 16) = 40.59, P < 0.0001). Three-way ANOVA with Tukey’s post hoc analysis for multiple comparisons; n = 3 separate experiments; data were presented as mean ± SEM. *P < 0.05, ***P < 0.0001.
Discussion
Formation of appropriate neural network is a prerequisite for proper brain function. Determining how MeCP2 deficiency impacts neuronal integration into the neural network is critical for understanding the pathogenesis and development of potential treatments for RTT. In the present study, we created a modified cell-type-specific RABV monosynaptic retrograde tracing method to show that targeted deletion of MeCP2 in adult-born DG neurons specifically impaired their long-range connectivity to cortical EC neurons. We then developed a novel in vitro neuronal connectivity assay for mouse hippocampal neurons and used a combination of RABV and MEA to show that MeCP2 knockdown from cultured hippocampal neurons led to alterations in neuronal connectivity and network excitability. We assessed the effect of (1–3) IGF-1, a clinically relevant reagent for RTT, and found that while (1–3) IGF-1 exhibited beneficial effect on neonatal hippocampal neurons in vitro, it showed no rescue effect on adult-born DG neurons in vivo. Our work demonstrates that MeCP2 is essential for immature neurons to establish appropriate network connectivity and (1–3) IGF-1 may exert differential effects depending on the types and developmental stages of neurons.
Previous studies have shown that MeCP2 deficiency leads to reduced maturation of both cultured hippocampal neurons (9,43) and adult new neurons (4,20). To uncover the impact of MeCP2 deficiency on first-order presynaptic partners of adult-born DG granule neurons, we utilized the retrovirus-based technique to selectively target new DG neurons for deletion of MeCP2 and primary RABV infection. We showed that DG new neurons with MeCP2 deletion resulted in a reduced long-range connection with EC, without significant effect on integration into local circuit or subcortical regions. It has been shown that newborn neurons in the adult DG incorporate stepwise into preexisting brain circuits. Newborn neurons are innervated initially by interneurons within the hippocampus, followed by modulatory cholinergic neurons in the MS/HDB and finally cortical neurons in the EC (34). The lateral EC is a major integration target for adult-born DG neurons (44). It is possible that MeCP2 may play a more important role in the later stage of the stepwise incorporation of adult-generated neurons into cortical circuits rather than local circuits.
We have previously shown that MeCP2 is expressed at all developmental stages of new adult DG neurons and in MeCP2 null mice, adult new neurons exhibit reduced dendritic complexity (4), which could be a deficit resulted from both intrinsic effect of MeCP2 on adult new neurons or extrinsic effect of MeCP2 in neurons and other cell types in the niche. The present data show that MeCP2 has significant intrinsic role in dendritic morphogenesis of adult new neurons which cannot be compensated by other WT cells residing in the niche. Future studies using MeCP2 conditional restoration mice may be used to evaluate the impact of MeCP2-deficient niche on MeCP2-expressing adult new neurons.
Since treatment with (1–3) IGF-1 peptide partially restores spine density and synaptic amplitude (30) and an analog of (1–3) IGF-1, NNZ-2566 has demonstrated efficacy in several Phase 2 trials on patients with RTT (21), we decided to investigate whether (1–3) IGF-1 could have therapeutic effects on MeCP2-deficient immature neurons. Surprisingly, we found that (1–3) IGF-1 treatment did not improve either connectivity or dendritic morphological deficits of MeCP2-deficient newborn DG neurons. On the contrary, (1–3) IGF-1 treatment rescued both connectivity and hyperactivities of cultured hippocampal neurons with acute MeCP2 knockdown. The differential effects of (1–3) IGF-1 on these neurons could be due to differences between DG granule neurons and cultured pyramidal neurons, or adult (8 weeks old) versus neonate (P0–P1) ages. On the other hand, previous studies have reported that the action of IGF-I signaling could be different depending on the experimental approach used, the developmental stage analyzed and cell type investigated (24).
Finally, we presented a novel observation on the excitability state of MeCP2-deficient primary hippocampal neurons. MEA allowed us to assess both excitability of neurons and synchronized activities of the network. It has been shown that MeCP2 deletion leads to elevated synchronization in the spontaneous firing activity of hippocampal CA1 neurons ex vivo and in vivo (45) and a hyperactive hippocampal network through aberrant excitation/inhibition of CA3 pyramidal neurons (46). The impaired neural circuit homeostasis probably contributes to limbic epileptiform seizures; a particularly severe co-morbidity affecting 60–80% of RTT patients (47). Therefore, MeCP2 seems to regulate the balance between synaptic excitation and inhibition. Previous research has shown that excitatory neurons depend on normal MeCP2 function to maintain the asynchronous state at baseline, whereas inhibitory interneurons require MeCP2 to enable the circuit to resist hypersynchrony in the face of perturbations to the excitation/inhibition balance (45). Interestingly, (1–3) IGF-1 treatment dampened the network hypersynchrony of MeCP2-deficient neurons, suggesting that (1–3) IGF-1 may repair neural circuit homeostasis by decreasing neuronal hyperexcitability (27). The mechanism underlying (1–3) IGF-1 therapeutic effect on RTT remains unclear. How (1–3) IGF-1 modulates the balance between synaptic excitation and inhibition is largely unexplored.
In summary, we developed a modified RABV dual-retrovirus monosynaptic retrograde tracing system to map the presynaptic partners of new DG neurons. This strategy allows for selective gene manipulation in starter cells and provides a method to assess intrinsic function of genes in regulating neuronal connectivity. In addition, although RABV has been used for a number of in vivo analysis (31,34,36), its application for in vitro analysis is limited because of high background resulting in a lack of sensitivity (37). Our in vitro RABV-mediated connectivity assay can be used to screen candidate drugs that may impact neuronal networks. Utilizing in vivo and in vitro RABV-mediated monosynaptic retrograde tracing systems combined with MEA technology, we provide direct evidence that MeCP2 is essential for immature neurons to establish appropriate structural and functional network connectivity.
Materials and Methods
Mice
All animal procedures were approved by the University of Wisconsin–Madison Care and Use Committee. WT C57BL/6 mice used for experiments and breeding were purchased from The Jackson Laboratory (Bar Harbor, ME). Conditioned knockout MeCP2-floxed (Mecp2f/y or cKO) mouse line used in this study has been published previously (48). Only 7- to 8-week-old male mice were used for experiments. Mice were group housed up to five per cage with the same gender and maintained on a 12 h light–dark cycle with food and water available ad libitum.
DNA plasmids
Retro-syn-nuclearGFP Cre and Retro-syn-double-floxed inverse open reading frame (DIO)-(TVA-oG) were cloned using Retro-CAG-red fluorescent protein (RFP) (49) as the backbone. An improved version of RABV glycoprotein, oG, was cloned from the plasmid pAAV-nEF-fDIO-oG, a generous gift from Dr Gallaway (Salk Institute) (50). Briefly, CAG-RFP coding sequence was replaced with syn-nuclear Cre or syn-DIO-(TVA-oG). SureSilencing shRNA vectors expressing shRNA against MeCP2 as well as GFP (shMeCP2) and negative control (shNC) were the same shRNA used in our previous work (20).
Production of lentivirus and retrovirus
Retrovirus production was performed as described in our previous publications (4,20,51,52). Briefly, retroviral vector DNA was co-transfected with packaging plasmids pCMV-gag-pol and pCMV-Vsvg into HEK293T cells using calcium phosphate-mediated method. The viral transfer vector DNA and packaging plasmid DNA were transfected in 5 × 15 cm dishes of cultured HEK293T cells using calcium phosphate. The medium containing retrovirus was collected at 36 and 60 h post-transfection, pooled, filtered through a 0.2 μm filter and concentrated using an ultracentrifuge at 19 400 rpm for 2 h at 4°C using a SW 32 Ti Swinging-Bucket Rotor (Beckman Coulter, Atlanta, Georgia). The virus was washed once and then resuspended in 100 μl phosphate buffered saline solution (PBS). We routinely obtained 1 × 108 infectious viral particles/ml for retrovirus.
Lentivirus was produced by polyethyleneimine (PEI)-mediated transfection. Lentiviral shRNA was co-transfected with packaging plasmids pMDL, REV and pCMV-Vsvg into HEK293T cells using PEI. HEK293T cells were plated a day ahead at 5 × 105 cells/10 ml in a 10 cm plate. The viral transfer shRNA and packaging plasmid DNA were mixed in 1 ml of Iscove’s Modified Dulbecco’s Medium (I3390, Sigma-Aldrich, St. Louis, MO). Next, 55 μl PEI (1 μg/μl) was added dropwise and mixed well. After 5 min, the DNA/PEI mix was added dropwise around the plate to the cells. The media was changed after 4–5 h. The medium containing lentivirus was collected at 36 and 60 h post-transfection and filtered through a 0.2 μm filter. The virus suspension was stored at 4°C for no more than 1 week.
In vivo virus grafting
In vivo viral grafting using stereotaxic surgery was performed as described (20,31). Briefly, MeCP2 WT and cKO mice were anesthetized with isoflurane and placed in a stereotactic instrument (David Kopf Instruments, Tujunga, CA). Microinjections were performed using custom-made injection 33 gauge needles (Hamilton, #776206, Reno, NV) connected to a 5 μl syringe (Hamilton, #87930). For dual-retroviruses injection, Retro-syn-nuclearGFP-Cre and Retro-syn-DIO-(TVA-oG) were mixed by 1:1 ratio. Then 1.5 μl retrovirus mix was stereotaxically injected into the DG using the following coordinates relative to bregma: caudal, −2.0 mm; lateral, +/−1.6 mm; ventral, −1.9 mm. From the end of first week to the end of the fourth week after retroviral injection, (1–3) IGF-1 was administered daily via i.p. injections (0.01 mg/g body weight, vehicle = saline, 0.01% BSA). At 35 days post-retroviral injections, the same mice were injected with 1.5 μl glycoprotein-gene-deleted pseudotyped RABV (RABV-EnvA-ΔG-mCh) at the same site. The RABV stock was a generous gift from Dr Gallaway (Salk Institute). The production of RABV was carried out as described previously (31,36). At 7 days post-RABV injection, mice were deeply anesthetized with ketamine and xylazine and transcardially perfused with saline followed by 4% paraformaldehyde (PFA). Finally, the brains were collected. The number of mice in different treatment groups is 18 (WT), 12 [WT+ (1–3) IGF-1], 14 (MeCP2 cKO) and 15 [MeCP2 cKO+ (1–3) IGF-1].
Brain tissue processing
Brain tissue processing was performed as described in our previous publication (53). The entire brains were sectioned into 125–135 40 μm thick successive horizontal sections through the dorsal–ventral extent of the brain using a sliding microtome (HM405, Thermo Scientific, Waltham, MA). Floating sections were stored in 96 well plates filled with cryoprotectant solution (glycerol, ethylene glycol and 0.1 M phosphate buffer, pH 7.4, 1:1:2 by volume) in a −20°C freezer, until needed.
Immunohistology
Immunohistology was performed based on published methods with modifications (20,54). Floating brain sections were first washed in PBS to remove cryoprotectant followed by incubation in PBS containing 10% normal goat serum and 0.3% Triton X-100 for 1 h at 37°C. Brain sections were then incubated overnight at 4°C with rabbit anti-MeCP2 monoclonal antibody (Cell Signaling Technology, Beverly, MA, #3456) diluted by 1:500 in a solution containing 2% normal goat serum and 0.3% Triton X-100. After three washes with PBS, the sections were incubated with secondary antibodies: Alexa Fluor® 647-conjugated goat anti-rabbit IgG (Invitrogen, Carlsbad, CA, #A21245) diluted by 1:1000 in a solution containing 2% normal goat serum and 0.3% Triton X-100 for 1 h at 37°C. The sections were washed three times with PBS and then counterstained with DAPI for 5 min at room temperature (RT). Finally, the sections were mounted with fluorescent mounting medium and stored at 4°C until analysis. Samples were observed using laser scanning confocal microscopy on a Nikon A1R-Si Confocal (Tokyo, Japan).
Neuronal integration analysis using recombinant RABV-mediated monosynaptic retrograde tracing method in vivo
At least one of every six serial horizontal sections (20–22 sections per brain totally in range of ventral: −1.5 to −7.0 mm relative to bregma) were mounted for quantification using an Olympus BX15 microscope (Tokyo, Japan). The numbers of GFP+ mCh+ DG ‘starter neurons’ and mCh+ ‘traced neurons’ in different brain regions were manually counted. The ratio of ‘starter neurons’ over ‘traced presynaptic neurons’ was used to determine the connectivity of newborn DG neurons. About 30–40 starter neurons were analyzed in each animal (Supplementary Material, Fig. S2). The brain regions analyzed include those known to connect with DG new neurons (34,36), including the subregions of the hippocampus (DG, ML, CA3 and the HL), subcortical regions (the nucleus of the HDB, MS) and the major long-range cortical input to the DG granule neurons, the EC. The locations of these brain regions were referred to mouse brain atlas on the Allen Mouse Brain Atlas website (http://mouse.brain-map.org/agea).
Morphological analysis of starter neurons
The 3D analyses of retroviral labeled adult-born DG granule neurons were performed as described in previous publications (20,31,52,55). Starter neurons were imaged on a Zeiss Apotome microscope (Oberkochen, Germany) with a 20× objective. Z-stacks of dendrites were captured at 1 μm intervals with the dendrites and the cell body of starter neurons analyzed by Neurolucida software with 3D module plug-in (MicroBrightField, Inc. Williston, VT, http://www.mbfbioscience.com). Roughly 30 neurons per DG were traced. Data were then extracted for sholl analysis.
Mouse primary hippocampus neuron isolation, transfection and immunocytochemistry
Hippocampal neurons were isolated from WT postnatal day 0 (P0) male mice and transfected as described previously (20,52). Briefly, hippocampal neurons isolated from WT P0 neonate mice were seeded on 12 mm Poly-D-lysine hydrobromide-coated glass coverslips in 24 well tissue culture plates in Neurobasal medium containing B27 supplement (ThermoFisher, Waltham, MA) and L-Glutame (ThermoFisher) at a density of 1 × 105 cells/well. Hippocampal neurons were transfected on day 4 DIVs as they were undergoing dendritic and axonal morphogenesis during this time. To avoid oversaturation of the ‘traced neurons’, we used a transfection method yielding sparsely transfected ‘starter neurons’. For each well, 1 μg of each of shRNA and synGToG plasmid DNA at a ratio of 1:1 was transfected into neurons using 2 μl Lipofectamine 2000 (ThermoFisher), as described (20). At 5–6 h after transfection, the medium was changed to fresh Neurobasal medium containing either (1–3) IGF-1 (10 ng/ml) or vehicle (PBS). Transfection efficiency in all our experiments was ∼1%. Fresh (1–3) IGF-1 (10 ng/ml) was then added every other day until the experiment ended. On 10 DIVs, neurons were infected with RABV. At 72 h after infection, the neurons were fixed.
Immunocytochemistry of cultured neurons was performed as described in previous publication (56). The coverslips were fixed with 4% PFA for 30 min at RT. For immunostaining, anti-MeCP2 monoclonal antibody (1:500), anti-MAP2 monoclonal antibody (1:500) and anti-GFAP monoclonal antibody (1:1000) were applied overnight at 4°C. Corresponding secondary antibodies, Alexa Fluor® 647-conjugated goat anti-rabbit IgG (1:1000) and Alexa Fluor® 568-conjugated goat anti-mouse IgG (1:1000), were used for 1 h at 37°C. Cells were then counterstained with DAPI for 5 min at RT. Finally, the coverslips were mounted with fluorescent mounting medium and stored at 4°C until analysis. Samples were observed using laser scanning confocal microscopy on a Nikon A1R-Si Confocal.
Neuronal integration analysis using recombinant RABV-mediated monosynaptic retrograde tracing method in vitro
For in vitro neuronal integration analysis, the numbers of ‘starter neurons’ and ‘traced neurons’ on the coverslips were quantified using Olympus BX15 microscope and Stereo Investigator software (MicroBrightField) as described (57). Similar to in vivo analysis, the ratio of ‘starter neurons’ over ‘traced neurons’ was used to determine the connectivity of primary hippocampal neurons. For each experiment, duplicate wells of cells were analyzed. Six independent biological replicates were used (n = 6) for statistical analyses.
MEA recording
MEA analysis was based on the previous work (58). MEA plates (MCS, Reutlingen, Germany) were composed of 24 wells, with each well containing an array of 12 embedded gold electrodes. The surface of the 24 well plate recording electrodes was first coated with 5 μl 0.1 mg/ml poly-D-lysine solution for 1 h at 37°C. Then, 5 μl of 100 000 cell suspension containing primary hippocampal neurons newly isolated from WT P0 neonatal mice and 1 ug/ml laminin were plated onto the electrode array located in the center of the well and incubated for 1 h at 37°C in a 5% CO2 tissue culture incubator. Then, 400 μl neuronal culture media was added to each well. On 4 DIVs, lentivirus-shMeCP2 or lentivirus-shNC was added to primary hippocampal neurons cultured in Neurobasal medium at a volume ratio of 1:1. At the same time, (1–3) IGF-1 (10 ng/ml) or vehicle control (PBS) was added and continued every other day until the end of the experiment. On 13 DIVs, MEA recording was performed. The plate was placed into a Multiwell-MEA recording system (MCS) and allowed at least 5 min to equilibrate, after which 5 min of spontaneous activity was recorded. Afterwards, 2 μl of 0.2 M KCl was added into 400 ul medium (final concentration of KCl is 1 mm) and another 5 min of evoked activity was recorded. For recordings, a band-pass filter (with a high-pass cutoff of 200 Hz and a low-pass cutoff of 3000 Hz) was applied, along with a variable threshold spike detector set at +/− 6 SD of the root mean squared of the background noise on each channel. All recordings were conducted at 37°C. Bursts were detected using a Multiwell-Analyzer, with the following threshold parameters: Max. Interval to Start Burst, 50 ms; Max. Interval to Start Burst, 50 ms; Min. Interval to between Burst, 100 ms; Min. duration of Burst, 50 ms; and minimum number of spikes in a burst, 4. Burst rate (Hz) and network burst rate (Hz) represent neural excitability and network synchronization, respectively. Each condition was done on two wells of neurons (experimental replicates). Neurons from three different pups were used as biological triplicates (n = 3).
Statistical analysis
Statistical analyses among multiple groups were carried out using two-way analysis of variance (ANOVA) or three-way ANOVA with Tukey’s post hoc analysis (Graphpad Prism 7). Sholl analysis was carried out using a multivariate analysis of variance (MANOVA) using SPSS statistical software (SPSS version 22, Armonk, New York). P-values < 0.05 were considered to indicate significant differences. The data bars and error bars indicate mean ± standard error of the mean (SEM).
Supplementary Material
Acknowledgements
We thank Y. Xing, M.E. Stockton and J. Lee, for technical assistance; R.D. Risgaard for editing; the Zhao Lab members for discussion; Dr G. Portugal (Harvard Apparatus) for helping with MEA; Dr Q. Chang for providing MeCP2-cKO mice; Dr E. Callaway (Salk Institute) for pseudotyped RABV and oG; Dr K. Knobel, J. Pinnow, D. Bollig and M. Eastwood at the Waisman IDDRC Model Core.
Conflict of Interest statement. None declared.
Funding
National Institutes of Health (R01MH116582, R01NS105200, R01MH078972, R56MH113146 and R21NS095632 to X.Z.); Waisman Center (U54HD090256); International Rett Syndrome Foundation, Brain Research Foundation, UW Vilas Trust (Kellett Mid-Career Award), Wisconsin Alumni Research Foundation and Jenni and Kyle Professorship to X.Z. and Grants-In-Aid from the State Scholarship Fund managed under China Scholarship Council (to Y.S.).
References
- 1. Leonard H., Cobb S. and Downs J. (2017) Clinical and biological progress over 50 years in Rett syndrome. Nat. Rev. Neurol., 13, 37–51. [DOI] [PubMed] [Google Scholar]
- 2. Jobe E.M. and Zhao X. (2017) DNA methylation and adult neurogenesis. Brain Plast., 3, 5–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fukuda T., Itoh M., Ichikawa T., Washiyama K. and Goto Y. (2005) Delayed maturation of neuronal architecture and synaptogenesis in cerebral cortex of Mecp2-deficient mice. J. Neuropathol. Exp. Neurol., 64, 537–544. [DOI] [PubMed] [Google Scholar]
- 4. Smrt R.D., Eaves-Egenes J., Barkho B.Z., Santistevan N.J., Zhao C., Aimone J.B., Gage F.H. and Zhao X. (2007) Mecp2 deficiency leads to delayed maturation and altered gene expression in hippocampal neurons. Neurobiol. Dis., 27, 77–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Palmer A., Qayumi J. and Ronnett G. (2008) MeCP2 mutation causes distinguishable phases of acute and chronic defects in synaptogenesis and maintenance, respectively. Mol. Cell. Neurosci., 37, 794–807. [DOI] [PubMed] [Google Scholar]
- 6. Chapleau C.A., Calfa G.D., Lane M.C., Albertson A.J., Larimore J.L., Kudo S., Armstrong D.L., Percy A.K. and Pozzo-Miller L. (2009) Dendritic spine pathologies in hippocampal pyramidal neurons from Rett syndrome brain and after expression of Rett-associated MECP2 mutations. Neurobiol. Dis., 35, 219–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kishi N. and Macklis J.D. (2010) MeCP2 functions largely cell-autonomously, but also non-cell-autonomously, in neuronal maturation and dendritic arborization of cortical pyramidal neurons. Exp. Neurol., 222, 51–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhou Z., Hong E.J., Cohen S., Zhao W.N., Ho H.Y., Schmidt L., Chen W.G., Lin Y., Savner E., Griffith E.C. et al. (2006) Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron, 52, 255–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ma D., Yoon S.I., Yang C.H., Marcy G., Zhao N., Leong W.Y., Ganapathy V., Han J., Van Dongen A.M., Hsu K.S. et al. (2015) Rescue of methyl-CpG binding protein 2 dysfunction-induced defects in newborn neurons by pentobarbital. Neurotherapeutics, 12, 477–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Goffin D. and Zhou Z.J. (2012) The neural circuit basis of Rett syndrome. Front. Biol., 7, 428–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Buckley M.J. (2005) The role of the perirhinal cortex and hippocampus in learning, memory and perception. Q. J. Exp. Psychol., 58, 246–268. [DOI] [PubMed] [Google Scholar]
- 12. Asaka Y., Jugloff D.G., Zhang L., Eubanks J.H. and Fitzsimonds R.M. (2006) Hippocampal synaptic plasticity is impaired in the Mecp2-null mouse model of Rett syndrome. Neurobiol. Dis., 21, 217–227. [DOI] [PubMed] [Google Scholar]
- 13. Calfa G., Hablitz J.J. and Pozzo-Miller L. (2011) Network hyperexcitability in hippocampal slices from Mecp2 mutant mice revealed by voltage-sensitive dye imaging. J. Neurophysiol., 105, 1768–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Moretti P., Levenson J.M., Battaglia F., Atkinson R., Teague R., Antalffy B., Armstrong D., Arancio O., Sweatt J.D. and Zoghbi H.Y. (2006) Learning and memory and synaptic plasticity are impaired in a mouse model of Rett syndrome. J. Neurosci., 26, 319–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nelson E.D., Kavalali E.T. and Monteggia L.M. (2006) MeCP2-dependent transcriptional repression regulates excitatory neurotransmission. Curr. Biol., 16, 710–716. [DOI] [PubMed] [Google Scholar]
- 16. Kempermann G., Song H. and Gage F.H. (2015) Neurogenesis in the adult hippocampus. Cold Spring Harb. Perspect. Biol., 7, a018812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lepousez G., Nissant A. and Lledo P.M. (2015) Adult neurogenesis and the future of the rejuvenating brain circuits. Neuron, 86, 387–401. [DOI] [PubMed] [Google Scholar]
- 18. Li H., Zhong X., Chau K.F., Santistevan N.J., Guo W., Kong G., Li X., Kadakia M., Masliah J., Chi J. et al. (2014) Cell cycle-linked MeCP2 phosphorylation modulates adult neurogenesis involving the Notch signalling pathway. Nat. Commun., 5, 5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen Z., Li X., Zhou J., Yuan B., Yu B., Tong D., Cheng C., Shao Y., Xia S., Zhang R. et al. (2017) Accumulated quiescent neural stem cells in adult hippocampus of the mouse model for the MECP2 duplication syndrome. Sci. Rep., 7, 41701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gao Y., Su J., Guo W., Polich E.D., Magyar D.P., Xing Y., Li H., Smrt R.D., Chang Q. and Zhao X. (2015) Inhibition of miR-15a promotes BDNF expression and rescues dendritic maturation deficits in MeCP2-deficient neurons. Stem Cells, 33, 1618–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vahdatpour C., Dyer A.H. and Tropea D. (2016) Insulin-like growth factor 1 and related compounds in the treatment of childhood-onset neurodevelopmental disorders. Front. Neurosci., 10, 450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. O'Kusky J. and Ye P. (2012) Neurodevelopmental effects of insulin-like growth factor signaling. Front. Neuroendocrinol., 33, 230–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bach M.A., Shen-Orr Z., Lowe W.L. Jr., Roberts C.T. Jr. and LeRoith D. (1991) Insulin-like growth factor I mRNA levels are developmentally regulated in specific regions of the rat brain. Brain Res. Mol. Brain Res., 10, 43–48. [DOI] [PubMed] [Google Scholar]
- 24. Nieto-Estevez V., Defterali C. and Vicario-Abejon C. (2016) IGF-I: a key growth factor that regulates neurogenesis and synaptogenesis from embryonic to adult stages of the brain. Front. Neurosci., 10, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Huat T.J., Khan A.A., Pati S., Mustafa Z., Abdullah J.M. and Jaafar H. (2014) IGF-1 enhances cell proliferation and survival during early differentiation of mesenchymal stem cells to neural progenitor-like cells. BMC Neurosci., 15, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tropea D., Kreiman G., Lyckman A., Mukherjee S., Yu H., Horng S. and Sur M. (2006) Gene expression changes and molecular pathways mediating activity-dependent plasticity in visual cortex. Nat. Neurosci., 9, 660–668. [DOI] [PubMed] [Google Scholar]
- 27. Bou Khalil R. (2017) Is insulin growth factor-1 the future for treating autism spectrum disorder and/or schizophrenia? Med. Hypotheses, 99, 23–25. [DOI] [PubMed] [Google Scholar]
- 28. Marchetto M.C., Carromeu C., Acab A., Yu D., Yeo G.W., Mu Y., Chen G., Gage F.H. and Muotri A.R. (2010) A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell, 143, 527–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Castro J., Garcia R.I., Kwok S., Banerjee A., Petravicz J., Woodson J., Mellios N., Tropea D. and Sur M. (2014) Functional recovery with recombinant human IGF1 treatment in a mouse model of Rett Syndrome. Proc. Natl. Acad. Sci. USA, 111, 9941–9946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tropea D., Giacometti E., Wilson N.R., Beard C., McCurry C., Fu D.D., Flannery R., Jaenisch R. and Sur M. (2009) Partial reversal of Rett Syndrome-like symptoms in MeCP2 mutant mice. Proc. Natl. Acad. Sci. USA, 106, 2029–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Guo W., Polich E.D., Su J., Gao Y., Christopher D.M., Allan A.M., Wang M., Wang F., Wang G. and Zhao X. (2015) Fragile X proteins FMRP and FXR2P control synaptic GluA1 expression and neuronal maturation via distinct mechanisms. Cell Rep., 11, 1651–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vivar C., Peterson B.D. and Praag H. (2016) Running rewires the neuronal network of adult-born dentate granule cells. Neuroimage, 131, 29–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wickersham I.R., Finke S., Conzelmann K.K. and Callaway E.M. (2007) Retrograde neuronal tracing with a deletion-mutant rabies virus. Nat. Methods, 4, 47–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Deshpande A., Bergami M., Ghanem A., Conzelmann K.K., Lepier A., Gotz M. and Berninger B. (2013) Retrograde monosynaptic tracing reveals the temporal evolution of inputs onto new neurons in the adult dentate gyrus and olfactory bulb. Proc. Natl. Acad. Sci. USA, 110, E1152–E1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li Y., Stam F.J., Aimone J.B., Goulding M., Callaway E.M. and Gage F.H. (2013) Molecular layer perforant path-associated cells contribute to feed-forward inhibition in the adult dentate gyrus. Proc. Natl. Acad. Sci. USA, 110, 9106–9111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vivar C., Potter M.C., Choi J., Lee J.Y., Stringer T.P., Callaway E.M., Gage F.H., Suh H. and Praag H. (2012) Monosynaptic inputs to new neurons in the dentate gyrus. Nat. Commun., 3, 1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sarkar A., Mei A., Paquola A.C.M., Stern S., Bardy C., Klug J.R., Kim S., Neshat N., Kim H.J., Ku M. et al. (2018) Efficient generation of CA3 neurons from human pluripotent stem cells enables modeling of hippocampal connectivity in vitro. Cell Stem Cell, 22, 684–697e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kirwan P., Turner-Bridger B., Peter M., Momoh A., Arambepola D., Robinson H.P. and Livesey F.J. (2015) Development and function of human cerebral cortex neural networks from pluripotent stem cells in vitro. Development, 142, 3178–3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Garcia I., Kim C. and Arenkiel B.R. (2013) Revealing neuronal circuitry using stem cell-derived neurons. Curr. Protoc. Stem Cell Biol., Chapter 2. Unit, 2D, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Garcia I., Huang L., Ung K. and Arenkiel B.R. (2012) Tracing synaptic connectivity onto embryonic stem cell-derived neurons. Stem Cells, 30, 2140–2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kapkaeva M.R., Popova O.V., Kondratenko R.V., Rogozin P.D., Genrikhs E.E., Stelmashook E.V., Skrebitsky V.G., Khaspekov L.G. and Isaev N.K. (2017) Effects of copper on viability and functional properties of hippocampal neurons in vitro. Exp. Toxicol. Pathol., 69, 259–264. [DOI] [PubMed] [Google Scholar]
- 42. Allio A., Calorio C., Franchino C., Gavello D., Carbone E. and Marcantoni A. (2015) Bud extracts from Tilia tomentosa Moench inhibit hippocampal neuronal firing through GABAA and benzodiazepine receptors activation. J. Ethnopharmacol., 172, 288–296. [DOI] [PubMed] [Google Scholar]
- 43. Xu X., Garcia J., Ewalt R., Nason S. and Pozzo-Miller L. (2017) The BDNF val-66-met polymorphism affects neuronal morphology and synaptic transmission in cultured hippocampal neurons from Rett syndrome mice. Front. Cell. Neurosci., 11, 203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vivar C. and Praag H. (2013) Functional circuits of new neurons in the dentate gyrus. Front. Neural Circuits, 7, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lu H., Ash R.T., He L., Kee S.E., Wang W., Yu D., Hao S., Meng X., Ure K., Ito-Ishida A. et al. (2016) Loss and gain of MeCP2 cause similar hippocampal circuit dysfunction that is rescued by deep brain stimulation in a Rett syndrome mouse model. Neuron, 91, 739–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Calfa G., Li W., Rutherford J.M. and Pozzo-Miller L. (2015) Excitation/inhibition imbalance and impaired synaptic inhibition in hippocampal area CA3 of Mecp2 knockout mice. Hippocampus, 25, 159–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Vignoli A., Savini M.N., Nowbut M.S., Peron A., Turner K., La Briola F. and Canevini M.P. (2017) Effectiveness and tolerability of antiepileptic drugs in 104 girls with Rett syndrome. Epilepsy Behav., 66, 27–33. [DOI] [PubMed] [Google Scholar]
- 48. Chen R.Z., Akbarian S., Tudor M. and Jaenisch R. (2001) Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat. Genet., 27, 327–331. [DOI] [PubMed] [Google Scholar]
- 49. Zhao C., Teng E.M., Summers R.G. Jr., Ming G.L. and Gage F.H. (2006) Distinct morphological stages of dentate granule neuron maturation in the adult mouse hippocampus. J. Neurosci., 26, 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kim E.J., Jacobs M.W., Ito-Cole T. and Callaway E.M. (2016) Improved monosynaptic neural circuit tracing using engineered rabies virus glycoproteins. Cell Rep., 15, 692–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Liu C., Teng Z.Q., Santistevan N.J., Szulwach K.E., Guo W., Jin P. and Zhao X. (2010) Epigenetic regulation of miR-184 by MBD1 governs neural stem cell proliferation and differentiation. Cell Stem Cell, 6, 433–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Smrt R.D., Szulwach K.E., Pfeiffer R.L., Li X., Guo W., Pathania M., Teng Z.Q., Luo Y., Peng J., Bordey A. et al. (2010) MicroRNA miR-137 regulates neuronal maturation by targeting ubiquitin ligase mind bomb-1. Stem Cells, 28, 1060–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gao Y., Wang F., Eisinger B.E., Kelnhofer L.E., Jobe E.M. and Zhao X. (2017) Integrative single-cell transcriptomics reveals molecular networks defining neuronal maturation during postnatal neurogenesis. Cereb. Cortex, 27, 2064–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sun Y., Wang Y., Zhao X. and Pu X. (2018) Nuclear translocation of DJ-1 protects adult neuronal stem cells in an MPTP mouse model of Parkinson's disease. Neuroreport, 29, 301–307. [DOI] [PubMed] [Google Scholar]
- 55. Giresi P.G., Kim J., McDaniell R.M., Iyer V.R. and Lieb J.D. (2007) FAIRE (formaldehyde-assisted isolation of regulatory elements) isolates active regulatory elements from human chromatin. Genome Res., 17, 877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Li S., Sun Y., Zhao X. and Pu X.P. (2012) Expression of the Parkinson's disease protein DJ-1 during the differentiation of neural stem cells. Brain Res., 1468, 84–93. [DOI] [PubMed] [Google Scholar]
- 57. Li Y., Stockton M.E., Bhuiyan I., Eisinger B.E., Gao Y., Miller J.L., Bhattacharyya A. and Zhao X. (2016) MDM2 inhibition rescues neurogenic and cognitive deficits in a mouse model of fragile X syndrome. Sci. Transl. Med., 8, 336ra361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. El Hady A., Afshar G., Broking K., Schluter O.M., Geisel T., Stuhmer W. and Wolf F. (2013) Optogenetic stimulation effectively enhances intrinsically generated network synchrony. Front. Neural Circuits, 7, 167. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.