

Abstract
LonP1 is crucial for maintaining mitochondrial proteostasis and mitigating cell stress. We identified a novel homozygous missense LONP1 variant, c.2282 C > T, (p.Pro761Leu), by whole-exome and Sanger sequencing in two siblings born to healthy consanguineous parents. Both siblings presented with stepwise regression during infancy, profound hypotonia and muscle weakness, severe intellectual disability and progressive cerebellar atrophy on brain imaging. Muscle biopsy revealed the absence of ragged-red fibers, however, scattered cytochrome c oxidase-negative staining and electron dense mitochondrial inclusions were observed. Primary cultured fibroblasts from the siblings showed normal levels of mtDNA and mitochondrial transcripts, and normal activities of oxidative phosphorylation complexes I through V. Interestingly, fibroblasts of both siblings showed glucose-repressed oxygen consumption compared to their mother, whereas galactose and palmitic acid utilization were similar. Notably, the siblings’ fibroblasts had reduced pyruvate dehydrogenase (PDH) activity and elevated intracellular lactate:pyruvate ratios, whereas plasma ratios were normal. We demonstrated that in the siblings’ fibroblasts, PDH dysfunction was caused by increased levels of the phosphorylated E1α subunit of PDH, which inhibits enzyme activity. Blocking E1α phosphorylation activated PDH and reduced intracellular lactate concentrations. In addition, overexpressing wild-type LonP1 in the siblings’ fibroblasts down-regulated phosphoE1α. Furthermore, in vitro studies demonstrated that purified LonP1-P761L failed to degrade phosphorylated E1α, in contrast to wild-type LonP1. We propose a novel mechanism whereby homozygous expression of the LonP1-P761L variant leads to PDH deficiency and energy metabolism dysfunction, which promotes severe neurologic impairment and neurodegeneration.
Introduction
Human mitochondrial Lon (LonP1) is a highly conserved protease belonging to the family of ATPases Associated with diverse cellular Activities (AAA+). LonP1 is critical for maintaining mitochondrial DNA (mtDNA) (1–3) and ensuring mitochondrial proteostasis by selectively degrading abnormal and oxidatively damaged proteins, as well as key rate limiting proteins (4–10). In response to various cellular stressors (e.g. hypoxia, oxidative stress and nutrient deprivation) (11–13), LonP1 functions to reprogram mitochondrial metabolism and energetics (2). LonP1 plays an essential role during embryogenesis, development and cellular lifespan (2).
Bi-allelic homozygous and compound heterozygous variants in LONP1 were first shown in 2015 to cause CODAS syndrome (MIM 600373) (14,15), which is defined by a constellation of cerebral, ocular, dental, auricular and skeletal abnormalities first reported by Shebib et al. in 1991 (16). Prior to this, only three additional individuals were described in the literature as having sufficiently overlapping features to receive the same clinical diagnosis (17–19). More recently, compound heterozygous LONP1 mutations that differ from those associated with classical CODAS syndrome have been reported (20,21). In one report, an individual who showed some CODAS-like symptoms (e.g. intellectual disability, congenital bilateral cataracts) also manifested a spectrum of neurological symptoms not observed in CODAS syndrome (e.g. spasticity, axial hypotonia, motor regression and progressive cerebellar atrophy) (20). In another study, an individual who had no CODAS-like features was found to have congenital lactic acidosis, muscle weakness and severe defects in oxidative phosphorylation (OXPHOS) and mtDNA depletion (21), which are characteristic of classical mitochondrial disease. Thus, different LONP1 mutations can cause a broad spectrum of disorders affecting different organ systems.
Little is known about the specific molecular mechanisms by which dysfunctional LonP1 promotes diverse clinical phenotypes. Here, we report the identification and characterization of a rare homozygous recessive missense variant in LONP1, c.2282 C > T, (p.Pro761Leu), which is associated with a severe neurodegenerative phenotype in two siblings from a consanguineous family with additional similarly affected members. Our report provides detailed clinical definition of this emerging neurological phenotype and provides novel insights into mechanisms by which LonP1 dysfunction may contribute to neurological disease. Using primary patient-derived fibroblasts expressing LonP1-P761L, we show that dysfunction of pyruvate dehydrogenase (PDH) is caused by increased phosphorylated E1α, which inhibits PDH and leads to elevated intracellular lactate. Our study demonstrates for the first time that wild-type LonP1 regulates the level of E1α phosphorylation, thereby modulating PDH activity.
Our findings expand the phenotypic spectrum of LONP1-related disorders to include individuals with severe neurologic impairment, dysregulation of energy metabolism and a neurodegenerative phenotype distinct from classical CODAS syndrome.
Results
Identification of an early-onset neurodegenerative disorder with distinctive muscle pathology
We describe two male siblings born to healthy parents of Afghani descent and family history significant for multiple levels of consanguinity (Fig. 1A). The siblings have three first cousins and a maternal uncle with similar profound developmental delays and neurodegeneration, and all died in childhood. Sibling 1, the proband, was born at term following an uncomplicated pregnancy and delivery. At 1 month of age, he underwent surgery for bilateral cataracts. At 5 months, developmental delays and hypotonia were noted, with persistent head lag. He rolled at 8 months and sat at 1 year. At 13 months, he had significant developmental regression following a three-day febrile illness and lost the ability to sit and cruise. At age 2 years, he had a second episode of regression during which he lost all of his developmental skills. At age 11, he had profound developmental delay with no head control and was non-verbal. He is exclusively gastric tube fed and dependent for all activities of daily living. Examination revealed brachycephaly and myopathic facies similar to his brother (Fig. 1B–G). Metabolic studies (lactate, quantitative plasma amino acids and urine organic acids) were normal, as were electromyography (EMG) and nerve conduction studies (NCS). Serial brain magnetic resonance imaging (MRI) demonstrated progressive cerebral and cerebellar atrophy (Fig. 1H); however, magnetic resonance spectroscopy (MRS) was normal.
Figure 1.
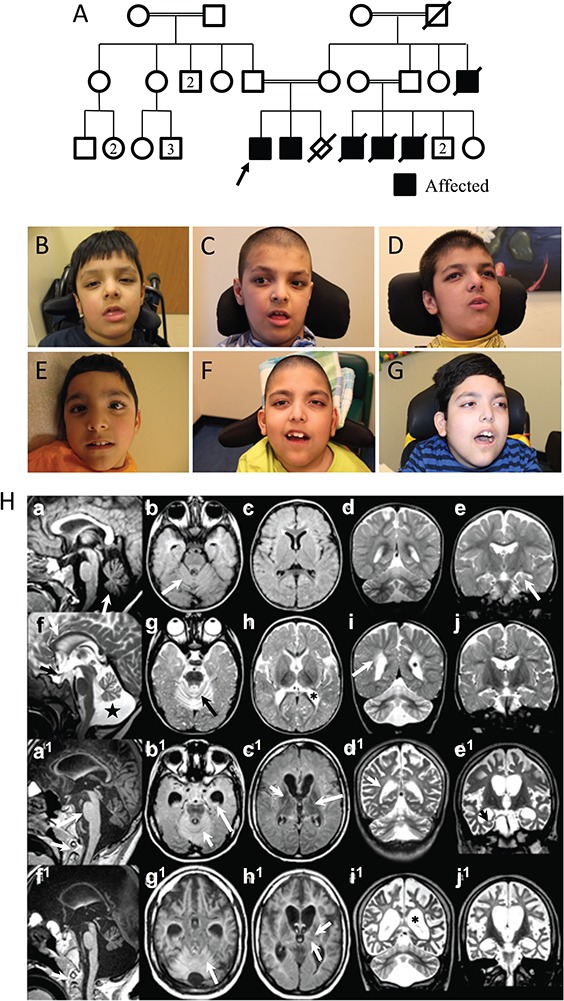
Pedigree, Clinical Photos and Brain Imaging. The pedigree is shown with affected family members shaded (A). The proband (Sibling 1) is shown with arrow. Clinical photographs of Sibling 1 (proband) at 4, 8 and 11 years (B, C and D) and Sibling 2 (his younger brother) at 3, 7 and 10 years (E, F and G). Common distinctive facial features include low anterior and posterior hairlines, synophyrus, deep-set eyes and a beaked nose with a bulbous tip. Serial MRI brain images (H) of Sibling 1 at 58 weeks and 7 years 10 months (a–e and a1–e1, respectively) and Sibling 2 at 11 months (9 months, corrected) and 7 years 4 months (f–j and f1–j1, respectively). Sagittal T1 (a) demonstrates mild vermian atrophy (arrow). Axial Fluid-attenuated inversion recovery (FLAIR) (b) reveals normal cerebellar cortex signal (arrow). Axial FLAIR (c) shows normal basal ganglia. Coronal T2 (d) identifies subtle cerebellar hemispheric volume loss. Coronal T2 (e) shows normal white matter tract signal and hippocampal volume (arrow). Sagittal T2 (f) shows thin corpus callosum (arrow) and optic chiasm (black arrow). The vermis is moderately atrophied, and the cisterna magna (star) is enlarged. Axial T2 (g) reveals cerebellar atrophy and bright cortex (black arrow). Axial T2 (h) demonstrates enlarged lateral ventricle trigones (*). Coronal T2 (i) confirms bright cerebellar cortex and abnormally increased periventricular white matter signal (arrow). Coronal T2 (j) shows mildly prominent sulci. Sagittal T1 (a1) shows marked corpus callosum thinning, mild progressive brainstem thinning (long arrow) and further loss of vermian volume when compared to initial study (top row, a). There is failure of odontoid tip ossification (short arrow). Axial FLAIR (b1) now shows bright cerebellar cortex (white arrow) and hippocampi (long white arrow) signal. The temporal horns have dilated. Axial FLAIR (c1) reveals low signal globi pallidi (arrow) and thalami. The internal capsule has lost normal myelin signal (long arrow). Coronal T2 (d1) demonstrates further loss of normal myelin signal (arrow). Coronal T2 (e1) shows marked hippocampal volume loss (black arrow) and further dilatation of ventricles and subarachnoid spaces. Sagittal T1 (f1) shows marked callosal thinning, mild progressive brainstem and further loss of vermian volume when compared to initial study (top row, f). There is failure of odontoid tip ossification (short arrow). Axial FLAIR (g1) demonstrates bright FLAIR signal of cerebellar cortex (arrow) and dilated temporal horns. Axial FLAIR (h1) reveals further atrophy of the basal ganglia and thalami. Internal capsule and internal medullary lamina white matter tracts show loss of normal myelin signal (arrows). Coronal T2 (i1) reveals severe atrophy, further loss of supratentorial myelin signal and marked ventriculomegaly (*). Coronal T2 (j1) shows marked loss of hippocampal volume and further dilatation of the subarachnoid spaces and ventricles.
Sibling 2, the proband’s brother, was born at 32 weeks gestational age. He was hypotonic at birth and required intubation for 2 weeks. He had significant difficulty swallowing and ultimately required gastrostomy tube placement. Ophthalmological examination was negative for cataracts. He obtained head control at 5 months and reached for objects at 6 months. He lost all these skills, and his hypotonia worsened at 8 months following a respiratory tract infection. At age 9, he functioned at the level of a newborn and had minimal head control. In contrast to his brother, he developed seizures, which were refractory to multiple medications. Serial brain MRI demonstrated progressive cerebellar and periventricular white matter volume loss, and MRS was normal (Fig. 1H). Detailed clinical histories for both siblings are available in Table 1 and as supplementary data.
Table 1.
Clinical features of individuals with bi-allelic mutations within the protease domain of LONP1
| Current study | Current study | Individual described by Inui et al. [ 20 ] | |
|---|---|---|---|
| Patient | Sibling 1 | Sibling 2 | Proband |
| Age at last review | 11 years | 9 years | 12 years |
| Gender | Male | Male | Male |
| Country of parental origin | Afghanistan | Afghanistan | Japan |
| Parental consanguinity | + | + | − |
| Birth weight/gestational age | 3400 g / 38 weeks | 1680 g / 32 weeks | 2324 g / 36 weeks |
| Axial hypotonia | +, noted at 5 months | +, present since birth | +, noted at 10 months |
| Appendicular hypertonia/spasticity | +, progressive | +, progressive | + |
| Feeding/swallowing difficulties | + | + | + |
| G-tube and age at dependency | 3.5 years | 2.5 years | 8 years |
| Current height (centile) | 50th centile | 80th centile | Not reported, −1.5 SD at birth |
| Current OFC (centile) | 25th centile | 80th centile | Not reported, +0.3 SD at birth |
| Ophthalmology | Bilateral congenital cataracts | Alternating esotropia, no cataracts | Bilateral congenital cataracts |
| Contractures | + | + | Not reported |
| Developmental regression | +, 13 months | +, 8 months | +, 2 years |
| Severe global developmental delay/intellectual disability | +, estimated current developmental age is 4 months | +, estimated current developmental age is 4 months | + |
| Communication | No speech | No speech | No speech |
| Gross motor | Unable to sit, non-ambulatory | Unable to sit, non-ambulatory | Unable to sit or crawl |
| Fine motor | No purposeful hand movements | No purposeful hand movements | Not reported |
| Muscle weakness | +, MRC grade 3/5 | +, MRC grade 3/5 | Not reported |
| Seizures, age of onset | − | +, 6 years | +, 2 years 10 months |
| Seizure types | N/A | Generalized tonic–clonic, myoclonic, atonic | Brief tonic seizures |
| Current anti-convulsive therapy | N/A | Lamotrigine, Levetiracetam, Clobazam, Lorazepam | Valproate |
| Dentition | Normal | Normal | Normal |
| Auricular system | Normal | Normal | Normal |
| Skeletal | Mild epiphyseal hypoplasia of distal femora | Mild epiphyseal hypoplasia of distal femora and lateral tibia | Bilateral epiphyseal dysplasia of knees |
| Native PDH (fibroblasts) (normal range 0.7–2.5 nmoles/min/mg protein) | 0.46 | 0.43 | Not reported |
| DCA activated PDH (fibroblasts) (normal range 0.9–2.5 nmoles/min/mg protein) | 1.03 | 1.07 | Not reported |
| Cellular lactate / Pyruvate ratio (fibroblasts) (normal range 10–25 nmoles/h/mg protein) | 38.16 | 36.12 | Not reported |
| Plasma lactate | Normal | Normal | Normal |
| Pyruvate carboxylase (fibroblasts) (normal range 0.3–3.5 nmoles/h/mg protein) | 1.22 | 1.40 | Not reported |
| Brain MRI | Progressive cerebral and cerebellar atrophy | Progressive cerebellar and periventricular volume loss | Progressive cerebellar and caudate atrophy, T2-hyperintensity of the cerebellar cortex |
| Electroencephalography | N/A | Encephalopathic background with frequent multifocal discharges | Rare spikes |
| EMG/NCS | Normal | Normal | Normal |
| LONP1 (NM_004793.3) | c.2282 C > T (homozygous) | c.2282 C > T (homozygous) | c.296dup; c.2356C > T |
| Predicted effect on protein | p. Pro761Leu | p. Pro761Leu | p.Ser100Glnfs*46; p.Arg786Trp |
| Type of mutation | Missense | Missense | Stop; missense |
The muscle biopsy findings were comparable for both Siblings 1 and 2 and are shown in Figure 2. Histochemical analysis using light microscopy revealed normal myofibrils with peripheral nuclei (Fig. 2A and B). Gomori trichrome staining showed the absence of ragged-red fibers (Fig. 2B), which are commonly observed in classical mtDNA diseases and other muscle disorders. ATPase and nicotinamide adenine dinucleotide hydride (NADH) staining showed normal fiber ratios; however, scattered cytochrome c oxidase (COX)-negative and lightly reactive succinate dehydrogenase fibers were observed (Fig. 2C and D). Glycogen and lipid staining were normal (data not shown). Transmission electron microscopy (TEM) of muscle biopsies showed abnormal mitochondria (Fig. 2E and F), and TEM of skin fibroblasts from both siblings demonstrated abnormally elongated mitochondria with electron dense spherical inclusions, which were not observed in their mother (Fig. 2G–I).
Figure 2.
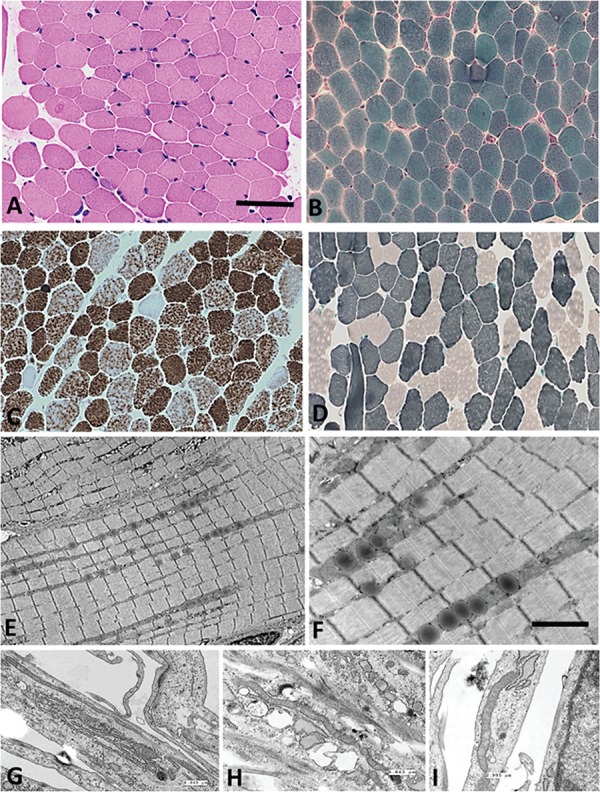
Muscle biopsy results including histochemistry and TEM. Representative histology of muscle biopsy from Sibling 1 is shown in A–D. (A) Haematoxylin and eosin, (B) modified Gomori trichrome (note the absence of ragged-red fibers) (C) COX histochemistry (note the scattered COX-deficient fibers) and (D) ATPase 9.6. The scale bar represents 260 microns in A–D. (E–F) TEM appearance of muscle oriented longitudinally. Scale bar represents 2000 nanometre in E and F. (G–I) TEM from fibroblasts. G (Sibling 1) and H (Sibling 2) show fibroblasts from both brothers, and I shows fibroblasts from Mother. In G, the measured mitochondrion is 6.945 microns in length; in H, it is 5.843 microns; and in I, the mitochondrion is 2.995 microns.
LonP1-Pro761Leu is not associated with mtDNA depletion or lactic acidosis
Using genomic DNA and total RNA isolated from cultured skin fibroblasts of Siblings 1 and 2 and their mother, we determined the relative levels of mtDNA, mtDNA-encoded transcripts and LONP1 transcripts, respectively (Fig. 3A and B). MtDNA copy number and gene expression were not depleted in either Sibling 1 or 2 compared to their mother. Metabolic studies showed normal levels of plasma lactate, pyruvate, quantitative plasma amino acids and urine organic acids.
Figure 3.
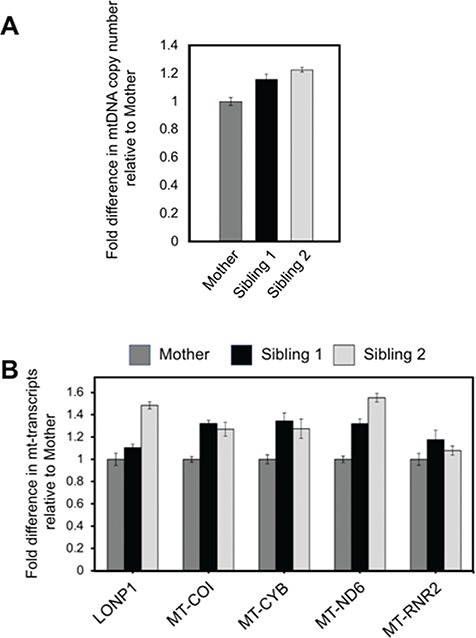
mtDNA copy number and mitochondrial RNA transcript levels. (A) Total DNA was isolated from cultured primary skin fibroblasts, and relative fold difference in mtDNA copy number was determined by qPCR of mtDNA-encoded MT-CYB, using the nuclear APP gene as an endogenous control. (B) Relative fold change in mitochondrial transcript levels was determined for LONP1 and the mtDNA-encoded genes MT-COI, MT-CYB, MT-ND6 and MT-RNR2. HPRT1 was used as an endogenous control.
Identification of a rare homozygous variant of LONP1 c.2282 C > T, [p.Pro761Leu]
Whole-exome sequencing was performed on Siblings 1 and 2, their Mother and Father, which identified a homozygous missense variant in the nuclear-encoded LONP1 gene (NM_004793.3) c.2282 C > T, [p.Pro761Leu]. This amino acid substitution is predicted to be located within the highly conserved protease domain (Fig. 4A). No other clinically significant sequence variants were identified in LONP1 or in other genes. Sanger sequencing for both Siblings 1 and 2 and their parents verified that each allele was inherited from their carrier parents (Fig. 4B). The variant is present in the Genome Aggregation Database GnomAD in the heterozygous state at a low frequency, <0.002%, and has been reported in five heterozygous individuals from two populations (African and European) in a total of 276 578 alleles (http://gnomad.broadinstitute.org). The variant is predicted to be damaging and deleterious to protein function by PolyPhen-2 and MutationTaster, respectively (Supplementary Material, Fig. S1).
Figure 4.
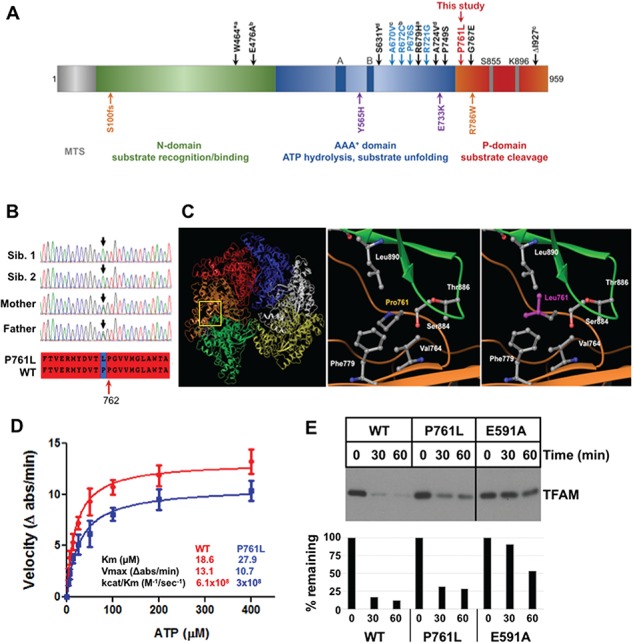
Lon-P761L mutant protein homology modeling and biochemical analysis. (A) Domain organization of Lon-P1 monomeric subunit representing the MTS, the N-, AAA+- and P domains. Indicated above the domain structure is the predicted location of the variant described in this study (red) [p.Pro761Leu]. The homozygous LONP1 mutations causing CODAS syndrome are shown in blue letters above the domain organization—[pAla670Val], [p.Arg672Cys], [p.Pro676Ser]), [p.Arg721Gly] compound heterozygous CODAS mutations are shown in black letters—superscript notations (a, b, c, d) indicate the compound heterozygous mutant alleles—a: [p. W464*/p.Arg679His], b: [p. Glu476Ala/p.Pro749Ser], c: [p.Ala670Val/p.I927del], d: [p.Ser631Tyr/p.Ala724Val]. Below the domain structure, the atypical CODAS syndrome mutation is shown in orange—([p.Ser100Glnfs*46/p. Arg786Trp]), and the compound heterozygous mutations associated with classical mtDNA disease are shown in purple—([p. Tyr565His/p.Glu733Lys]). (B) Chromatograms demonstrating the LONP1 variant c. 2282 C > T [p. Pro761Leu] in the homozygous state in affected Siblings 1 and 2 and heterozygous state in their unaffected parents. The aligned amino acid sequence of the wild-type and mutant Lon proteins surrounding position 761 is also shown (red highlight). (C) Left and middle panels: homology model of the hexameric human mitochondrial Lon protease was generated using the crystal structure of B. subtilis Lon (PDB 3M6A) as a template. Yellow box: topological position of P761. This region has six proline residues close to P761, which are located at the interface of adjacent LonP1 subunits (green and brown) in the complex. Right panel: replacement of P761 to L761 does not alter the bend in this unstructured region as there is another proline at position 762. However, P761L is expected to increase hydrophobic interactions between two subunits that involve residues V764 and F779 from the same subunit, and T886 and L890 from the adjacent subunit. (D) ATPase and protease activities using purified recombinant wild-type LonP1 and LonP1-P761L. ATPase activity was determined using an enzyme-linked assay, in which LonP1 proteins (400 nM) were incubated in buffer (50-mm HEPES KOH pH 7.5, 5-mm Mg(OAc)2, 75-mm KOAc) with phosphoenolpyruvate (3 mm), NADH (300 μM), pyruvate kinase/lactate dehydrogenase (12–20 U/ml) and ATP (0–400 μM). The change in NADH absorbance (ϵ340 nm = 6220 M−1 cm−1) was monitored in a Spectramax plate reader for 5 min. (E) Wild-type and mutant LonP1 degradation of TFAM. LonP1 (1 μM, monomer) with TFAM (3 μM) were incubated in buffer (50-mm Hepes-KOH pH 8.0, 150-mm NaCl, 10-mm MgCl2) and ATP-4 mm. Reactions were conducted at 37°C for the indicated times, then terminated by addition of reducing sample buffer and visualized by SDS PAGE and chemiluminescence imaging. Data are representative of two independent experiments.
LonP1-Pro761Leu mutation impairs enzymatic activity and substrate-specific proteolysis
The LonP1 holoenzyme is composed of six identical subunits forming a ring-shaped complex (Fig. 4A and C). The monomeric subunit is synthesized in the cytosol with an N-terminal mitochondrial-targeting sequence (MTS), which directs its translocation into the matrix, where the MTS is cleaved off (Fig. 4A). The mature processed protein has the following three domains: a weakly conserved N-terminal domain, involved in substrate recognition and binding; a AAA+ domain that is conserved in the superfamily of ATPases Associated with various cellular Activities (22); and a highly conserved proteolytic (P) domain present in all Lon-like proteases from archaea and eubacteria to mammals (Fig. 4A).
Of the reported LONP1 variants, the predicted LonP1- Pro761Leu mutant protein is the only one resulting from a homozygous mutation within the protease domain (Fig. 4A, noted above the domains in red). There are 17 other homozygous and compound heterozygous variants within the LonP1 protein that have been identified (14,15,23) (Fig. 4A). Twelve variants are associated with CODAS syndrome (Fig. 4, noted above the domains in blue homozygous and in black compound heterozygous mutations). The majority of these mutations clusters within the AAA+ domain. Variants associated with atypical CODAS syndrome (20), classical mitochondrial disease (21) and infantile cataract (23) have also been reported (Fig. 4A, noted below the domains in orange and purple, respectively).
To address whether the LonP1- Pro761Leu substitution alters protease structure and/or function, we employed a homology model of the human mitochondrial LonP1 that we generated previously (Fig. 4C) (10). Structural analyses showed that Pro761Leu in each Lon monomer is located on a loop at the interface of adjacent subunits (Fig. 4C, left and middle panels). Mutant modeling of Pro761 to Leu761 did not affect loop structure (Fig. 4C, right panel) as this loop is maintained by an adjacent proline at position 762 (Pro762) (Fig. 4B, red arrow). The Pro761Leu substitution most likely enhances the hydrophobic interactions contributed by Val764 and Phe779 (on the same subunit) and Thr886 and Leu890 on the adjacent subunit (Fig. 4C, right panel). This increased hydrophobic interaction likely reduces the flexibility of the LonP1 hexamer, which may negatively impact the efficiency of ATP-dependent proteolysis.
As predicted by homology modeling, reduced catalytic efficiency was observed upon analyzing the ATPase and protease activities of the purified recombinant LonP1-P761L mutant protein. LonP1-P761L had lower affinity for ATP and a lower rate of ATP hydrolysis (Fig. 4D). For LonP1-P761L, the catalytic efficiency, as defined by kcat/Km, was 50% lower than the wild-type enzyme (Fig. 4D). Notably, the missense mutation P761L within the protease domain reduces the ATPase activity of the mutant protein. We have previously shown that replacement of the highly conserved serine at position 855 to an alanine in the wild-type enzyme also has impaired ATPase activity (24).
We examined the proteolytic activity of LonP1-P761L compared to wild-type Lon (LonP1-WT) and a Walker Box B mutant (LonP1-E591A), which has severely impaired ATPase and protease activities (Supplementary Material, Fig. S2). Our previous work has shown that mitochondrial transcription factor A (TFAM) is an endogenous substrate of LonP1 when it is not bound to DNA, either because mtDNA is absent or severely depleted, or because TFAM is phosphorylated, which impairs DNA binding (7,21). Purified recombinant LonP1-WT efficiently degraded TFAM, such that only 17% and 12% remained after 30 and 60 min of incubation, respectively (Fig. 4E). In contrast, the Walker B mutant LonP1-E591A showed defective TFAM degradation; 90% of the substrate remained after 30 min, and 53% after 60 min. Of note, LonP1-P761L showed intermediate protease activity as compared to LonP1-WT and LonP1-E591A. When incubated with LonP1-P761L, 31% and 28% of TFAM remained after 30 and 60 min, respectively. We also examined the degradation of an unstructured protein substrate—fluorescently labeled casein (FITC-casein) (Supplementary Material, Fig. S2). We found that LonP1-P761L degraded this unfolded substrate almost as efficiently as LonP1-WT (albeit with a lag period), such that β-casein was completely degraded by 15–30 min. In contrast, LonP1-E591A showed markedly impaired degradation of this substrate. These data suggest that defects in the proteolytic activity of LonP1-P761L may be substrate-specific.
Homozygous LonP1-P761L mitochondria and fibroblasts show normal OXPHOS activities
The scattered Complex IV COX-negative staining observed on muscle biopsy (Fig. 2C) prompted us to examine the activities of OXPHOS complexes in mitochondria isolated from the primary skin fibroblasts of Siblings 1 and 2. Compared to mitochondria from a normal (unrelated) control, mitochondria from Siblings 1 and 2 showed normal activities of all OXPHOS complexes—Complexes I and III (combined), II and III (combined), IV and V (Table 2). We also compared the activity of Complex IV of Siblings 1 and 2 and their mother using kinetic and endpoint assays with protein extracts from primary fibroblasts (Fig. 5A and B, respectively). Complex IV activity was indistinguishable between both siblings and their clinically normal heterozygous mother. In addition, we also examined the relative protein levels of Complex IV in cells from both siblings and their mother and showed that equivalent amounts of this complex were present (Fig. 5C). These results also provide controls for the activity assays (Fig. 5A and B), by demonstrating that equal amounts of Complex IV protein were used to measure COX activity.
Table 2.
OXPHOS activities in fibroblast mitochondria (nmol/min/mg)
| Complex I and III | Complex II and III | Complex IV | Complex V | |
|---|---|---|---|---|
| Sibling 1 | 27.92 | 32.98 | 41.69 | 478.82 |
| Sibling 2 | 28.31 | 31.92 | 38.10 | 428.04 |
| Control | 25.58 | 23.93 | 37.96 | 382.29 |
Figure 5.
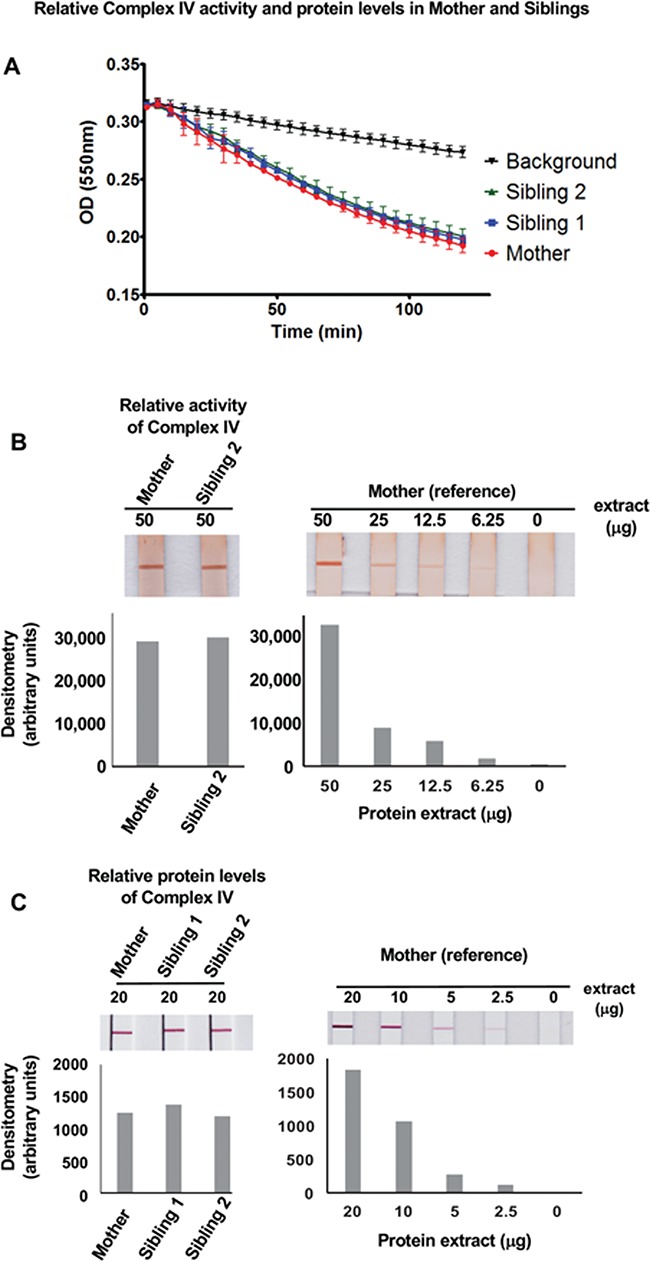
Relative Complex IV activity and quantity in fibroblasts of Mother and affected Siblings. (A) Kinetic comparison of Complex IV activities in protein extracts from Siblings 1 and 2 and Mother fibroblasts. (B) Endpoint assay analyzing Complex IV activity in protein extracts (50 μg) from Sibling 2 and Mother fibroblasts. The latter was serially diluted from 50 to 6.25 μg and used as a reference control. Corresponding densitometric analysis is shown below. (C) Complex IV protein quantity analysis in protein extracts (20 μg) from Siblings 1and 2 and Mother fibroblasts. The latter was serially diluted from 20 to 2.5 μg and used as a reference control. Corresponding image densitometry analysis is shown below.
Homozygous LonP1-P761L fibroblasts show defects in the oxidative metabolism of glucose but not galactose or palmitic acid
We sought to determine if there were differences in the utilization of different energetic substrates in the primary skin fibroblasts from Siblings 1 and 2 and their mother. These cells showed comparable levels of LonP1 protein (see Fig. 7A), co-localization of LonP1 with mitochondria and similar mitochondrial morphology when cultured with glucose- or galactose-containing medium (Supplementary Material, Figs S3 and S4, respectively). Using these fibroblasts, we simultaneously measured oxygen consumption rate (OCR), and extracellular acidification rate (ECAR) in real time, to determine their respective electron transport chain and glycolytic activities. Cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 10-mm glucose supplemented with 10% (v/v) fetal bovine serum (FBS) for 5 days. Complex V-F1Fo ATP synthase was active, as mitochondrial OCR was blocked by the Fo inhibitor oligomycin. Fibroblasts from Siblings 1 and 2 (Fig. 6A, blue and green, respectively) showed substantially reduced spare respiratory capacity (SRC) compared to that of their mother (Fig. 6A, red). SRC is the difference between basal OCR and maximal OCR induced by the mitochondrial membrane potential uncoupler carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP), which is a measure of the cell’s ability to boost mitochondrial ATP production in response to increased energy demand (25). The addition of rotenone similarly inhibited Complex I in the cells of both siblings and their mother. Interestingly, the affected siblings’ cells that had been cultured in medium containing 10-mm galactose for 5 days did not show a deficit in SRC as compared to their mother’s cells cultured under the same conditions (Fig. 6B). When FCCP was added to cells cultured in galactose-containing medium, the maximal OCR for the mother and siblings was the same (Fig. 6B). The relatively slow conversion of galactose to glucose-1-phosphate, which is then converted to pyruvate, forces cells to have greater reliance on OXPHOS. These results show that in galactose-containing medium, the SRC capacity of the siblings’ fibroblasts is comparable to that of their mother. We also observed that the glycolytic activity of fibroblasts from both siblings and their mother did not significantly differ when the cells were cultured in medium containing either glucose or galactose (Supplementary Material, Fig. S5).
Figure 7.
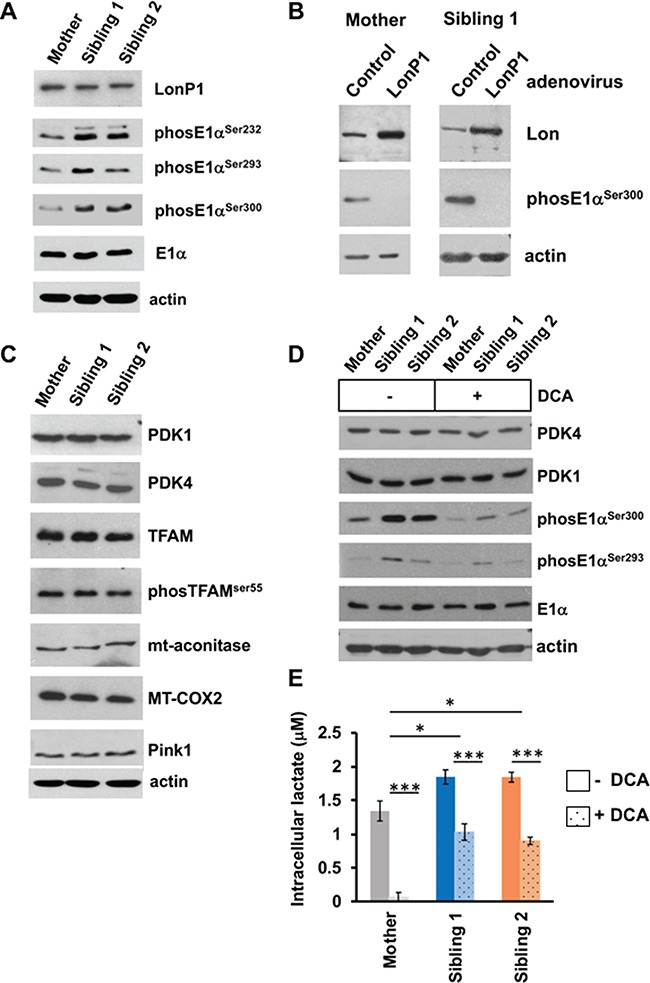
LonP1-P761L upregulates phosphorylated E1α in the affected Siblings’ fibroblasts. Immunoblotting of cell extracts (40 μg) from Mother and Sibling fibroblasts cultured in AMEM supplemented with 10% FBS. (A and C) Baseline protein levels in untreated cells. (B) Mother and Sibling 1 fibroblasts transduced with control (LacZ) or LonP1-WT using an adenovirus delivery system for 2 days. (D) Mother and Sibling fibroblasts treated with DCA (5 mm) for 12 h. (E) Lactate concentration in fibroblasts of Siblings and Mother, which had been cultured with and without DCA (5 mM) for 24 h.
Figure 6.
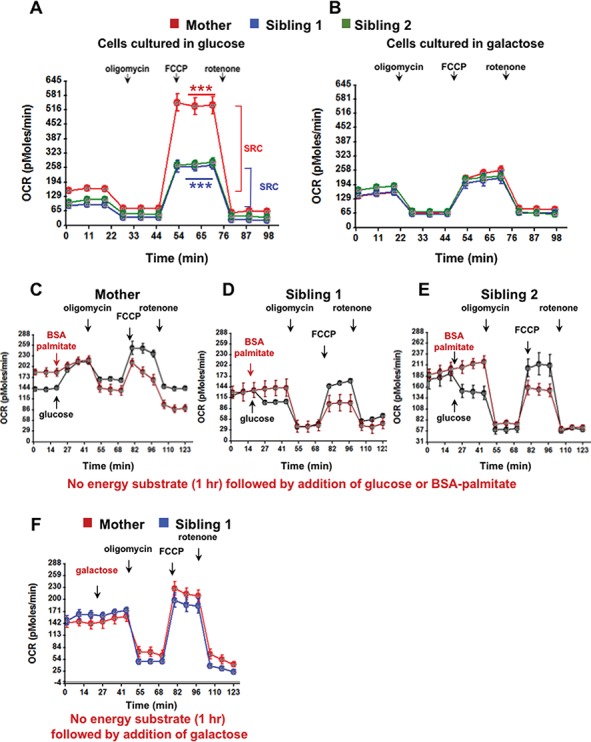
Mitochondrial bioenergetics. OCR of cultured fibroblasts (4 × 104 cells per well) from Sibling 1 (blue), Sibling 2 (green) and Mother (red) was measured using a Seahorse XF24 analyzer. (A and B) Baseline OCR was measured, after which the following reagents were sequentially injected into the wells—oligomycin (1 μM), FCCP (3 μM) and antimycin A + rotenone (1 μM each). In A, the significant difference in SRC between Siblings 1 and 2 (blue) and Mother (red) is denoted by the blue asterisks. **P < 0.001, ***P < 0.0001 determined by one-way ANOVA, followed by post-test. In B, no significant difference in SRC was observed. The data are representative of four independent experiments. (C–E) Cultured primary fibroblasts were glucose-deprived for 1 h at 37°C. Baseline OCR was measured, after which the following compounds were sequentially injected—glucose (10 mm) or BSA–palmitate (100 μM), oligomycin (1 μM), FCCP (3 μM) and rotenone (1 μM) as performed in A and B. The data are representative of two independent experiments. (F) Cultured primary fibroblasts were glucose-deprived for 1 h at 37°C. Baseline OCR was measured, after which the following compounds were sequentially injected—galactose (10 mm), oligomycin (1 μM), FCCP (3 μM) and rotenone (1 μM) as performed in A and B. The data are representative of two independent experiments.
We next examined OCR in acute response to different energetic substrates (Fig. 6C–E). Fibroblasts that had been in culture with Minimum Essential Medium Eagle - Alpha Modification (AMEM) medium (5.6-mm glucose) were cultured for 1 h in the absence of any carbon source, after which basal OCR was measured. When glucose (10 mm) was introduced into the wells containing the mother’s cells, OCR rapidly increased. In contrast, glucose strikingly suppressed OCR in fibroblasts from Sibling 1 and Sibling 2 (Fig. 6C, D and E). However, when bovine serum albumin (BSA)-conjugated palmitate was injected, OCR was not suppressed and was stimulated in fibroblasts of the mother and both siblings (Fig. 6C, D and E). The oxidation of palmitic acid in mitochondria produces acetyl CoA, NADH and FADH2, which directly fuels the TCA cycle and OXPHOS, thus bypassing glycolysis and pyruvate conversion to acetyl CoA. As with BSA–palmitate, galactose did not suppress OCR when added to fibroblasts of Sibling 1 and mother (Fig. 6F), in contrast to glucose, which down-regulated OCR in both Siblings 1 and 2 (Fig. 6D and E).
LonP1-P761L fibroblasts demonstrate PDH deficiency caused by increased levels of the phosphorylated PDH-E1α subunit
To determine whether the suppression of oxygen consumption by glucose in the siblings’ fibroblasts might be associated with altered PDH function, PDH activity was measured by the production of 14CO2 from [1-14C] pyruvate. Fibroblasts from both siblings showed reduced native PDH activity of 0.46 and 0.43 nmol/min/mg protein (normal range 0.7–2.5), which was 33% and 31% of the control value (1.40 nmol/min/mg protein), respectively. However, if the cells were treated with dichloroacetate (DCA), an inhibitor of PDH kinases (PDKs), Siblings 1 and 2 fibroblasts showed normal PDH activity of 1.03 and 1.07 nmol/min/mg protein, respectively (Table 1). Notably, although plasma lactate was normal, the fibroblasts from both Siblings 1 and 2 had elevated cellular lactate/pyruvate ratios of 38.16 and 36.12 nmol/h/mg protein, respectively (normal range 9–25 nmol/h/mg protein).
The PDH complex (PDC) consists of five enzymes: E1, pyruvate decarboxylase composed of α and β subunits (E1α and E1β); E2, dihydrolipoamide acetyltransferase; E3, dihydrolipoamide dehydrogenase; PDK; and PDH phosphatase (PDP). PDC is regulated by phosphorylation–dephosphorylation and is inhibited by phosphorylation of E1α at any one of three serine residues at position 232, 293 or 300 (p232, p293 or p300) (26). We suspected phosphoE1α was upregulated in the siblings’ fibroblasts, and indeed we observed increased p232, p293 and p300 in fibroblasts from Sibling 1 and increased p232 and p300 in Sibling 2 compared to their mother (Fig. 7A) (Supplementary Material, Fig. S6A shows all loading controls). There was no difference in total protein levels of E1α (Fig. 7A). To demonstrate that the accumulation of phosphoE1α was LonP1-dependent, we overexpressed wild-type LonP1 using an adenovirus delivery system and found that the level of p300 E1α was decreased in fibroblasts from both Sibling 1 and mother (Fig. 7B). Collectively, these results suggest that LonP1-P761L promotes PDH deficiency by up-regulating phosphoE1α.
As previous studies have shown that LonP1 mediates the proteolytic turnover of PDK4 (but not PDK1 or PDK2) in mouse heart mitochondria (5), we examined the steady state protein levels of PDKs, as well as other reported substrates of human LonP1. No difference in PDK1 or PDK4 was observed in fibroblasts from either sibling compared to their mother (Fig. 7C) (Supplementary Material, Fig. S6B shows all loading controls). We were not able to determine the levels of PDK2 and PDK3 due to poor antibody specificity. In addition, no differences in the protein levels of other endogenous substrates of human LonP1 were observed, specifically for phosphorylated TFAM (7), mitochondrial aconitase (4), COX subunit 1 (MT-CO1) (15) and PTEN-induced putative kinase (Pink1) (27) (Fig. 7C). Previous work has suggested that DCA inhibition of PDK4 induces its release from the PDC complex and promotes degradation by LonP1. However, we did not observe any change in the levels of either PDK4 or PDK1 when the cells were treated with DCA (5 mm) for 12 h (Fig. 7D). As expected, DCA treatment reduced the levels of phosphoE1α (p300 and p293) in both the mother’s and siblings’ cells. However, levels of phosphoE1α remained higher in both siblings’ cells compared to their mother even in the presence of DCA. There was no effect on the total cellular levels of E1α (Fig. 7D).
We next investigated whether PDH inhibition was responsible for the metabolic defects observed in the siblings’ fibroblasts. We found that restoration of PDH activity by DCA treatment normalized the elevated intracellular lactate levels in cultured fibroblasts from Siblings 1 and 2. In the absence of DCA, Siblings 1 and 2 had significantly higher intracellular lactate levels than their mother under the same conditions (P < 0.05) (Fig. 7E). In contrast, when the siblings’ fibroblasts were cultured with DCA (5 mm) for 24 h, these cells showed significantly reduced intracellular lactate levels (P < 0.001), which were lower than those measured in untreated mother’s cells without DCA (Fig. 7E).
Finally, to determine whether wild-type and mutant LonP1 directly degraded phosphorylated E1α, we used an in vitro system with recombinant LonP1-WT and mutant proteins, LonP1-P761L and Walker Box B mutant LonP1-E591A (Fig. 8). Unassembled E1α (i.e. not assembled with the E1β subunit) was phosphorylated in vitro using recombinant PDK4 (Supplementary Material, Fig. S6). PhosphoE1α was incubated with LonP1-WT, LonP1-P761L or LonP1-E591A. During the 60-min incubation period, LonP1-P761L failed to degrade phosphoE1α, in contrast to LonP1-WT (Fig. 8A). LonP1-P761L showed impaired degradation equivalent to that observed for LonP1-E591A, which is a defective ATPase and protease (Fig. 4E and Supplementary Material, Fig. S2). We observed that LonP1-P761L also failed to degrade unphosphorylated and unassembled E1α, in contrast to the wild-type protease (Fig. 8B). These results provide strong evidence that LonP1-P761L contributes to PDH deficiency in Siblings 1 and 2 by failing to degrade both phosphoE1α and unphosphorylated, unassembled E1α. Our findings suggest that LonP1 directly mediates the regulation of PDH activity and lactate metabolism and that this is disrupted by LonP1-P761L. This dysregulation of PDH function and lactate metabolism may contribute to the neurologic impairment observed in the patients.
Figure 8.
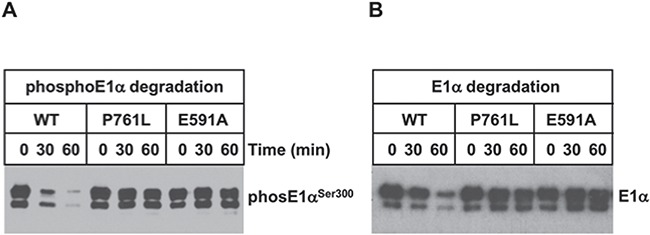
LonP1-P761L fails to degrade E1α and phosphoE1α compared to LonP1-WT. LonP1 -WT, -P761L or E591A (0.5 μM) were incubated with (A) phosphorylated E1α or (B) E1α (1 μM) in reaction buffer (50-mm HEPES KOH pH 8.0, 150-mm NaCl, 10-mm MgCl2) with ATP (4 mm) at 37°C and aliquots were removed at the times as indicated and immunoblotted.
Discussion
We report two siblings, born to a healthy consanguineous couple of Afghani descent, with profound hypotonia and muscle weakness, severe intellectual disability, developmental regression and progressive cerebellar atrophy associated with pathogenic variants in LONP1 identified by whole-exome sequencing. The degree of profound neurological involvement of these siblings is in striking contrast to patients with bi-allelic LONP1 variants associated with CODAS syndrome who have neurological involvement ranging from normal development and intelligence to moderate developmental delay, with nearly all attaining the ability to walk without support (14–19). In addition, the siblings we describe do not manifest the cardinal symptoms of CODAS syndrome (14,15).
Similar clinical features as those we report were described in an individual with bilateral congenital cataracts and progressive atrophy of the cerebellar cortex and caudate, who lacked the typical features of CODAS syndrome (20). The individual had compound heterozygous frameshift and missense LONP1 mutations (c.296dup/c.2356C > T, p.Ser100Glnfs*46/p.Arg786Trp) (20). This frameshift predicts a short, truncated protein that is unlikely to assemble with the protease domain of mutant protein LonP1-Arg786Trp (Table 1). The majority of pathogenic LONP1 variants associated with CODAS syndrome clusters in its AAA+ domain (14,15), which has been shown to mediate the unfolding and stepwise translocation of bound substrate into the proteolytic chamber (28). Our functional data suggest a genotype–phenotype correlation with mutations in the protease domain of LonP1 causing severe neurological impairment.
We demonstrate that the affected siblings’ fibroblasts have decreased PDH activity caused by increased phosphorylation of E1α as a result of LonP1-P761L (Table 1 and Fig. 7A and D), which can be normalized by DCA, a pan-inhibitor of PDKs (Table 1 and Fig. 7A and D). DCA treatment decreased the levels of E1α p232, p293 and p300 in Sibling 1 and decreased the levels of p232 and p300 in Sibling 2 (Fig. 7D). We also showed that DCA treatment of both siblings’ fibroblasts significantly reduced the elevated intracellular lactate levels (Fig. 7E), demonstrating that the phosphorylation-dependent activity of PDH regulates intracellular lactate concentration. DCA-activated PDH activity has also been reported in individuals with PDP 1 (PDP1) deficiency (MIM 608782) (29), which is caused by mutations in PDP1. However, PDP1-deficient individuals exhibit transient lactic acidosis and a lactate doublet present on brain MRS, a variable developmental delay without neurodegeneration (29,30), which are distinctly different symptoms than those of the patients described here. Our findings further show that LonP1 overexpression down-regulated the level of phosphoE1α (Fig. 7B). Moreover, purified recombinant LonP1-WT degrades phosphoE1α, as well as unphosphorylated E1α, whereas LonP1-P761L does not (Fig. 7A and B), thus establishing the E1α subunit of PDH as a proteolytic substrate of LonP1. These data demonstrate that LonP1 is directly associated with regulation of PDH activity and lactate metabolism.
Our in vitro studies show that the LonP1-P761L mutant exhibits substrate-specific defects in its proteolytic activity. Purified LonP1-P761L does not degrade phosphorylated E1α or unphosphorylated and unassembled E1α (Fig. 8), although it is able to degrade TFAM (Fig. 4E) and β-casein (Supplementary Material, Fig. S2), albeit with lower efficiency than LonP1-WT. This suggests that LonP1-P761L in vivo not only fails to degrade inhibitory phosphoE1α, but may also fail to mediate the quality control of unphosphorylated E1α in its assembly with E1β and of the PDC, which includes PDKs and PDPs. We speculate that LonP1-P761L may have different pathophysiological effects in different cell types and tissues depending upon the protein targets and regulators of these targets, which are normally degraded by fully functional LonP1-WT. For example, it is possible that in neurons and muscle, LonP1-P761L dysfunction promotes reduced Complex IV activity and may also have global and tissue-specific effects on protein quality control and regulation of anabolic and catabolic pathways.
Progressive neurodegeneration and systemic lactic acidosis are the most common clinical features of PDH deficiency disorders (31). One study reported PDH-deficient individuals with profound neurologic impairment and minimal systemic acidosis; however, cerebral lactic acidosis was observed (32). PDH is a central gatekeeper of glucose oxidation, converting glycolytically-produced pyruvate to acetyl CoA, which fuels mitochondrial ATP synthesis. Neuronal function depends almost exclusively on energy derived from glucose oxidation, which is dependent upon the conversion of pyruvate to acetyl CoA by PDH. It is possible that PDH deficiency in patients with the LonP1-P761L mutation contributes significantly to their severe neurologic impairment. PDH deficiency is associated with mutations in the genes encoding proteins within the PDC, such as the catalytic E1α and E1β subunits, the E3 binding protein and the PDP1 phosphatase (31). We propose that pathogenic LONP1 variants encoding mutant proteins such as LonP1-P761L, which fail to degrade phosphoE1α, represent a new class of genetic mutations causing PDH deficiency.
In non-neuronal cells that are metabolically more flexible, it is possible that the siblings’ cells utilize energetic substrates and pathways other than those required for glucose oxidation, such as fatty acid oxidation or the anaplerotic conversion of pyruvate and aspartate to oxaloacetate or glutamate to α-ketoglutarate. Our results support this hypothesis as the siblings’ cells show stimulation of OXPHOS by the fatty acid BSA–palmitate and also show normal pyruvate carboxylase (PC) activity, which is required to convert pyruvate to acetyl CoA (Table 1). As galactose can be used as a carbohydrate source for oxidative metabolism by the siblings’ cells with comparable efficiency as that of their mother, this suggests that there is little or no defect in pyruvate entry into the mitochondrial matrix. Indeed, fibroblasts from patients with relatively mild forms of PDH deficiency have been shown to survive on galactose-containing medium (33). However, in these PDH-deficient cells, normal cellular lactate:pyruvate ratios are observed (33), which is in contrast to elevated lactate:pyruvate ratios observed in the affected siblings’ fibroblasts (Table 1). Further studies are required to elucidate the specific mechanisms underlying glucose suppression of mitochondrial oxygen consumption, which we observed in the fibroblasts from both affected siblings.
In summary, we report the identification of a novel homozygous missense mutation in LONP1, c.2282 C > T, (p.Pro761Leu), which was identified in two siblings born to a healthy consanguineous couple of Afghani descent. Both children lacked the key clinical features of CODAS syndrome or classical mitochondrial disease but instead showed stepwise regression during infancy followed by progressive and profound neurologic impairment. Skin fibroblasts from both siblings demonstrated increased lactate:pyruvate ratio, increased phosphorylation of PDH E1α, decreased PDH activity and glucose-repressed oxygen consumption compared to their clinically normal heterozygous mother. In vitro studies revealed that the recombinant LonP1-P761L mutant protein failed to degrade phosphoE1α, in contrast to wild-type LonP1, which rapidly degrades this substrate. These data clearly identify the E1α subunit of PDH as a new LonP1 substrate and demonstrate that defective degradation of phosphoE1α by LonP1-P761L dysregulates PDH, a novel mechanism for the pathogenesis of PDH deficiency leading to severe neurologic impairment and neurodegeneration.
Materials and Methods
Subjects
The parents provided written informed consent for study participation for themselves and their children, as well as for publication of clinical information, molecular findings and photographs. This study was approved by the Research Ethics Board of the Hospital for Sick Children (REB #1000009004).
Molecular studies
Whole-exome sequencing was performed at The Centre for Applied Genomics (Toronto, Canada) using genomic DNA extracted from whole blood. The affected Siblings’ DNA was sequenced to a mean depth of coverage of 71× and 81×, with >90% of targeted bases reaching at least 20× coverage. Paired end sequencing (2 × 100 bp) was performed using the Illumina HiSeq2500 following enrichment with the Agilent SureSelect Human All Exon V4 kit. After base calling with CASAVA v1.8.2, reads were mapped to the hg19 reference sequence using the BWAbacktrack algorithm from BWA v0.5.9. Duplicate reads were removed using MarkDuplicates from Picard v1.79. Local read realignment around indels, base quality score recalibration and variant calling with UnifiedGenotyper were performed using GATK v1.128. Indel calls were discarded if they overlapped repeat masked regions and hard filtered by variant call annotations QualByDepth (QD < 10.0), ReadPosRankSumTest (ReadPosRankSum < 20.0) and Strand Bias (SB > 0.01). Resulting variant calls were annotated with population frequency information, variant effect on protein and in silico prediction programs using a custom pipeline based on ANNOVAR. The variant LONP1 c.2282 C > T, (p.Pro761Leu), was submitted to the LOVD database and the data can be accessed using the URL https://databases.lovd.nl/shared/individuals/00163815.
Determination of mtDNA copy number
Genomic DNA (100 ng) from cultured skin fibroblasts was used for amplification of both the mtDNA-encoded MT-CYB target gene and the nuclear-DNA–encoded APP reference control gene. Genomic DNA was amplified in reactions (20 μl; Applied Biosystems; universal polymerase chain reaction (PCR) master mix) with the following Taqman primers (160 nM). Forward: APP, 5′-TTTTTGTGTGCTCTCCCAGGTCT-3′; and MT-CYB, 5′-GCCTGCCTGATCCTCCAAAT-3′. Reverse: APP 5′-TGGTCACTGGTTGGTTGGC-3′; and MT-CYB, 5′-AAGGTAGCGGATGATTCAGCC-3′. TaqMan probes: APP 5′-[6FAM]CCCTGAACTGCAGATCACCAATGTGGTAG[TAM]-3′; and MT-CYB, 5′-[6FAM]CACCAGACGCCTCAACCGCCTT [TAM]-3′. An AB 7500 RT-PCR system (Applied Biosystems) and standard quantitative PCR (qPCR) conditions were employed at 95°C for 10 min, 40 cycles at 95°C for 15 s and 60°C for 1 min. All reactions were performed in triplicate. Relative quantitation of the mtDNA copy number was calculated by the ΔΔCt method.
Determination of mitochondrial RNA transcript levels
Cultured fibroblasts were harvested and centrifuged at 3000 rpm for 5 min at room temperature. Total RNA was isolated (RNeasy kit) as described in the manufacturer’s protocol for both cultured cells and tissues. RNA (100–500 ng) was converted into cDNA by using high-capacity cDNA reverse transcription kit (Applied Biosystems). cDNA (50 ng) was used in triplicate for relative quantification using TaqMan gene expression assays for the gene products as indicated. HPRT was used as an endogenous control. The assay was performed in Bio-Rad CFX96 real-time PCR machine. The relative quantification of the expression was calculated using a ΔΔCt method by the Bio-Rad CFX96 software.
Molecular modeling
A homology model of the human mitochondrial LonP1 hexamer was generated using the crystal structure of Bacillus subtilis Lon (PDB 3M6A) as a template (10). Briefly, we generated a homology model of a single LonP1 subunit that lacks its N-terminal MTS (Fig. 4C), using three independent molecular modeling software tools: ‘Prime’ (Schrodinger Inc., NY), Modellar (salilab.org) and I-TASSER online server at University of Michigan (http://zhanglab.ccmb.med.umich.edu/I-TASSER/). The final model of the LonP1 subunit was obtained by averaging three models. The LonP1 hexamer was obtained by superimposing the structure of one subunit onto the crystal structure of B. subtilis Lon (PDB 3M6A) as a guide.
Lon purification
Wild-type and mutant LonP1 lacking their predicted MTSs were fused to an N-terminal hexa-histidine affinity tag were engineered using PCR-based QuikChange® Site-Directed Mutagenesis Kit and cloned into ProEx bacterial expression vector. The proteins were expressed in Rosetta 2 Escherichia coli and purified by immobilized metal affinity chromatography using a nickel agarose as previously described (24).
ATPase assay
LonP1 protein (400 nM) was incubated in reaction buffer (50-mm HEPES KOH pH 7.5, 10-mm MgOAC2, 0.1-mg/ml BSA). The reactions were initiated by the addition of phosphoenolpyruvate (3 mm), NADH (300 μM), pyruvate kinase/lactate dehydrogenase (12–20 U/ml) and ATP (0–400 μM). The absorbance at 340 nM was monitored for 5 min at room temperature to obtain reaction velocities. Data were analyzed by Michaelis–Menten curve fitting using GraphPad Prism 5.
In vitro assays with recombinant wild-type and mutant LonP1, TFAM and E1α
LonP1 protein (0.5 or 1 μM) was incubated with protein substrate (1 or 3 μM) in reaction buffer (50 mm HEPES KOH pH 8.0, 150 mm NaCl, 10 mm MgCl2) with or without ATP (4 mm). Reactions were incubated at 37°C, and aliquots were removed at the time period as indicated and added to Laemmli reducing sample buffer (1× final concentration) and immunoblotted with substrate-specific antibodies as specified and visualized by autoradiography or ChemFluor imaging (ThermoFisher, Waltham, MA). Fluorescently labeled casein was visualized by SDS-PAGE and Typhoon phosphorimaging (GE Healthcare Life Sciences, Marlborough, MA). Phosphorylated E1α was produced by incubating E1α (1.6 μM) and PDK4 (1.6 μM) in buffer (50-mm HEPES KOH pH 8.0, 150-mm NaCl, 10-mm MgCl2) with ATP (250 μM) at 30°C for 2 h.
Bioenergetic studies
Fibroblasts were cultured to 80% confluency in 10-cm plates in AMEM (containing 5.6-mm glucose) or DMEM (containing 25-mm glucose or 24-mm galactose) supplemented with 10% FBS as indicated. The cells were trypsinized and plated (4 × 104 cells) in Seahorse XF-24 microplates and incubated overnight at 37°C (5% CO2). On the following day before the assay, the medium was replaced with unbuffered DMEM Base (8.3 g/L, pH 7.4) supplemented with or without glucose or galactose and incubated for 1 h at 37°C (without CO2).
Basal OCR and ECAR were measured, followed by the sequential addition of the following: (1) oligomycin (1 μM final), a Complex V ATP synthase inhibitor used to measure the ATP-dependent oxygen consumption; (2) FCCP (3 μM final), an uncoupler carbonyl cyanide 4-trifluoromethoxyphenylhydrazone used to measure maximum respiratory capacity; and (3) antimycin A plus rotenone, which are Complex I and III inhibitors, respectively (each 1 μM final). The calculation of basal OCR, % SRC, proton leak, coupling efficiency, non-mitochondrial respiration and ECAR were carried out using Mito Stress Report generator (Seahorse, Agilent technologies, Billerica, MA). Experiments measuring OCR and ECAR in response to glucose (10 mM) and BSA–palmitate (100 μM) were performed using cells cultured in AMEM (containing 5.6-mm glucose), which had been starved for 1 h without carbohydrate addition. After baseline OCR/ECAR measurements, glucose (10 mm) or BSA–palmitate (100 μM) were injected into each well, followed by the addition of oligomycin, FCCP and rotenone (at concentrations as indicated above). The SRC was calculated by subtracting the basal respiration OCR from the maximal respiration OCR. The statistical significance was calculated by one-way analysis of variance (ANOVA), and P < 0.001 was considered as significant.
PDC and PC activity analysis
PDC and PC activities in fibroblasts from Siblings and normal (non-Mother) controls were measured using procedures established by the Mitochondrial Disease Laboratory, Department of Paediatric Laboratory Medicine at the Hospital for Sick Children, Toronto, CA. Briefly, PDH activity was measured by [1- 14C]-pyruvate decarboxylation as described previously (34), and PC activity was measured using 1-14C-sodium bicarbonate in a protocol adapted from Utter et al. (35).
OXPHOS complex activity analysis
The activities of OXPHOS complexes—Complexes I and III (combined), II and III (combined), IV and V—were measured in mitochondria isolated from Siblings 1 and 2 and normal (non-Mother) fibroblasts; Complex II and III (combined) and Complex IV activities were also measured in extracts from Siblings 1 and 2 and normal (non-Mother) fibroblasts, using protocols established in the Mitochondrial Disease Laboratory, Department of Paediatric Laboratory Medicine at the Hospital for Sick Children, Toronto, CA.
Relative Complex IV activity and protein quantification in cultured primary fibroblasts from the Mother and Siblings were determined using COX kinetic and dipstick assays (Abcam ab109909 and ab109876, respectively). In the kinetic assay, COX activity was measured as the change in substrate absorbance following the procedure recommended by the manufacturer. Briefly, proteins extracted from the primary fibroblasts were incubated in a 96 well plate coated with an anti-Complex IV–specific antibody, followed by washing to remove non-specific proteins. COX activity was determined by following the oxidation of reduced cytochrome c, which was measured as a decrease in absorbance at 550 nm every 1 min for 2 h. Absorbance versus time was plotted to obtain the kinetic graph. In the dipstick assay, protein extracts (50 μg) from Sibling 2 and Mother control fibroblasts were employed following the manufacturer’s procedures. Signal intensities were determined by densitometric quantification.
The relative protein levels of Complex IV were quantified using Complex IV Human Protein Quantity Dipstick Assay Kit (Abcam ab109877), following the manufacturer’s protocol. Protein extracts (20 μg) from the cultured primary fibroblasts were employed, and the signal intensity was determined by densitometric analysis.
Immunoblotting
Cultured primary fibroblasts were grown to 80–90% confluency, and cellular proteins extracted with lysis buffer (0.5% Triton X-100, 50-mm Tris, pH 7.5, 300-mm NaCl) containing 1X Halt protease and phosphatase cocktail inhibitor (Thermo Fisher). Protein extracts (40 μg) were immunoblotted with antibodies recognizing LonP1 (custom-produced) (36), phosphorylated E1α p232, p293 and p300 (Millipore AP1063, AP1062, AP1064, respectively); PDH (Millipore, ABS2082); PDK1 (Abcam, ab207450), PDK4 (Abcam, ab71240), MT-COX2 (Abcam, ab91317), Pink1 (Abcam, ab23707); TFAM (a generous gift from Dr Daniel Bogenhagen) and phosphoTFAMser55 [custom-made by Genscript (7)] and mt-aconitase (a generous gift from Dr Luke Szweda).
Adenovirus transduction
Mother and Sibling 1 fibroblasts were seeded with complete AMEM containing 10% FBS and were 70% confluent the day before adenovirus transduction. After 24 h, cells were transduced with Lacz (control) or LonP1 adenovirus with reduced FBS (5%) for 24 h, followed by replacing the transduction medium with the complete AMEM. After 2 days of transduction, cells were harvested and proteins were extracted for immunoblot analysis.
Intracellular lactate measurements
Intracellular lactate concentrations were determined using Lactate-Glo™ Assay (Promega J5021, Madison, WI), following the manufacturer’s instructions. In this luciferase-linked assay, the amount of luciferin produced is proportional to the amount of lactate in the sample. Briefly, fibroblasts from Mother and Siblings 1 and 2 were cultured in the presence or absence of DCA (5 mm) in 384 well plates. Luciferin production was detected in a luciferase reaction, and light emission was measured. Lactate standard curves were used to calculate the lactate concentration in the respective samples.
Supplementary Material
Acknowledgements
We would like to express our gratitude to our patients and their family for their generosity and for participating in this study. We also thank The Centre for Applied Genomics (Toronto) for technical expertise.
Conflict of Interest statement. None declared.
Funding
This work was supported by the University of Toronto McLaughlin Centre: Next Generation Diagnostics: Advancing the Clinical Application of Genomic Analysis (grant ID MC-2012-13A to G.Y.) and funded in part by National Institutes of Health (1R21HD008391601) and a generous gift from M. Maholtra to C.K.S.
References
- 1. Suzuki C.K., Suda K., Wang N. and Schatz G. (1994) Requirement for the yeast gene LON in intramitochondrial proteolysis and maintenance of respiration. Science, 264, 891. [DOI] [PubMed] [Google Scholar]
- 2. Quiros P.M., Espanol Y., Acin-Perez R., Rodriguez F., Barcena C., Watanabe K., Calvo E., Loureiro M., Fernandez-Garcia M.S., Fueyo A. et al. (2014) ATP-dependent Lon protease controls tumor bioenergetics by reprogramming mitochondrial activity. Cell Rep., 8, 542–556. [DOI] [PubMed] [Google Scholar]
- 3. Dyck L., Neupert W. and Langer T. (1998) The ATP-dependent PIM1 protease is required for the expression of intron-containing genes in mitochondria. Genes Dev., 12, 1515–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bota D.A. and Davies K.J. (2002) Lon protease preferentially degrades oxidized mitochondrial aconitase by an ATP-stimulated mechanism. Nat. Cell Biol., 4, 674–680. [DOI] [PubMed] [Google Scholar]
- 5. Crewe C., Schafer C., Lee I., Kinter M. and Szweda L.I. (2017) Regulation of pyruvate dehydrogenase kinase 4 in the heart through degradation by the Lon protease in response to mitochondrial substrate availability. J. Biol. Chem., 292, 305–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Granot Z., Kobiler O., Melamed-Book N., Eimerl S., Bahat A., Lu B., Braun S., Maurizi M.R., Suzuki C.K., Oppenheim A.B. et al. (2007) Turnover of mitochondrial steroidogenic acute regulatory (StAR) protein by Lon protease: the unexpected effect of proteasome inhibitors. Mol. Endocrinol., 21, 2164–2177. [DOI] [PubMed] [Google Scholar]
- 7. Lu B., Lee J., Nie X., Li M., Morozov Y.I., Venkatesh S., Bogenhagen D.F., Temiakov D. and Suzuki C.K. (2013) Phosphorylation of human TFAM in mitochondria impairs DNA binding and promotes degradation by the AAA+ Lon protease. Mol. Cell, 49, 121–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ngo J.K., Pomatto L.C., Bota D.A., Koop A.L. and Davies K.J. (2011) Impairment of lon-induced protection against the accumulation of oxidized proteins in senescent wi-38 fibroblasts. J. Gerontol. A. Biol. Sci. Med. Sci., 66, 1178–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tian Q., Li T., Hou W., Zheng J., Schrum L.W. and Bonkovsky H.L. (2011) Lon peptidase 1 (LONP1)-dependent breakdown of mitochondrial 5-aminolevulinic acid synthase protein by heme in human liver cells. J. Biol. Chem., 286, 26424–26430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Venkatesh S., Lee J., Singh K., Lee I. and Suzuki C.K. (2012) Multitasking in the mitochondrion by the ATP-dependent Lon protease. Biochim. Biophys. Acta, 1823, 56–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fukuda R., Zhang H., Kim J.W., Shimoda L., Dang C.V. and Semenza G.L. (2007) HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell, 129, 111–122. [DOI] [PubMed] [Google Scholar]
- 12. Hori O., Ichinoda F., Tamatani T., Yamaguchi A., Sato N., Ozawa K., Kitao Y., Miyazaki M., Harding H.P., Ron D. et al. (2002) Transmission of cell stress from endoplasmic reticulum to mitochondria: enhanced expression of Lon protease. J. Cell Biol., 157, 1151–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sepuri N.B.V., Angireddy R., Srinivasan S., Guha M., Spear J., Lu B., Anandatheerthavarada H.K., Suzuki C.K. and Avadhani N.G. (2017) Mitochondrial LON protease-dependent degradation of cytochrome c oxidase subunits under hypoxia and myocardial ischemia. Biochim. Biophys. Acta Bioenerg., 1858, 519–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dikoglu E., Alfaiz A., Gorna M., Bertola D., Chae J.H., Cho T.J., Derbent M., Alanay Y., Guran T., Kim O.H. et al. (2015) Mutations in LONP1, a mitochondrial matrix protease, cause CODAS syndrome. Am. J. Med. Genet. A, 167, 1501–1509. [DOI] [PubMed] [Google Scholar]
- 15. Strauss K.A., Jinks R.N., Puffenberger E.G., Venkatesh S., Singh K., Cheng I., Mikita N., Thilagavathi J., Lee J., Sarafianos S. et al. (2015) CODAS syndrome is associated with mutations of LONP1, encoding mitochondrial AAA+ Lon protease. Am. J. Hum. Genet., 96, 121–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shebib S.M., Reed M.H., Shuckett E.P., Cross H.G., Perry J.B. and Chudley A.E. (1991) Newly recognized syndrome of cerebral, ocular, dental, auricular, skeletal anomalies: CODAS syndrome—a case report. Am. J. Med. Genet., 40, 88–93. [DOI] [PubMed] [Google Scholar]
- 17. Almeida J.C., Vargas F.R., Barbosa-Neto J.G. and Llerena J.C. Jr. (1995) CODAS syndrome: a new distinct MCA/MR syndrome with radiological changes of spondyloepiphyseal dysplasia. Another case report. Am. J. Med. Genet., 55, 19–20. [DOI] [PubMed] [Google Scholar]
- 18. Innes A.M., Chudley A.E., Reed M.H., Shuckett E.P., Hildes-Ripstein G.E. and Greenberg C.R. (2001) Third case of cerebral, ocular, dental, auricular, skeletal anomalies (CODAS) syndrome, further delineating a new malformation syndrome: first report of an affected male and review of literature. Am. J. Med. Genet., 102, 44–47. [DOI] [PubMed] [Google Scholar]
- 19. Marlin S., Ducou Le Pointe H., Le Merrer M., Portnoi M.F., Chantot S., Jonard L., Mantel-Guiochon A., Siffroi J.P., Garabedian E.N. and Denoyelle F. (2010) Fourth case of cerebral, ocular, dental, auricular, skeletal syndrome (CODAS), description of new features and molecular analysis. Am. J. Med. Genet. A, 152A, 1510–1514. [DOI] [PubMed] [Google Scholar]
- 20. Inui T., Anzai M., Takezawa Y., Endo W., Kakisaka Y., Kikuchi A., Onuma A., Kure S., Nishino I., Ohba C. et al. (2017) A novel mutation in the proteolytic domain of LONP1 causes atypical CODAS syndrome. J. Hum. Genet., 62, 653–655. [DOI] [PubMed] [Google Scholar]
- 21. Peter B., Waddington C.L., Olahova M., Sommerville E.W., Hopton S., Pyle A., Champion M., Ohlson M., Siibak T., Chrzanowska-Lightowlers Z.M.A. et al. (2018) Defective mitochondrial protease LonP1 can cause classical mitochondrial disease. Hum. Mol. Genet., 27, 1743–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Iyer L.M., Leipe D.D., Koonin E.V. and Aravind L. (2004) Evolutionary history and higher order classification of AAA+ ATPases. J. Struct. Biol., 146, 11–31. [DOI] [PubMed] [Google Scholar]
- 23. Khan A.O. and AlBakri A. (2018) Clinical features of LONP1-related infantile cataract. J. AAPOS, 22, 229–231. [DOI] [PubMed] [Google Scholar]
- 24. Liu T., Lu B., Lee I., Ondrovicova G., Kutejova E. and Suzuki C.K. (2004) DNA and RNA binding by the mitochondrial lon protease is regulated by nucleotide and protein substrate. J. Biol. Chem., 279, 13902–13910. [DOI] [PubMed] [Google Scholar]
- 25. Brand M.D. and Nicholls D.G. (2011) Assessing mitochondrial dysfunction in cells. Biochem. J., 435, 297–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rardin M.J., Wiley S.E., Naviaux R.K., Murphy A.N. and Dixon J.E. (2009) Monitoring phosphorylation of the pyruvate dehydrogenase complex. Anal. Biochem., 389, 157–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jin S.M. and Youle R.J. (2013) The accumulation of misfolded proteins in the mitochondrial matrix is sensed by PINK1 to induce PARK2/Parkin-mediated mitophagy of polarized mitochondria. Autophagy, 9, 1750–1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sauer R.T. and Baker T.A. (2011) AAA+ proteases: ATP-fueled machines of protein destruction. Annu. Rev. Biochem., 80, 587–612. [DOI] [PubMed] [Google Scholar]
- 29. Maj M.C., MacKay N., Levandovskiy V., Addis J., Baumgartner E.R., Baumgartner M.R., Robinson B.H. and Cameron J.M. (2005) Pyruvate dehydrogenase phosphatase deficiency: identification of the first mutation in two brothers and restoration of activity by protein complementation. J. Clin. Endocrinol. Metab., 90, 4101–4107. [DOI] [PubMed] [Google Scholar]
- 30. Cameron J.M., Maj M., Levandovskiy V., Barnett C.P., Blaser S., Mackay N., Raiman J., Feigenbaum A., Schulze A. and Robinson B.H. (2009) Pyruvate dehydrogenase phosphatase 1 (PDP1) null mutation produces a lethal infantile phenotype. Hum. Genet., 125, 319–326. [DOI] [PubMed] [Google Scholar]
- 31. Patel K.P., O'Brien T.W., Subramony S.H., Shuster J. and Stacpoole P.W. (2012) The spectrum of pyruvate dehydrogenase complex deficiency: clinical, biochemical and genetic features in 371 patients. Mol. Genet. Metab., 106, 385–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Brown G.K., Haan E.A., Kirby D.M., Scholem R.D., Wraith J.E., Rogers J.G. and Danks D.M. (1988) `Cerebral’ lactic acidosis: defects in pyruvate metabolism with profound brain damage and minimal systemic acidosis. Eur. J. Pediatr., 147, 10–14. [DOI] [PubMed] [Google Scholar]
- 33. Robinson B.H., Petrova-Benedict R., Buncic J.R. and Wallace D.C. (1992) Nonviability of cells with oxidative defects in galactose medium: a screening test for affected patient fibroblasts. Biochem. Med. Metab. Biol., 48, 122–126. [DOI] [PubMed] [Google Scholar]
- 34. Sheu K.F., Hu C.W. and Utter M.F. (1981) Pyruvate dehydrogenase complex activity in normal and deficient fibroblasts. J. Clin. Invest., 67, 1463–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Utter M.F. and Keech D.B. (1963) Pyruvate Carboxylase. I. Nature of the Reaction. J. Biol. Chem., 238, 2603–2608. [PubMed] [Google Scholar]
- 36. Lu B., Liu T., Crosby J.A., Thomas-Wohlever J., Lee I. and Suzuki C.K. (2003) The ATP-dependent Lon protease of Mus musculus is a DNA-binding protein that is functionally conserved between yeast and mammals. Gene, 306, 45–55. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.