

Abstract
Infantile hypertrophic pyloric stenosis (IHPS) is a disorder of young infants with a population incidence of ∼2/1000 live births, caused by hypertrophy of the pyloric sphincter smooth muscle. Reported genetic loci associated with IHPS explain only a minor proportion of IHPS risk. To identify new risk loci, we carried out a genome-wide meta-analysis on 1395 surgery-confirmed cases and 4438 controls, with replication in a set of 2427 cases and 2524 controls. We identified and replicated six independent genomic loci associated with IHPS risk at genome wide significance (P < 5 × 10−8), including novel associations with two single nucleotide polymorphisms (SNPs). One of these SNPs, rs6736913 [odds ratio (OR) = 2.32; P = 3.0 × 10−15], is a low frequency missense variant in EML4 at 2p21. The second SNP, rs1933683 (OR = 1.34; P = 3.1 × 10−9) is 1 kb downstream of BARX1 at 9q22.32, an essential gene for stomach formation in embryogenesis. Using the genome-wide complex trait analysis method, we estimated the IHPS SNP heritability to be 30%, and using the linkage disequilibrium score regression method, we found support for a previously reported genetic correlation of IHPS with lipid metabolism. By combining the largest collection of IHPS cases to date (3822 cases), with results generalized across populations of different ancestry, we elucidate novel mechanistic avenues of IHPS disease architecture.
Introduction
Infantile hypertrophic pyloric stenosis (IHPS), first described by the Danish pediatrician Dr Harald Hirschsprung in 1888 (1), is a serious condition of young infants caused by hypertrophy of the pyloric sphincter smooth muscle, which can lead to obstruction of the gastric outlet and consequent projectile vomiting. The only definitive treatment is pyloromyotomy, a surgical procedure first described by Ramstedt in 1912 (2), in which an incision of the pyloric muscle is made to relieve its constriction and allow the contents of the stomach to pass to the duodenum. IHPS has an incidence of 2 to 5 per 1000 live births among individuals of European ancestry, with a 4- to 5-fold higher risk in boys than in girls (3). Several studies have suggested that exposure to medications and perinatal factors may be associated with the development of IHPS, such as use of macrolide antibiotics (4), being first-born, delivery by cesarean section, preterm birth and bottle feeding (5). Moreover, sharp changes in reported incidence of IHPS in Europe (6) emphasize the importance of modifiable environmental exposures on the risk of IHPS. Nevertheless, its familial aggregation of 20-fold increased risk among siblings, heritability estimates of 87% (7) and co-occurrence with multiple genetic syndromes (8) suggest a strong genetic component for IHPS.
Two genome-wide association studies (GWAS) of IHPS have been published, both by our group, in which we identified four genome-wide significant risk loci near the muscleblind-like splicing regulator 1 gene, the NK2 homeobox 5 gene (NKX2-5) and the apolipoprotein A-I (APOA1) gene (9,10). However, these four loci only explain a modest proportion of IHPS heritability.
In the present study, we report the largest investigation to date of the influence of common and low frequency genetic variation on IHPS risk by doing a genome-wide meta-analysis of our previous GWAS cohort (9,10), with an additional 427 surgery-confirmed cases and 2031 controls, all from Denmark. The results were replicated in 1794 cases and 1875 controls of European ancestry and further confirmed in 633 cases and 633 controls from a Hispanic population.
Results
Study subjects
In order to identify IHPS susceptibility loci we did a genome-wide meta-analysis of two IHPS case-control cohorts, all of Danish ancestry, in which the IHPS cases were surgery-confirmed singletons without major congenital malformations (see Materials and Methods).
Genome-wide meta-analysis confirms four known loci and detects two additional independent loci
After applying quality control measures and performing HRC imputation (20), the meta-analysis included a total of 1395 cases and 4438 controls at 6 576 307 autosomal single nucleotide polymorphisms (SNPs) (MAF > 1%, r2 imputation info score ≥ 0.8). Our meta-analysis showed a genomic inflation of 1.10 (Supplementary Material, Fig. S1). However, the linkage disequilibrium (LD) score regression intercept was 1.02, suggesting that the majority of the inflation in test statistics was attributable to the polygenic architecture of IHPS, rather than population stratification or cryptic relatedness (25,36).
In the discovery phase analysis, we identified eight genome-wide significant loci (P < 5 × 10−8) (Fig. 1; Table 1; Supplementary Material, Table S1 and 2; Figs S2–9), and conditioning on the top SNP at each locus did not reveal secondary signals (Supplementary Material, Figs S2–9). No heterogeneity was found between the two IHPS discovery phase cohorts (Cochran’s Q test, P > 0.2; Table 1) for the genome-wide significant loci. Among the eight loci that were genome-wide significant, four on chromosomes 3q25.1, 3q25.2, 5q35.1 and 11q23.3 were previously associated with IHPS (9,10), whereas the remaining four on chromosomes 2p21, 3p14.3, 9q22.32 and 13q21.31 have not previously been reported (Table 1; Fig. 1; Supplementary Material, Tables S1 and 2; Figs S2–9).
Figure 1.
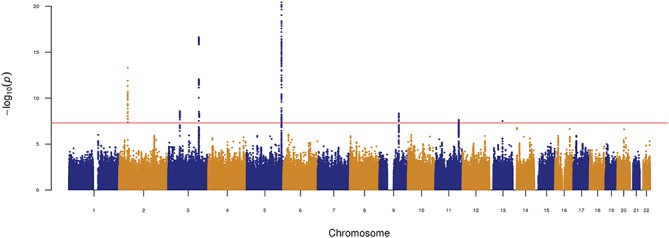
Manhattan plot of the IHPS genome-wide association meta-analysis in the discovery cohort (1395 cases and 4438 controls; Denmark).
Table 1.
Association results with IHPS in discovery (Denmark), replication (Sweden and US—non-Hispanic Whites) and combined analysis
| Alleles | Frequency | Number | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| SNP | Position | A1 | A2 | Cohort | Cases | Controls | Cases | Controls | OR (95% CI) | P | Q |
| rs6736913 | 2:42510018 | A | G | Discovery | 0.045 | 0.019 | 1395 | 4438 | 2.42 (2.17–2.68) | 4.79E-12 | 0.6215 |
| United States | 0.024 | 0.012 | 1671 | 1685 | 1.98 (1.59–2.37) | 5.82E-04 | 0.0532 | ||||
| Sweden | 0.031 | 0.006 | 113 | 177 | 5.78 (4.19–7.37) | 3.06E-02 | - | ||||
| Combined | 0.033 | 0.017 | 3179 | 6300 | 2.32 (2.11-2.53) | 3.00E-15 | 0.1992 | ||||
| rs7644486 | 3:57869963 | C | T | Discovery | 0.319 | 0.260 | 1395 | 4438 | 1.32 (1.23–1.41) | 5.34E-09 | 0.4966 |
| United States | 0.303 | 0.287 | 1677 | 1693 | 1.08 (0.98–1.19) | 1.44E-01 | 0.3867 | ||||
| Sweden | 0.292 | 0.297 | 113 | 177 | 0.98 (0.61–1.35) | 9.05E-01 | - | ||||
| Combined | 0.309 | 0.268 | 3185 | 6308 | 1.20 (1.13–1.27) | 1.83E-07 | 0.0370 | ||||
| rs11712066 | 3:151830309 | G | A | Discovery | 0.171 | 0.252 | 1395 | 4438 | 0.62 (0.52–0.73) | 3.73E-17 | 0.7899 |
| United States | 0.148 | 0.209 | 1671 | 1676 | 0.65 (0.53–0.78) | 8.29E-11 | 0.9453 | ||||
| Sweden | 0.161 | 0.231 | 112 | 175 | 0.62 (0.16–1.07) | 3.54E-02 | - | ||||
| Combined | 0.159 | 0.240 | 3178 | 6289 | 0.64 (0.55–0.72) | 2.57E-27 | 0.9840 | ||||
| rs573872 | 3:153472163 | G | T | Discovery | 0.280 | 0.225 | 1395 | 4438 | 1.33 (1.23–1.43) | 7.74E-09 | 0.2193 |
| United States | 0.241 | 0.203 | 1671 | 1684 | 1.24 (1.12–1.35) | 2.90E-04 | 0.0522 | ||||
| Sweden | 0.336 | 0.214 | 113 | 175 | 1.88 (1.49–2.26) | 1.42E-03 | - | ||||
| Combined | 0.261 | 0.219 | 3179 | 6297 | 1.31 (1.24–1.38) | 4.96E-13 | 0.0455 | ||||
| rs6556059 | 5:172645766 | T | C | Discovery | 0.452 | 0.355 | 1395 | 4438 | 1.50 (1.41–1.59) | 4.24E-19 | 0.5220 |
| United States | 0.426 | 0.343 | 1667 | 1680 | 1.42 (1.32–1.52) | 4.27E-12 | 0.9360 | ||||
| Sweden | 0.474 | 0.334 | 113 | 175 | 1.79 (1.44–2.14) | 1.10E-03 | - | ||||
| Combined | 0.439 | 0.351 | 3175 | 6293 | 1.48 (1.41–1.54) | 1.49E-31 | 0.6910 | ||||
| rs1933683 | 9:96712771 | G | C | Discovery | 0.131 | 0.094 | 1395 | 4438 | 1.50 (1.36–1.64) | 4.58E-09 | 0.8138 |
| United States | 0.139 | 0.120 | 1671 | 1685 | 1.22 (1.10–1.34) | 1.17E-03 | 0.5543 | ||||
| Sweden | 0.170 | 0.127 | 112 | 177 | 1.40 (0.93–1.87) | 1.58E-01 | - | ||||
| Combined | 0.136 | 0.102 | 3178 | 6300 | 1.34 (1.24–1.43) | 3.05E-09 | 0.1920 | ||||
| rs12721025 | 11:116706047 | A | G | Discovery | 0.087 | 0.058 | 1395 | 4438 | 1.58 (1.42–1.74) | 4.12E-08 | 0.8483 |
| United States | 0.087 | 0.069 | 1667 | 1671 | 1.27 (1.09–1.45) | 9.39E-03 | 0.0318 | ||||
| Sweden | 0.097 | 0.051 | 113 | 175 | 2.03 (1.37–2.69) | 3.56E-02 | - | ||||
| Combined | 0.087 | 0.061 | 3175 | 6284 | 1.45 (1.33–1.57) | 1.07E-09 | 0.0691 | ||||
SNP, dbSNP v146 variant ID; Position, hg19 human genome assembly; A1, effect allele; A2, alternative allele; Frequency, effect allele frequency; odds ratio [OR; 95% confidence interval (CI)], OR of the effect allele (95% CI); P, fixed-effects meta-analysis P-value; Q, P-value for Cochrane's Q statistic (measure of heterogeneity). All specimens are of European ancestry.
At the known locus on 5q35.1 near NKX2-5, we identified a more significantly associated SNP (Supplementary Material, Table S2). While the previously reported top SNP rs29784, downstream of BNIP1, had a meta-analysis P = 1.37 × 10−15, this study’s top SNP rs9313619, intergenic, has a meta-analysis P = 3.80 × 10−21. Moreover, rs9313619 remained genome-wide significant when conditioning on rs29784 (conditional P = 3.50 × 10−8), while if conditioning on rs9313619, the previously reported top SNP rs29784 remained only nominally significant (P = 0.02). Of note, rs9313619 is in a repeat region with significant homology to other regions of the genome, and we, therefore, selected the SNP rs6556059 (r2 = 0.92 with rs9313619) for replication phase genotyping. The SNP rs6556059 is an expression quantitative trait loci (eQTL) for NKX2-5 in skin and heart (atrial appendage), as assessed in adult tissues from the Genotype-Tissue Expression (GTEx) Consortium project (35), with the IHPS risk allele associated with increased expression of NKX2-5. Conditional analyses at 3q25.1, 3q25.2 and 11q23.3 suggest that the previously reported top SNPs remain as such in this study.
Two new IHPS loci on chromosomes 2p21 and 9q22.32 were confirmed in the replication phase analyses, while the loci on 3p14.3 and 13q21.31 were not (Table 1; Supplementary Material, Tables S1 and 2). The new IHPS locus with the strongest effect size was located on chromosome 2p21. Its top SNP rs34216221, intronic in MTA3, had an odds ratio (OR) of 2.53 (P = 4.97 × 10−14, Fig. 2A) in our discovery phase meta-analysis and a risk allele frequency of 0.01 in the 1000 Genomes European population (37). The GWAS significant SNPs on this locus span the genes EML4, COX7A2L, KCNG3, MTA3, OXER1 and HAAO, of which EML4 and MTA3 show the highest levels of Histone H3 tri-methyl K36 (H3K36me3) marks in fetal and adult stomach and small intestine (Supplementary Material, Fig. S10), which are marks known to be associated with actively transcribed regions (33). If the top choice variant for replication was in a repeat region or had non-uniquely mapped flanking sequence to the human genome, we chose an alternative variant with high LD (r2 > 0.8) with the top one (see Materials and Methods). In meta-analyzed replication results, we genotyped the SNP rs6736913, a missense variant in the EML4 gene on chromosome 2p21 (NM_019063.4:c.847G>A; NP_061936.2:p.Glu283Lys), and observed an OR for IHPS of 2.10 (P = 1.15 × 10−4) (Table 1; Supplementary Material, Table S1) . When the results from the discovery and replication phases were meta-analyzed, the summary OR for IHPS was 2.32 (P = 3.00 × 10−15) for rs6736913 on 2p21 EML4-MTA3 locus (Table 1; Supplementary Material, Table S1).
Figure 2.
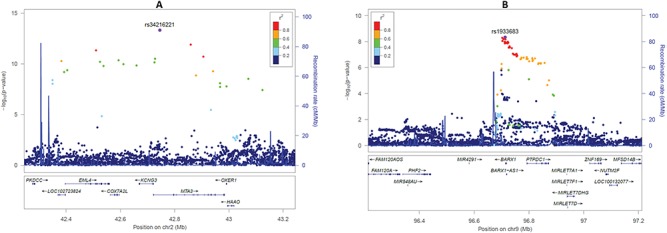
Regional association plots of the new genome-wide significant loci for IHPS. (A) Association results at the 2p21 EML4-MTA3 locus. (B) Association results at the 9q22.32 BARX1 locus. Color-coded LD is shown for the top SNP at each locus (LD determined with our discovery phase cohorts). The x-axis represents the genomic region (hg19 assembly) surrounding 1 Mb of the top SNP, while the y-axis represents the strength of the association in −log10(P-value).
The other novel IHPS locus was found on chromosome 9q22.32. Here, the lead SNP rs1933683, downstream of BARX1, had an OR of 1.50 (P = 4.58 × 10−9, Fig. 2B) in our discovery phase meta-analysis and a risk allele frequency of 0.13 in the 1000 Genomes European population (37). The GWAS significant SNPs at this locus cover the BARX1 (BARX homeobox 1) gene encoding a homeodomain transcription factor. Among the adult GTEx tissues (37), BARX1 is predominantly expressed in the stomach and the gastroesophageal junction (Supplementary Material, Fig. S11). In the developing fetus (34), BARX1 is highly enriched in stomach expression, when compared to the heart, intestine, kidney, lung and adrenal gland (Supplementary Material, Fig. S12). Moreover, BARX1 also has H3K36me3 marks in the fetal and adult stomach (Supplementary Material, Fig. S13). For this BARX1 locus, we genotyped the top SNP rs1933683 and observed an OR for IHPS of 1.19 (P = 1.20 × 10−2) in the replication phase (Table 1; Supplementary Material, Table S1). When the results from the discovery and replication phases were meta-analyzed, the summary OR for IHPS was 1.34 (P = 3.05 × 10−9) (Table 1; Supplementary Material, Table S1).
We were not able to create a functional assay on the 13q21.31 locus for the US replication phase cohorts. Moreover, its high missingness (0.03), non-significant P-value (P = 0.44) and opposite direction of effect in the Swedish replication cohort compared with the discovery phase cohorts (Supplementary Material, Table S1), allied to the unavailability of good proxies (high r2 and low P-value) for the top SNP, rs12870105, (Supplementary Material, Table S2; Fig. S9) precluded us from evaluating this signal further.
In our meta-analysis constrained to European ancestry samples, we applied a fixed-effects model assuming that the underlying allelic effect is the same in each population (Table 1; Supplementary Material, Table S1). Adding the Hispanic Whites, we may no longer assume that each population has the same underlying allelic effect. The trans-ethnic meta-analysis, where we allow a random effect to account for differences in allelic effect between Hispanic Whites and European ancestry samples, show that we have decreased power to detect effects for variants with substantial heterogeneity between the two ethnic groups, namely the variants at 3q25.1 and 5q35.1 (Supplementary Material, Table S1).
Genetic correlation
As a way to identify possible biomarker(s) associated with IHPS, we sought to determine the genetic correlation of the IHPS genome-wide meta-analysis results versus results for GWAS of circulating metabolites (38) by using the LD score regression method (25,26,30). We detected a nominally significant (P < 0.05) positive genetic correlation with high-density lipoprotein (HDL) cholesterol and APOA1, and a negative correlation with verylow-density lipoprotein cholesterol (Supplementary Material,Table S3). To address the possibility that these genetic correlations were mainly driven by the APOA gene cluster locus, we removed the IHPS association results from this locus (chr11:116.5–117.5 Mb, hg19 assembly). However, the results did not change significantly (Supplementary Material, Table S4). Although these findings were not statistically significant when corrected for the number of metabolites tested (N = 79), a link between lipid levels and risk of IHPS is consistent with previous results. More specifically, characteristics of the IHPS locus on chromosome 11q23.3 near APOA1 (10) suggest a functional mechanism related to cholesterol levels, and allied to our reported inverse relationship between levels of circulating total cholesterol in neonates and IHPS risk (10).
Heritability
In the US non-Hispanic Whites of European ancestry replication cohort, the six confirmed IHPS SNPs jointly explained 5.2% of the variance in liability to IHPS, with children in the top 5% of the polygenic risk score (PRS) distribution being at 8.45-fold increased risk compared with the bottom 5%. Using the LD score regression method (25,26) on our IHPS discovery phase genome-wide meta-analysis results, we estimated the IHPS-SNP heritability to be 30% ± standard error 6%. A similar SNP heritability estimate was determined with genome-wide complex trait analysis (GCTA) (25% ± 4%) (27).
Discussion
In what we believe is the largest genetic study of IHPS so far, we identified and replicated six independent genomic loci, out of which two, on chromosomes 2p21 and 9q22.32, are reported here for the first time (Table 1).
The new locus on chromosome 2p21 showed the strongest IHPS association with an OR = 2.32. Among the genes at the locus, EML4 (echinoderm microtubule-associated protein like-4) and MTA3 (metastasis associated 1 family member 3) are actively transcribed in the fetal and adult stomach. Mutations in both of these genes have been associated with tumor progression and metastasis in various cancers. Notably, knockdown of MTA3 expression has been associated with gastroesophageal junction cancer (39) and, therefore, one might speculate whether aberrant regulation of MTA3 could also cause congenital malformations of the esophagus. A higher reported incidence of hypertrophic pyloric stenosis in esophageal atresia patients (40) supports this hypothesis. Moreover, reports on the association between congenital malformations and increased cancer risk suggest a shared etiology (41–45).
The gene BARX1 on chromosome 9q22.32 encodes a homeobox transcription factor BARX1, which is specifically expressed in both the adult and fetal human stomach. The mouse homolog, Barx1, is essential to the formation of the stomach in the developing mouse embryo (46). The pyloric sphincter is absent in Barx1 null mice (47), while ectopic expression of Barx1 in the developing intestine induces smooth muscle of the stomach type (48). Our association signal at the BARX1 locus suggests that this homeobox transcription factor also plays an essential role in the development of the gastrointestinal tract in humans. Further support is found in familial syndromes involving duplications of the 9q22.1-9q31 region. This region harbors the BARX1 gene and these syndromes often include IHPS as part of the clinical presentation (49,50). This is a good example of a GWAS investigating isolated cases helping to identify the gene involved in a specific condition in a syndrome associated with a larger genomic region. As a way forward in the understanding of the pathophysiology of IHPS, GWAS can give robust results that are in line with observations from various syndromic cases (8).
NKX2-5 still remains the most obvious candidate causal gene at the 5q35.1 IHPS locus due to being an eQTL gene for the IHPS-associated SNPs and due to its causal role in the development of the pyloric sphincter (51–53).
A positive genetic correlation of IHPS with HDL cholesterol and APOA1, and a negative correlation with low-density lipoprotein (LDL) cholesterol were detected in this study, while our previous study (10) found that umbilical cord blood from newborns who later presented with IHPS had lower levels of total cholesterol compared with matched controls. Other observations from the literature suggest cholesterol as an important factor in IHPS risk. For example, children with Smith–Lemli–Opitz syndrome, an inborn defect of cholesterol biosynthesis, have markedly increased risk of IHPS (54,55). IHPS risk is also known to be associated with bottle feeding (7,56,57), and bottle feeding is associated with decreased total and LDL cholesterol levels in infancy (58). However, we should stress that our results do not demonstrate cholesterol as a causal risk factor for IHPS nor we can answer at what stage of development this putative risk is created. Also, our genetic correlation results must be interpreted with caution, since the lipid genetics studies on which the correlations are based on used adult participants, whereas relatively little is known about the genetic regulation of cholesterol levels in fetal life or in newborns. Nonetheless, further studies addressing the possible mechanistic influence of cholesterol levels on IHPS physiopathology are warranted. These studies could, for example, be focused on the role of cholesterol in nervous system development (59), since pyloric sphincter smooth muscle tissue from IHPS patients has been found to be deficiently innervated (60).
In this study, we estimated the heritability to be 30%. Even though the SNP heritability estimate is about one-third of a previous family based estimate of IHPS heritability (87%) on the same background population (7), it is within its 95% confidence interval (CI) in the model with a shared environmental component. Nevertheless, there may still be a genetic component of the disease that is not captured by the genotyped and well-imputed variants used in our meta-analysis. Whether IHPS genetic architecture is composed of common variants with small effect sizes not detected with our current sample size, and/or rare variants not captured by genotyped arrays used, remains to be answered (61).
This genetic study included the largest sample of IHPS cases to date, with cohorts from three European ancestry populations and one from Hispanics, amounting to a total of 3708 cases and 6769 controls. By imputing our data to the Haplotype Reference Consortium (HRC) reference panel, we were able to analyze IHPS association for all low frequency and common variants detected in European ancestry populations. Further studies including, e.g. fine mapping by targeted sequencing and/or by trans-ethnic analysis, are needed to pinpoint the causative variant at each locus. In addition, more research is needed to dissect possible shared pathological mechanisms between IHPS and other diseases.
Taken together, our genome-wide meta-analysis of IHPS establishes two new loci for the disease, explains an estimated 30% of disease heritability and supports a previously reported link to lipid metabolism (10). Moreover, we were able to generalize the replicated IHPS loci to populations of different ancestry and admixture. These results elucidate novel mechanistic avenues of IHPS disease architecture, which warrant further investigation.
Materials and Methods
Study design
We report on a discovery phase of genome-wide meta-analysis of two IHPS GWAS cohorts comprising 1395 cases and 4438 controls. This was followed by a replication phase in which we genotyped the genome-wide significant loci of the discovery phase in two independent cohorts, amounting to a total of 1794 cases and 1875 controls of European ancestry, and further confirmed in 633 cases and 633 controls from a Hispanic population.
Subjects
The discovery phase comprised two case-control IHPS cohorts of Danish ancestry. IHPS surgery-confirmed cases were identified through the Danish National Patient Register, which includes all hospital diagnoses and operations since 1977 (11,12). The first cohort consists of 968 cases and 2407 controls, details of which have been previously published (9). Briefly, eligible IHPS cases comprised children that had a pyloromyotomy in their first year of life, were singletons born in Denmark with parents and grandparents born in Northwestern Europe, were born with no severe pregnancy complications and did not have any other major congenital malformations. The second IHPS cohort comprised 427 IHPS cases, with the same eligibility criteria as the first cohort described above, and 2031 controls. All controls in the discovery phase were defined as samples with the same selection criteria as for the cases, but without any IHPS diagnosis or surgery code. All samples from the discovery phase were drawn from the Danish Neonatal Screening Biobank (13) or from the biobank of the Danish National Birth Cohort (14), both of which are part of the Danish National Biobank.
For the replication phase, we used 2313 IHPS cases and 2331 controls from the USA and 114 cases and 177 controls from Sweden. The US cohort comprised de-identified DNA extracted from archived newborn blood spots of babies delivered to New York State residents during 1998–2005 and was divided into three subsamples, due to differences in sampling time and maternal race/ethnicity (Supplementary Material, Table S1). Replication cohorts US 1 and US 2 included non-Hispanic Whites of European ancestry sampled at different times, and US 3 included only Hispanic Whites. Cases were identified from the population-based New York State Congenital Malformations Registry. Controls were a random sample of all New York State live births frequency-matched by birth year and race/ethnicity to cases. The Swedish cohort consisted of whole blood specimens or skin biopsies from cases that had pyloromyotomy at a pediatric surgery clinic and placenta specimens from non-malformed controls born in 2006. This study was approved by the Research Ethics Committee of the Capital Region (Copenhagen) and the Danish Data Protection Agency. The Danish Scientific Ethics Committee also granted exemption from obtaining informed consent from participants as this research project was based on genotyping samples from biobank material (H-3-2009-093). For the replication cohorts, the ethics committees at the Karolinska Institutet approved the use of the Swedish samples, while the institutional review board at the New York State Department of Health and National Institutes of Health Office of Human Subjects Research Protection approved the use of the US samples.
Genotyping and quality control
The first (9) and second cohorts of the discovery phase were genotyped using the Illumina Human660W-Quad_v1_A and Multi-Ethnic Global_v2_A2 arrays, respectively. We applied the following quality control steps in each cohort separately, in order to sequentially remove (1) variant probes not uniquely aligned to the genome, (2) variants not present in dbSNP v.146, (3) variants with conflicting reference alleles between Illumina and dbSNP, (4) variants with missingness >5%, (5) variants that failed Hardy–Weinberg test at P < 1 × 10−6, (6) variants with P < 0.01 in tests of differences in missingness between cases and controls, (7) variants with batch effect at P-value < 1e−5 (we had more than one genotyped batch of specimens per cohort), (8) variants with minor allele frequency <1%, (9) specimens with discordant sex between registry records and genotypes and (10) specimens with missingness >2%. These quality controls steps were done with PLINK v1.90b3o (15). We also removed related individuals with an estimated kinship coefficient Phi > 0.05, within and between the two discovery phase cohorts, using KING (16) as implemented in VCFtools (17). European ancestry outliers were removed using the R package SNPRelate v1.2.0 (18), based on principal component analysis (PCA) values for all our cases and controls when analyzed together with the European samples from the Human Genome Diversity Project (19). Outliers were defined as specimens with PCA1 or PCA2 values further than six times the interquartile range from the mean PCA1 (or PCA2) values. The PCA outliers removal procedure was repeated until no more specimens were removed (two rounds sufficed). After the quality control steps, we ended up with 968 cases and 2407 controls genotyped at 521 521 variants for the first cohort and 427 cases and 2031 controls genotyped at 779 286 variants for the second cohort, with a total genotyping success rate of 99.8%.
Genotype imputation
After genotyping QC, we imputed unobserved genotypes with phased haplotypes from the HRC panel (version r1.0) (20). Imputation was done separately for the two discovery phase cohorts on the imputation server at the Wellcome Trust Sanger Institute (https://imputation.sanger.ac.uk/) using the positional Burrows–Wheeler transform algorithm (21). Prior to imputation, phasing at the server was done using SHAPEIT2 v2.r790 (22).
Association analysis
Association analysis of imputed autosomal SNPs with IHPS was performed separately in each discovery phase cohort using logistic regression on imputed SNP dosages that had an imputation info score ≥ 0.8 and MAF > 1%, under an additive genetic model using PLINK (15). After filtering, we meta-analyzed the 6 576 307 SNPs present in both discovery phase cohorts with an inverse variance-based fixed effects model, as implemented in PLINK (15). Regional association plots were created with LocusZoom (23). To test for the presence of additional independent IHPS-associated SNPs at each of the genome-wide significant loci, we conditioned on the imputed allelic dose of each top SNP per locus in each discovery phase cohort and meta-analyzed the results.
GWAS replication
The replication phase cohorts from the USA and Sweden were genotyped at LGC Genomics using PCR-based KASP genotyping chemistry assays. For the four new loci that reached genome-wide significance in the discovery phase, we prioritized the top variant at each locus for genotyping. However, if the top variant was in a repeat region and/or had significant homology to many regions in the human genome, we chose an alternative variant that: (1) was typed on the array in at least one of the discovery phase cohorts, (2) had high LD (r2 > 0.8) to the top variant and (3) was genome-wide significant in the meta-analysis of the discovery phase. Association analysis of the GWAS replication SNPs was performed separately in each replication phase cohort using logistic regression on SNP genotypes, under an additive genetic model using PLINK (15). Results obtained in the replication cohorts of European ancestry were meta-analyzed via an inverse variance-based fixed effects model, as implemented in PLINK (15). Two SNPs were not genotyped in the US Hispanic cohort (rs6736913 and rs12721025) because power to detect an association was low based on expected allele frequencies in the 1000 Genomes AMR population (37). GWAS replicated loci were defined as (1) having P < 5 × 10−8 in the discovery phase and discovery plus replication phases, (2) nominally significant associations in the meta-analysis of the replication cohorts of European ancestry (P < 0.05) and (3) having the same direction of effects in all replication cohorts of European ancestry. Heterogeneity of the SNP associations across cohorts was tested with the Cochran’s Q statistic (24). Of note, the US Hispanic cohort was not used for replication per se but for confirming, or not, the GWAS replication SNPs in a population other than just European ancestry. We also report a trans-ethnic inverse variance-based meta-analysis with a random effect accounting for the potential difference in underlying allelic effect between the US Hispanic cohort and the European ancestry samples (Supplementary Material, Table S1).
Heritability and genetic correlation
Heritability estimation of our IHPS meta-analysis GWAS was done with two methods: the LD score regression method (25,26) on the meta-analysis summary statistics and the GCTA-GREML method (27) (GCTA v1.24.735) on the individual-level genotyped QC variants in each discovery phase cohort and inverse-variance meta-analyzed. The heritability on observed scale was transformed to the liability scale using gear (28) (https://sourceforge.net/p/gbchen/wiki/Hong%20Lee's%20Transformation%20for%20Heritability/), with prevalence of IHPS set to 0.002 (3). The proportion of variance explained by the top replicated SNPs was calculated with the Nagelkerke’s coefficient of determination (29) on the US non-Hispanic Whites of European ancestry replication cohort, as implemented in the RsqGLM function in https://modtools.wordpress.com/2014/10/30/rsqglm/. The IHPS PRS was computed as ∑ log(w)*SNP, with w being the OR for each of the replicated variants in the discovery phase meta-analysis, while SNP is the genotype of the replicated variants (coded 0 to 2) in the samples from the US replication cohort. Missing genotypes were imputed to the mean US cohort value for each SNP.
Genetic correlation of our IHPS meta-analysis GWAS with GWAS of circulating metabolites (38) was done with LD score regression method (25,26), as implemented in LD hub (30).
Histone marks in fetal and adult stomach and small intestine
H3K36me3 marks in the fetal and adult stomach and small intestine were obtained from the National Institute of Health (NIH) Roadmap Epigenomics Mapping Consortium datasets of observed ChIP-seq P-value signal tracks (31), available at http://epigenomegateway.wustl.edu/browser/ (32). We chose H3K36me3 as this was the only available histone mark tagging the entire active transcribed regions (33) for our relevant tissues. Genes were defined as having H3K36me3 marks if these overlapped its RefSeq gene coordinates with P-value signal < 5 × 10−8.
Fetal and adult tissue-specific gene expression
Fetal tissue-specific gene expression data (34) were downloaded from NCBI Bioproject PRJNA270632 on January 9, 2018. Fetal expression data were available from at least three specimens and up to seven specimens for each tissue. Adult tissue-specific gene expression data were obtained from the GTEx Portal on January 9, 2018, GTEx Analysis Release V7 (35). Adult expression data were available from at least five specimens and up to 564 samples for each tissue.
Supplementary Material
Supplementary Material includes Tables S1–4, and Figures S1–13. The genome-wide meta-analysis summary statistics are available at http://danishnationalbiobank.com/gwas.html.
Supplementary Material
Acknowledgements
This study was funded by the Danish Medical Research council (DFF 4004-00512) and has been conducted using the Danish National Biobank resource. The Danish National Biobank is supported by the Novo Nordisk Foundation. B.F. was supported by an Oak Foundation fellowship. Funding was also provided by the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development (Contract numbers HHSN275201100001I and HHSN27500005), the Swedish Research Council, the Foundation Frimurare (Stockholm) as well as support from Stockholm City Council.
Conflict of Interest statement. None declared.
References
- 1. Hirschsprung H. (1888) Fälle von angeborener Pylorusstenose, beobachtet bei Säuglingen. Jahrb der Kinderh., 28, 61–68. [Google Scholar]
- 2. Ramstedt C. (1912) Zur Operation der angeborenen Pylorusstenose. Med. Klin., 8, 1702–1705. [Google Scholar]
- 3. MacMahon B. (2006) The continuing enigma of pyloric stenosis of infancy: a review. Epidemiology, 17, 195–201. [DOI] [PubMed] [Google Scholar]
- 4. Lund M., Pasternak B., Davidsen R.B., Feenstra B., Krogh C., Diaz L.J., Wohlfahrt J. and Melbye M. (2014) Use of macrolides in mother and child and risk of infantile hypertrophic pyloric stenosis: nationwide cohort study. BMJ, 348, 1908–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhu J., Zhu T., Lin Z., Qu Y. and Mu D. (2017) Perinatal risk factors for infantile hypertrophic pyloric stenosis: a meta-analysis. J. Pediatr. Surg., 52, 1389–1397. [DOI] [PubMed] [Google Scholar]
- 6. Pedersen R.N., Garne E., Loane M., Korsholm L., Husby S. and EUROCAT Working Group (2008) Infantile hypertrophic pyloric stenosis: a comparative study of incidence and other epidemiological characteristics in seven European regions. J. Matern. Fetal Neonatal Med., 21, 599–604. [DOI] [PubMed] [Google Scholar]
- 7. Krogh C., Fischer T.K., Skotte L., Biggar R.J., Øyen N., Skytthe A., Goertz S., Christensen K., Wohlfahrt J. and Melbye M. (2010) Familial aggregation and heritability of pyloric stenosis. JAMA, 303, 2393–2399. [DOI] [PubMed] [Google Scholar]
- 8. Peeters B., Benninga M.A. and Hennekam R.C. (2012) Infantile hypertrophic pyloric stenosis—genetics and syndromes. Nat. Rev. Gastroenterol. Hepatol., 9, 646–660. [DOI] [PubMed] [Google Scholar]
- 9. Feenstra B., Geller F., Krogh C., Hollegaard M.V., Gørtz S., Boyd H.A., Murray J.C., Hougaard D.M. and Melbye M. (2012) Common variants near MBNL1 and NKX2-5 are associated with infantile hypertrophic pyloric stenosis. Nat. Genet., 44, 334–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Feenstra B., Geller F., Carstensen L., Romitti P.A., Körberg I.B., Bedell B., Krogh C., Fan R., Svenningsson A., Caggana M. et al. (2013) Plasma lipids, genetic variants near APOA1, and the risk of infantile hypertrophic pyloric stenosis. JAMA, 310, 714–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lynge E., Sandegaard J.L. and Rebolj M. (2011) The Danish National Patient Register. Scand. J. Public Health, 39, 30–33. [DOI] [PubMed] [Google Scholar]
- 12. Pedersen C.B. (2011) The Danish Civil Registration System. Scand. J. Public Health, 39, 22–25. [DOI] [PubMed] [Google Scholar]
- 13. Nørgaard-Pedersen B. and Hougaard D.M. (2007) Storage policies and use of the Danish Newborn Screening Biobank. J. Inherit. Metab. Dis., 30, 530–536. [DOI] [PubMed] [Google Scholar]
- 14. Olsen J., Melbye M., Olsen S.F., Sørensen T.I., Aaby P., Andersen A.M., Taxbøl D., Hansen K.D., Juhl M., Schow T.B. et al. (2001) The Danish National Birth Cohort—its background, structure and aim. Scand. J. Public Health, 29, 300–307. [DOI] [PubMed] [Google Scholar]
- 15. Chang C.C., Chow C.C., Tellier L.C., Vattikuti S., Purcell S.M. and Lee J.J. (2015) Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience, 4, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Manichaikul A., Mychaleckyj J.C., Rich S.S., Daly K., Sale M. and Chen W.M. (2010) Robust relationship inference in genome-wide association studies. Bioinformatics, 26, 2867–2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Danecek P., Auton A., Abecasis G., Albers C.A., Banks E., DePristo M.A., Handsaker R.E., Lunter G., Marth G.T., Sherry S.T. et al. (2011) The variant call format and VCFtools. Bioinformatics, 27, 2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zheng X., Levine D., Shen J., Gogarten S.M., Laurie C. and Weir B.S. (2012) A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics, 28, 3326–3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jakobsson M., Scholz S.W., Scheet P., Gibbs J.R., VanLiere J.M., Fung H.C., Szpiech Z.A., Degnan J.H., Wang K., Guerreiro R. et al. (2008) Genotype, haplotype and copy-number variation in worldwide human populations. Nature, 451, 998–1003. [DOI] [PubMed] [Google Scholar]
- 20. McCarthy S., Das S., Kretzschmar W., Delaneau O., Wood A.R., Teumer A., Kang H.M., Fuchsberger C., Danecek P., Sharp K. et al. (2016) A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet., 48, 1279–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Durbin R. (2014) Efficient haplotype matching and storage using the positional Burrows–Wheeler transform (PBWT). Bioinformatics, 30, 1266–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Delaneau O., Zagury J.F. and Marchini J. (2013) Improved whole-chromosome phasing for disease and population genetic studies. Nat. Methods, 10, 5–6. [DOI] [PubMed] [Google Scholar]
- 23. Pruim R.J., Welch R.P., Sanna S., Teslovich T.M., Chines P.S., Gliedt T.P., Boehnke M., Abecasis G.R. and Willer C.J. (2010) LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics, 26, 2336–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cochran W.G. (1950) The comparison of percentages in matched samples. Biometrika, 37, 256–266. [PubMed] [Google Scholar]
- 25. Bulik-Sullivan B.K., Loh P.R., Finucane H.K., Ripke S., Yang J., Schizophrenia Working Group of the Psychiatric Genomics Consortium, Patterson N., Daly M.J., Price A.L. and Neale B.M. (2015) LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet., 47, 291–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bulik-Sullivan B., Finucane H.K., Anttila V., Gusev A., Day F.R., Loh P.R., ReproGen Consortium, Psychiatric Genomics Consortium, Genetic Consortium for Anorexia Nervosa of the Wellcome Trust Case Control Consortium 3, Duncan L. et al. (2015) An atlas of genetic correlations across human diseases and traits. Nat. Genet., 47, 1236–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yang J., Lee S.H., Goddard M.E. and Visscher P.M. (2011) GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet., 88, 76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lee S.H., Wray N.R., Goddard M.E. and Visscher P.M. (2011) Estimating missing heritability for disease from genome-wide association studies. Am. J. Hum. Genet., 88, 294–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nagelkerke N.J.D. (1991) A note on a general definition of the coefficient of determination. Biometrika, 78, 691–692. [Google Scholar]
- 30. Zheng J., Erzurumluoglu A.M., Elsworth B.L., Kemp J.P., Howe L., Haycock P.C., Hemani G., Tansey K., Laurin C., Early Genetics and Lifecourse Epidemiology (EAGLE) Eczema Consortium et al. (2017) LD Hub: a centralized database and web interface to perform LD score regression that maximizes the potential of summary level GWAS data for SNP heritability and genetic correlation analysis. Bioinformatics, 33, 272–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Roadmap Epigenomics Consortium, Kundaje A., Meuleman W., Ernst J., Bilenky M., Yen A., Heravi-Moussavi A., Kheradpour P., Zhang Z., Wang J. et al. (2015) Integrative analysis of 111 reference human epigenomes. Nature, 518, 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhou X., Li D., Zhang B., Lowdon R.F., Rockweiler N.B., Sears R.L., Madden P.A., Smirnov I., Costello J.F. and Wang T. (2015) Epigenomic annotation of genetic variants using the Roadmap Epigenome Browser. Nat. Biotechnol., 33, 345–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Barski A., Cuddapah S., Cui K., Roh T.Y., Schones D.E., Wang Z., Wei G., Chepelev I. and Zhao K. (2007) High-resolution profiling of histone methylations in the human genome. Cell, 129, 823–837. [DOI] [PubMed] [Google Scholar]
- 34. Szabo L., Morey R., Palpant N.J., Wang P.L., Afari N., Jiang C., Parast M.M., Murry C.E., Laurent L.C. and Salzman J. (2015) Statistically based splicing detection reveals neural enrichment and tissue-specific induction of circular RNA during human fetal development. Genome Biol., 16, 126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. GTEx Consortium, Laboratory, Data Analysis & Coordinating Center (LDACC)—Analysis Working Group, Statistical Methods groups—Analysis Working Group, Enhancing GTEx (eGTEx) groups, NIH Common Fund, NIH/NCI, NIH/NHGRI, NIH/NIMH, NIH/NIDA, Biospecimen Collection Source Site—NDRI et al. (2017) Genetic effects on gene expression across human tissues. Nature, 550, 204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yang J., Weedon M.N., Purcell S., Lettre G., Estrada K., Willer C.J., Smith A.V., Ingelsson E., O'Connell J.R., Mangino M. et al. (2011) Genomic inflation factors under polygenic inheritance. Eur. J. Hum. Genet., 19, 807–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. 1000 Genomes Project Consortium, Auton A., Brooks L.D., Durbin R.M., Garrison E.P., Kang H.M., Korbel J.O., Marchini J.L., McCarthy S., McVean G.A. et al. (2015) A global reference for human genetic variation. Nature, 526, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kettunen J., Demirkan A., Würtz P., Draisma H.H., Haller T., Rawal R., Vaarhorst A., Kangas A.J., Lyytikäinen L.P., Pirinen M. et al. (2016) Genome-wide study for circulating metabolites identifies 62 loci and reveals novel systemic effects of LPA. Nat. Commun., 7, 11122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dong H., Guo H., Xie L., Wang G., Zhong X., Khoury T., Tan D. and Zhang H. (2013) The metastasis-associated gene MTA3, a component of the Mi-2/NuRD transcriptional repression complex, predicts prognosis of gastroesophageal junction adenocarcinoma. PLoS One, 8, e62986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Beelen N.W., Mous D.S., Brosens E., Klein A., Ven C.P., Vlot J., Ijsselstijn H. and Wijnen R. (2014) Increased incidence of hypertrophic pyloric stenosis in esophageal atresia patients. Eur. J. Pediatr. Surg., 24, 20–24. [DOI] [PubMed] [Google Scholar]
- 41. Vaidya A., Flores S.K., Cheng Z.M., Nicolas M., Deng Y., Opotowsky A.R., Lourenço D.M. Jr., Barletta J.A., Rana H.Q., Pereira M.A. et al. (2018) EPAS1 mutations and paragangliomas in cyanotic congenital heart disease. N. Engl. J. Med., 378, 1259–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Norwood M.S., Lupo P.J., Chow E.J., Scheurer M.E., Plon S.E., Danysh H.E., Spector L.G., Carozza S.E., Doody D.R. and Mueller B.A. (2017) Childhood cancer risk in those with chromosomal and non-chromosomal congenital anomalies in Washington State: 1984–2013. PLoS One, 12, e0179006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Casagrande A. and Pederiva F. (2016) Association between congenital lung malformations and lung tumors in children and adults: a systematic review. J. Thorac. Oncol., 11, 1837–1845. [DOI] [PubMed] [Google Scholar]
- 44. Carozza S.E., Langlois P.H., Miller E.A. and Canfield M. (2012) Are children with birth defects at higher risk of childhood cancers? Am. J. Epidemiol., 175, 1217–1224. [DOI] [PubMed] [Google Scholar]
- 45. Altmann A.E., Halliday J.L. and Giles G.G. (1998) Associations between congenital malformations and childhood cancer. A register-based case-control study. Br. J. Cancer, 78, 1244–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kim B.M., Buchner G., Miletich I., Sharpe P.T. and Shivdasani R.A. (2005) The stomach mesenchymal transcription factor Barx1 specifies gastric epithelial identity through inhibition of transient Wnt signaling. Dev. Cell, 8, 611–622. [DOI] [PubMed] [Google Scholar]
- 47. Kim B.M., Miletich I., Mao J., McMahon A.P., Sharpe P.A. and Shivdasani R.A. (2007) Independent functions and mechanisms for homeobox gene Barx1 in patterning mouse stomach and spleen. Development, 134, 3603–3613. [DOI] [PubMed] [Google Scholar]
- 48. Jayewickreme C.D. and Shivdasani R.A. (2015). Control of stomach smooth muscle development and intestinal rotation by transcription factor BARX1. Dev. Biol., 405(1), 21–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yamamoto Y., Oguro N., Nara T., Horita H., Niitsu N. and Imaizumi S. (1988) Duplication of part of 9q due to maternal 12;9 inverted insertion associated with pyloric stenosis. Am. J. Med. Genet., 31, 379–384. [DOI] [PubMed] [Google Scholar]
- 50. Heller A., Seidel J., Hübler A., Starke H., Beensen V., Senger G., Rocchi M., Wirth J., Chudoba I., Claussen U. and Liehr T. (2000) Molecular cytogenetic characterisation of partial trisomy 9q in a case with pyloric stenosis and a review. J. Med. Genet., 37, 529–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Theodosiou N.A. and Tabin C.J. (2005) Sox9 and Nkx2.5 determine the pyloric sphincter epithelium under the control of BMP signaling. Dev. Biol., 279, 481–490. [DOI] [PubMed] [Google Scholar]
- 52. Udager A.M., Prakash A., Saenz D.A., Schinke M., Moriguchi T., Jay P.Y., Lim K.C., Engel J.D. and Gumucio D.L. (2014) Proper development of the outer longitudinal smooth muscle of the mouse pylorus requires Nkx2-5 and Gata3. Gastroenterology, 146, 157–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Prakash A., Udager A.M., Saenz D.A. and Gumucio D.L. (2014) Roles for Nkx2-5 and Gata3 in the ontogeny of the murine smooth muscle gastric ligaments. Am. J. Physiol. Gastrointest. Liver Physiol., 307, 430–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Schechter R., Torfs C.P. and Bateson T.F. (1997) The epidemiology of infantile hypertrophic pyloric stenosis. Paediatr. Perinat. Epidemiol., 11, 407–427. [DOI] [PubMed] [Google Scholar]
- 55. Ryan A.K., Bartlett K., Clayton P., Eaton S., Mills L., Donnai D., Winter R.M. and Burn J. (1998) Smith–Lemli–Opitz syndrome: a variable clinical and biochemical phenotype. J. Med. Genet., 35, 558–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Krogh C., Biggar R.J., Fischer T.K., Lindholm M., Wohlfahrt J. and Melbye M. (2012) Bottle-feeding and the risk of pyloric stenosis. Pediatrics, 130, 943–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. McAteer J.P., Ledbetter D.J. and Goldin A.B. (2013) Role of bottle feeding in the etiology of hypertrophic pyloric stenosis. JAMA Pediatr., 167, 1143–1149. [DOI] [PubMed] [Google Scholar]
- 58. Owen C.G., Whincup P.H., Odoki K., Gilg J.A. and Cook D.G. (2002) Infant feeding and blood cholesterol: a study in adolescents and a systematic review. Pediatrics, 110, 597–608. [DOI] [PubMed] [Google Scholar]
- 59. Mauch D.H., Nägler K., Schumacher S., Göritz C., Müller E.C., Otto A. and Pfrieger F.W. (2001) CNS synaptogenesis promoted by glia-derived cholesterol. Science, 294, 1354–1357. [DOI] [PubMed] [Google Scholar]
- 60. Langer J.C., Berezin I. and Daniel E.E. (1995) Hypertrophic pyloric stenosis: ultrastructural abnormalities of enteric nerves and the interstitial cells of Cajal. J. Pediatr. Surg., 30, 1535–1543. [DOI] [PubMed] [Google Scholar]
- 61. Manolio T.A., Collins F.S., Cox N.J., Goldstein D.B., Hindorff L.A., Hunter D.J., McCarthy M.I., Ramos E.M., Cardon L.R., Chakravarti A.. et al. (2009). Finding the missing heritability of complex diseases. Nature, 461, 747–753. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.