

Pioneering work by, among others, the groups of Hunt and Mann at the start of this century opened up the era of mass spectrometry (MS)-based phosphoproteomics.1−3 Through advances in sample preparation, phosphopeptide enrichment, mass spectrometric detection, peptide sequencing, and dedicated database search algorithms, this field has matured and it is nowadays possible to monitor thousands of protein phosphorylation events qualitatively and quantitatively.4−8 Such phosphoproteomics studies are important, as reversible protein phosphorylation is regulating nearly all biological processes and the deregulation of phosphorylation has been associated with the onset of various pathologies.9−15 Notwithstanding this maturation and broad acceptance of mass spectrometry based phosphoproteomics by the research community, the mass spectrometric analysis of phosphorylation remains demanding, mainly due to the often severe substoichiometric levels of protein phosphorylation and the intrinsic lability of the phosphate group, hampering both enrichment and unambiguous sequencing analysis of phosphopeptides. Here we review the increasing knowledge gathered about the mechanisms behind the fragmentation of phosphopeptide ions and describe additionally recent advances made to improve and facilitate the identification and site localization of phosphorylated peptides. Since our earlier related review in 2009,16 several fragmentation methods have been introduced and successfully applied to phosphopeptides sequencing, such as electron capture (ECD), electron transfer induced dissociation (ETD), and hybrid fragmentation techniques such as EThcD (a combination of ETD and higher energy collisional dissociation (HCD)) and AI-ETD (a combination of ETD with infrared photoactivation). A plethora of data has been gathered on endogenous Ser, Thr, Tyr phosphorylation but also on large synthetic phosphopeptide libraries, providing new insight into the mechanisms behind phosphopeptide fragmentation. Moreover, data on phosphopeptides harboring phosphorylated histidine, arginine, or lysine have become available, showing some distinct fragmentation behaviors. Through all this acquired knowledge on phosphopeptide fragmentation and site localization, MS-based phosphoproteomics has developed into a respectable and valuable tool for the broader life science research community.
Sequencing Phosphopeptides Using Collision Induced Dissociation
Basic Principles of Collision Induced Dissociation Applied to Peptide Ions
During collision induced dissociation (CID), gas-phase peptide ions are subjected to collisions with inert gas atoms or molecules, leading to the incremental increase of the peptide ions internal energies and ultimately to their unimolecular dissociation into fragment neutrals and ions, whereby the ions are used to elucidate the peptide sequence or to pinpoint the modification sites following data analysis. CID methods are the most prominent ion activation methods in the field of bottom-up proteomics, due to their high efficiency and ease of implementation. However, this ease of implementation cannot conceal the complexity of energy transfer and dissociation mechanisms involved, discussed in detail in various recent reviews.17−19 When considering applications of CID to (phospho)proteomics, one refers almost exclusively to the so-called low-energy CID, during which collision energies in the range of 1–200 eV are used. Nonelastic collisions with the inert gas molecules lead to the transfer of translational into internal energy, mainly in the form of vibrational energy. Low-energy CID is a stepwise process and multiple collisions (tens to hundreds) are necessary to accumulate sufficient internal energy to reach a threshold energy necessary to overcome for the gas-phase unimolecular dissociation reaction to happen.20,21 For this reason, CID presents relatively long activation times in comparison with alternative dissociation techniques discussed later. During this process, the internal energy is quickly (<10–12 s) equilibrated through the vibrational degrees of freedom of the peptide ions. This statistical redistribution of internal energy implies that following CID, low-energy dissociation pathways are preferably accessed.
The main dissociation pathways occurring during CID are charge-directed. In positive ion mode, the mobile proton model, introduced by Gaskell, Wysocki, and co-workers,22−27 provides a generally accepted model to explain CID fragmentation behavior of protonated peptides. Peptide gas-phase ions formed by soft ionization techniques (i.e., electrospray ionization) are characterized by their relatively low level of internal energy: ionizing protons are thus localized at the energetically favored positions, mainly at basic residues side chains (histidine, lysine, and arginine) and/or at the N-terminal amine. Increase of the internal energy of the peptide upon CID facilitates the mobilization of the protons to alternative, less favored sites along the backbone. Protonation of the nitrogen atom of the peptide backbone amide bond results in the weakening of the amide bond, and the subsequent oxazolone pathway results preferentially in the formation of the well-known complementary b and y fragment ions17 (Figure 1).
Figure 1.

Peptide ion fragmentation ladder with the complementary a/x, b/y, and c/z ion series displayed. Upon CID, the oxazolone pathway, resulting in the preferential formation of the b/y ion series is energetically favored after protonation of the nitrogen atom of the amide bond.
Neutral Losses of Serine and Threonine Phosphorylated Peptides
Particular for phosphorylated peptides, low-energy relaxation pathways can additionally lead to the loss of the rather labile phosphate group, either as neutral phosphoric (H3PO4) or meta-phosphoric (HPO3) acids, competing with the above-described backbone fragmentation.6,16 We will first focus on the mechanisms of neutral losses, while the impacts of such neutral losses on phosphopeptides identification and phosphosites localization are discussed later. As CID dissociation pathways are mainly charge-directed, the mobility of the ionizing protons bears a crucial importance in the type of gas-phase reactions involved. Three major classes of proton mobility conditions have been defined,28 depending on the number of ionizing protons (i.e., the peptide’s charge state) and the number of basic residues present in the peptide sequence. If the number of charges is higher to the number of basic residues, the conditions are considered as mobile. Limited mobility corresponds to cases, wherein the number of basic residues is higher than or equal to the number of protons. Because of the very high gas-phase basicity of arginine residues, large amounts of energy are necessary to mobilize a proton initially sequestered at such an arginine side-chain. Conditions are thus considered as nonmobile if the number of arginine residues is higher than or equal to the number of charges carried by the peptide.
It is well established that upon CID fragmentation, phosphoserine and phosphothreonine (pST) peptides mainly fragment through elimination of H3PO4.6,16 This phosphoric acid loss can originate from a direct loss of H3PO4 or combined losses of HPO3 and a water molecule from a residue’s side chain containing a hydroxyl group. It was also observed that the propensity of pST phosphopeptides to undergo neutral loss is inversely correlated with the proton mobility (i.e., neutral loss is more likely to occur under limited proton mobility conditions).29 Over the years, several alternative mechanisms have been proposed for the observed phosphate neutral losses, including β-elimination, E2 eliminations, and nucleophilic substitutions involving diverse nucleophiles displacing the phosphate group. Gronert et al. originally obtained similar activation barriers for the different dissociation mechanisms, implying that neutral loss could occur via multiple pathways.30 Based on computational and experimental evidence, it is now accepted that under mobile proton conditions, H3PO4 neutral loss via a SN2 mechanism is favored, initiated by the mobilization of a proton to one of the oxygen atoms of the phospho-group.29,31,32 This protonation makes the β-carbon linked to the charged phospho-group electrophilic and thus suitable for a subsequent nucleophilic attack from a neighboring nucleophile group. Neutral loss then preferably occurs via formation of a five-membered oxazoline ion after nucleophilic attack from the N-terminal carbonyl oxygen, expelling the phospho-group (Figure 2).
Figure 2.

Neutral H3PO4 elimination from phosphopeptides under high (A) and low (B) proton mobility conditions. Note that alternative SN2 mechanisms have been proposed, depending on the nature of the nucleophilic attacking group. Here, the energetically favored formation of the five-membered oxazoline ion is displayed. The energetically favored HPO3 neutral loss mechanism is displayed in part C. Reproduced from Modeling of the gas-phase phosphate group loss and rearrangement in phosphorylated peptides, Rožman, M., J. Mass Spectrom.2011, Vol. 46, pp. 949–955 (ref (31)). Copyright 2011 Wiley.
It was initially hypothesized that neutral losses via charge-remote mechanisms were favored in case of nonmobile or limited mobility conditions, with a β-elimination reaction leading to the formation of dehydroalanine or dehydrobutyrine in the case of phosphoserine or phosphothreonine peptides, respectively.33 However, Palumbo et al. showed that charge-directed mechanisms were favored even under nonmobile conditions.29 This observation was rationalized by the formation of a hydrogen bond between the arginine guanidinium group and the phosphate group. By withdrawing electrons, this interaction creates an electrophilic group suitable for the SN2 reaction, can assist the phosphate leaving group, and the basic group can act as a charge neutralizer by transferring its proton to the phosphate group to form H3PO4. Rožman confirmed by a combination of quantum mechanics and RRKM modeling that neutral loss via charge-directed mechanisms are indeed also energetically favored in limited proton mobility conditions, through interaction of positively charged guanidinium with the phospho-group (Figure 2).31
This indicates that the proton mobility conditions do not significantly influence the neutral loss mechanism but rather affect the competition between the neutral loss and backbone fragmentation channels. Under high mobile proton conditions, energetic and kinetic properties of the neutral loss pathway match those of the classical oxazolone pathway. Backbone fragmentation mechanisms, which are primarily charge-directed, necessitate a larger amount of activation energy when the proton mobility is limited, while Rožman calculated that activation barriers for neutral loss mechanisms under both high and low mobile proton conditions (displayed in Figure 2A,B) were somewhat similar.31 Hence, under limited proton mobility conditions, the neutral loss reaction can become energetically and kinetically favored in comparison with peptide backbone fragmentation. To illustrate this point, Lanucara et al. were able to reduce the extent of neutral loss and thus increase backbone fragmentation by increasing the proton mobility of phosphopeptides via enzymatic removal of the basic C-term residue.34
Laskin et al. recently showed that the phosphoric acid neutral loss is likely a two-step process under limited proton mobility conditions, whereby the phosphate abstraction is followed by the dissociation of the ion–molecule complex, the phosphopeptide sequence influencing the overall observed reaction kinetics.35 They concluded that phosphate neutral loss under mobile conditions exhibited a relatively high dissociation barrier with a loose transition state, while neutral loss under nonmobile conditions is characterized by a lower dissociation barrier with a tight transition state.
A bias toward higher neutral loss abundance during CID of serine phosphorylated peptides in comparison with threonine phosphorylated peptides has been documented, especially under high mobile proton conditions, and can be explained by a difference in fragmentation dynamics.29,31 Meta-phosphoric (HPO3) neutral loss can also occur (Figure 2C), albeit much less frequently than the phosphoric acid neutral loss, potentially leading to the gas-phase rearrangement of the phosphate group. This meta-phosphate loss can be accompanied by a water loss from residues containing a hydroxyl moiety (in particular, nonmodified serine and threonine residues) or carboxyl groups, cumulatively resulting in an apparent phosphoric acid loss.36,37 This implies that the dehydration site is not indicative of the position of the phosphorylation site.
Cui et al. showed that the proton mobility conditions also influence the competition between the direct loss (i.e., SN2 mechanism) and the combined loss, the probabilities of the latter increasing under limited proton mobility and nonmobile proton conditions.36 This is in accordance with the calculations made by Rožman, revealing that HPO3 loss can compete with H3PO4 loss under limited proton mobility conditions.31 In an effort to limit the neutral loss extent, a chemical derivatization strategy mitigating charge-directed mechanisms leading to phosphate neutral loss was developed,38 while addition of a dinuclear gallium complex improved the stability of the phospho-ester bond during CID.39
In an extensive study, factors influencing the occurrence and abundance of neutral losses under CID conditions were analyzed by Brown et al.40 They verified that the extent of neutral loss is highly sequence-dependent, whereby the proton mobility alone cannot explain the differences observed from peptide to peptide sequence. For this study, they compiled more than 30 000 ion trap based CID fragmentation spectra originating from tryptic phosphopeptides. They confirmed the correlation between neutral losses and proton mobility (Figure 3A) and showed that neutral loss was on average 3-fold higher for b-ions in comparison with y-ions, indicating that proper phosphosite localization should rely more on y-ions series.
Figure 3.
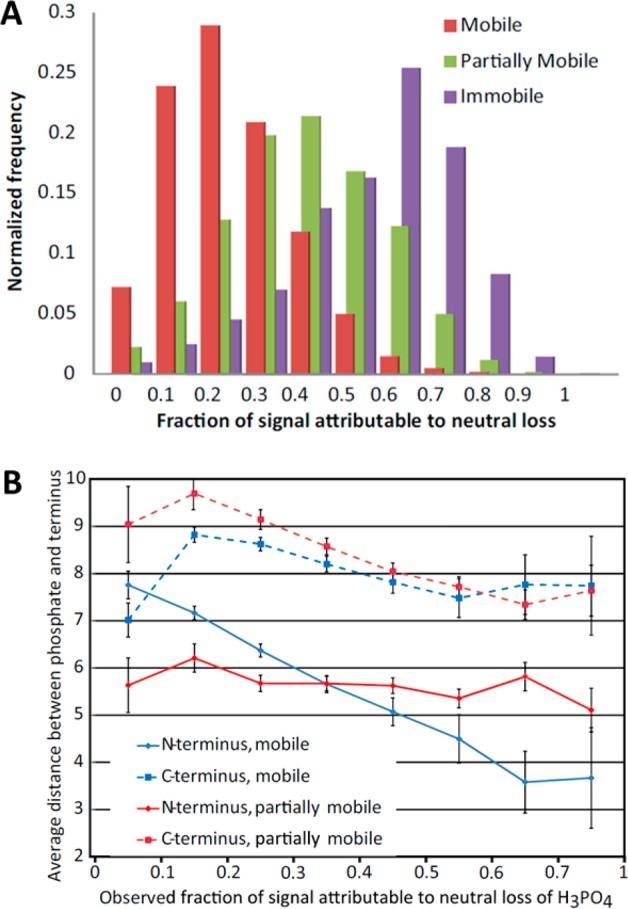
Fraction of signal attributable to neutral loss observed in spectra derived from 5749 monophosphorylated pS and pT tryptic peptides using ion trap CID fragmentation (A). The extent of neutral loss correlates with proton mobility but does not fully explain the observed behavior. The observed fraction of signal intensity attributable to H3PO4 neutral loss, as a function of the proximity of the phosphorylation site to both peptide termini, is displayed in part B. Reprinted by permission from Springer. J. Am. Soc. Mass Spectrom., Large-Scale Examination of Factors Influencing Phosphopeptide Neutral Loss during Collision Induced Dissociation. Brown, R., Stuart, S. S., Houel, S., Ahn, N. G., Old, W. M. Vol. 26, 2015, pp. 1128–1142 (ref (40)). Copyright 2015.
The study of Brown et al. also elucidated that the phosphosite proximity to the predominantly protonated N-terminus enhances the magnitude of the neutral loss (Figure 3B) in mobile conditions. To explain this behavior, the authors proposed a mechanism in which the formation of a hydrogen bond between the N-terminus, which is the least basic group protonated with high occupancy, and the phospho-group is favoring the SN2 reaction leading to neutral loss. Such effect is not observed in limited conditions as the N-terminus is less likely to be protonated.
Surprisingly, basic residues, which are supplying the protons under mobile conditions and promoting neutral loss reaction in limited and nonmobile proton conditions, suppress neutral loss when directly adjacent to the phosphosite, which the authors explained by steric hindrance due to the formation of a strong hydrogen bond preventing the formation of the oxazoline ring. More generally, under mobile conditions, strong hydrogen bonds between the phospho-group and proximal basic residues impede the phospho-group protonation or interaction with the less stable charge donor protonated N-terminus, which, in fine, favors backbone fragmentation following the mobile proton model. Additionally, Brown et al. determined that neutral water and ammonia loss can compete with phosphate neutral loss and that the presence of aspartic and glutamic acids promote neutral loss, while proline, by reducing the backbone flexibility, can hamper neutral loss by limiting distal interaction between the phospho-group and basic residues.
Neutral Losses of Tyrosine Phosphorylated Peptides
Tyrosine phosphorylated peptides exhibit some specific fragmentation characteristics. Upon CID, tyrosine phosphorylated (pY) peptide ions can occasionally exhibit a meta-phosphoric (HPO3) neutral loss but to a markedly lower extent when compared to neutral losses observed during CID of serine and threonine phosphorylated peptides.6,16 The SN2 reaction resulting in the H3PO4 elimination is hampered due to steric hindrance from the aromatic group, which also stabilizes the C–O bond cleaved during H3PO4 loss via resonance. HPO3 loss occurs via a charge-remote mechanism, while only limited H3PO4 loss is observed, which can only result from a concomitant loss of HPO3 and water. In accordance with an early report that neutral loss of the phosphate moiety from pY peptides is charge dependent,41 Everley et al. recently reported that phosphate neutral loss from tyrosine phosphorylated peptides drastically increases after isobaric labeling (Figure 4), during which a primary amine is replaced by a tertiary amine of high gas-phase basicity, thus substantially affecting the proton mobility.42 In fact, 97% of peptides for which the neutral loss was observed were of low proton mobility, highlighting the fact that phosphotyrosine neutral loss should be seriously considered during database searches of iTRAQ- and TMT-labeled phosphotyrosine peptides. While intense neutral losses after CID of labeled pST peptides have also been reported,43,44 as far as we know the impact of isobaric labeling on the extent of neutral loss during CID fragmentation of pST peptides has yet to be quantified, but a similar trend can be expected.
Figure 4.
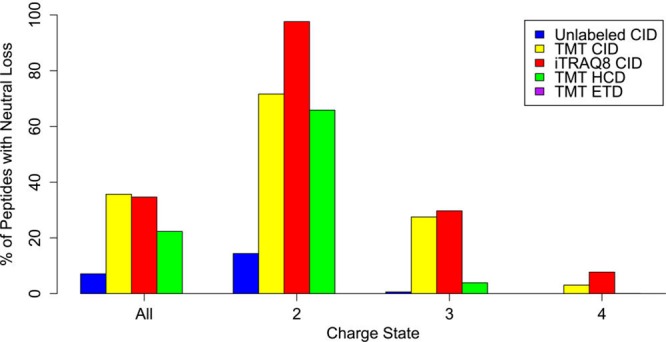
Isobaric labeling enhances neutral loss in phosphotyrosine peptides. Tyrosine phosphorylated peptides were reported as exhibiting neutral loss when the neutral loss intensity was >33% of the base peak. A substantial increase in neutral loss is observed upon isobaric labeling, which is caused by the lowering of the proton mobility. Note that under ETD conditions (purple) no neutral loss was observed. Reproduced from Everley, R. A.; Huttlin, E. L.; Erickson, A. R.; Beausoleil, S. A.; Gygi, S. P. J. Proteome Res.2017, 16, 1069–1076 (ref (42)). Copyright 2017 American Chemical Society.
Resonance Excitation versus Beam-Type CID
Low-energy CID is achieved through either resonance excitation or beam-type collisional dissociation.20 While basically the same fragmentation rules apply and both techniques are well suited for the analysis of nonmodified peptides, several factors make beam-type CID superior to resonance excitation in the analysis of phosphopeptides.45 Ion trap CID (IT-CID) corresponds to the resonance excitation of trapped peptide ions by applying a supplemental voltage matching the precursor secular frequency. The depth of the trapping potential well is such that, to prevent the ejection of ions, the precursor can only be excited to a few electron volts of kinetic energy. Hence, hundreds of low-energy collisions, over long activation times (>10 ms), are necessary to build up enough internal energy to enable fragmentation. Because of these long activation times, IT-CID is considered as a slow-heating technique, during which extensive gas-phase rearrangements can occur prior to dissociation. In the case of the fragmentation of phosphorylated peptides, evidence for gas-phase rearrangement of a phosphate moiety to another nonmodified serine or threonine residue has been reported,46 albeit with limited impact on the phosphosite localization.47,48 Activation times in beam-type CID (including higher energy collisional dissociation, HCD49) are shorter, as precursor ions are not excited by resonance but instead are accelerated into the neutral gas bath of a collision cell. The high focusing power of radiofrequency (rf) multipole collision cells enables higher energy collisions with the neutral gas without significant ion loss, decreasing the activation time to approximately 0.1 ms. This reduces potential gas-phase rearrangement of the phosphate moiety,50 for which the fastest reaction occurs on a millisecond time-scale.31 More importantly, if neutral losses are common upon beam-type CID, HCD has been shown to generate less phosphate neutral loss in comparison with IT-CID and is thus better suited to pinpoint more accurately phosphorylation sites.36 In addition, Diedrich et al. demonstrated that the use of a stepped HCD collision energy (fragmentation at multiple collision energies) can result in an increase of identified phosphopeptides and phosphosites localization confidence.51
During IT-CID, only precursor ions are excited by resonance while fragments fall off-resonance. For phosphopeptides this can result in the generation of high abundant noninformative ions corresponding to the phosphate neutral loss(es) from the precursor ion without further backbone fragmentation. To address this issue, multistage activation (MSA) has been applied to increase phosphopeptide identification through subsequent activation of the neutral loss product ions.52 In contrast, during beam-type CID, all ions are activated and fragments can undergo multiple sequential fragmentation events, resulting in the generation of richer fragmentation spectra.53 Fragments resulting from neutral loss can thus be subsequently dissociated into sequence-informative b and y-ions, facilitating phosphopeptide identification. As such, HCD fragmentation of phosphopeptides has been reported to yield in general better identification scores than IT-CID based fragmentation.45,54,55 In some cases, the generation of an x-ion resulting from the fragmentation of an ion that underwent phosphoric neutral loss (Figure 5) can aid the phosphosite localization.50 Similarly, even if neutral loss via elimination mechanisms is a minor dissociation channel, Pilo et al. recently showed that CID fragmentation of a dehydroalanine-containing product yields atypical c/z ions after cleavage of the N–Cα bond of the dehydroalanine residue. These low abundance c/z ions were used to pinpoint the phosphosite within a serine phosphorylated peptide56 but do not constitute absolute diagnostic ions as dehydroalanine can also originate from serine dehydration.
Figure 5.

Formation of the atypical 4,5-dihydrooxazolyl x ion, resulting from the fragmentation of an ion that underwent a phosphoric acid loss via the SN2 mechanism but cannot fragment following the classic oxazolone pathway. Note that the formation of an x-ion of the same mass is possible (albeit infrequent) during beam type fragmentation of nonphosphorylated peptides, but a quality filtering based on the higher intensity of the x-ions over y-ions enables one to filter out nonphosphorylated peptides. According to two studies, the x-ion was present in 24 and 36% of spectra deriving from HCD fragmentation of pST peptides.50,53 Reproduced from Kelstrup, C. D.; Hekmat, O.; Francavilla, C.; Olsen, J. V. J. Proteome Res.2011, 10, 2937–2948 (ref (50)). Copyright 2011 American Chemical Society.
Beam-type CID (e.g., HCD) spectra are also characterized by the presence of abundant informative immonium ions in the low-mass region.53,57,58 Immonium ions result from two sequential cleavages and are particularly abundant in the case of residues presenting both a heteroatom and an aromatic ring that can provide charge stabilization. While commonly observed after beam-type CID, such ions are usually difficult to detect after IT-CID due to the lack of sequential fragmentation and low-mass cutoff applied, which is inherent to the use of the rf trapping field. Immonium ions bear a particular importance in phosphoproteomics, as the phosphotyrosine immonium ion possesses a unique m/z value and can thus serve as a diagnostic tool. Diedrich et al. showed that the pY immonium ion abundances scale up with the HCD normalized energy,51 and in addition to serve diagnostic purposes, such ions have been used in analytical strategies to scan for phosphotyrosine precursors.59 The chance to observe the diagnostic pY immonium ion is also directly correlated with the abundance of the tyrosine phosphorylated peptides. In our laboratory, at normal HCD collision energy, the immonium ion is observed for ∼90% of the pY phosphopeptides after pY immunoprecipitation, while only for ∼20% of the low abundant pY phosphopeptides present in samples in which pSTY are coenriched. Everley et al. also recently showed that the low proton mobility resulting of isobaric labeling alters the probability of observing the pY diagnostic immonium ion,42 which is in line with the fact that the immonium ion is only observed at high collision energy in the case of iTRAQ labeled pY peptides.43
Another significant difference between the two techniques lies in the fact that beam-type dissociation processes are usually coupled with high-resolution detection of fragments by time-of-flight (TOF) or Fourier transform (FT) Orbitrap mass analyzers, where IT-CID is often coupled with low-resolution ion trap detection. While ion traps exhibit higher sensitivity and often faster duty cycles, measuring the fragment ions with high mass accuracy and high signal-to-noise ratio is essential to achieve confident phosphosite localization. In the early days of HCD, CID-IT was reported to outperform beam-type CID in term of identification numbers (but not in terms of scoring), which was imputed to the slower duty cycle time of HCD.54 Today, thanks to instrumental progress over the past decade, beam-type CID (and more particularly HCD) outperforms CID-IT for the identification of phosphopeptides and subsequent phosphosite localization.45
Electron Transfer/Capture Dissociation of Phosphopeptides
Principles of Electron Transfer/Capture Dissociation
In MS-based proteomics, electron capture/transfer induced dissociation (ExD) techniques have become viable alternatives to the collisional activation techniques discussed above.60 Introduced in 1998, electron capture dissociation (ECD) for peptide and protein sequencing relies on the capture of a low-energy electron (generally below 1 eV) by a multicharged gas-phase cation.61 Electron capture forms an electronically excited charge-reduced radical cation, initiating radical-ion dissociation reactions. Quadrupole ion traps cannot efficiently trap electrons due to the low mass cutoff of the rf field, and for this reason, ECD was initially performed in FT ion cyclotron resonance (FTICR) instruments, as the strong magnetic fields applied enable simultaneous trapping of analytes and electrons. Introduced in 2004, electron transfer dissociation (ETD) is an alike technique involving an ion/ion reaction during which an anion reagent with low electron affinity (usually fluoranthene) transfer an electron to a polycharged peptide cation.62 By applying unbalanced rf in an ion trap, it is possible to obtain charge-sign independent trapping, i.e., it is possible to efficiently trap both anionic reagent and cationic peptides. The widespread availability, low cost, and robustness of ion trap instruments as well as its compatibility with chromatographic time scales largely helped to popularize ETD, making it nowadays the preferred ExD technique in the field of bottom-up proteomics. Notably, a compact ECD cell that can be implemented on Orbitrap and Q-TOF platforms may rejuvenate ECD in the near future.63,64
Besides the differences in electron capture/transfer, ECD and ETD hold much in common from a mechanistic point of view. Different dissociation mechanisms have been proposed and are discussed in several recent reviews.65−67 Very briefly, the two well-accepted mechanisms (named Cornell61 and Utah–Washington68 mechanisms) result in the formation of an unstable aminoketyl radical, leading ultimately to the cleavage of the peptide backbone bond between the amide nitrogen atom and the Cα atom of the neighboring residue (N–Cα bond). This preferred fragmentation pathway involves random cleavage along the peptide backbone and produces complementary even-electron c-type and odd-electron z•-type ions series. When considering its application to phosphoproteomics, ExD-based fragmentations possess several advantageous features. First, the phosphate group does not possess bound anionic states and exhibits negative electron and hydrogen affinity;69 hence, phosphorylation of side chain residues are virtually nonreactive upon ExD. ExD also enables, in theory, to achieve better sequence coverage through the generation of more complete ion series, which is an important factor for confident phosphorylation site localization, as discussed later. Finally, after electron capture/transfer, backbone cleavages occur faster than the rate of internal vibrational energy redistribution, diminishing the undesired neutral losses of the phosphate moieties. This ability of cleaving strong backbone bonds while preserving weak phosphoester bonds, which are labile upon vibrational activation, was reported early on and still constitutes one of the major advantages of ExD dissociation over CID-based methods, especially for phosphopeptides fragmentation.70−73
However, ExD techniques are hampered by two major bottlenecks. The first one is the strong dependency of the ExD efficiency on the precursor charge density, as inter alia electron capture cross sections and ion/ion reaction rates are highly dependent on the peptide charge state.74,75 As a result, ExD of doubly protonated peptides generally suffers from lower fragmentation efficiency when compared to CID/HCD.76 During ETD, the competition between undesirable proton transfer from the peptide to the anion reagent and electron transfer is also influenced by the peptide charge state, the detrimental proton transfer being more prominent in the case of doubly charged peptides.77 The second major bottleneck is that ExD necessitates longer reaction times in comparison with CID activation times, especially when considering beam-type CID. Indeed, while ExD are fast processes, they suffer from low fragmentation frequency and require long reaction times to achieve extensive precursor ion dissociation. For example, ETD reaction times to reach optimal fragmentation are in the order of 100 ms for doubly charged precursor and less than 50 ms for triply charged peptides.78 These longer cycle times (compared to CID) have an important impact on the number of identifiable (phospho)peptides, as discussed later, and as such limits the dynamic range and depth of ETD experiments.
The capture or transfer of low-energy electrons does not always disrupt noncovalent interactions, which can result in the product ions staying together, termed nondissociative ExD. The prevalence of these phenomena, termed sometimes ECnoD/ETnoD, during which such complexes appear as nondissociated charge reduced precursor ions, is also directly related to the precursor charge density.79 Indeed, Coulombic repulsion between positively charged side chains favors dissociation, and for the same charge state, a higher m/z increases the chance of noncovalent interactions. Remarkably, detected tryptic phosphopeptides have been reported to present a higher average charge state when compared to nonmodified tryptic peptides,80,81 which would favor dissociation of ExD products. However, this increase of average charge state is caused by an increase of trypsin miscleavages due to strong in-solution interactions between arginine/lysine side chains and phosphate groups,82,83 and in reality phosphopeptides exhibit on average lower charge density than nonmodified tryptic peptides.80 Because the ETD reaction is performed in a linear ion trap at relatively high pressure (∼10–3 Torr) in comparison with ECD (∼10–10 Torr), internal energies of the ETD fragment ions are reduced through collisional cooling, which further hampers their subsequent dissociation. Phosphopeptide chemical derivatization, or the addition of a dinuclear zinc complex, which selectively binds to the phosphate group thus increasing the phosphopeptide charge state, notably led to improved ExD.84−86
Moreover, ECnoD/ETnoD is notably problematic for phosphorylated peptides because of the strong intramolecular noncovalent interactions in the gas phase between the phospho-group and the side chains of basic amino acid residues, which have been reported to be able to survive ExD87 and even low-energy CID.88 Cooper and co-workers showed that such gas-phase interactions, identified as being salt bridges and ionic hydrogen bonds, influence the observed fragmentation patterns as well as fragment abundances and overall have deleterious effects on ECD, which can be mitigated by performing ECD on precursors of higher charge state.89,90 Using synthetic model phosphopeptides, they later demonstrated that the structures of phosphopeptides, and not only their sequences influence ECD fragmentation behaviors.91 Moss et al. reported an unusual ECD behavior resulting in the preferred neutral loss of phosphoric acid after ECD (but not ETD) of a phosphorylated pentapeptide, which the authors explained by a dipole-guided electron capture at the arginine side chain.69 While the presence of a phosphate group can influence ExD fragmentation of phosphopeptides, Chen et al. demonstrated that it has little effect on the ECD behavior of phosphoproteins, as fragmentation patterns of phosphorylated and nonphosphorylated proteins were highly similar.92
ExD with Supplemental Activation to Extend Sequence Coverage
In ExD, supplemental activation enables to circumvent some of the above-described issues through the introduction of additional energy to the precursor ions, increasing the efficiency of ExD-based techniques, especially for the fragmentation of lower charge peptides. This extra energy is mainly supplied as vibrational energy via collisional activation or infrared photoexcitation. In contrast with ECD, the collisional cooling occurring during the ETD reaction mitigates the effects of preactivation. CID activation during ECD is nearly impossible due to the high vacuum restrictions of FTICR instruments, while resonance excitation during the ETD reaction has undesirable effects, as it would increase the precursor velocity and impair good spatial overlap between the two ion clouds, thus negatively impacting the ion/ion reaction rate. For this reason, CID activation can either occur before or after ECD or after ETD. ETcaD, during which the charge-reduced precursor is activated by resonance to disrupt noncovalent interactions was introduced in 2007 and greatly alleviated ETnoD issues.93
In 2012, the Heck lab introduced EThcD fragmentation, which corresponds to an all-ions dual fragmentation, i.e., both unreacted species and product ions resulting from ETD are subjected to subsequent HCD fragmentation.94 This resulted in extensive backbone fragmentation via the concomitant generation of both c/z and b/y ion series. EThcD was later applied to phosphoproteomics (Figure 6), leading to significant increases in sequence coverage when compared to HCD or ETD alone, along with an increase in confidence for site localization.95 In addition, EThcD proved to be less time-consuming than ETcaD, due to the shorter activation times of beam-type CID.
Figure 6.

EThcD fragmentation spectrum of a doubly phosphorylated peptide. Complete sequence coverage is achieved while neutral losses are minimal, enabling unambiguous localization of both phosphosites. Reproduced from Frese, C. K.; Zhou, H.; Taus, T.; Altelaar, A. F. M.; Mechtler, K.; Heck, A. J. R.; Mohammed, S. J. Proteome Res.2013, 12, 1520–1525 (ref (95)). Copyright 2013 American Chemical Society.
The Coon lab recently introduced activated ion ETD (AI-ETD) for phosphoproteomics (Figure 7A).80 During AI-ETD, peptides are activated during the ETD reaction by infrared photoactivation, as photon irradiation does not impede the ETD process. Therefore, in contrast with ETcaD and EThcD, the application of supplemental energy does not lead to an increase in duty cycle time. During infrared multiphoton dissociation (IRMPD), analytes are irradiated by low energy photons (λ = 10.6 μm; corresponding to an energy of ∼0.1 eV per photon). Absorption of hundreds of photons over long activation times is necessary to trigger dissociation, thus enabling intramolecular vibrational energy redistribution. For that reason, the stepwise activation process can be compared to the one of slow-heating CID and preferably induces the formation of b/y ions series. Notably, the P–O stretch presents a remarkably high infrared absorption cross-section, making IRMPD of phosphopeptides a particularly efficient process.96,97 As a consequence, AI-ETD of phosphopeptides resulted in the generation of more b/y ions when compared with the dissociation of nonmodified peptides.98
Figure 7.

Comparison of ETD and AI-ETD of a singly phosphorylated, doubly protonated peptide (A). AI-ETD enables complete sequence coverage and confident phosphosite localization. In contrast, during ETD, the ETnoD process is preventing the achievement of full sequence coverage and site localization. The extent of neutral phosphate moiety loss observed upon ETD, ETcaD, EThcD, AI-ETD (at 12 W and 15 W laser power), AI-ETD+ (AI-ETD at 15 W followed by additional IRMPD) and HCD is displayed in parts B and C. Reproduced from Riley, N. M.; Hebert, A. S.; Dürnberger, G.; Stanek, F.; Mechtler, K.; Westphall, M. S.; Coon, J. J. Anal. Chem.2017, 89, 6367–6376 (ref (80)). Copyright 2017 American Chemical Society.
Extent of Neutral Losses Following ETD-Based Activation
Naturally, supplying additional activation energy deposited as vibrational energy can lead to the neutral losses of the phosphate moiety, which could mitigate the benefits of ETD fragmentation. Riley et al. recently investigated neutral loss extent upon fragmentation of endogenous phosphopeptides using different ETD-based techniques.80 As expected, ETD fragmentation did not induce significant neutral losses (Figure 7B,C). ETcaD and EThcD exhibited low phosphate neutral losses, to a similar extent. Observed neutral losses mainly resulted from phosphate loss of b-ions (which is consistent with previous report that b-ions are more prone to neutral losses40). AI-ETD exhibited the highest magnitude of neutral losses among all ETD-based techniques, which can be explained by the slow-heating of all ions and high efficiency of infrared photons absorption by phosphopeptides. These neutral losses however did not impede phosphopeptide identification and localization, as AI-ETD led to the identification of the highest number of localized phosphopeptides.
Noncanonical Phosphorylations
Besides the well-studied pSTY phosphorylations, it is established that phosphorylation can also occur on six other residues (i.e., His, Arg, Lys, Cys, Asp, and Glu).99 The amount of information on the gas-phase fragmentation behavior of these less common phosphorylation events is however still limited in comparison with the STY phosphorylation. Because histidine phosphorylation occurs on the aromatic imidazole side-chain, direct H3PO4 neutral loss via SN2 or elimination mechanisms is impossible and can thus only occur via combined losses of HPO3 and a water molecule. Still, in contrast with CID of tyrosine phosphorylated peptides, a prominent H3PO4 loss is observed upon IT-CID,100,101 alongside less abundant HPO3 and H5PO5 ancillary neutral losses (Figure 8A).102 The high magnitude of neutral losses can be attributed to the lower energy required to break the N–P bond in comparison with the activation threshold of the O–P bond.103 Oslund et al. investigated the source of the water molecules in the combined neutral loss pathway and deduced that the water loss mainly originated from the carboxylic moieties at the C-terminal residue and from aspartate/glutamate side chains.102 When comparing the fragmentation behavior of pHis peptides with those of pSTY peptides, they concluded that the neutral loss triplet was preferentially observed during fragmentation of pHis peptides (∼40% of pHis peptides against less than 5% for pSTY peptides) and used the presence of this specific neutral loss to trigger additional subsequent fragmentation.
Figure 8.

(A) IT-CID fragmentation spectra of phosphopeptides of sequence TSHYSIMAR, phosphorylated at the histidine (top) or adjacent tyrosine residue (bottom). Reproduced from Oslund, R. C.; Kee, J.-M.; Couvillon, A. D.; Bhatia, V. N.; Perlman, D. H.; Muir, T. W. J. Am. Chem. Soc.2014, 136, 12899–12911 (ref (102)). Copyright 2014 American Chemical Society. (B) HCD spectra of a pHis peptide. The phosphosite is unambiguously localized and the neutral loss triplet as well as the low mass diagnostic phosphohistidine immonium ion are observed. Reprinted by permission from Macmillan Publishers Ltd.: NATURE. Widespread bacterial protein histidine phosphorylation revealed by mass spectrometry-based proteomics. Potel, C. M., Lin, M.-H., Heck, A. J. R., Lemeer, S. Nat. Methods2018, Vol. 15, pp. 187–190 (ref (107)). Copyright 2018.
Arginine phosphorylated peptides also exhibited intense H3PO4 neutral loss from the precursor ions upon CID,104,105 sometimes accompanied by H5PO5 loss but not HPO3. Notably, a pArg peptide exhibited the same neutral loss pattern as a pHis peptide.104 As for pHis peptides, the additional water loss originates from the C-terminus or residue side chains containing carboxyl and hydroxyl functions, the extent of observed neutral loss(es) is dependent on the proton mobility and size of the pArg peptide.104
The extent of the phosphate group neutral loss, combined with the fact that it involves losses from at least two distinct functional groups within the peptide complicates accurate localization of N-phosphosites. Moreover, gas-phase rearrangements of N-phosphorylation to the C-terminus or other acceptors upon CID fragmentation have been reported.100,104,106 Low resolution IT-CID and HCD fragmentation of synthetic pArg peptides led to a false localization rate of 10–20%.104 It however has to be noted that such false localization constituted false negative identification of N-phosphorylation (i.e., mislocalization of the N-phosphosite at another residue). Several groups have compared the fragmentation patterns of synthetic pHis peptides with those of identified endogenous pHis peptides to achieve confident identification/localization of pHis peptides,101,102,107 while a pArg spectral library was built to study arginine phosphorylation in a bacterium.108 ETD was also reported to improve confident phosphosite localization of both pArg104,105 and pHis,100 and it was demonstrated that the pHis immonium ion of specific m/z 190.0367 can be used as a diagnostic ion107 (Figure 8B).
Lysine phosphorylation is the least studied of the three possible N-phosphorylations, and its analysis by mass spectrometry is somehow more delicate. While CID was successfully used for the study of pHis/pArg, CID of pLys peptides resulted in the complete loss of the phosphate group, mainly as phosphoric acid.109,110 ECD and ETD enabled the identification of lysine phosphorylated peptides,109,110 but in contrast with pArg, gas-phase scrambling of the phosphate group was observed even upon ETD.111 Concerning cysteine phosphorylation, intense H3PO4 and HPO3 neutral losses were reported upon vibrational activation.112,113 Bertran-Vicente et al. reported that while HCD resulted in complete loss of the phosphate group for some pCys peptides, EThcD permitted the unambiguous localization of cysteine phosphosites.113
We could not find any information concerning the fragmentation behavior of pAsp and pGlu peptides, which can be explained by the fact that no chemical approach to synthesize pAsp and pGlu peptides exists as well as no tailored enrichment method.114 Finally, pyrophosphorylations of Ser and Thr were confidently identified and localized by EthcD fragmentation, following a triggering approach when a diagnostic neutral loss doublet of 98 and 178 Da was observed upon CID fragmentation.115
Identification of Phosphopeptides
Different peptide sequence matching search engines and algorithms coexist, such as Mascot,116 Sequest,117 Andromeda,118 MSAmanda,119 and MS-GF+,120 that all can be used to match experimental tandem mass spectra to peptide sequences contained in a database. In addition to computing a score reflecting the reliability of peptide identification, all these search engines utilize a target-decoy strategy to estimate the false discovery rate (FDR).121 During the decoy search, MS2 spectra are searched against decoy sequences derived from the randomization or reversal of the original target database’s sequences, enabling an estimation of the number of random matches (false positives) in the target search and to control the number of false discoveries, which constitutes a second reliability measure for peptide identification. It could be logical to think that unambiguous identification of phosphopeptides by CID is more difficult than identification of their nonphosphorylated counterparts. Indeed, the fact that neutral losses compete with backbone fragmentation (i.e., there are less b and y ions susceptible to match theoretical fragment masses) combined with the increase of the number of fragments obtainable due to the increase of accessible dissociation channels could hinder the identification of phosphopeptides by standard search engines and give rise to more spurious matches. This would be particularly true in the case of low-resolution ion trap CID, during which substantial nonsequence informative neutral loss from the precursor ion can occur. However, by producing richer spectra due to the possibility of sequential fragmentation events at higher energies, combined mostly with high-resolution determination of fragment masses, beam-type CID/HCD largely circumvents these issues. This was clearly illustrated in the analysis of two large libraries of synthetic phosphopeptides and their nonphosphorylated counterparts (>100 000 peptides each) by HCD fragmentation, which surprisingly revealed that phosphopeptides were in fact easier to identify122 (i.e., the FDR values were significantly lower in comparison with their nonphosphorylated peptides and this at any score value, Figure 9).
Figure 9.
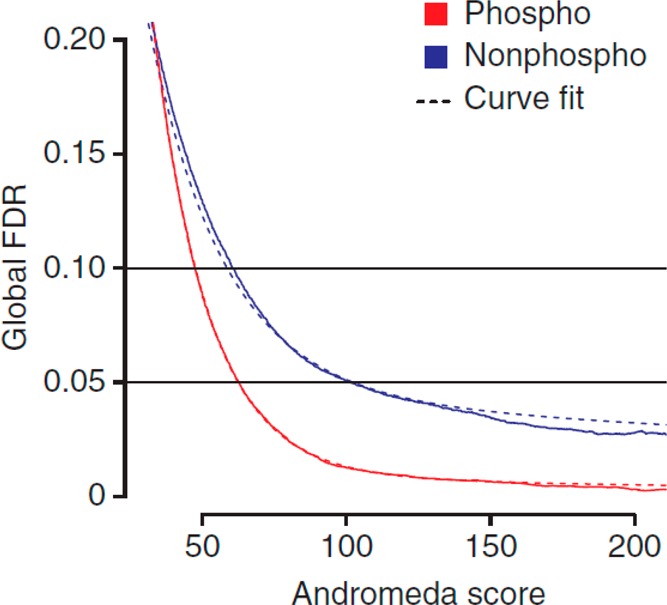
Identification of phosphopeptides using HCD fragmentation seems to be simpler than the identification of their nonphosphorylated counterparts. Here, the false discovery rate (FDR) is plotted as a function of the Andromeda score. Similar trends were observed when the Mascot search engine was used. Reprinted by permission from A large synthetic peptide and phosphopeptide reference library for mass spectrometry-based proteomics. Marx, H., Lemeer, S., Schliep, J. E., Matheron, L., Mohammed, S., Cox, J., Mann, M., Heck, A. J. R., Kuster, B. Nat. Biotechnol.2013, Vol. 31, pp. 557–564 (ref (122)). Copyright Springer Nature, Nature Biotechnology 2013.
Moreover, no significant biases in favor of identification of pS, pT, or pY peptides as well as any biases toward amino acids sequence motifs were observed. The latter has some important implications, as it signifies that observed statistically enriched phosphorylation motifs derive from specific kinase activities and not from enhanced CID fragmentation due to the presence of proline and aspartic acid residues.17 This study, also revealed that in terms of number of identified phosphopeptides, HCD outperformed ETD fragmentation, once again underpinning that neutral losses observed during HCD fragmentation do not significantly impair the identification of phosphopeptides. Surprisingly, in this study, ETD did not surpass HCD fragmentation in identifying highly charged precursors. If the overlap between phosphopeptides identified by both techniques is relatively high (∼70%), it however appears that HCD and ETD remain complementary dissociation methods. It should be noted that the synthetic library was made by sequential permutation of amino acids around well-known phospho-motifs, generating many in sequence (and chemical nature) alike monophosphorylated peptides, which may not fully represent the authentic phosphoproteome.
ETD accompanied by supplemental activation usually gives rise to a higher confidence in phosphopeptide identification, notably through the generation of dual ion series, resulting in higher scores. Riley et al. reported that EThcD and AI-ETD both resulted in an ∼15% increase of the median identification score in comparison with HCD, while ETcaD yields significantly lower scores (∼17% decrease of the median score when compared to HCD).80 In the original EThcD paper, Frese et al. reported an ∼28% increase of the average score using EThcD fragmentation in comparison with HCD.95 Still, as extracted from different available studies comparing different fragmentation techniques, it appears that as of today, HCD remains the gold standard dissociation technique in phosphoproteomics, due to its higher speed and efficiency.80,95,122,123 Alternative dissociation techniques can yield more confident identification scores but come often at the expense of the total number of identified phosphopeptides (Figure 10). Possibly in the future, ExD methods could compete with HCD, if the activation times could be diminished, giving similar duty cycles.
Figure 10.

Comparison and overlap of the number of phosphopeptides identified by using different fragmentation methods. Due to its shorter cycle time, HCD outperforms all ETD-based techniques. Between the ETD-based techniques, AI-ETD yields the highest number of phosphopeptide identifications, followed by EThcD, ETcaD, and finally ETD. Reproduced from Riley, N. M.; Hebert, A. S.; Dürnberger, G.; Stanek, F.; Mechtler, K.; Westphall, M. S.; Coon, J. J. Anal. Chem.2017, 89, pp 6367–6376 (ref (80)). Copyright 2017 American Chemical Society.
Phosphorylation Sites Localization
Although the identification of phosphopeptides by MS-based proteomics has become more facile, a serious challenge in today’s phosphoproteomics remains the unambiguous localization of the phosphosites(s) within the identified phosphopeptides, which is essential to understand the roles of phosphorylation events. While as described above, neutral losses do not impair phosphopeptide identification, it can seriously hamper the correct identification of the localization of the phosphosite, as fragments resulting from a neutral loss and nonmodified fragments (or nonmodified fragments associated with commonly observed water loss in the case of phosphoric acid loss) have the same mass and are thus undistinguishable. Moreover, confident phosphosite localization often requires achieving complete peptide sequence coverage, as several candidate phosphorylation sites (i.e., Ser, Thr, Tyr, His, etc.) are often present within the same peptide. The assessment of phosphosite localization confidence usually requires dedicated additional computational approaches, as phosphosite localizations reported by commonly used search engines can be quite unreliable. To do so, two main paradigms exist: (i) computing the probability of an incorrect match for each phosphopeptide isoforms or (ii) using the difference between scores of the different phosphopeptide isoforms. The former category regroups algorithms such as A-score,124 PhosphoRS (or PtmRS),125 the PTM-score of Andromeda,118 Slomo,126 while the latter corresponds to the Mascot delta score,127 the SLIP score in Protein Prospector,128 and Luciphor.129 Most of these approaches are relatively similar in term of basic principles (the score calculation of most search engines being also probability based), but several factors could explain the reported discrepancies between localization strategies. For example, (i) the type of fragments considered, (ii) the peak depth, (iii) whether the phosphate group neutral losses are used (iv) whether the algorithm was designed for low or high mass resolution and accuracy measurements, (v) how the localization probability is derived from the calculated probabilities of a random match for each candidate.
The pioneering A-score method124 will, for example, only consider site-determining fragments (i.e., only ions enabling to discriminate two candidates), while other algorithms consider all fragments in the localization probability calculation. For the PTM-score, neutral loss fragments will be automatically considered if they result in an increase of the score while in phosphoRS, the user can choose to use such fragments. In the case of fragmentation techniques inducing substantial neutral losses, considering such peaks is resulting in a significant increase of the number of localized phosphosites.80 Nevertheless, one has to keep in mind that such fragments possess the same mass than nonmodified fragments that suffered water loss, which is commonly observed upon collisional activation and as such it has been demonstrated that considering phosphate neutral losses can lead to an increase of the false localization rate.130
In the following, we will focus on two of the most commonly used algorithms, phosphoRS and the PTM-score of Andromeda to demonstrate how localization probabilities are calculated and to illustrate potential differences between localization algorithms. The peak depth, i.e., how many of the most intense peaks per m/z windows are considered for score calculation influences the probability calculation, as the more peaks are considered, the more the probability for a match to be random match increases. Peak picking enables to eliminate low intensity peaks that can originate from electric or chemical noise or coisolation of other precursor ions, both of which can interfere with correct localization. The way of determining the peak depth for each spectrum is different between the two algorithms: it will be determined as the peak depth value yielding the higher score for the PTM-score or as the higher score difference between the different phosphopeptide isoforms if site-determining fragments are present in phosphoRS. For the calculation of the PTM-score, the peak depth is uniform across all m/z windows, while it is individually determined for each m/z window in phosphoRS. After peak picking, a value of n matching theoretical fragments out of the N total number of peaks contained in the spectra is obtained. The probability of each phosphopeptide isoform match to be random is then determined by calculation of the probability of n matched peaks out of a total of N peaks to be random (in other words, the smaller the probability, the more confident the match), by applying a cumulative binomial distribution:
The calculation of the probability p of a random peak to be matched is also different between the two algorithms. For the PTM-score calculation, p is equal to the peak depth divided by the m/z window (100 m/z). This derives from the fact that the PTM-score was first introduced for ion trap detection, for which a mass tolerance of 0.5 Da is common (i.e., there is 4% of chance of a peak to be a random match if 4 peaks are picked in a 100 m/z window). The PTM-score is thus using a conservative way of calculating p and significantly overestimates its value in the case of high-resolution data. In phosphoRS, p is defined as
N corresponds to the total number of picked peaks, d to the fragment mass tolerance, and w to the full mass range of the spectrum. Hence, the value of p is adapted to the mass accuracy of the mass analyzer. Finally, in both cases localization probabilities are inferred from the differences of the values of P calculated for the different isoforms (Figure 11).
Figure 11.

Workflows for the calculation of phosphosite localization probabilities by phosphoRS (left), and MaxQuant/Andromeda (PTM-score, right). Reproduced from Taus, T.; Köcher, T.; Pichler, P.; Paschke, C.; Schmidt, A.; Henrich, C.; Mechtler, K.J. Proteome Res.2011, 10, 5354–5362 (ref (125)). Copyright 2011 American Chemical Society. Reproduced from Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R. A.; Olsen, J. V.; Mann, M. J. Proteome Res.2011, 10, 1794–1805 (ref (118)). Copyright 2011 American Chemical Society. Reprinted from Cell Rep., Vol. 8, Sharma, K., D’Souza, R. C. J., Tyanova, S., Schaab, C., Wiśniewski, J. R., Cox, J., Mann, M. Ultradeep Human Phosphoproteome Reveals a Distinct Regulatory Nature of Tyr and Ser/Thr-Based Signaling. pp. 1583–1594. (ref (131)). Copyright 2014, with permission from Elsevier.
False Localization Rate Estimation
Most of the localization algorithms score the confidence of the localization but do not estimate the false localization rate (FLR), and instead arbitrary score cut-offs are used, such as the 0.75 cutoff recommended for the Andromeda-PTMscore, corresponding to so-called class I phosphosites.1 Indeed, the estimation of the FLR is less straightforward than the FDR estimation as a similar target-decoy approach is no longer valid, as for an identified phosphopeptide with a given sequence an incorrect localization of a phosphosite does not correspond to a random match as many fragments will match both correct and incorrect localizations.132 Therefore, reversal/randomization of sequences contained in the database will not provide an accurate estimation of false localizations. However, the addition of noncanonical phosphorylations into the equation, combined with the continuous increase in the number of MS2 spectra generated per experiment obviously increases the risk of false localization, making FLR control more and more essential, as it enables the user to make an informed decision about localization score cutoff. One elegant strategy, used in the SLIP approach,128 is to perform searches during which decoy phosphorylations on glutamic acid (E) and proline (P) residues are allowed. There are two rationales behind the choice of these two amino acids: the combined occurrence of E and P matches the one of S and T (∼16%), and E and P residues are present in some of the most dominant phosphorylation motifs, ensuring the presence of decoy sites in the vicinity of the real phosphosites. Luciphor also uses a target decoy approach to estimate the FLR, in which decoy phosphopeptides are generated by placing the phosphorylation on each residue present in the sequence.129 As the number of decoy sites is usually higher than the number of target sites, this constitutes a more conservative approach.
Performance Comparison between Different Phosphosite Localization Tools
While all localization tools perform adequately (i.e., present a low FLR), comparing their performance can be complicated by the fact that most localization tools are tied to specific search engines. This is for example the case for two of the most popular localization tools, ptmRS and the PTM-score.133 It is thus difficult to determine if observed discrepancies in confidently localized phosphosites originate from the difference between search engines or from the localization tools. Such differences between the different search engines will not be discussed here. Analyzing synthetic phosphopeptide libraries, for which the correct phosphorylation sites are known is the only situation in which the true FLR can be determined. However, one has to keep in mind that analyzing synthetic peptides often does not reflect the complexity of real biological samples, which present higher dynamic range and/or coisolation of peptides, both resulting in the potential generation of lower quality spectra.
Marx et al. compared the PTM-score, phosphoRS, and the Mascot delta score performances on correctly localizing phosphosites from a large phosphopeptides library.122 Keeping both the FDR and FLR at 1%, the Andromeda-PTM-score and Mascot-phosphoRS score proved to be significantly more sensitive than the Mascot delta score and exhibited similar numbers of correctly identified and localized phosphopeptides. However, phosphosites localized by the two search engine/localization tools presented a low overlap of around 50% after analysis of both the synthetic phosphopeptides libraries and a biological sample. Similarly, phosphoRS, A-score, and MD-score only presented an overlap of 50% when considering unique unambiguously localized phosphosites from a biological sample after database search with the same search algorithm,125 indicating that there is still much room for improvement in computational strategies for confident phosphosite localization. Notably, all localization tools slightly underestimated the false localization rate at high localization probability scores. In contrast, at a 1% FDR at the phosphopeptides level, false discovery rates were significantly overestimated at lower localization probability,125,134 illustrating that this discussion is still not closed. Finally, to our knowledge, the robustness of these search algorithms has not been tested against large libraries of multiple phosphorylated peptides.
Besides peak-probability based approaches, simulating HCD spectra of phosphopeptides of enzymatically dephosphorylated peptides, while laborious, enabled accurate phosphosite localization and presented a lower FLR than A-score and ptmRS.130 Recently, Yang et al. developed a semi supervised learning algorithm to assess the confidence of each amino acids obtained by de novo sequencing.135 Interestingly, this algorithm was used to calculate localization probabilities of phosphorylation sites within phosphopeptides identified by either de novo sequencing or database search and significantly outperformed the peak-probability based algorithms A-score and ptmRS both in terms of number of identifications and localization accuracy after a database search of synthetic phosphopeptides libraries.
Comparison of Different Fragmentation Techniques
While we concluded that HCD performs very well in term of identification of phosphopeptides, it has been demonstrated that confident localization of a phosphosite becomes increasingly difficult when the number of putative phosphosites increase, and the closer another phosphorylation site candidate is from the actual phosphosite.122,134 Interestingly, this issue is much less observed with ETD fragmentation, underlining that both the lack of neutral loss during ETD and the more comprehensive coverage achieved via ETD random cleavages are beneficial for correct phosphosite localization.122 More generally, localization probability cutoff to reach a 1% FLR is significantly higher in the case of HCD when compared to ETD-based fragmentation methods.122,125 By using the Marx et al. data set, Wiese et al. demonstrated that in the case of ETD fragmentation, a localization probability cutoff of 0.55 (lowest reported localization probability threshold) was sufficient to fall below 1% FLR after analyzing the data with the combination the Andromeda-PTM-score at a 1% FDR.134 In contrast, the authors reported that a cutoff of 1 (maximum localization probability) was necessary to reach a FLR below 1% in the case of HCD fragmentation. Ferries et al. recently reported similar results with a different, albeit significantly smaller synthetic phosphopeptide library.123 They showed that with EThcD fragmentation, the lowest localization probabilities calculated correspond to a FLR below 1% (Figure 12). This is in agreement with the initial report on EThcD of Frese et al., who reported that EThcD fragmentation led to a significant increase of the proportion of correct phosphosite localizations when compared to HCD.95
Figure 12.
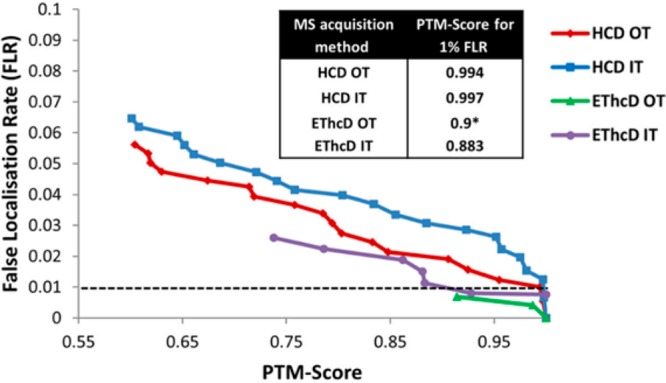
False localization rate determination in four different analytical peptide fragmentation strategies: HCD and EThcD fragmentation with high-resolution (Orbitrap, OT) or low-resolution (Ion trap, IT) fragment ion detection. Reproduced from Ferries, S.; Perkins, S.; Brownridge, P. J.; Campbell, A.; Eyers, P. A.; Jones, A. R.; Eyers, C. E. J. Proteome Res.2017, Vol. 16, pp 3448–3459 (ref (123)). Copyright 2017 American Chemical Society.
In addition, Ferries et al. also reported that high-resolution measurements of fragments (i.e., within the Orbitrap) strongly aids to reduce false localizations. The Olsen group showed that increasing Orbitrap resolution to 60k after HCD fragmentation as well as the maximum injection time resulted in an increase in localization probabilities through the increase of spectra quality (at the cost of longer cycle times).136 Both Marx et al. and Wiese et al. concluded that tyrosine phosphorylation is easier to localize by HCD than serine and threonine phosphorylation.122,134 This can be explained by the low tendency of pY peptides to undergo neutral loss. Wiese et al. reported that a PTM-score localization probability cutoff of 0.86 was sufficient to reach a FLR value below 1%, indicating that in the case of pY peptides, HCD is the preferred fragmentation technique. Finally, Ferries et al. showed that localization confidence logically decreases with the number of phosphorylation present on a peptide, ETD-based techniques being more suited for the accurate localization of multiple phosphorylation.123 This is of importance as multiphosphorylated peptides represented between 20 and 40% of identified phosphopeptides in recent phosphoproteomics studies on mammalian cells.81,137
Concluding Remarks
The field of MS-based phosphoproteomics has seriously matured over the past decade and has been adopted by dozens of research groups worldwide. Notwithstanding the achieved successes and wide adaptation by the community, one should not forget key remaining challenges, which can only be tackled by introducing new technologies. To make such further advances in MS-based phosphoproteomics, a deep understanding of peptide fragmentation mechanisms is essential, whereby we hope that this review provides the interested researcher a good starting point by summarizing the current knowledge. With new peptide activation methods in mass spectrometry also come new search algorithms and more advanced algorithms to determine with higher accuracy the exact amino-acid being modified by the phosphorylation. Such information is critical for the understanding of the role of that particular event. Our review of the literature clearly illustrates that especially confident site localization is a remaining challenge, whereby current approaches still disagree with each other too much.
Several new technologies are on the horizon for MS-based phosphoproteomics that we intently, and due to space limitations, did not discuss here. Alternative radical-induced dissociation techniques presenting different fragmentation patterns when compared to classic ExD have recently shown interesting preliminary results when applied to the fragmentation of phosphopeptides,138−140 but it remains unsure if they will be compatible with high-throughput phosphoproteomics. Among the different photoactivation-based dissociation methods applied to phosphopeptides sequencing,141−144 UV-photodissociation (UV-PD) has emerged as a credible alternative to CID/ExD. UV-PD has been exploited to phosphopeptides to identify and site-localize the phosphate moiety. Early results indicate that UV-PD is compatible with the high-throughput identification of phosphopeptides while the propensity of the phosphate group to remain bound to the peptide during fragmentation increases when compared to HCD.145−147 Top-down and middle-down phosphoproteomics approaches, which have greatly benefited from recent technological advances in alternative fragmentation techniques may improve the identification and site localization of protein phosphorylations and be ideally suited to not only map multiple modifications on a peptide or protein at once but also determine phospho-isoform abundance, reaction kinetics and to study functional cross-talk between post-translational modifications.80,148−155
While ionization in negative ion mode still suffers from a lack of efficiency when compared with the positive ion mode, negative ETD, UVPD, and AI-ETD showed some promising results and could help to study acid-labile phosphorylation events.146,156−158 Ion mobility separation coupled to MS can provide and additional layer of separation, allowing potentially the separation of phosphopeptide isomers.159−161 The availability of such synthetic phosphopeptide isomers can also be used in targeted MS approaches, whereby the retention time of the peptide and the comparison with the isotopically labeled standard aid the confidence in identification, site localization, and quantification.162,163 In short, a lot of effort is made by the proteomics research community to tackle important persisting issues in MS based phosphoproteomics. It will be interesting to see in a decade from now what the field will have further achieved. For sure a lot of novel biological new insights, hopefully based on well-reproducible and confident phosphoproteomics data.
Acknowledgments
We thank all group members for their helpful contributions. We acknowledge financial support by the large-scale proteomics facility Proteins@Work (Project 184.032.201) embedded in The Netherlands Proteomics Centre and supported by The Netherlands Organization for Scientific Research (NWO). Additional support came through the European Union Horizon 2020 program FET-OPEN Project MSmed (Project 686547), and the European Union Horizon 2020 Program INFRAIA Project Epic-XS (Project 823839). S.L. acknowledges support from The Netherlands Organization for Scientific Research (NWO) through a VIDI Grant (Project 723.013.008) and A.J.R.H. through the Spinoza Award SPI.2017.028.
Biographies
Clement M. Potel (1987) is currently a postdoctoral researcher at the EMBL (Heidelberg, Germany). He obtained his Master’s degree in analytical chemistry from the University of Strasbourg, France, and his Ph.D. from the University of Utrecht, The Netherlands, in 2018. His research focuses on the development of new analytical strategies in the field of phosphoproteomics, with a focus on the study of the elusive histidine phosphorylation and bacterial signaling.
Simone Lemeer (1978) received her Ph.D. in 2008 from Utrecht University, The Netherlands. Until 2014, she worked as a postdoctoral researcher and later junior group leader at the Chair of Bioanalytics at the Technical University Munich, Germany. In 2014 she became Associate Professor in Biomolecular Mass Spectrometry and Proteomics at Utrecht University, The Netherlands. Her research focuses on the development of methods for the enrichment and analysis of phosphopeptides and the correct site localization. In addition, she uses this technology to study phosphorylation mediated signaling in health and disease.
Albert J. R. Heck (1964) received his Ph.D. in 1993 from the University of Amsterdam. He worked until 1996 as a postdoctoral researcher in the Department of Chemistry at Stanford University and Sandia National Laboratories (USA). In 1996, he became senior research fellow and later lecturer at the University of Warwick (U.K.). Since 1998 he is professor in Biomolecular Mass Spectrometry and Proteomics at Utrecht University, The Netherlands. Heck is recipient of the ACS Frank H. Field and Joe L. Franklin Award and the NWO Spinoza Prize. He is member of EMBO and the Royal Netherlands Academy of Sciences and Arts. His research focuses on the development and applications of mass spectrometry-based proteomics. Heck introduced technologies for phospho-enrichment and the use of alternative proteases and hybrid peptide fragmentation techniques. Besides, Heck is also known for his expertise in structural biology and a pioneer in native mass spectrometry and cross-linking mass spectrometry.
The authors declare no competing financial interest.
References
- Olsen J. V.; et al. Global, In Vivo, and Site-Specific Phosphorylation Dynamics in Signaling Networks. Cell 2006, 127, 635–648. 10.1016/j.cell.2006.09.026. [DOI] [PubMed] [Google Scholar]
- Ficarro S. B.; et al. Phosphoproteome analysis by mass spectrometry and its application to Saccharomyces cerevisiae.. Nat. Biotechnol. 2002, 20, 301–305. 10.1038/nbt0302-301. [DOI] [PubMed] [Google Scholar]
- Chi A.; et al. Analysis of phosphorylation sites on proteins from Saccharomyces cerevisiae by electron transfer dissociation (ETD) mass spectrometry. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 2193–2198. 10.1073/pnas.0607084104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen J. V.; Mann M. Status of Large-scale Analysis of Post-translational Modifications by Mass Spectrometry. Mol. Cell. Proteomics 2013, 12, 3444–3452. 10.1074/mcp.O113.034181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley N. M.; Coon J. J. Phosphoproteomics in the Age of Rapid and Deep Proteome Profiling. Anal. Chem. 2016, 88, 74–94. 10.1021/acs.analchem.5b04123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palumbo A. M.; et al. Tandem mass spectrometry strategies for phosphoproteome analysis. Mass Spectrom. Rev. 2011, 30, 600–625. 10.1002/mas.20310. [DOI] [PubMed] [Google Scholar]
- White F. M.; Wolf-Yadlin A. Methods for the Analysis of Protein Phosphorylation–Mediated Cellular Signaling Networks. Annu. Rev. Anal. Chem. 2016, 9, 295–315. 10.1146/annurev-anchem-071015-041542. [DOI] [PubMed] [Google Scholar]
- Lemeer S.; Heck A. J. The phosphoproteomics data explosion. Curr. Opin. Chem. Biol. 2009, 13, 414–420. 10.1016/j.cbpa.2009.06.022. [DOI] [PubMed] [Google Scholar]
- Deribe Y. L.; Pawson T.; Dikic I. Post-translational modifications in signal integration. Nat. Struct. Mol. Biol. 2010, 17, 666–672. 10.1038/nsmb.1842. [DOI] [PubMed] [Google Scholar]
- Lemmon M. A.; Schlessinger J. Cell Signaling by Receptor Tyrosine Kinases. Cell 2010, 141, 1117–1134. 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishi H.; Hashimoto K.; Panchenko A. R. Phosphorylation in Protein-Protein Binding: Effect on Stability and Function. Structure 2011, 19, 1807–1815. 10.1016/j.str.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kholodenko B. N. Cell-signalling dynamics in time and space. Nat. Rev. Mol. Cell Biol. 2006, 7, 165–176. 10.1038/nrm1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P. The role of protein phosphorylation in human health and disease. The Sir Hans Krebs Medal Lecture. Eur. J. Biochem. 2001, 268, 5001–5010. 10.1046/j.0014-2956.2001.02473.x. [DOI] [PubMed] [Google Scholar]
- Blume-Jensen P.; Hunter T. Oncogenic kinase signalling. Nature 2001, 411, 355. 10.1038/35077225. [DOI] [PubMed] [Google Scholar]
- Brognard J.; Hunter T. Protein kinase signaling networks in cancer. Curr. Opin. Genet. Dev. 2011, 21, 4–11. 10.1016/j.gde.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boersema P. J.; Mohammed S.; Heck A. J. R. Phosphopeptide fragmentation and analysis by mass spectrometry. J. Mass Spectrom. 2009, 44, 861–878. 10.1002/jms.1599. [DOI] [PubMed] [Google Scholar]
- Paizs B.; Suhai S. Fragmentation pathways of protonated peptides. Mass Spectrom. Rev. 2005, 24, 508–548. 10.1002/mas.20024. [DOI] [PubMed] [Google Scholar]
- Mayer P. M.; Poon C. The mechanisms of collisional activation of ions in mass spectrometry. Mass Spectrom. Rev. 2009, 28, 608–639. 10.1002/mas.20225. [DOI] [PubMed] [Google Scholar]
- Shukla A. K.; Futrell J. H. Tandem mass spectrometry: dissociation of ions by collisional activation. J. Mass Spectrom. 2000, 35, 1069–1090. 10.1002/1096-9888(200009)35:9<1069::AID-JMS54>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Mitchell Wells J.; McLuckey S. A. Collision-Induced Dissociation (CID) of Peptides and Proteins. In Methods in Enzymology; Academic Press, 2005; Vol. 402, pp 148–185. [DOI] [PubMed] [Google Scholar]
- McLuckey S. A.; Goeringer D. E. SPECIAL FEATURE: TUTORIAL Slow Heating Methods in Tandem Mass Spectrometry. J. Mass Spectrom. 1997, 32, 461–474. 10.1002/(SICI)1096-9888(199705)32:5<461::AID-JMS515>3.0.CO;2-H. [DOI] [Google Scholar]
- Dongré A. R.; Jones J. L.; Somogyi Á.; Wysocki V. H. Influence of Peptide Composition, Gas-Phase Basicity, and Chemical Modification on Fragmentation Efficiency: Evidence for the Mobile Proton Model. J. Am. Chem. Soc. 1996, 118, 8365–8374. 10.1021/ja9542193. [DOI] [Google Scholar]
- Wysocki V. H.; Tsaprailis G.; Smith L. L.; Breci L. A. Mobile and localized protons: a framework for understanding peptide dissociation. J. Mass Spectrom. 2000, 35, 1399–1406. 10.1002/1096-9888(200012)35:12<1399::AID-JMS86>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Jones J. L.; Dongre A. R.; Somogyi A.; Wysocki V. H. Sequence Dependence of Peptide Fragmentation Efficiency Curves Determined by Electrospray Ionization/Surface-Induced Dissociation Mass Spectrometry. J. Am. Chem. Soc. 1994, 116, 8368–8369. 10.1021/ja00097a055. [DOI] [Google Scholar]
- Tsaprailis G.; et al. Influence of Secondary Structure on the Fragmentation of Protonated Peptides. J. Am. Chem. Soc. 1999, 121, 5142–5154. 10.1021/ja982980h. [DOI] [Google Scholar]
- Cox K. A.; Gaskell S. J.; Morris M.; Whiting A. Role of the site of protonation in the low-energy decompositions of gas-phase peptide ions. J. Am. Soc. Mass Spectrom. 1996, 7, 522–531. 10.1016/1044-0305(96)00019-0. [DOI] [PubMed] [Google Scholar]
- Summerfield S. G.; Whiting A.; Gaskell S. J. Intra-ionic interactions in electrosprayed peptide ions. Int. J. Mass Spectrom. Ion Processes 1997, 162, 149–161. 10.1016/S0168-1176(96)04512-0. [DOI] [Google Scholar]
- Kapp E. A.; et al. Mining a Tandem Mass Spectrometry Database To Determine the Trends and Global Factors Influencing Peptide Fragmentation. Anal. Chem. 2003, 75, 6251–6264. 10.1021/ac034616t. [DOI] [PubMed] [Google Scholar]
- Palumbo A. M.; Tepe J. J.; Reid G. E. Mechanistic Insights into the Multistage Gas-Phase Fragmentation Behavior of Phosphoserine- and Phosphothreonine-Containing Peptides. J. Proteome Res. 2008, 7, 771–779. 10.1021/pr0705136. [DOI] [PubMed] [Google Scholar]
- Gronert S.; Li K. H.; Horiuchi M. Manipulating the fragmentation patterns of phosphopeptides via gas-phase boron derivatization: Determining phosphorylation sites in peptides with multiple serines. J. Am. Soc. Mass Spectrom. 2005, 16, 1905–1914. 10.1016/j.jasms.2005.07.018. [DOI] [PubMed] [Google Scholar]
- Rožman M. Modelling of the gas-phase phosphate group loss and rearrangement in phosphorylated peptides. J. Mass Spectrom. 2011, 46, 949–955. 10.1002/jms.1974. [DOI] [PubMed] [Google Scholar]
- Lanucara F.; et al. Kinetic control in the CID-induced elimination of H3PO4 from phosphorylated serine probed using IRMPD spectroscopy. Chem. Commun. 2014, 50, 3845–3848. 10.1039/C4CC00877D. [DOI] [PubMed] [Google Scholar]
- Tholey A.; Reed J.; Lehmann W. D. Electrospray tandem mass spectrometric studies of phosphopeptides and phosphopeptide analogues. J. Mass Spectrom. 1999, 34, 117–123. 10.1002/(SICI)1096-9888(199902)34:2<117::AID-JMS769>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Lanucara F.; Lee D. C.H.; Eyers C. E. Unblocking the Sink: Improved CID-Based Analysis of Phosphorylated Peptides by Enzymatic Removal of the Basic C-Terminal Residue. J. Am. Soc. Mass Spectrom. 2014, 25, 214–225. 10.1007/s13361-013-0770-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskin J.; Kong R. P. W.; Song T.; Chu I. K. Effect of the basic residue on the energetics and dynamics of dissociation of phosphopeptides. Int. J. Mass Spectrom. 2012, 330–332, 295–301. 10.1016/j.ijms.2012.09.013. [DOI] [Google Scholar]
- Cui L.; Yapici I.; Borhan B.; Reid G. E. Quantification of Competing H3PO4 Versus HPO3 + H2O Neutral Losses from Regioselective 18O-Labeled Phosphopeptides. J. Am. Soc. Mass Spectrom. 2014, 25, 141–148. 10.1007/s13361-013-0744-4. [DOI] [PubMed] [Google Scholar]
- Cui L.; Reid G. E. Examining factors that influence erroneous phosphorylation site localization via competing fragmentation and rearrangement reactions during ion trap CID-MS/MS and -MS3. Proteomics 2013, 13, 964–973. 10.1002/pmic.201200384. [DOI] [PubMed] [Google Scholar]
- Cotham V. C.; McGee W. M.; Brodbelt J. S. Modulation of Phosphopeptide Fragmentation via Dual Spray Ion/Ion Reactions Using a Sulfonate-Incorporating Reagent. Anal. Chem. 2016, 88, 8158–8165. 10.1021/acs.analchem.6b01901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svane S.; Kryuchkov F.; Lennartson A.; McKenzie C. J.; Kjeldsen F. Overcoming the Instability of Gaseous Peptide Phosphate Ester Groups by Dimetal Protection. Angew. Chem., Int. Ed. 2012, 51, 3216–3219. 10.1002/anie.201108481. [DOI] [PubMed] [Google Scholar]
- Brown R.; Stuart S. S.; Houel S.; Ahn N. G.; Old W. M. Large-Scale Examination of Factors Influencing Phosphopeptide Neutral Loss during Collision Induced Dissociation. J. Am. Soc. Mass Spectrom. 2015, 26, 1128–1142. 10.1007/s13361-015-1109-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeGnore J.; Qin J. Fragmentation of phosphopeptides in an ion trap mass spectrometer. J. Am. Soc. Mass Spectrom. 1998, 9, 1175–1188. 10.1016/S1044-0305(98)00088-9. [DOI] [PubMed] [Google Scholar]
- Everley R. A.; Huttlin E. L.; Erickson A. R.; Beausoleil S. A.; Gygi S. P. Neutral Loss Is a Very Common Occurrence in Phosphotyrosine-Containing Peptides Labeled with Isobaric Tags. J. Proteome Res. 2017, 16, 1069–1076. 10.1021/acs.jproteome.6b00487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linke D.; Hung C.-W.; Cassidy L.; Tholey A. Optimized Fragmentation Conditions for iTRAQ-labeled Phosphopeptides. J. Proteome Res. 2013, 12, 2755–2763. 10.1021/pr400113n. [DOI] [PubMed] [Google Scholar]
- Erickson B. K.; et al. Evaluating Multiplexed Quantitative Phosphopeptide Analysis on a Hybrid Quadrupole Mass Filter/Linear Ion Trap/Orbitrap Mass Spectrometer. Anal. Chem. 2015, 87, 1241–1249. 10.1021/ac503934f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaraj N.; D’Souza R. C. J.; Cox J.; Olsen J. V.; Mann M. Feasibility of Large-Scale Phosphoproteomics with Higher Energy Collisional Dissociation Fragmentation. J. Proteome Res. 2010, 9, 6786–6794. 10.1021/pr100637q. [DOI] [PubMed] [Google Scholar]
- Palumbo A. M.; Reid G. E. Evaluation of Gas-Phase Rearrangement and Competing Fragmentation Reactions on Protein Phosphorylation Site Assignment Using Collision Induced Dissociation-MS/MS and MS3. Anal. Chem. 2008, 80, 9735–9747. 10.1021/ac801768s. [DOI] [PubMed] [Google Scholar]
- Aguiar M.; Haas W.; Beausoleil S. A.; Rush J.; Gygi S. P. Gas-Phase Rearrangements Do Not Affect Site Localization Reliability in Phosphoproteomics Data Sets. J. Proteome Res. 2010, 9, 3103–3107. 10.1021/pr1000225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mischerikow N.; Altelaar A. F. M.; Navarro J. D.; Mohammed S.; Heck A. J. R. Comparative Assessment of Site Assignments in CID and Electron Transfer Dissociation Spectra of Phosphopeptides Discloses Limited Relocation of Phosphate Groups. Mol. Cell. Proteomics 2010, 9, 2140–2148. 10.1074/mcp.M900619-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen J. V.; et al. Higher-energy C-trap dissociation for peptide modification analysis. Nat. Methods 2007, 4, 709–712. 10.1038/nmeth1060. [DOI] [PubMed] [Google Scholar]
- Kelstrup C. D.; Hekmat O.; Francavilla C.; Olsen J. V. Pinpointing Phosphorylation Sites: Quantitative Filtering and a Novel Site-specific x-Ion Fragment. J. Proteome Res. 2011, 10, 2937–2948. 10.1021/pr200154t. [DOI] [PubMed] [Google Scholar]
- Diedrich J. K.; Pinto A. F.M.; Yates J. R. Energy dependence of HCD on peptide fragmentation: stepped collisional energy finds the sweet spot. J. Am. Soc. Mass Spectrom. 2013, 24, 1690–1699. 10.1007/s13361-013-0709-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder M. J.; Shabanowitz J.; Schwartz J. C.; Hunt D. F.; Coon J. J. A Neutral Loss Activation Method for Improved Phosphopeptide Sequence Analysis by Quadrupole Ion Trap Mass Spectrometry. Anal. Chem. 2004, 76, 3590–3598. 10.1021/ac0497104. [DOI] [PubMed] [Google Scholar]
- Michalski A.; Neuhauser N.; Cox J.; Mann M. A Systematic Investigation into the Nature of Tryptic HCD Spectra. J. Proteome Res. 2012, 11, 5479–5491. 10.1021/pr3007045. [DOI] [PubMed] [Google Scholar]
- Jedrychowski M. P. Evaluation of HCD- and CID-type Fragmentation Within Their Respective Detection Platforms For Murine Phosphoproteomics. Mol. Cell. Proteomics 2011, 10, M111.009910. 10.1074/mcp.M111.009910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frese C. K.; et al. Improved Peptide Identification by Targeted Fragmentation Using CID, HCD and ETD on an LTQ-Orbitrap Velos. J. Proteome Res. 2011, 10, 2377–2388. 10.1021/pr1011729. [DOI] [PubMed] [Google Scholar]
- Pilo A. L.; Peng Z.; McLuckey S. A. The dehydroalanine effect in the fragmentation of ions derived from polypeptides. J. Mass Spectrom. 2016, 51, 857–866. 10.1002/jms.3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papayannopoulos I. A. The interpretation of collision-induced dissociation tandem mass spectra of peptides. Mass Spectrom. Rev. 1995, 14, 49–73. 10.1002/mas.1280140104. [DOI] [Google Scholar]
- Medzihradszky K. F.; Chalkley R. J. Lessons in de novo peptide sequencing by tandem mass spectrometry. Mass Spectrom. Rev. 2015, 34, 43–63. 10.1002/mas.21406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steen H.; Küster B.; Fernandez M.; Pandey A.; Mann M. Detection of Tyrosine Phosphorylated Peptides by Precursor Ion Scanning Quadrupole TOF Mass Spectrometry in Positive Ion Mode. Anal. Chem. 2001, 73, 1440–1448. 10.1021/ac001318c. [DOI] [PubMed] [Google Scholar]
- Sleno L.; Volmer D. A. Ion activation methods for tandem mass spectrometry. J. Mass Spectrom. 2004, 39, 1091–1112. 10.1002/jms.703. [DOI] [PubMed] [Google Scholar]
- Zubarev R. A.; Kelleher N. L.; McLafferty F. W. Electron Capture Dissociation of Multiply Charged Protein Cations. A Nonergodic Process. J. Am. Chem. Soc. 1998, 120, 3265–3266. 10.1021/ja973478k. [DOI] [Google Scholar]
- Syka J. E. P.; Coon J. J.; Schroeder M. J.; Shabanowitz J.; Hunt D. F. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 9528–9533. 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voinov V. G.; Bennett S. E.; Beckman J. S.; Barofsky D. F. ECD of tyrosine phosphorylation in a triple quadrupole mass spectrometer with a radio-frequency-free electromagnetostatic cell. J. Am. Soc. Mass Spectrom. 2014, 25, 1730–1738. 10.1007/s13361-014-0956-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fort K. L.; et al. Exploring ECD on a Benchtop Q Exactive Orbitrap Mass Spectrometer. J. Proteome Res. 2018, 17, 926–933. 10.1021/acs.jproteome.7b00622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tureček F.; Julian R. R. Peptide Radicals and Cation Radicals in the Gas Phase. Chem. Rev. 2013, 113, 6691–6733. 10.1021/cr400043s. [DOI] [PubMed] [Google Scholar]
- Zhurov K. O.; Fornelli L.; Wodrich M. D.; Laskay Ü. A.; Tsybin Y. O. Principles of electron capture and transfer dissociation mass spectrometry applied to peptide and protein structure analysis. Chem. Soc. Rev. 2013, 42, 5014–5030. 10.1039/c3cs35477f. [DOI] [PubMed] [Google Scholar]
- Qi Y.; Volmer D. A. Electron-based fragmentation methods in mass spectrometry: An overview. Mass Spectrom. Rev. 2017, 36, 4–15. 10.1002/mas.21482. [DOI] [PubMed] [Google Scholar]
- Anusiewicz I.; Skurski P.; Simons J. Refinements to the Utah–Washington Mechanism of Electron Capture Dissociation. J. Phys. Chem. B 2014, 118, 7892–7901. 10.1021/jp5004819. [DOI] [PubMed] [Google Scholar]
- Moss C. L.; et al. Dipole-Guided Electron Capture Causes Abnormal Dissociations of Phosphorylated Pentapeptides. J. Am. Soc. Mass Spectrom. 2011, 22, 731–751. 10.1007/s13361-011-0083-2. [DOI] [PubMed] [Google Scholar]
- Shi S. D.-H.; et al. Phosphopeptide/Phosphoprotein Mapping by Electron Capture Dissociation Mass Spectrometry. Anal. Chem. 2001, 73, 19–22. 10.1021/ac000703z. [DOI] [PubMed] [Google Scholar]
- Stensballe A.; Jensen O. N.; Olsen J. V.; Haselmann K. F.; Zubarev R. A. Electron capture dissociation of singly and multiply phosphorylated peptides. Rapid Commun. Mass Spectrom. 2000, 14, 1793–1800. 10.1002/1097-0231(20001015)14:19<1793::AID-RCM95>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Molina H.; Horn D. M.; Tang N.; Mathivanan S.; Pandey A. Global proteomic profiling of phosphopeptides using electron transfer dissociation tandem mass spectrometry. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 2199–2204. 10.1073/pnas.0611217104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweet S. M. M.; Bailey C. M.; Cunningham D. L.; Heath J. K.; Cooper H. J. Large Scale Localization of Protein Phosphorylation by Use of Electron Capture Dissociation Mass Spectrometry. Mol. Cell. Proteomics 2009, 8, 904–912. 10.1074/mcp.M800451-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zubarev R. A.; et al. Electron Capture Dissociation for Structural Characterization of Multiply Charged Protein Cations. Anal. Chem. 2000, 72, 563–573. 10.1021/ac990811p. [DOI] [PubMed] [Google Scholar]
- McLuckey S. A.; Stephenson J. L. Ion/ion chemistry of high-mass multiply charged ions. Mass Spectrom. Rev. 1998, 17, 369–407. 10.1002/(SICI)1098-2787(1998)17:6<369::AID-MAS1>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Good D. M.; Wirtala M.; McAlister G. C.; Coon J. J. Performance Characteristics of Electron Transfer Dissociation Mass Spectrometry. Mol. Cell. Proteomics 2007, 6, 1942–1951. 10.1074/mcp.M700073-MCP200. [DOI] [PubMed] [Google Scholar]
- Liu J.; McLuckey S. A. Electron transfer dissociation: Effects of cation charge state on product partitioning in ion/ion electron transfer to multiply protonated polypeptides. Int. J. Mass Spectrom. 2012, 330–332, 174–181. 10.1016/j.ijms.2012.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose C. M.; et al. A Calibration Routine for Efficient ETD in Large-Scale Proteomics. J. Am. Soc. Mass Spectrom. 2015, 26, 1848–1857. 10.1007/s13361-015-1183-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitteri S. J.; Chrisman P. A.; McLuckey S. A. Electron-Transfer Ion/Ion Reactions of Doubly Protonated Peptides: Effect of Elevated Bath Gas Temperature. Anal. Chem. 2005, 77, 5662–5669. 10.1021/ac050666h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley N. M.; et al. Phosphoproteomics with Activated Ion Electron Transfer Dissociation. Anal. Chem. 2017, 89, 6367–6376. 10.1021/acs.analchem.7b00212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batth T. S.; Francavilla C.; Olsen J. V. Off-Line High-pH Reversed-Phase Fractionation for In-Depth Phosphoproteomics. J. Proteome Res. 2014, 13, 6176–6186. 10.1021/pr500893m. [DOI] [PubMed] [Google Scholar]
- Dickhut C.; Feldmann I.; Lambert J.; Zahedi R. P. Impact of Digestion Conditions on Phosphoproteomics. J. Proteome Res. 2014, 13, 2761–2770. 10.1021/pr401181y. [DOI] [PubMed] [Google Scholar]
- Woods A. S.; Ferré S. Amazing Stability of the Arginine–Phosphate Electrostatic Interaction. J. Proteome Res. 2005, 4, 1397–1402. 10.1021/pr050077s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asakawa D.; Osaka I. High-Confidence Sequencing of Phosphopeptides by Electron Transfer Dissociation Mass Spectrometry Using Dinuclear Zinc(II) Complex. Anal. Chem. 2016, 88, 12393–12402. 10.1021/acs.analchem.6b03645. [DOI] [PubMed] [Google Scholar]
- Chamot-Rooke J.; van der Rest G.; Dalleu A.; Bay S.; Lemoine J. The Combination of Electron Capture Dissociation and Fixed Charge Derivatization Increases Sequence Coverage for O-Glycosylated and O-Phosphorylated Peptides. J. Am. Soc. Mass Spectrom. 2007, 18, 1405–1413. 10.1016/j.jasms.2007.04.008. [DOI] [PubMed] [Google Scholar]
- Lu Y.; Zhou X.; Stemmer P. M.; Reid G. E. Sulfonium ion derivatization, isobaric stable isotope labeling and data dependent CID- and ETD-MS/MS for enhanced phosphopeptide quantitation, identification and phosphorylation site characterization. J. Am. Soc. Mass Spectrom. 2012, 23, 577–593. 10.1007/s13361-011-0190-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson S. N.; Dutta S.; Woods A. S. The use of ECD/ETD to identify the site of electrostatic interaction in noncovalent complexes. J. Am. Soc. Mass Spectrom. 2009, 20, 176–179. 10.1016/j.jasms.2008.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson S. N.; Wang H.-Y. J.; Yergey A.; Woods A. S. Phosphate Stabilization of Intermolecular Interactions. J. Proteome Res. 2006, 5, 122–126. 10.1021/pr0503578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creese A.; Cooper H. The Effect of Phosphorylation on the Electron Capture Dissociation of Peptide Ions. J. Am. Soc. Mass Spectrom. 2008, 19, 1263–1274. 10.1016/j.jasms.2008.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D.; et al. Probing the Electron Capture Dissociation Mass Spectrometry of Phosphopeptides with Traveling Wave Ion Mobility Spectrometry and Molecular Dynamics Simulations. J. Am. Soc. Mass Spectrom. 2015, 26, 1004–1013. 10.1007/s13361-015-1094-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Clavijo A. F.; Duque-Daza C. A.; Creese A. J.; Cooper H. J. Electron capture dissociation mass spectrometry of phosphopeptides: Arginine and phosphoserine. Int. J. Mass Spectrom. 2015, 390, 63–70. 10.1016/j.ijms.2015.07.024. [DOI] [Google Scholar]
- Chen B.; et al. The Impact of Phosphorylation on Electron Capture Dissociation of Proteins: A Top-Down Perspective. J. Am. Soc. Mass Spectrom. 2017, 28, 1805–1814. 10.1007/s13361-017-1710-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaney D. L.; et al. Supplemental Activation Method for High-Efficiency Electron-Transfer Dissociation of Doubly Protonated Peptide Precursors. Anal. Chem. 2007, 79, 477–485. 10.1021/ac061457f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frese C. K.; et al. Toward Full Peptide Sequence Coverage by Dual Fragmentation Combining Electron-Transfer and Higher-Energy Collision Dissociation Tandem Mass Spectrometry. Anal. Chem. 2012, 84, 9668–9673. 10.1021/ac3025366. [DOI] [PubMed] [Google Scholar]
- Frese C. K.; et al. Unambiguous phosphosite localization using electron-transfer/higher-energy collision dissociation (EThcD). J. Proteome Res. 2013, 12, 1520–1525. 10.1021/pr301130k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flora J. W.; Muddiman D. C. Gas-Phase Ion Unimolecular Dissociation for Rapid Phosphopeptide Mapping by IRMPD in a Penning Ion Trap: An Energetically Favored Process. J. Am. Chem. Soc. 2002, 124, 6546–6547. 10.1021/ja0261170. [DOI] [PubMed] [Google Scholar]
- Crowe M. C.; Brodbelt J. S. Infrared multiphoton dissociation (IRMPD) and collisionally activated dissociationof peptides in a quadrupole ion trapwith selective IRMPD of phosphopeptides. J. Am. Soc. Mass Spectrom. 2004, 15, 1581–1592. 10.1016/j.jasms.2004.07.016. [DOI] [PubMed] [Google Scholar]
- Riley N. M.; Westphall M. S.; Hebert A. S.; Coon J. J. Implementation of Activated Ion Electron Transfer Dissociation on a Quadrupole-Orbitrap-Linear Ion Trap Hybrid Mass Spectrometer. Anal. Chem. 2017, 89, 6358–6366. 10.1021/acs.analchem.7b00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter T. Why nature chose phosphate to modify proteins. Philos. Trans. R. Soc., B 2012, 367, 2513–2516. 10.1098/rstb.2012.0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinnijenhuis A. J.; Kjeldsen F.; Kallipolitis B.; Haselmann K. F.; Jensen O. N. Analysis of Histidine Phosphorylation Using Tandem MS and Ion–Electron Reactions. Anal. Chem. 2007, 79, 7450–7456. 10.1021/ac0707838. [DOI] [PubMed] [Google Scholar]
- Kee J.-M.; Oslund R. C.; Perlman D. H.; Muir T. W. A pan-specific antibody for direct detection of protein histidine phosphorylation. Nat. Chem. Biol. 2013, 9, 416–421. 10.1038/nchembio.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oslund R. C.; et al. A phosphohistidine proteomics strategy based on elucidation of a unique gas-phase phosphopeptide fragmentation mechanism. J. Am. Chem. Soc. 2014, 136, 12899–12911. 10.1021/ja507614f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kee J.-M.; Muir T. W. Chasing Phosphohistidine, an Elusive Sibling in the Phosphoamino Acid Family. ACS Chem. Biol. 2012, 7, 44–51. 10.1021/cb200445w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt A.; Ammerer G.; Mechtler K. Studying the fragmentation behavior of peptides with arginine phosphorylation and its influence on phospho-site localization. Proteomics 2013, 13, 945–954. 10.1002/pmic.201200240. [DOI] [PubMed] [Google Scholar]
- Fuhrmann J.; et al. McsB is a protein arginine kinase that phosphorylates and inhibits the heat-shock regulator CtsR. Science 2009, 324, 1323–1327. 10.1126/science.1170088. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Sanchez M.-B.; Lanucara F.; Hardman G. E.; Eyers C. E. Gas-phase intermolecular phosphate transfer within a phosphohistidine phosphopeptide dimer. Int. J. Mass Spectrom. 2014, 367, 28–34. 10.1016/j.ijms.2014.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potel C. M.; Lin M.-H.; Heck A. J.R.; Lemeer S. Widespread bacterial protein histidine phosphorylation revealed by mass spectrometry-based proteomics. Nat. Methods 2018, 15, 187–190. 10.1038/nmeth.4580. [DOI] [PubMed] [Google Scholar]
- Junker S.; et al. Spectral Library Based Analysis of Arginine Phosphorylations in Staphylococcus aureus. Mol. Cell. Proteomics 2018, 17, 335–348. 10.1074/mcp.RA117.000378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertran-Vicente J.; et al. Site-Specifically Phosphorylated Lysine Peptides. J. Am. Chem. Soc. 2014, 136, 13622–13628. 10.1021/ja507886s. [DOI] [PubMed] [Google Scholar]
- Kowalewska K.; et al. Electron capture dissociation mass spectrometric analysis of lysine-phosphorylated peptides. Biosci. Rep. 2010, 30, 433–443. 10.1042/BSR20090167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertran-Vicente J.; Schümann M.; Hackenberger C. P.R.; Krause E. Gas-Phase Rearrangement in Lysine Phosphorylated Peptides During Electron-Transfer Dissociation Tandem Mass Spectrometry. Anal. Chem. 2015, 87, 6990–6994. 10.1021/acs.analchem.5b01389. [DOI] [PubMed] [Google Scholar]
- Sun F.; et al. Protein cysteine phosphorylation of SarA/MgrA family transcriptional regulators mediates bacterial virulence and antibiotic resistance. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 15461–15466. 10.1073/pnas.1205952109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertran-Vicente J.; et al. Chemoselective synthesis and analysis of naturally occurring phosphorylated cysteine peptides. Nat. Commun. 2016, 7, 12703. 10.1038/ncomms12703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser A.; Penkert M.; Hackenberger C. P. R. Chemical Approaches to Investigate Labile Peptide and Protein Phosphorylation. Acc. Chem. Res. 2017, 50, 1883–1893. 10.1021/acs.accounts.7b00170. [DOI] [PubMed] [Google Scholar]
- Penkert M.; et al. Unambiguous Identification of Serine and Threonine Pyrophosphorylation Using Neutral-Loss-Triggered Electron-Transfer/Higher-Energy Collision Dissociation. Anal. Chem. 2017, 89, 3672–3680. 10.1021/acs.analchem.6b05095. [DOI] [PubMed] [Google Scholar]
- Perkins D. N.; Pappin D. J. C.; Creasy D. M.; Cottrell J. S. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 1999, 20, 3551–3567. 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Eng J. K.; McCormack A. L.; Yates J. R. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 1994, 5, 976–989. 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- Cox J.; et al. Andromeda: A Peptide Search Engine Integrated into the MaxQuant Environment. J. Proteome Res. 2011, 10, 1794–1805. 10.1021/pr101065j. [DOI] [PubMed] [Google Scholar]
- Dorfer V.; et al. MS Amanda, a Universal Identification Algorithm Optimized for High Accuracy Tandem Mass Spectra. J. Proteome Res. 2014, 13, 3679–3684. 10.1021/pr500202e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.; Pevzner P. A. MS-GF+ makes progress towards a universal database search tool for proteomics. Nat. Commun. 2014, 5, 5277. 10.1038/ncomms6277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong K.; Kim S.; Bandeira N. False discovery rates in spectral identification. BMC Bioinf. 2012, 13, S2. 10.1186/1471-2105-13-S16-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx H.; et al. A large synthetic peptide and phosphopeptide reference library for mass spectrometry-based proteomics. Nat. Biotechnol. 2013, 31, 557–564. 10.1038/nbt.2585. [DOI] [PubMed] [Google Scholar]
- Ferries S.; et al. Evaluation of Parameters for Confident Phosphorylation Site Localization Using an Orbitrap Fusion Tribrid Mass Spectrometer. J. Proteome Res. 2017, 16, 3448–3459. 10.1021/acs.jproteome.7b00337. [DOI] [PubMed] [Google Scholar]
- Beausoleil S. A.; Villén J.; Gerber S. A.; Rush J.; Gygi S. P. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol. 2006, 24, 1285–1292. 10.1038/nbt1240. [DOI] [PubMed] [Google Scholar]
- Taus T.; et al. Universal and Confident Phosphorylation Site Localization Using phosphoRS. J. Proteome Res. 2011, 10, 5354–5362. 10.1021/pr200611n. [DOI] [PubMed] [Google Scholar]
- Bailey C. M.; et al. SLoMo: Automated Site Localization of Modifications from ETD/ECD Mass Spectra. J. Proteome Res. 2009, 8, 1965–1971. 10.1021/pr800917p. [DOI] [PubMed] [Google Scholar]
- Savitski M. M.; et al. Confident Phosphorylation Site Localization Using the Mascot Delta Score. Mol. Cell. Proteomics 2011, 10, M110.003830. 10.1074/mcp.M110.003830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker P. R.; Trinidad J. C.; Chalkley R. J. Modification Site Localization Scoring Integrated into a Search Engine. Mol. Cell. Proteomics 2011, 10, M111.008078. 10.1074/mcp.M111.008078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fermin D.; Walmsley S. J.; Gingras A.-C.; Choi H.; Nesvizhskii A. I. LuciPHOr: Algorithm for Phosphorylation Site Localization with False Localization Rate Estimation Using Modified Target-Decoy Approach. Mol. Cell. Proteomics 2013, 12, 3409–3419. 10.1074/mcp.M113.028928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suni V.; Imanishi S. Y.; Maiolica A.; Aebersold R.; Corthals G. L. Confident Site Localization Using a Simulated Phosphopeptide Spectral Library. J. Proteome Res. 2015, 14, 2348–2359. 10.1021/acs.jproteome.5b00050. [DOI] [PubMed] [Google Scholar]
- Sharma K.; et al. Ultradeep Human Phosphoproteome Reveals a Distinct Regulatory Nature of Tyr and Ser/Thr-Based Signaling. Cell Rep. 2014, 8, 1583–1594. 10.1016/j.celrep.2014.07.036. [DOI] [PubMed] [Google Scholar]
- Chalkley R. J.; Clauser K. R. Modification Site Localization Scoring: Strategies and Performance. Mol. Cell. Proteomics 2012, 11, 3–14. 10.1074/mcp.R111.015305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalkley R. J. Modification Site Localization in Peptides. In Modern Proteomics–Sample Preparation, Analysis and Practical Applications; Springer: Cham, Switzerland, 2016; pp 243–247, 10.1007/978-3-319-41448-5_13. [DOI] [PubMed] [Google Scholar]
- Wiese H.; et al. Comparison of Alternative MS/MS and Bioinformatics Approaches for Confident Phosphorylation Site Localization. J. Proteome Res. 2014, 13, 1128–1137. 10.1021/pr400402s. [DOI] [PubMed] [Google Scholar]
- Yang H.; et al. pSite: Amino Acid Confidence Evaluation for Quality Control of De Novo Peptide Sequencing and Modification Site Localization. J. Proteome Res. 2018, 17, 119–128. 10.1021/acs.jproteome.7b00428. [DOI] [PubMed] [Google Scholar]
- Kelstrup C. D.; et al. Rapid and Deep Proteomes by Faster Sequencing on a Benchtop Quadrupole Ultra-High-Field Orbitrap Mass Spectrometer. J. Proteome Res. 2014, 13, 6187–6195. 10.1021/pr500985w. [DOI] [PubMed] [Google Scholar]
- Potel C. M.; Lin M.-H.; Heck A. J.R.; Lemeer S. Defeating Major Contaminants in Fe3+- Immobilized Metal Ion Affinity Chromatography (IMAC) Phosphopeptide Enrichment. Mol. Cell. Proteomics 2018, 17, 1028–1034. 10.1074/mcp.TIR117.000518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook S. L.; et al. Comparison of CID, ETD and metastable atom-activated dissociation (MAD) of doubly and triply charged phosphorylated tau peptides. J. Mass Spectrom. 2012, 47, 786–794. 10.1002/jms.3023. [DOI] [PubMed] [Google Scholar]
- Takahashi H.; et al. Hydrogen Attachment/Abstraction Dissociation (HAD) of Gas-Phase Peptide Ions for Tandem Mass Spectrometry. Anal. Chem. 2016, 88, 3810–3816. 10.1021/acs.analchem.5b04888. [DOI] [PubMed] [Google Scholar]
- Borotto N. B.; Ileka K. M.; Tom C. A.T.M.B.; Martin B. R.; Håkansson K. Free Radical Initiated Peptide Sequencing for Direct Site Localization of Sulfation and Phosphorylation with Negative Ion Mode Mass Spectrometry. Anal. Chem. 2018, 90, 9682–9686. 10.1021/acs.analchem.8b02707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodbelt J. S. Photodissociation mass spectrometry: New tools for characterization of biological molecules. Chem. Soc. Rev. 2014, 43, 2757–2783. 10.1039/C3CS60444F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halim M. A.; et al. Ultraviolet, Infrared, and High-Low Energy Photodissociation of Post-Translationally Modified Peptides. J. Am. Soc. Mass Spectrom. 2018, 29, 270–283. 10.1007/s13361-017-1794-9. [DOI] [PubMed] [Google Scholar]
- Smith S. A.; et al. Enhanced Characterization of Singly Protonated Phosphopeptide Ions by Femtosecond Laser-induced Ionization/Dissociation Tandem Mass Spectrometry (fs-LID-MS/MS). J. Am. Soc. Mass Spectrom. 2010, 21, 2031–2040. 10.1016/j.jasms.2010.08.016. [DOI] [PubMed] [Google Scholar]
- Shaffer C. J.; Slováková K.; Tureček F. Near-UV photodissociation of phosphopeptide cation-radicals. Int. J. Mass Spectrom. 2015, 390, 71–80. 10.1016/j.ijms.2015.05.009. [DOI] [Google Scholar]
- Fort K. L.; et al. Implementation of Ultraviolet Photodissociation on a Benchtop Q Exactive Mass Spectrometer and Its Application to Phosphoproteomics. Anal. Chem. 2016, 88, 2303–2310. 10.1021/acs.analchem.5b04162. [DOI] [PubMed] [Google Scholar]
- Robinson M. R.; Taliaferro J. M.; Dalby K. N.; Brodbelt J. S. 193 nm Ultraviolet Photodissociation Mass Spectrometry for Phosphopeptide Characterization in the Positive and Negative Ion Modes. J. Proteome Res. 2016, 15, 2739–2748. 10.1021/acs.jproteome.6b00289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayfield J. E.; et al. Mapping the Phosphorylation Pattern of Drosophila melanogaster RNA Polymerase II Carboxyl-Terminal Domain Using Ultraviolet Photodissociation Mass Spectrometry. ACS Chem. Biol. 2017, 12, 153–162. 10.1021/acschembio.6b00729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y.; et al. Top-down Proteomics Reveals Concerted Reductions in Myofilament and Z-disc Protein Phosphorylation after Acute Myocardial Infarction. Mol. Cell. Proteomics 2014, 13, 2752–2764. 10.1074/mcp.M114.040675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabrouskov V.; Ge Y.; Schwartz J.; Walker J. W. Unraveling Molecular Complexity of Phosphorylated Human Cardiac Troponin I by Top Down Electron Capture Dissociation/Electron Transfer Dissociation Mass Spectrometry. Mol. Cell. Proteomics 2008, 7, 1838–1849. 10.1074/mcp.M700524-MCP200. [DOI] [PubMed] [Google Scholar]
- Brunner A. M.; et al. Benchmarking Multiple Fragmentation Methods on an Orbitrap Fusion for Top-down Phospho-Proteoform Characterization. Anal. Chem. 2015, 87, 4152–4158. 10.1021/acs.analchem.5b00162. [DOI] [PubMed] [Google Scholar]
- Tamara S.; Scheltema R. A.; Heck A. J.R.; Leney A. C. Phosphate Transfer in Activated Protein Complexes Reveals Interaction Sites. Angew. Chem., Int. Ed. 2017, 56, 13641–13644. 10.1002/anie.201706749. [DOI] [PubMed] [Google Scholar]
- Chen Y.; et al. Quantitative Mass Spectrometry Reveals that Intact Histone H1 Phosphorylations are Variant Specific and Exhibit Single Molecule Hierarchical Dependence. Mol. Cell. Proteomics 2016, 15, 818–833. 10.1074/mcp.M114.046441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleland T. P.; et al. High-throughput analysis of intact human proteins using UVPD and HCD on an Orbitrap mass spectrometer. J. Proteome Res. 2017, 16, 2072–2079. 10.1021/acs.jproteome.7b00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lössl P.; et al. Deciphering the Interplay among Multisite Phosphorylation, Interaction Dynamics, and Conformational Transitions in a Tripartite Protein System. ACS Cent. Sci. 2016, 2, 445–455. 10.1021/acscentsci.6b00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B.; et al. Coupling functionalized cobalt ferrite nanoparticle enrichment with online LC/MS/MS for top-down phosphoproteomics. Chem. Sci. 2017, 8, 4306–4311. 10.1039/C6SC05435H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen J. A.; Kaoud T. S.; Dalby K. N.; Brodbelt J. S. 193-nm photodissociation of singly and multiply charged peptide anions for acidic proteome characterization. Proteomics 2011, 11, 1329–1334. 10.1002/pmic.201000565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley N. M.; et al. The Negative Mode Proteome with Activated Ion Negative Electron Transfer Dissociation (AI-NETD). Mol. Cell. Proteomics 2015, 14, 2644–2660. 10.1074/mcp.M115.049726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlister G. C.; et al. Analysis of the Acidic Proteome with Negative Electron-Transfer Dissociation Mass Spectrometry. Anal. Chem. 2012, 84, 2875–2882. 10.1021/ac203430u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shvartsburg A. A.; Creese A. J.; Smith R. D.; Cooper H. J. Separation of peptide isomers with variant modified sites by high-resolution differential ion mobility spectrometry. Anal. Chem. 2010, 82, 8327–8334. 10.1021/ac101878a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shvartsburg A. A.; Singer D.; Smith R. D.; Hoffmann R. Ion Mobility Separation of Isomeric Phosphopeptides from a Protein with Variant Modification of Adjacent Residues. Anal. Chem. 2011, 83, 5078–5085. 10.1021/ac200985s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xuan Y.; Creese A. J.; Horner J. A.; Cooper H. J. High-field asymmetric waveform ion mobility spectrometry (FAIMS) coupled with high-resolution electron transfer dissociation mass spectrometry for the analysis of isobaric phosphopeptides. Rapid Commun. Mass Spectrom. 2009, 23, 1963–1969. 10.1002/rcm.4101. [DOI] [PubMed] [Google Scholar]
- Schmidlin T.; et al. Assessment of SRM, MRM3, and DIA for the targeted analysis of phosphorylation dynamics in non-small cell lung cancer. Proteomics 2016, 16, 2193–2205. 10.1002/pmic.201500453. [DOI] [PubMed] [Google Scholar]
- Kennedy J. J.; et al. Immobilized Metal Affinity Chromatography Coupled to Multiple Reaction Monitoring Enables Reproducible Quantification of Phospho-signaling. Mol. Cell. Proteomics 2016, 15, 726–739. 10.1074/mcp.O115.054940. [DOI] [PMC free article] [PubMed] [Google Scholar]